

Abstract
HIF-1α activates genes under hypoxia and was hypothesized to regulate bone regeneration. Surprisingly, HIF-1α+/− fracture calluses are larger, stronger, and stiffer than HIF-1α+/+ calluses because of decreased apoptosis. These data identify apoptosis inhibition as a means to enhance bone regeneration.
Introduction
Bone regeneration subsequent to fracture involves the synergistic activation of multiple signaling pathways. Localized hypoxia after fracture activates hypoxia-inducible factor 1α (HIF-1α), leading to increased expression of HIF-1 target genes. We therefore hypothesized that HIF-1α is a key regulator of bone regeneration.
Materials and Methods
Fixed femoral fractures were generated in mice with partial HIF-1α deficiency (HIF-1α+/−) and wildtype littermates (HIF-1α+/+). Fracture calluses and intact contralateral femurs from postfracture days (PFDs) 21 and 28 (N = 5–10) were subjected to μCT evaluation and four-point bending to assess morphometric and mechanical properties. Molecular analyses were carried out on PFD 7, 10, and 14 samples (N = 3) to determine differential gene expression at both mRNA and protein levels. Finally, TUNEL staining was performed on PFD 14 samples (N = 2) to elucidate differential apoptosis.
Results
Surprisingly, fracture calluses from HIF-1α+/− mice exhibited greater mineralization and were larger, stronger, and stiffer. Microarray analyses focused on hypoxia-induced genes revealed differential expression (between genotypes) of several genes associated with the apoptotic pathway. Real-time PCR confirmed these results, showing higher expression of proapoptotic protein phosphatase 2a (PP2A) and lower expression of anti-apoptotic B-cell leukemia/lymphoma 2 (BCL2) in HIF-1α+/+ calluses. Subsequent TUNEL staining showed that HIF-1α+/+ calluses contained larger numbers of TUNEL+ chondrocytes and osteoblasts than HIF-1α+/− calluses.
Conclusions
We conclude that partial HIF-1α deficiency results in decreased chondrocytic and osteoblastic apoptosis, thereby allowing the development of larger, stiffer calluses and enhancing bone regeneration. Furthermore, apoptosis inhibition may be a promising target for developing new treatments to accelerate bone regeneration.
Keywords: bone, fracture, regeneration, hypoxia, inducible factor 1α, apoptosis
INTRODUCTION
Successful skeletal fracture repair occurs as a series of inter-related processes that begins with inflammation and proceeds to intramembranous ossification, chondrogenesis, endochondral ossification, and finally, remodeling resulting in scarless repair. During each process, numerous signaling pathways are activated, leading to transcriptional responses that recapitulate aspects of embryonic skeletal development combined with responses to acute tissue injury.(1–5) Specifically, fracture repair begins with the formation of a hematoma, which initiates the inflammatory response(6) that in turn activates several components of the reparative process including intramembranous ossification and chondrogenesis.(3) The hematoma also isolates the fracture site from perfusion, leading to low oxygen tension and regional hypoxia.(7) Chondrocytes and osteoblasts responsible for formation of the soft and hard callus, respectively, must therefore proliferate and differentiate, as well as begin cartilage and bone deposition under conditions of hypoxia. In addition, timely blood vessel in-growth must be initiated to restore physiological perfusion and promote endochondral ossification.(8–10)
Hypoxia-inducible factor 1α (HIF-1α) is an integral transcriptional regulator of anaerobic metabolism and angiogenesis(11) that is functionally active in both osteoblasts and chondrocytes. Osteoblastic HIF-1α protein levels increase in response to hypoxia concomitant with elevation of vascular endothelial growth factor (VEGF) and proliferating cell nuclear antigen (PCNA) levels, as well as attendant higher rates of proliferation and differentiation.(12) Chondrocyte-specific HIF-1α–null mice exhibit perinatal lethality, attributed to tracheal defects, as well as gross skeletal malformations, including shortened limbs and abnormally wide and malformed ribcages.(13) Furthermore, chondrocytes isolated from these mice lack VEGF induction when exposed to hypoxia and show lower rates of proliferation under nonhypoxic conditions and decreased matrix synthesis and free ATP and lactate under both hypoxic and non-hypoxic culture conditions.(14,15)
Previously, our laboratory showed temporal and spatial upregulation of HIF-1α and several of its target genes during fracture repair.(2,16) This study was designed to further elucidate the mechanistic role of HIF-1α in fracture repair. Using an established fixed femoral fracture model, we performed tissue, cellular, and molecular analyses of fracture repair in mice with partial HIF-1α deficiency (HIF-1α+/−) and compared their response directly to that of their wildtype littermates (HIF-1α+/+). The results of these experiments establish HIF-1 as a key regulator of bone regeneration.
