

Abstract
The majority of clinical trials evaluating replication-selective oncolytic adenoviruses utilized mutants with immunomodulatory E3B genes deleted, likely contributing to the attenuated efficacy. We investigated whether an intact immune response could contribute to the observed improved efficacy in response to combinations with chemotherapeutics. Seven carcinoma cell lines were evaluated by combining viral mutants; dl309 (ΔE3B), dl704 (ΔE3gp19K), dl312 (ΔE1A) or wild-type Ad5 with the commonly used clinical drugs cisplatin and paclitaxel. Synergistic effects on cell death were determined by generation of combination indexes in cultured cells. In vivo tumor growth inhibition was achieved by virotherapy alone and was most efficacious with wild-type virus and least with the ΔE3B mutant. Significantly higher efficacy was observed when the viruses were combined with drugs. The greatest enhancement of tumor inhibition was in combination with the ΔE3B mutant restoring potency to that of Ad5 wild-type levels, observed only in animals with intact immune response. Increases in infectivity, viral gene expression and replication were identified as potential mechanisms contributing to the synergistic effects. Our results suggest that the attenuation of ΔE3B mutants can be overcome by low doses of chemotherapeutics only in the presence of an intact immune response indicating a role for T-cell-mediated functions.
Keywords: adenovirus, combination therapy, cytotoxic, synergy, immune response
Introduction
Single agent treatment is rarely successful in eliminating most advanced cancers, hence multimodality therapies including surgery, radiotherapy and chemotherapy regimens are frequently necessary. Even where there is early response to cytotoxic therapies, progression with metastatic disease often develops resulting in poor survival rates. Cross-resistance to treatment occurs with all currently available cytotoxic anticancer treatments including taxane and platinum drugs,1,2 fluoropyrimidine prodrugs,3 irinotecan (CPT-11),4 gemcitabine5 and targeted therapies such as signal transduction inhibitors.6 Consequently, novel cancer therapies that lack this cross-resistance are needed for use in conjunction with standard treatment regimens.
Replication-selective oncolytic adenoviruses, represent a novel, targeted anticancer therapy (virotherapy) that lacks cross-resistance with therapies currently used in the clinic.7 Oncolytic mutants have been constructed to target tumor tissue for selective replication and amplification at the tumor site with limited replication in normal cells, minimizing toxic side effects. The development of an oncolytic E1B55kD-deleted adenovirus (dl1520 also known as Onyx-015) was the first clinical application based on this concept8,9 and a modified version of this mutant is currently licensed for therapy of head and neck cancers in China (H101; Shanghai Sunway Biotech). The dl1520 mutant was attenuated in normal cells unable to inhibit p53-induced apoptosis and regulate viral nuclear mRNA export, while in the majority of human cancers viral replication could proceed.8,10-12 Mutants based on the dl1520 deletion have generated extensive data from clinical trials including over 300 patients proving both tumor selectivity and overall safety of virotherapy.7,13 Although low efficacy was documented with most deletion-mutants as a single agent therapy favorable interactions with both dl1520 or H101 and the cytotoxic drugs cisplatin and 5-fluorouracil (5FU) were reported in phase II and III trials with recurrent squamous cell carcinoma of the head and neck (SCCHN).14,15 The low single agent efficacy could partly be explained by deletion of the entire E1B gene resulting in elimination of functions essential for the viral life cycle.10,11 In addition, most oncolytic mutants to date, including dl1520 and H101, have the E3B genes deleted, a deletion later identified to cause the observed faster clearance by immune factors.16 Consequently, the E3B deletion likely contributed further to the low single agent efficacy.
Several improved oncolytic mutants have now been developed for clinical testing, for example, the dl922–947 and Ad5Δ24 mutants targeting pRb-pathway alterations;17,18 tissue-specific promoter-driven viruses targeting prostate19,20 and the majority of solid tumors.21-23 One prostate-specific mutant with intact E3 region (CG7870)20 was demonstrated to enhance cell death synergistically in combination with taxanes or radiotherapy, both in cells in culture and in xenografts in vivo.24 Enhanced efficacy was also demonstrated for Ad5Δ24 mutants in combination with TRAIL,25 RAD001 and Temozolomide,26 irinotecan27 and 5FC/5FU.28
We previously established murine syngeneic xenograft models with intact immune responses.16,29 Our findings demonstrated that deletion of the E3B genes attenuated viral efficacy due to faster clearance at the tumor site by macrophage infiltration, while deletion of the E3gp19K gene resulted in higher levels of viral replication and enhanced antitumor response, caused by activation of cytotoxic T-lymphocytes. The E3gp19K gene was therefore suggested as a better candidate for E3 deletions in future oncolytic mutants.
In this study we have further explored these models to test our hypothesis that the potency of attenuated E3B-deleted mutants could be improved if combined with the cytotoxic drugs cisplatin and paclitaxel in the presence of an intact immune system. We report strong synergistic effects on cell death with both drugs in combination with either wild-type virus or E3-deleted mutants. In cultured cells enhancement of cell death was variously due to changes in cellular uptake of virus, gene expression and viral replication. The greatest enhancement of tumor growth inhibition in vivo was with the E3B-deleted mutant in combination with the drugs although when given alone this mutant was the least potent. Combination treatment resulted in the efficacy of the E3B-mutant being restored to that of wild type. The improvement of efficacy with combination therapy was only observed in animals with intact immune system. Thus our data demonstrate that the poor antitumor efficacy of E3B-deleted mutants in immune-intact hosts could be significantly enhanced if combined with cytotoxic drugs.
Materials and methods
Cancer cell lines and adenoviruses
CMT-93 (murine rectal adenocarcinoma), CMT-TK (murine colorectal adenocarcinoma), CMT-64 (murine nonsmall cell lung carcinoma) and HEK293 (human embryonic kidney cells) were provided by Cell Services, Cancer Research UK (CRUK, London, UK). JC (murine mammary adenocarcinoma), LNCaP (human prostate epithelial carcinoma), H460 (human nonsmall-cell lung carcinoma) and HCT116 (human colorectal adenocarcinoma) were obtained from ATCC (Manassas, VA). All cell lines were grown under standard cell culture conditions in Dulbecco's Modified Eagle Media (D-MEM) supplemented with 10% fetal calf serum. The following viruses and mutants were used: Ad5 (adenovirus type 5 wild type), dl312 (ΔE1A, ΔE3B), AdGFP (ΔE1), dl309 (ΔE3B) and dl704 (ΔE3gp19kD). Inactivation of the dl312 mutant was performed using psoralen and UV (PUV dl312).29 All viruses used in the study had a particle to infectious unit ratio (vp/PFU) of 10–50.
Cell killing assay and synergistic interactions
Cells were plated at 1 × 104 cells per well in 96-well plates under standard conditions. One day later, cells were either infected with viruses, or paclitaxel (Taxol) (Calbiochem, San Diego, CA) or cisplatin (Sigma-Aldrich, Dorset, UK) were added as previously described.30 Cell death was determined 7 days after seeding using the MTS assay kit (Promega, Madison, WI). Dose–response curves were generated and concentrations killing 50% of cells (EC50-values) were determined using untreated cells as reference. The dose ranges were as follows: paclitaxel 3 × 10−8–3 × 104 nm, cisplatin 8 × 10−7–2 × 102 μm and viruses 1 × 10−6–1 × 105 particle per cell (ppc). Four different dilution ratios were included in each experiment keeping virus/drug ratios constant for each dose–response curve: virus/paclitaxel ratios; (1) 3, (2) 33, (3) 333, (4) 3333, and virus/cisplatin ratios; (1) 50, (2) 250, (3) 1250, (4) 6250. Isobolograms were generated and combination indexes (CI) established to determine synergistic or antagonistic effects on cell death in response to each combination ratio as previously described.30 Each data point was generated from triplicate samples, repeated 3–5 times.