MATERIALS AND METHODS
Animal model
All methods and animal procedures were reviewed and approved by the University’s Institutional Animal Care and Use Committee and met or exceeded all federal guidelines for the humane use of animals in research. Fixed fractures were generated in the left femurs of adult female HIF-1α+/−and HIF-1α+/+ mice by the procedure first described by Bonnarens and Einhorn(17) and routinely used in our laboratory.(2,4,5,16,18) At predetermined postfracture days (PFDs), mice were killed by CO2 inhalation, and the fractured and contralateral (intact) femurs were harvested and dissected free of soft tissue. Tail and liver samples were additionally harvested for genotype verification. Samples used for imaging and mechanical testing were harvested on PFDs 21 and 28 and stored in saline at −20°C until testing. Samples for RNA and protein extraction were harvested on PFDs 7, 10, and 14 and were stored in RNAlater (Ambion) at −80°C until extraction, as previously described.(16) Samples for histology and TUNEL staining were harvested on PFD 14 and immediately fixed in formalin (Fischer), followed by decalcification as previously reported.(16) Samples were embedded in OCT (Tissue Tek), and serial 8-μm longitudinal sections were cut using a cryostat (Leica) equipped with a CryoJane tape-transfer system (Instrumedics). All analyses were carried out by experimenters blinded to genotype.
μCT
μCT analyses were performed on intact (HIF-1α+/+, N = 20; HIF-1α+/−, N = 24) and fractured femurs (PFD 21 HIF-1α+/+, N = 5; PFD 21 HIF-1α+/−, N = 5; PFD 28 HIF-1α+/+, N = 9; PFD 28 HIF-1α+/−, N = 6) using a μCT 40 (Scanco Medical) scanner at a slice resolution of 12 μm. Samples were first evaluated in Scout Mode to determine femur lengths and regions of interest (ROIs). Intact ROIs consisted of 212 slices centered at the mid-diaphysis, and fracture callus ROIs ranged from 410 to 509 slices, centered at the calluses midpoint. For the intact analyses, contour lines were applied to periosteal and endosteal surfaces, and bone volume (BV) and total volume (TV) were computed at threshold settings of 1000 upper and 350 lower, with sigma of 0.5 and support of 1.0. Endosteal (endosteal TV–endosteal BV), cortical (periosteal BV), and periosteal (endosteal + cortical) volumes were computed and converted to cross-sectional areas. Calluses were assessed for total and mineral volume. Contour lines were applied to periosteal surfaces of three stacks of 42 slices (proximal, central, and distal), and BV and TV were computed at thresholds of 460 upper and 170 lower (same sigma and support). TV (new bone, old cortical bone, soft tissue) and BV (newly formed bone) are reported as total and mineral volume, respectively. All results are reported as average ± SD.