Adenovirus replication assay
Cells were seeded at 1 × 105–106 cells per dish in 60 mm culture dishes. One day after seeding cells were either infected at 10, 100 or 1000 ppc or treated with paclitaxel at 2 and 10 nm, or cisplatin at 2.5, 10 and 50 μm. Cells and media were collected 24–96 h postinfection and analyzed by the limiting dilution method (tissue culture inhibitory dose at 50%: TCID50) as previously described.16 Each sample was determined in triplicate and data from 2–3 separate studies were averaged and expressed as PFU per milliliter or fold change compared to control (virus alone at each time point). An internal Ad5 standard was included as reference.
Flow cytometry analysis
For infectivity studies, cells were seeded at 2 × 104 cells per well in 24-well plates and 24 h later treated with 2 and 10 nm paclitaxel or 2.5 and 10 μm cisplatin. After an additional 24 h cells were infected with AdGFP at 100 and 1000 ppc followed by trypsinisation and resuspension in FACS buffer (PBS, 2% BSA, 1 mm EDTA) 48 h postinfection. Flow cytometry was performed on a Becton Dickinson FACS-Calibur instrument, acquiring 10 000 events per sample from duplicate wells using propidium iodide (PI) to exclude dead cells. Each experiment was repeated 2–3 times and analyzed with the CellQuest software.
Immunoblot analysis
Subconfluent cells were infected with viruses in the presence of paclitaxel or cisplatin under identical conditions to the synergy studies described above. Cell lysates were prepared 24–72 h postinfection, 10 or 20 μg of total protein were separated on SDS–PAGE under reducing conditions and transferred to nitrocellulose membranes. Viral and cellular proteins were detected by rabbit anti-Ad2 E1A at 1:1000 (SC-430; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-penton at 1:2500 (Novartis-GTI, Gaithersburg, MD) and goat anti-actin at 1:4000 (SC-1615; Santa Cruz Biotechnology). Detection was by horseradish peroxidase-conjugated secondary goat anti-rabbit IgG, goat anti-mouse IgG, rabbit anti-goat IgG (Dako, Glostrup, Denmark) and chemiluminescence reagent followed by autoradiography (Amersham-Pharmacia, Buckinghamshire, UK).
In vivo tumor growth
Tumors were grown in one flank of C57Bl/6 or athymic (nu/nu) mice by subcutaneous implantation of CMT-64 and CMT-93 cells at 1 × 105 and 1 × 106, respectively. Dose responses to viral mutants were determined by intratumoral administration of 1 × 108, 1 × 109 and 1 × 1010 viral particle (vp) per injection three times at 48 h intervals. Cisplatin was tested at 0.2, 1.0, 2.0 and 5.0 mg kg−1 and paclitaxel at 1.0, 5.0, 15.0 and 22.0 mg kg−1, both administered intraperitoneally 2–3 times from 2–8 days after the first virus injection. A suboptimal concentration and treatment interval for each agent given alone was selected to enable detection of additive/synergistic effects on tumor growth inhibition in the combination studies: cisplatin 2 mg kg−1 and paclitaxel 15 mg kg−1. Tumor volumes were determined using the formula: volume = (length × width2 × π)/6. Treatments were initiated when tumors were at least 100 μl with tumor growth and progression followed until tumors reached 1.44 cm2 or until symptomatic tumor ulceration occurred (according to UK Home Office Regulations). Treatment groups were balanced by tumor size at the time of treatment initiation in all cases (t-test for tumor volumes, P>0.8). Survival analysis expressed as time to progression (tumor volume ≥500 μl) was performed according to the method of Kaplan–Meier (log rank test for statistical significance).
Assessment of in vivo replication
Following two intratumoral injections of CMT-64 tumors on days 1 and 3 at 1 × 1010 vp per injection and drug treatment on days 2 and 4, tumors were harvested and flash frozen 5 and 8 days after the last virus administration (n = 3 per time point). Thawed tumors were physically dispersed and tissue lysates prepared followed by TCID50 analysis as described.16
Results
Sensitivity to virus and cytotoxic drug-induced cell death varies between cell lines
Dose–response curves for paclitaxel, cisplatin and viral mutants were established in murine and human carcinoma cell lines. The murine CMT-93 cells were as sensitive to adenovirus-induced cell death (EC50 = 1.1 ± 1.3 ppc) as the three tested human cell lines H460, HCT116 and LNCaP (EC50 = 1–10 ppc) (Supplementary Table S1). Other murine cell lines were less sensitive with EC50-values for CMT-64 and JC cells 100–1000 times higher (Supplementary Table S1). The E3B-deleted mutant (dl309) was up to 3 times more potent than wild-type virus with EC50 values for CMT-93 cells of 0.4 ± 0.3 while an E3gp19K-gene-deleted mutant (dl704) was less potent at 51.2 ± 10 ppc. The same order of potency was observed in all cell lines (Supplementary Table S1).
To determine whether E1A and viral gene expression rather than viral replication were essential for cell death, the replication-defective mutant dl312 and its nonprotein expressing inactivated particle control PUV dl312 were also tested. Limited cell death, <20% was seen with dl312 at 1 × 106 ppc with no cell death at 1 × 105 ppc in any cell line. The inactivated particle did not induce cell death at concentrations up to 1 × 106 ppc. These findings demonstrated that the expression of E1A and/or other viral genes were essential for virus-induced cell death while the contribution of the infective particle alone was negligible.
The sensitivity to cytotoxic drugs was less variable than to viral mutants (Supplementary Table S1). For paclitaxel, the human H460 and HCT116 cells had EC50-values close to 100 nm while those of all other cell lines were 10–100 times lower, with CMT-64 being the most sensitive at 0.5 ± 0.1 nm. Most cell lines showed similar responses to cisplatin with EC50-values of 5–20 μm, while CMT-TK and HCT116 cells were less sensitive at 30 and 64 μm, respectively.
Combination treatment with viral mutants and drugs act synergistically to enhance cell death
Various conditions for combination treatments were tested to optimize cell death in selected carcinoma cell lines. When wild-type virus was combined with paclitaxel or cisplatin, more than additive effects on cell death were demonstrated in CMT-93, CMT-TK and LNCaP cells independent of order of addition, while in CMT-64 cells the response was sequence-dependent, with synergistic effects only when drugs were added prior to virus, but not vice versa (Figures 1a-d, Supplementary Table S2). Simultaneous additions in the CMT-64 cells resulted in synergy only with cisplatin (Figure 1d). The JC cells displayed mixed responses with antagonistic, synergistic and additive effects (Supplementary Table S2). The effects in the human cell lines H460 and HCT116 were clearly antagonistic with both paclitaxel and cisplatin, while in the LNCaP cells all combinations resulted in synergistic increases in cell death (Figure 1c, Supplementary Table S2).
Figure 1.

Representative isobolograms from combination treatment of wild-type virus and paclitaxel or cisplatin in murine and human cell lines. (a–c) Cells were infected with virus either 24 h prior to (V–T), (V–C) or 24 h after (T–V), (C–V) addition of paclitaxel (T) or cisplatin (C) at predetermined concentration ratios as in Materials and methods, (a) CMT-93, (b) CMT-64, (c) LNCaP. (d) Paclitaxel and cisplatin were added simultaneously with virus to the CMT-93 and CMT-64 cells. EC50 values for each combination ratio were determined from dose–response curves and represented in isobolograms. The straight line connects the EC50 values for each agent alone and illustrates the theoretical values resulting in additive effects. Data points below the line represent synergistic (CI≤0.8) and above the line antagonistic (CI≥1.2) interactions. Data are from one representative experiment in triplicate.
To determine if E3- or E1A deletions could modify the synergistic responses the dl309, dl704 and dl312 mutants were tested under optimal synergy conditions (drug 24 h prior to virus). As expected for cells in culture, the results for the dl309 and dl704 mutants paralleled those for wild-type virus while no enhanced effects on cell death with dl312 were seen (Table 1). The synergistic effects with the E3-mutants, quantified as CI-values, were identical to those of wild-type virus combined with both paclitaxel and cisplatin in the CMT-64, CMT-93 and LNCaP cells. The greatest synergy was observed in the CMT-93 and LNCaP cells with CI values ≤0.5 in 16 and 12 out of 24 combinations, respectively, with overall synergy in all 24 (CMT-93) and 23 (LNCaP) data points (illustrated for dl309; Supplementary Table S3).
Table 1.
Synergy (S) and antagonism (A) in murine and human cell lines
| Viral mutant |
CMT-64 |
CMT-93 |
JC |
LNCaP |
||||
|---|---|---|---|---|---|---|---|---|
| T | C | T | C | T | C | T | C | |
| Ad5 | S | S | S | S | A | A | S | S |
| dl309 | S | S | S | S | A | A | S | S |
| dl704 | S | S | S | S | A | A | S | S |
| dl312 | ne | ne | ne | ne | ne | ne | ne | ne |
Abbreviations: C, cisplatin; ne, no effect; T, paclitaxel.
T or C were given 24 h prior to infection with viruses.
S or A in at least 3/4 data points with no clear antagonism or synergy respectively; ne is no sensitization to virus and no effect on drug induced cell death; data represent 4–8 combinations for each condition.
Changes in viral replication are involved in the synergistic response to combination treatment
Despite the strong enhancement of cell death in the murine cell lines in response to combination treatments only small changes in viral replication rates were observed (Figure 2a). In CMT-64 cells replication increased over time with highest levels at 72 h in the presence of paclitaxel but not cisplatin. In the human LNCaP cells replication increased >10-fold at 48–72 h with paclitaxel and fourfold with cisplatin at 72 h. Replication rate in other cell lines were not affected by drug addition, as shown for the JC cells that responded antagonistically to combination treatments (Figure 2a, Supplementary Table S2). The addition of drugs to the nonpermissive CMT-93 and CMT-TK cells did not induce replication under any condition (data not shown).
Figure 2.

Effects of paclitaxel and cisplatin on viral replication, infectivity and gene expression. (a) Cells were treated with paclitaxel at 10 nm and cisplatin at 10 μm 24 h prior to Ad5 infection at 100 ppc (H460 and LNCaP) and 1000 ppc (CMT-64 and JC) and harvested 24 h (white bars), 48 h (striped bars) and 72 h (black bars) postinfection. Replication rate was determined by limiting dilution assays. Results are expressed as percent increase in replication in each cell line compared to cells treated with virus alone at the respective time points. Averages are from two experiments in triplicate ± s.e.m. (b) Cells were pretreated with 10 nm paclitaxel (black bars) and 10 μm cisplatin (white bars) 24 h prior to infection with nonreplicating AdGFP at 100 ppc (H460) and 1000 ppc (CMT-64, CMT-93 and JC). Changes in infectivity are expressed as fold increase compared to cells treated with virus alone, presented as averages of two experiments ± s.e.m. (a, b) P-values were determined by the t-test for treated versus untreated cells, P<0.05 (*). (c) CMT-64 and CMT-93 cells were infected at 1000 ppc 24 h after treatment with 10 nm paclitaxel or 10 μm cisplatin and harvested 24 and 48 h postinfection. Immunoblotting was performed for detection of penton and E1A using β-actin as a loading control (U = uninfected cells, ctrl = HEK293 cells infected with Ad5wt). (d) CMT-64 cells and tumors, in athymic (nu/nu) and immunocompetent (C57Bl/6) animals were infected with Ad5 and dl309 and harvested as described in Materials and methods. Viral burst was determined by the TCID50 assay and replication expressed as ratios of dl309 to Ad5. Data presented as averages from six tumors in each group ± s.d. Significance was tested with t-test, P<0.005 (*) when compared to initial time points (24 h and 2 days, respectively).
Uptake of virus is strongly enhanced by cytotoxic drug treatment
Cells were infected with a nonreplicating GFP-expressing mutant 24 h after drug addition at 100 and 1000 ppc. In both the replication-permissive CMT-64 and nonpermissive CMT-93 murine cells increased GFP expression was observed with cisplatin and virus at 1000 ppc (five- and twofold, respectively) (Figure 2b). With paclitaxel a fivefold increase was determined in the CMT-64 cells only, with a similar trend when cells were infected at lower doses (Supplementary Figure S1). No significant changes in infectivity were observed in other cell lines (Figure 2b). An increase in both viral infectivity and cellular uptake receptors was also determined in the LNCaP cells in response to taxanes (data not shown).
Infected cells pretreated with paclitaxel or cisplatin express high levels of E1A
Numerous reports have suggested that E1A expression can sensitize carcinoma cells to cytotoxic drugs.31-33 To further explore whether E1A would contribute to the synergistic effects with paclitaxel and cisplatin, E1A-expression levels were determined under conditions for optimal synergy. In the CMT-64 cells, higher E1A expression was seen after 24 and 48 h with both cisplatin and paclitaxel, while in CMT-93 cells stimulated E1A expression was only observed with cisplatin (Figure 2c). The increases were highest in both cell lines with 10 μm cisplatin after 48 h. Parallel increases in late protein expression after 48 h in the CMT-64 but not the CMT-93 cells were also observed, demonstrated by penton expression (Figure 2c). Taken together, the in vitro data demonstrate that the higher levels of infection, viral protein expression and replication clearly played a role even though the specific changes were cell line- and drug-dependent. Paclitaxel appeared to more potently stimulate viral replication while cisplatin induced higher levels of E1A expression. The deletion of the E3B gene did not change the magnitude of the synergistic responses with drugs and paralleled that of Ad5 in replication, infectivity and gene expression in combination with drugs.
The E3B-deleted mutant replicates at higher levels in CMT-64 cells and in xenografts in immune deficient animals
To compare the replication rates of Ad5 and dl309 in cells in culture, CMT-64 cells were infected at 1000 ppc and harvested after 24–96 h. Replication was higher with the dl309 mutant at all time points, 2.5-fold after 48 h (Figure 2d, left panel). However, when tumor bearing athymic and intact mice were treated with the viruses only replication in athymic mice paralleled that of cells in culture with higher levels for the dl309 mutant (Figure 2d, middle and right panels). In animals with intact immune response there was no increase in replication for the mutant at any time point after treatment.