Biomechanical testing
Intact (HIF-1α+/+, N = 20; HIF-1α+/−, N = 21) and fractured femurs (PFD 21 HIF-1α+/+, N = 5; PFD 21 HIF-1α+/−, N = 5; PFD 28 HIF-1α+/+, N = 10; PFD 28 HIF-1α+/−, N = 7) were subjected to four-point bending tests using an MTS machine (858 Mini Bionix II; MTS) equipped with a 100N load cell (SMT1–100N; Interface) and a custom designed loading jig. Samples were centered in the jig and loaded anterior-posterior under displacement control at 0.1 mm/s. Displacement, force, and time were digitally sampled at 204.8 Hz. Stiffness was determined by fitting linear regression lines to the elastic regions of the loading curves, whereas strength was determined by finding the maximum force applied before failure. Callus results were normalized to their contralateral intact femurs. Results are reported as average ± SD.
Microarrays analyses
Microarray analyses were performed using focused cDNA microarrays (GEArray Q Series Mouse Hypoxia and Angiogenesis Array; SuperArray). For each microarray, 3 μg of pooled callus RNA (N = 3 animals/genotype and time-point) was labeled with biotin-16-dUTP (Roche) and amplified using the AmpoLabeling-LPR kit (SuperArray), according to the manufacturer’s instructions. After overnight hybridization, membranes were washed, blocked, incubated with binding buffer, washed again, and incubated in enhanced chemiluminescence (ECL) solution. The membranes were exposed to film (BioMax MR; Kodak), and the resulting signals were digitally scanned (HP ScanJet 5370C). Analyses were performed using the GEArray Expression Analysis Suite software package (SuperArray). After aligning the spots, the signal intensities were computed subject to the following dataset parameters: background correction, Minimum Value; summation type, Average Density; normalization, Selected Genes (β-actin); clover mode, Off. The normalized intensity values for each gene were used to calculate percent differences in expression between genotypes. This calculation was performed by dividing the difference of the normalized HIF-1α+/+ and HIF-1α+/− values by their average value and multiplying the result by 100. Consequently, positive results indicate higher expression in HIF-1α+/+ calluses and negative results indicate higher expression in HIF-1α+/− samples. These percent differences were subsequently subjected to a threshold that eliminated genes not showing percent differences >100% for at least one time-point. MIAME compliant data from these experiments have been submitted to the NCBI GEO database (Accession no. GSE6091).
Quantitative real-time RT-PCR
Quantitative real-time RT-PCR (qPCR) was performed on pooled RNA samples (N = 3 animals/genotype and time-point) from intact bone and fracture calluses. The genes, accession numbers, primer sequences, and amplicon sizes are presented in Table 1. All experiments were performed with an annealing temperature of 58°C and RNA concentrations of 5 ng/reaction, as previously described.(16) All experiments were performed three times, and the results are reported as average ± SD.
Table 1.
qPCR PRIMERS
| Gene | Accession no. | Primer sequences | Size (bp) |
|---|---|---|---|
| HIF-1α | NM_010431 | 5′-GCTGAAGACACAGAGGCAAA-3′5′-ATACTTGGAGGGCTTGGAGA-3′ | 158 |
| PP2A | NM_017374.2 | 5′-GGAAATCACCAGATACGAACTACC-3′5′-GCACTCATCATAAAAGCCATACAC-3′ | 182 |
| BCL2 | NM_009741.2 | 5′-ATGTGTGTGGAGAGCGTCAA-3′5′-AGAGACAGCCAGGAGAAATCAA-3′ | 182 |
| β-Actin | NM_007393.1 | 5′-ACTGGGACGACATGGAGAAG-3′5′-GAGGCATACAGGGACAGCA-3′ | 202 |
The first column lists the name of each of the genes used in the qPCR analysis, the second presents the accession number of the sequence used in construction of the primers, the third lists the sequences of the primers used (forward primer on top, reverse primer on bottom), and the final column indicates the sizes of the resulting amplicons.