In vivo administration of Ad5 dl309 and drugs alone inhibit tumor xenograft growth
To further explore the synergistic effects in vivo, subcutaneous tumors were grown and suboptimal doses for growth reduction to 5–40% of control tumors were determined for each agent alone. CMT-64 tumors grew at a slower rate in athymic mice, and treatment consistently resulted in smaller tumors compared to animals with an intact immune response treated identically (Figures 3a,c and Supplementary Figure S2). While no differences in efficacy were observed between the dl309 and wild-type viruses in the athymic animals (Figure 3c, left panel), a trend toward higher efficacy for wild-type virus was detected in intact animals at these dose levels (Figure 3a, left panel). In the CMT-93 models, significant virus-induced tumor inhibition was only observed in mice with an intact immune system (Figure 3b). On day 15 after Ad5 treatment, tumors were 75% of controls while in athymic mice no significant growth inhibition was detected at any time point (not shown). No efficacy was observed with the dl312 mutant in any model.
Figure 3.
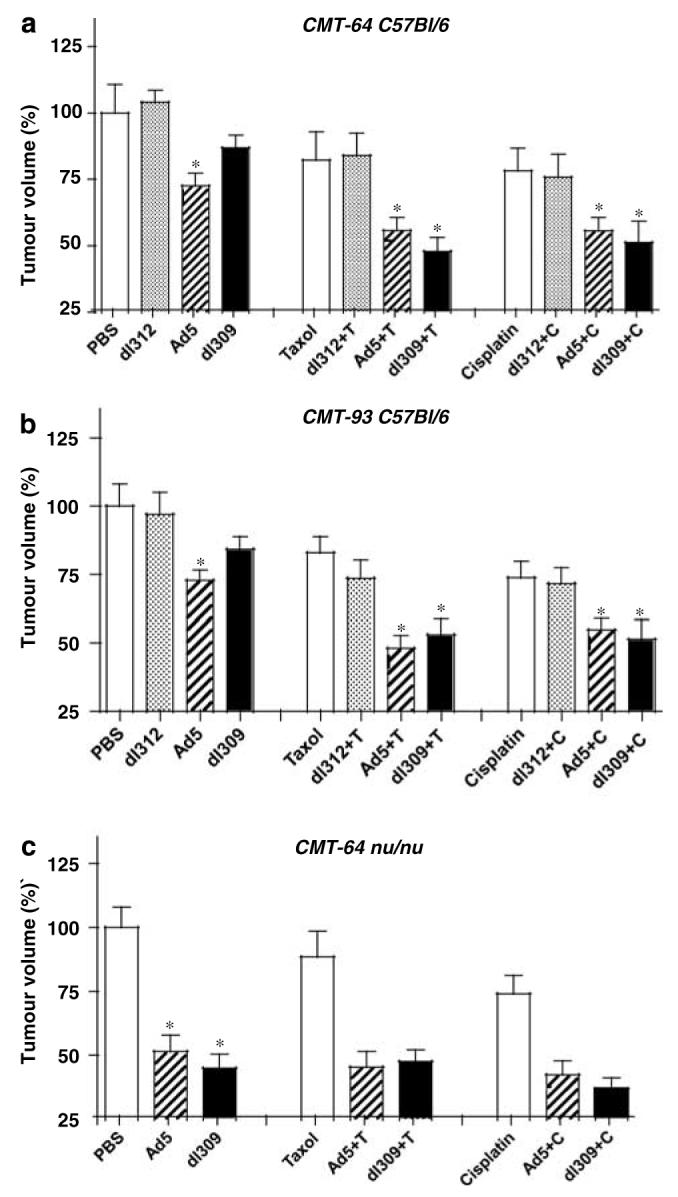
Growth inhibition in response to single and combination treated CMT-64 and CMT-93 tumors in animals with normal and defective immune responses. Animals with intact (a, b) and defective immune response (c) bearing CMT-64 (a, c) and CMT-93 (b) tumors treated with viral mutants and paclitaxel (T) or cisplatin (C). Tumor ratios were calculated comparing the averages for each treatment group to untreated tumors (PBS) on day 15 after inoculation. Average tumor volumes ± s.d. were calculated for each group (n = 10 animals per group), data were analyzed by t-test and are representative of three separate studies. P-values <0.05 (*) when single agent treatment was compared to PBS or dl312 and combination treatments compared to both single agent treatments.
Combination therapy significantly enhance antitumor efficacy in vivo
In intact animals with CMT-93 tumors, tumor growth inhibition was greater with both Ad5 and dl309 when combined with cisplatin or paclitaxel compared to each agent alone (Figure 3b, middle and right panels). The greatest reduction in tumor size was with dl309 in combination with either drug. Similar tumor growth inhibition was observed for Ad5 with both drugs while combinations with the nonreplicating dl312 mutant did not result in significant growth inhibition. Growth reductions were more than additive for the dl309 mutant with either drug and for wild-type virus with paclitaxel while all other combinations were either additive or less than additive. Identical studies in athymic mice resulted in no additional effects of combination treatments than that induced by drugs alone (data not shown).
In contrast, combination treatments in the replication-permissive CMT-64 model resulted in greatest tumor reductions in athymic mice rather than immunocompetent animals (Figures 3a and c, middle and right panels). While growth inhibition in immunocompetent mice was small with single virus or drug treatments (5–25%), the inhibition in athymic mice was greater in response to virus than to drugs, 45–60 and 20–40%, respectively, with no significant increase in response to combination therapy (Figure 3c). In intact mice, the most significant enhancement of tumor growth inhibition was with dl309 together with either cisplatin or paclitaxel and to a lesser degree for Ad5 with both drugs. The combined effects were more than additive for the dl309 mutant with both drugs; cisplatin alone reduced tumors by 24%, paclitaxel by 20%, dl309 by 12%, while combinations resulted in 50% with either drug. All other combinations were either additive or less than additive.
Combination therapy prolong the time to tumor progression for the ΔE3B mutant
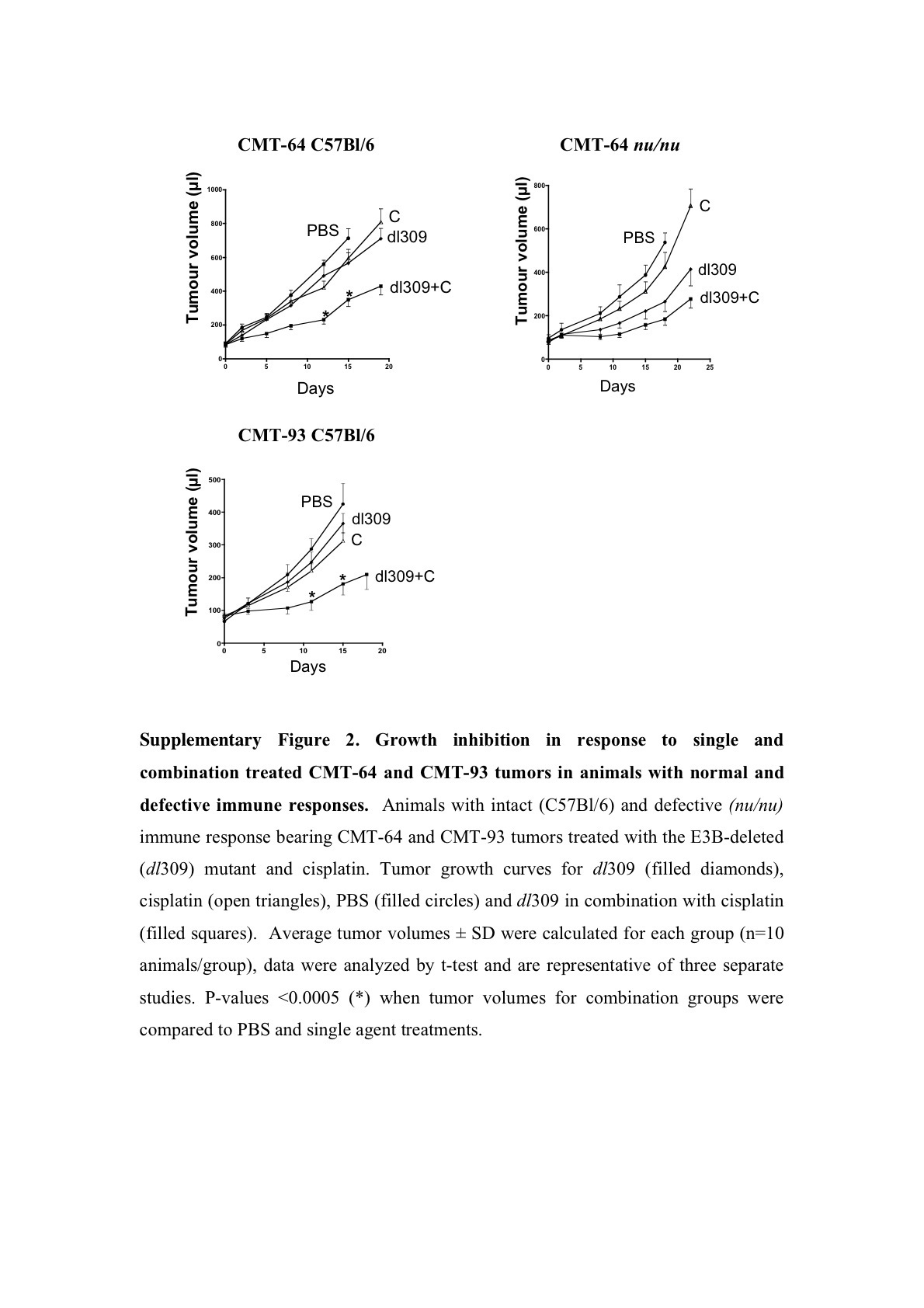
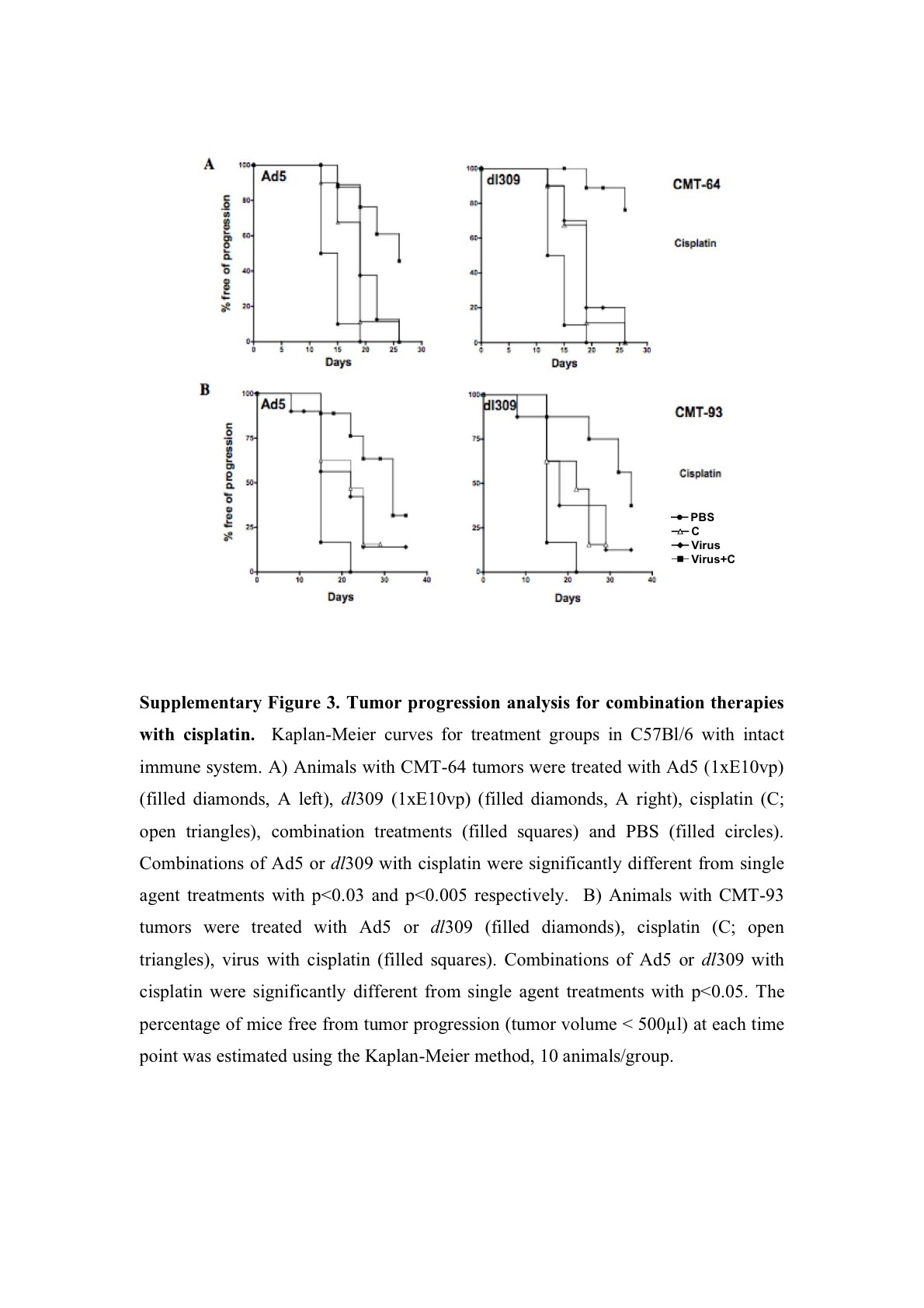
Tumor growth inhibition was paralleled by prolonged survival, measured as time to tumor progression, in both CMT-93 and CMT-64 models (Figure 4). The effect was greatest for the dl309 mutant combined with paclitaxel or cisplatin in immunocompetent mice with CMT-64 tumors (Figure 4a and Supplementary Figure S3A). In combination with paclitaxel 90 and 50% of the dl309 and Ad5 treated animals, respectively, were alive on day 22 post-treatment as opposed to 20% in the single agent groups (Figure 4a). When the cisplatin study was stopped at day 26 post-treatment, all animals treated with single agents had been killed due to tumor burden while 70% of animals in the dl309 combination treated groups were still alive with tumors <400 μl (Supplementary Figure S3A). Similar effects were seen in animals treated with Ad5 with 50% of animals alive on day 26 (Supplementary Figure S3A). In athymic mice, the combination therapies were less efficacious, while virus treatment alone resulted in significantly prolonged time to progression compared to control treated groups. On day 18 post-treatment, drugs alone resulted <20% of animals alive while Ad5 or dl309 treatment resulted in 90–100% of animals still alive with all animals alive in the combination treated groups (data not shown). A similar trend was observed for CMT-93 tumor progression in intact mice (Figure 4b and Supplementary Figure S3B). On day 30 post-treatment, 70% of animals were alive when dl309 was combined with both drugs and 50% of animals in the Ad5 combination groups. Single agent treatment resulted in only 10–40% of animals surviving at this time point. None of the models were suitable for long-term survival studies due to the frequent ulceration of tumors during regression in response to viral administration. On days 20–26 post-treatment, all studies had to be stopped due to animal welfare regulations (UK Home Office Regulations).