Immunoblot assay
Immunoblots were performed using pooled protein samples (N = 3 animals/genotype and time-point) from intact bone and fracture calluses. For each blot, 50 μg of each protein pool was fractionated by SDS-PAGE (7% acrylamide) and processed as previously described.(16) Primary antibodies for HIF-1α (NB100–105; Novus) and β-actin (A5441; Sigma), as well as horseradish peroxidase (HRP)-conjugated secondary antibodies (AP308P; Chemicon), were used. Quantification of signal intensity was performed using the Kodak 1-D software package (Kodak). The intensity values for HIF-1α were divided by their corresponding β-actin values, yielding results in the unitless term of expression relative to β-actin. Blots were performed three times, and results are reported as average ± SD.
TUNEL staining and histology
TUNEL staining was carried out on PFD 14 callus sections (N = 2 sections/animal and 2 animals/genotype) using an in situ apoptosis kit (Trevigen) in accordance with the manufacturer’s protocol. Negative controls were generated on adjacent sections by omitting TdT in the labeling solution. To visualize regions of bone and cartilage, Safranin O/Fast Green staining was also performed by standard histological methodology. Sections were analyzed under brightfield microscopy (Axiovert; Carl Zeiss), and images were captured with a CCD camera (AxioCam MRc; Carl Zeiss). To quantify apoptosis, the number of positive cells from ×200 images containing the highest number of positive cells within the regions of cartilage and woven bone were counted by four independent individuals. Results are reported as average number of positive cells ± SD.
RESULTS
Intact HIF-1α+/− femurs and fracture calluses are larger
Intact femurs from HIF-1α+/+ and HIF-1α+/− mice were assessed by μCT for differences in femoral geometry. Measurements of femoral length yielded virtually identical results (16.6 ± 0.6 and 16.7 ± 0.6 mm for the HIF-1α+/+ and HIF-1α+/− mice, respectively). However, when the cross-sectional endosteal, cortical, and periosteal areas for the mid-diaphyseal ROI (Figs. 1A and 1B) were compared, a significant increase in periosteal cross-sectional area was revealed for the HIF-1α+/− femurs (1.43 ± 0.17 mm2) compared with the HIF-1α+/+ femurs (1.32 ± 0.15 mm2; Fig. 1C).
FIG. 1.
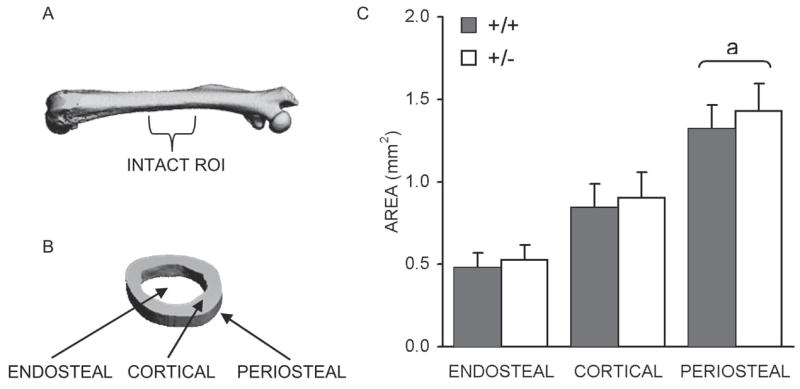
Intact femoral μCT analysis. (A) μCT image of intact femur with approximate ROI used for areal analysis labeled. (B) Section of analyzed ROI with endosteal, cortical, and periosteal regions labeled. (C) Bar graph showing average endosteal, cortical, and periosteal area cross-sectional areas ± SD (mm2), as determined for HIF-1α+/+ and HIF-1α+/− mice. ap < 0.03, significant difference between genotypes (Mann Whitney).
HIF-1α+/+ and HIF-1α+/− fracture calluses ROIs (Fig. 2A) were also analyzed, and visual observation of the resultant images (Figs. 2B–2E) revealed several qualitative differences between genotypes and time-points. At PFD 21, HIF-1α+/− calluses appear wider and more porous than HIF-1α+/+ calluses (Figs. 2B and 2C). By PFD 28, calluses from both genotypes show evidence of remodeling, characterized by reduced porosity and smoothed and condensed periosteal surfaces (Figs. 2D and 2E). This is more apparent in HIF-1α+/− calluses (Fig. 2E), suggesting that fracture repair may be proceeding in an accelerated manner in these mice. In agreement with these qualitative assessments, quantitated total and mineral volumetric measurements also revealed that HIF-1α+/− calluses are larger (Figs. 2F–2H). Significant increases were identified for mineral volume in the central region on PFD 21 (0.96 ± 0.18 mm3 for HIF-1α+/+ and 1.51 ± 0.43 mm3 for HIF-1α+/−; Fig. 2G) and total volume in the distal region on PFD 21 (1.65 ± 0.30 mm3 for HIF-1α+/+ and 2.97 ± 1.15 mm3 for HIF-1α+/−; Fig. 2H).
FIG. 2.
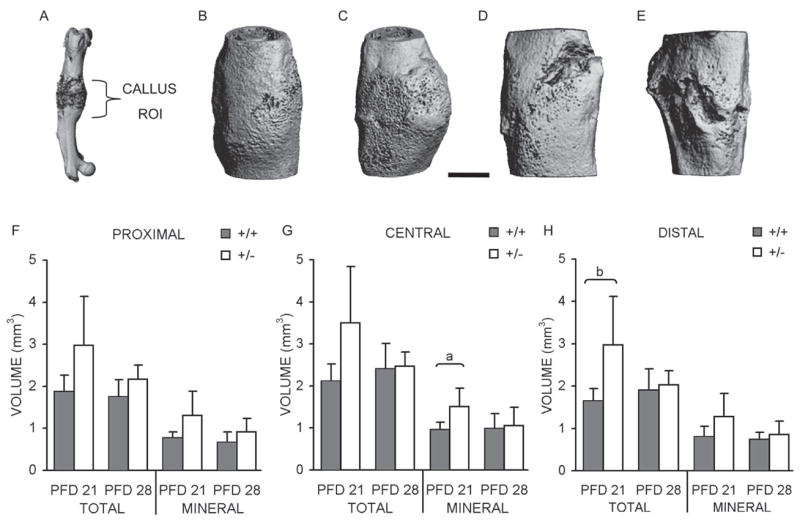
Fracture callus μCT analysis. (A) μCT image of fractured femur with approximate ROI used for volumetric analysis labeled. Representative images of calluses from (B) PFD 21 HIF-1α+/+ mouse; (C) PFD 21 HIF-1α+/− mouse; (D) PFD 28 HIF-1α+/+ mouse; and (E) PFD 28 HIF-1α+/− mouse. Scale bar represents 1 mm. Average total (including mineralized and unmineralized tissue) and mineral volume (only newly formed bone; mm3) were quantified, and bar graphs showing these averages ± SD for (F) proximal; (G) central; and (H) distal regions are presented. ap < 0.03 and bp < 0.04, significant difference between genotypes (Mann Whitney).
Intact HIF-1α+/− femurs and fracture calluses are biomechanically superior
Subsequent to μCT analysis, biomechanical testing was conducted on the HIF-1α+/+ and HIF-1α+/− intact and fractured femurs to determine stiffness and strength. No differences in stiffness are present in the intact femurs (Fig. 3A). However, consistent with the observed increase in cross-sectional area, a significant 15% increase in strength was observed for intact HIF-1α+/− femurs (Fig. 3B). Similarly, testing of fracture calluses identified a significant increase of 240% at PFD 21 for stiffness, along with a significant 88% increase in strength at PFD 28 for the HIF-1α+/− samples (Figs. 3C and 3D). Together with the μCT results, these data indicate that bone regeneration is enhanced in HIF-1α+/− mice.
FIG. 3.