Figure 4.

Tumor progression analysis for combination therapies with paclitaxel. Kaplan–Meier curves for treatment groups in C57Bl/6 with intact immune system. (a) Animals with CMT-64 tumors were treated with Ad5 (1 × 1010 vp) (filled diamonds, (a) left), dl309 (1 × 1010 vp) (filled diamonds, (a) right), paclitaxel (T; open triangles), combination treatments (filled squares) and PBS (filled circles). Combinations of Ad5 or dl309 with paclitaxel were significantly different from single agent treatments with P<0.05. (b) Animals with CMT-93 tumors were treated with Ad5 or dl309 (filled diamonds), paclitaxel (T; open triangles), virus with paclitaxel (filled squares). Combinations of dl309 with paclitaxel were significantly different from single agent treatments with P<0.05. The percentage of mice free from tumor progression (tumor volume <500 μl) at each time point was estimated using the Kaplan–Meier method, 10 animals per group.
Cytotoxic drugs improve viral replication in the presence of an intact immune response
To investigate further the enhancement of efficacy in response to dl309 and combination treatments, we determined replication levels in CMT-64 tumors from athymic and immunocompetent mice (Figure 2d) including those treated with paclitaxel (Figure 5). Treatment with both paclitaxel and cisplatin led to higher levels of replication for Ad5 and dl309 viruses in animals with intact immune response as demonstrated for paclitaxel (Figure 5), while no significant increases were seen in athymic animals (data not shown). The enhancement of replication resulted in an overall improvement of the poorly replicating dl309 mutant to levels similar to that of wild-type virus under the same conditions (Figure 5).
Figure 5.
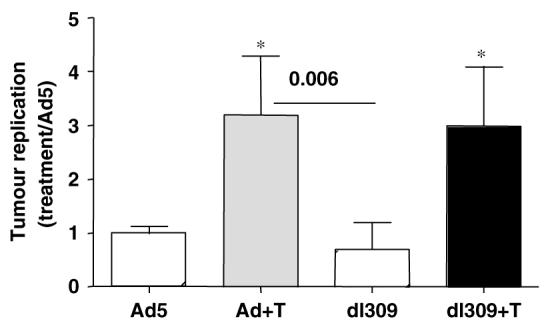
Replication of Ad5 and the ΔE3B mutant in CMT-64 tumor xenografts. Replication ratios in tumors grown in intact mice treated with virus and combinations of virus and paclitaxel as described in Materials and methods. Tumors were harvested five days post-inoculation with virus. Data are expressed as change in replication compared to treatment with wild-type virus (Ad5) alone. Tumors were from six animals analyzed in triplicate. P-values were determined by t-test, P<0.005 when combination treatments were compared to each virus alone (*) and with dl309 alone compared to Ad5 combined with paclitaxel.
Discussion
Establishment of efficient combination therapies with replication-selective oncolytic adenoviral mutants is an area of great interest. Virotherapy has raised high expectations partly because of lack of cross-resistance with current clinical drugs. Most oncolytic mutants evaluated in clinical trials to date have deletions in the immunomodulatory E3B region including dl1520 and CG7060 (CV706).8,19 Here, we have demonstrated that the reduced efficacy of E3B-deleted mutants in vivo could be improved when administered together with cisplatin or paclitaxel and also synergistically enhance drug-induced cell killing. While combination treatments with cytotoxic drugs resulted in additional growth inhibition with all viruses, the effect was greater with the ΔE3B mutant resulting in restored potency in two immunocompetent models, one permissive (CMT64) and one nonpermissive (CMT-93), for viral replication. These effects were not detected in immune defective animals.
While in the replication-permissive models viral efficacy was reduced in normal mice due to the host immune defense,16 in combination with cytotoxic drugs viral efficacy was restored to the levels observed in athymic animals. Chemodrugs can modify the functions of immune cells such as macrophages, NK-cells, cytotoxic and regulatory T-cells. Both paclitaxel and cisplatin have been reported to promote T-cell and macrophage activities through cytokine induction.34-37 For example, paclitaxel was shown to stimulate activated macrophages to release TNFα, NO, IL-12 and other chemokines, which in turn induced apoptosis in tumor cells.37 Consequently paclitaxel not only kills tumor cells by directly inducing apoptosis through p53-related mechanisms but also indirectly by induction of higher local concentrations of released immune effectors such as NO and TNFα. Oncolytic mutants with E3B deletions were recently demonstrated to be defective in the prevention of TNF-induced apoptosis due to absence of the 14.7K protein.38 In addition, another cytotoxic drug, cyclophosphamide, was reported to selectively eliminate regulatory T-cells (Treg) prolonging viral gene expression in tumors.39,40 It was suggested that virus and drug combinations exert several complex effects not only on the tumor cells and local immune effectors but also on remote immune cells, perhaps affecting suppressive functions of the innate immune response. The specific enhancement of our ΔE3B mutant was significant only in mice with an intact thymus, implying that the response was dependent on T-cell activities.
The higher efficacy with viruses alone in nude mice with CMT-64 tumors might be due the slower growth rate in athymic animals allowing for more efficient viral replication and spread within the tumor. Viral replication data also indicate that replication was a major reason for the better efficacy when mutants were given alone in athymic mice, while virus-infected cells were cleared at a faster rate in the presence of T-cells. We suggest that the greater enhancement of viral efficacy in the presence of drugs in immunocompetent animals was caused by a local decrease in activity of the innate immune response and an increased infiltration of tumor-specific T-cells, perhaps allowing for prolonged viral replication, preventing the rapid clearance seen with the dl309 mutant alone. Studies are in progress to investigate this hypothesis. In addition, we found that a potent replicating mutant was required to achieve significant enhancement. Replication and efficacy of the dl1520 mutant (E1B55k- and E3B-deleted) is severely attenuated in CMT-64 cells, and when tested in our in vivo model only a slight enhancement was observed (data not shown).
Another important factor for enhancement of the antitumor response was viral E1A-gene expression. The E1A 12S and 13S proteins have been demonstrated to sensitize tumor cells to NK-cells, activated macrophages, T-cell-mediated apoptosis and effector molecules such as TNFα, NO, TRAIL and FasL in addition to direct sensitization to cytotoxic drugs including paclitaxel and cisplatin.32,41-43 Numerous investigators have demonstrated that the E1A gene is essential for sensitization to cytotoxic agents by adenoviral mutants.24,44 The E1A gene alone has also been evaluated for local delivery in several gene therapy trials targeting breast, ovarian and head and neck cancers and was demonstrated to have potential for future cytotoxic drug combination trials.45,46 The sensitization of tumor cells to E1A expression could be caused by a number of reported mechanisms such as induction of procaspases, activation of p38 and modifications of Her2/neu, p21, p19ARF, p300/CBP, pRb and p21 activities, allowing for multiple effects dependent on the microenvironment.32,37,47,48 Our results indicate dependence on E1A expression with good efficacy in tumor cells not supporting viral replication but expressing E1A and late viral proteins both in culture and in vivo. In addition, E1A levels were elevated in response to chemotherapeutics possibly further activating any of these mechanisms. We conclude that efficacy in response to combination therapy might be dependent on several factors such as viral E1A expression and replication as well as the recruitment of T-cell-derived immune effector cells and molecules. The lack of efficacy in athymic mice with CMT-93 tumors was likely due to the limited spread of virus (and E1A expression) within the tumor in the absence of viral replication, in contrast to the more efficient spread of virus in cells in culture.