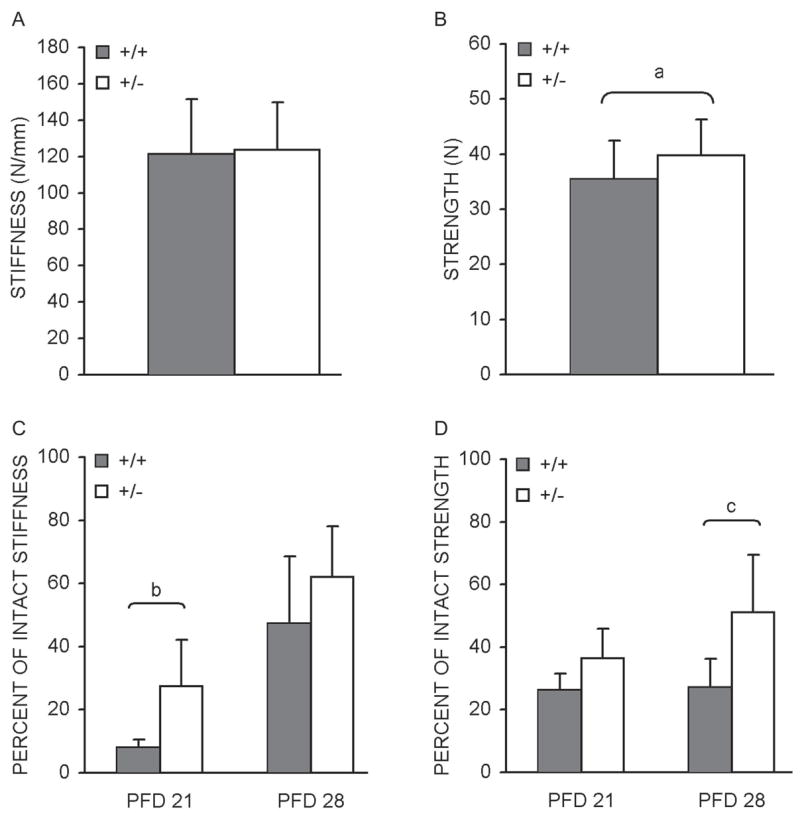
Intact and fracture callus biomechanical properties Bar graphs denoting average intact femoral (A) stiffness (N/mm) and (B) strength ± SD (N), as determined by four-point bending of samples from HIF-1α+/+ and HIF-1α+/− mice. Test results for HIF-1α+/+ and HIF-1α+/− fracture calluses were normalized to their contralateral (intact) controls and are presented as bar graphs indicating percent of intact: (C) stiffness and (D) strength ± SD. ap < 0.05, bp < 0.03, and cp < 0.01, significant difference between genotypes (Mann Whitney).
HIF-1α expression is reduced in HIF-1α+/− mice
Although HIF-1α is predominately regulated at the protein level, prior research conducted in our laboratory indicated that HIF-1α mRNA levels increase during the course of bone regeneration.(16) As such, the expression levels of HIF-1α mRNA in the fracture calluses of both genotypes were determined by qPCR analysis. As expected, the expression of HIF-α mRNA in HIF-1α+/− calluses was lower than in HIF-1α+/+ calluses, with significant decreases of 26% and 50% seen on PFDs 10 and 14, respectively (Fig. 4A). Furthermore, although HIF-1α mRNA expression increased relative to intact bone in calluses from both genotypes, the pattern and duration of induction varied. HIF-1α+/+ mice showed significantly elevated HIF-1α expression at all three time-points, with peak expression of 6.6-fold (compared with intact) on PFD 10 (Fig. 4A). In contrast, expression in HIF-1α+/− calluses peaked on PFD 7, with a similar magnitude, and steadily diminished on PFDs 10 and 14, with significant induction seen only at PFDs 7 and 10 (Fig. 4A).
FIG. 4.