We demonstrated that conditions to achieve synergistic and additive effects on death in cultured cells in response to combination treatments were strongly dependent on cell line and the specific concentration ratios of drug and virus. We also demonstrated that the higher levels of infection, viral protein expression and replication clearly played a role in inducing synergistic cell death. In the CMT-64 cells elevated E1A-levels induced by paclitaxel were paralleled by increased infectivity and replication while cisplatin did not affect replication. In CMT-93 cells cisplatin exposure produced elevated E1A-levels and enhanced infectivity while no changes were detected in response to paclitaxel. In addition, deletions of either E3B or E3gp19kD genes did not change the magnitude of the responses when compared to wild-type virus. We anticipate that it is possible to achieve synergistic or additive effects in all carcinoma cell lines by carefully adjusting experimental conditions.
Our data suggest that the choice of tumor cell lines and model systems need consideration when evaluating conditions for combination treatments. To achieve optimal antitumor efficacy with oncolytic adenoviral mutants it is crucial to identify gene regions that can be deleted without decreasing potency. Our data demonstrate that efficacy of a less potent replicating mutant with deletions in the E3B genes can be significantly enhanced when administered together with cytotoxic drugs. Consequently, when using replication-selective deletion mutants as single agent treatment it would be advantageous to include the E3B genes, while in combination with cytotoxic drugs this region could be deleted, enabling the insertion of larger transgenes. On the other hand, deletion of the smaller E3gp19kD region could enhance viral potency both as a single agent and in combination with cytotoxic drugs and would be sufficient for accommodation of smaller transgenes. Further studies elucidating the T-cell-mediated effects responsible for the enhancement of efficacy with the combination therapies are in progress to enable more efficacious combination regimens to be developed in the future.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We want to thank Gary Martin (CRUK, London, UK) and colleagues at Clare Hall for excellent experimental assistance and Jennelle Francis (Molecular Oncology Unit) for production and characterization of all viral mutants. We also wish to thank Lynda Hawkins, Patricia Ryan and Jingping Yang (GTI-Novartis, Gaithersburg, MD) for helpful and insightful discussions.
Footnotes
Supplementary Information accompanies the paper on Cancer Gene Therapy website (http://www.nature.com/cgt)
References
- 1.Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of Taxol resistance related to microtubules. Oncogene. 2003;22:7280–7295. doi: 10.1038/sj.onc.1206934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 3.Schoffski P. The modulated oral fluoropyrimidine prodrug S-1, and its use in gastrointestinal cancer and other solid tumors. Anticancer Drugs. 2004;15:85–106. doi: 10.1097/00001813-200402000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Chun JH, Kim HK, Kim E, Kang HC, Park JH, Bae JM, et al. Increased expression of metallothionein is associated with irinotecan resistance in gastric cancer. Cancer Res. 2004;64:4703–4706. doi: 10.1158/0008-5472.CAN-04-1063. [DOI] [PubMed] [Google Scholar]
- 5.Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minogushi M, et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007;96:457–463. doi: 10.1038/sj.bjc.6603559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin E, Nemunaitis J. Oncolytic viral therapies. Cancer Gene Ther. 2004;11:643–664. doi: 10.1038/sj.cgt.7700733. [DOI] [PubMed] [Google Scholar]
- 8.Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 9.Ganly I, Kirn D, Eckhardt G, Rodriguez GI, Soutar DS, Otto R, et al. A phase I study of Onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin Cancer Res. 2000;6:798–806. [PubMed] [Google Scholar]
- 10.Harada JN, Berk AJ. p53-Independent and -dependent requirements for E1B-55K in adenovirus type 5 replication. J Virol. 1999;73:5333–5344. doi: 10.1128/jvi.73.7.5333-5344.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 12.O'Shea CC, Soria C, Bagus B, McCormick F. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell. 2005;8:61–74. doi: 10.1016/j.ccr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Reid T, Warren R, Kirn D. Intravascular adenoviral agents in cancer patients: lessons from clinical trials. Cancer Gene Ther. 2002;9:979–986. doi: 10.1038/sj.cgt.7700539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khuri FR, Nemunaitis J, Ganly I, Arseneau J, Tannock IF, Romel L, et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000;6:879–885. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- 15.Xia ZJ, Chang JH, Zhang L, Jiang WQ, Guan ZZ, Liu JW, et al. Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Ai Zheng. 2004;23:1666–1670. [PubMed] [Google Scholar]
- 16.Wang Y, Hallden G, Hill R, Arnand A, Liu TC, Francis J, et al. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat Biotechnol. 2003;21:1328–1335. doi: 10.1038/nbt887. [DOI] [PubMed] [Google Scholar]
- 17.Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6:1134–1139. doi: 10.1038/80474. [DOI] [PubMed] [Google Scholar]
- 18.Stolarek R, Gomez-Manzano C, Jiang H, Suttle G, Lemoine MG, Fueyo J. Robust infectivity and replication of Delta-24 adenovirus induce cell death in human medulloblastoma. Cancer Gene Ther. 2004;11:713–720. doi: 10.1038/sj.cgt.7700731. [DOI] [PubMed] [Google Scholar]
- 19.DeWeese TL, van der Poel H, Li S, Mikhak B, Drew R, Goemann M, et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001;61:7464–7472. [PubMed] [Google Scholar]
- 20.Small EJ, Carducci MA, Burke JM, Rodriguez R, Fong L, van Ummersen L, et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol Ther. 2006;14:107–117. doi: 10.1016/j.ymthe.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 21.Ryan PC, Jakubczak JL, Stewart DA, Hawkins LK, Cheng C, Clarke LM, et al. Antitumor efficacy and tumor-selective replication with a single intravenous injection of OAS403, an oncolytic adenovirus dependent on two prevalent alterations in human cancer. Cancer Gene Ther. 2004;11:555–569. doi: 10.1038/sj.cgt.7700735. [DOI] [PubMed] [Google Scholar]
- 22.Schepelmann S, Hallenbeck P, Ogilvie LM, Hedley D, Friedlos F, Martin J, et al. Systemic gene-directed enzyme prodrug therapy of hepatocellular carcinoma using a targeted adenovirus armed with carboxypeptidase G2. Cancer Res. 2005;65:5003–5008. doi: 10.1158/0008-5472.CAN-05-0393. [DOI] [PubMed] [Google Scholar]
- 23.Johnson L, Shen A, Boyle L, Kunich J, Pandey K, Lemmon M, et al. Selectively replicating adenoviruses targeting deregulated E2F activity are potent, systemic antitumor agents. Cancer Cell. 2002;1:325–337. doi: 10.1016/s1535-6108(02)00060-0. [DOI] [PubMed] [Google Scholar]
- 24.Yu DC, Chen Y, Dilley J, Li Y, Embry M, Zhang H, et al. Antitumor synergy of CV787, a prostate cancer-specific adenovirus, and paclitaxel and docetaxel. Cancer Res. 2001;61:517–525. [PubMed] [Google Scholar]
- 25.Guo W, Zhu H, Zhang L, Davis J, Teraishi F, Roth JA, et al. Combination effect of oncolytic adenovirotherapy and TRAIL gene therapy in syngeneic murine breast cancer models. Cancer Gene Ther. 2006;13:82–90. doi: 10.1038/sj.cgt.7700863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alonso MM, Gomez-Manzano C, Jiang H, Bekele NB, Piao Y, Yung WKA, et al. Combination of the oncolytic adenovirus ICOVIR-5 with chemotherapy provides enhanced anti-glioma effect in vivo. Cancer Gene Ther. 2007;14:756–761. doi: 10.1038/sj.cgt.7701067. [DOI] [PubMed] [Google Scholar]
- 27.Gomez-Manzano C, Alonso MM, Yung WK, McCormick F, Curiel DT, Lang FF, et al. Delta-24 increases the expression and activity of topoisomerase I and enhances the antiglioma effect of irinotecan. Clin Cancer Res. 2006;12:556–562. doi: 10.1158/1078-0432.CCR-05-1892. [DOI] [PubMed] [Google Scholar]
- 28.Conrad C, Miller CR, Ji Y, Gomez-Manzano C, Bharara S, McMurrey JS, et al. Delta24-hyCD adenovirus suppresses glioma growth in vivo by combining oncolysis and chemosensitization. Cancer Gene Ther. 2005;12:284–294. doi: 10.1038/sj.cgt.7700750. [DOI] [PubMed] [Google Scholar]
- 29.Hallden G, Hill R, Wang Y, Anand A, Liu TC, Lemoine NR, et al. Novel immunocompetent murine tumor models for the assessment of replication-competent oncolytic adenovirus efficacy. Mol Ther. 2003;8:412–424. doi: 10.1016/s1525-0016(03)00199-0. [DOI] [PubMed] [Google Scholar]
- 30.Hallden G, Thorne S, Yang J, Kirn DK. Replication-selective oncolytic adenoviruses: methods and protocols. In: Springer C, editor. Suicide Gene Therapy: Methods and Protocols for Cancer. Totowa, NJ, USA: Humana Press; 2003. [Google Scholar]
- 31.Lee WP, Tai DI, Tsai SL, Yeh CT, Chao Y, Lee SD, et al. Adenovirus type 5 E1A sensitizes hepatocellular carcinoma cells to gemcitabine. Cancer Res. 2003;63:6229–6236. [PubMed] [Google Scholar]
- 32.Liao Y, Zou YY, Xia WY, Hung MC. Enhanced paclitaxel cytotoxicity and prolonged animal survival rate by a nonviral-mediated systemic delivery of E1A gene in orthotopic xenograft human breast cancer. Cancer Gene Ther. 2004;11:594–602. doi: 10.1038/sj.cgt.7700743. [DOI] [PubMed] [Google Scholar]
- 33.Zhou Z, Jia SF, Hung MC, Kleinerman ES. E1A sensitizes HER2/neu-overexpressing Ewing's sarcoma cells to topoisomerase II-targeting anticancer drugs. Cancer Res. 2001;61:3394–3398. [PubMed] [Google Scholar]
- 34.Mullins DW, Burger CJ, Elgert KD. Paclitaxel enhances macrophage IL-12 production in tumor-bearing hosts through nitric oxide. J Immunol. 1999;162:6811–6818. [PubMed] [Google Scholar]
- 35.Bhushan A, Kupperman JL, Stone JE, Kimberley PJ, Calman NS, Hacker MP, et al. Drug resistance results in alterations in expression of immune recognition molecules and failure to express Fas (CD95) Immunol Cell Biol. 1998;76:350–356. doi: 10.1046/j.1440-1711.1998.00758.x. [DOI] [PubMed] [Google Scholar]
- 36.Shishodia S, Sodhi A, Shrivastava A. Involvement of Ras and MAP kinase (ERK-1) in cisplatin-induced activation of murine bone marrow-derived macrophages. Biochem Mol Biol Int. 1998;45:527–534. doi: 10.1080/15216549800202912. [DOI] [PubMed] [Google Scholar]
- 37.Lanni JS, Lowe SW, Licitra EJ, Liu JO, Jacks T. p53-independent apoptosis induced by paclitaxel through an indirect mechanism. Proc Natl Acad Sci USA. 1997;94:9679–9683. doi: 10.1073/pnas.94.18.9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider-Brachert W, Tchikov V, Merkel O, Jakob M, Hallas C, Kruse ML, et al. Inhibition of TNF receptor 1 internalization by adenovirus 14.7K as a novel immune escape mechanism. J Clin Invest. 2006;116:2901–2913. doi: 10.1172/JCI23771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Paolo NC, Tuve S, Ni S, Hellstrom KE, Hellstrom I, Lieber A. Effect of adenovirus-mediated heat shock protein expression and oncolysis in combination with low-dose cyclophosphamide treatment on antitumor immune responses. Cancer Res. 2006;66:960–969. doi: 10.1158/0008-5472.CAN-05-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamfers ML, Fulci G, Gianni D, Tang Y, Kurozumi K, Kaur B, et al. Cyclophosphamide increases transgene expression mediated by an oncolytic adenovirus in glioma-bearing mice monitored by bioluminescence imaging. Mol Ther. 2006;14:779–788. doi: 10.1016/j.ymthe.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miura TA, Morris K, Ryan S, Cook JL, Routes JM. Adenovirus E1A, not human papillomavirus E7, sensitizes tumor cells to lysis by macrophages through nitric oxide- and TNF-alpha-dependent mechanisms despite up-regulation of 70-kDa heat shock protein. J Immunol. 2003;170:4119–4126. doi: 10.4049/jimmunol.170.8.4119. [DOI] [PubMed] [Google Scholar]
- 42.Cook JL, Miura TA, Ikle DN, Lewis AM, Jr, Routes JM. E1A oncogene-induced sensitization of human tumor cells to innate immune defenses and chemotherapy-induced apoptosis in vitro and in vivo. Cancer Res. 2003;63:3435–3443. [PubMed] [Google Scholar]
- 43.Routes JM, Ryan S, Clase A, Miura T, Kuhl A, Potter TA, et al. Adenovirus E1A oncogene expression in tumor cells enhances killing by TNF-related apoptosis-inducing ligand (TRAIL) J Immunol. 2000;165:4522–4527. doi: 10.4049/jimmunol.165.8.4522. [DOI] [PubMed] [Google Scholar]
- 44.Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3:639–645. doi: 10.1038/nm0697-639. [DOI] [PubMed] [Google Scholar]
- 45.Hortobagyi GN, Ueno NT, Xia W, Zhang S, Wolf JK, Putnam JB, et al. Cationic liposome-mediated E1A gene transfer to human breast and ovarian cancer cells and its biologic effects: a phase I clinical trial. J Clin Oncol. 2001;19:3422–3433. doi: 10.1200/JCO.2001.19.14.3422. [DOI] [PubMed] [Google Scholar]
- 46.Madhusudan S, Tamir A, Bates N, Flanagan E, Gore ME, Barton DP, et al. A multicenter phase I gene therapy clinical trial involving intraperitoneal administration of E1A-lipid complex in patients with recurrent epithelial ovarian cancer overexpressing HER-2/neu oncogene. Clin Cancer Res. 2004;10:2986–2996. doi: 10.1158/1078-0432.ccr-03-0291. [DOI] [PubMed] [Google Scholar]
- 47.Liao Y, Hung MC. Regulation of the activity of p38 mitogen-activated protein kinase by Akt in cancer and adenoviral protein E1A-mediated sensitization to apoptosis. Mol Cell Biol. 2003;23:6836–6848. doi: 10.1128/MCB.23.19.6836-6848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frisch SM, Mymryk JS. Adenovirus-5 E1A: paradox and paradigm. Nat Rev Mol Cell Biol. 2002;3:441–452. doi: 10.1038/nrm827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.