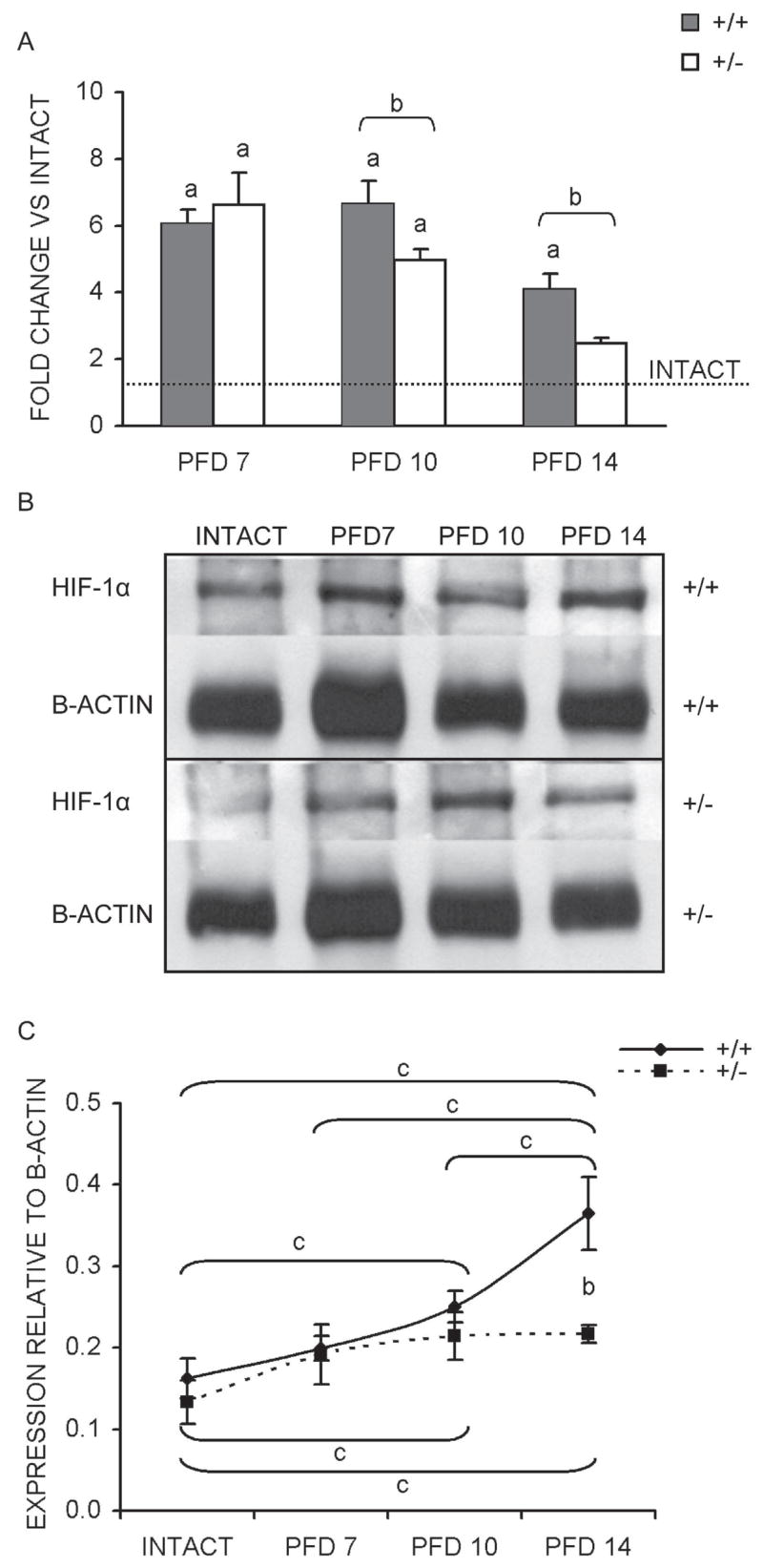
Temporal mRNA and protein expression of HIF-1α. (A) Results of qPCR determined mRNA expression levels (average of three runs ± SD) are presented as a bar graph showing fold change vs. intact bone (dashed line) for each time-point assessed. (B) Representative images of immunoblots performed to assess HIF-1α protein levels with bands corresponding to HIF-1α (120 kDa) and β-actin (42 kDa) labeled for each genotype and sample. (C) Line graph of quantified signal intensities from HIF-1α immunoblots after normalization to β-actin (average of three blots ± SD) for each time-point. ap < 0.01, significant difference between intact and callus (Mann Whitney); bp < 0.02, significant difference between genotypes (Mann Whitney); cp < 0.03, significant difference between samples within each genotype (Kruskal-Wallis with Tukey posthoc).
Because the availability of HIF-1α to form the functional HIF-1 transcriptional complex is controlled post-translationally, immunoblot assays for HIF-1α were performed (Fig. 4B). Quantitation of signal intensities revealed a significant 59% decrease in HIF-1α protein from HIF-1α+/− calluses compared with HIF-1α+/+ calluses on PFD 14 (Fig. 4C). Analogous to mRNA expression, the patterns of HIF-1α protein induction relative to intact bone also vary by genotype. HIF-1α+/+ calluses are characterized by a steady rise in HIF-1α protein, with significant increases (compared with intact) seen on PFDs 10 and 14. In addition, the PFD 14 peak of 2.25-fold relative to intact was significantly higher than the expression levels at PFDs 7 and 10 (Fig. 4C). In contrast, HIF-1α expression levels in HIF-1α+/− calluses showed significant elevation (compared with intact) at PFDs 10 and 14, but plateaued at ~1.6-fold relative to intact and showed no differences between the sampled callus time-points (Fig. 4C). These findings are entirely consistent with the expectation that partial HIF-1α deficiency should result in decreased expression of HIF-1α.
Molecular mechanism for enhanced fracture repair
We next sought to study the molecular basis of the observed effects by identifying the gene(s) responsible for enhanced bone regeneration in the HIF-1α+/− mice. This was accomplished by probing microarrays spotted with a subset of hypoxia-induced genes with RNA isolated from HIF-1α+/+ and HIF-1α+/− calluses on PFDs 7, 10, and 14. Eighteen of these genes were identified as differentially regulated between genotypes (Table 2). These genes include ribosomal and mitochondrial genes (RPL137A, RPS2, UCP2), growth factor–related genes (ADD1, ECE1, IGF2, IL6), transcriptional regulatory genes (AGTPBP1, BHLHB2, HIF-1α), and genes related to apoptosis (BCL2, IGF2, IL6, PP2A). The identification of differential expression of apoptosis-related genes in HIF-1α+/+ and HIF-1α+/−calluses presented a possible mechanistic explanation for the observed differences in bone regeneration. Therefore, more rigorous qPCR analyses of the proapoptotic gene protein phosphatase 2a (PP2A)(19,20) and the anti-apoptotic gene B-cell leukemia/lymphoma 2 (BCL2)(19,21,22) were conducted. Consistent with the HIF-1α expression analyses, these experiments included intact RNA samples, allowing for comparison of expression levels to intact bone and between genotypes.
Table 2.
Hypoxia Microarray Results
| Gene name | Accession no. | Symbol | PFD 7 | PFD 10 | PFD 14 |
|---|---|---|---|---|---|
| Adducin 1 (α) | NM_013457 | Add1 | 0.00 | 0.00 | 188.56 |
| RIKEN cDNA A230056J06 gene | NM_023328 | Agtpbp1 | 0.00 | −105.94 | 0.00 |
| B-cell leukemia/lymphoma 2 | NM_009741 | Bcl2 | 0.00 | −135.56 | 0.00 |
| Basic helix-loop-helix domain containing, class B2 | NM_011498 | Bhlhb2 | 0.00 | 85.90 | −143.05 |
| Catalase | NM_009804 | Cat | −140.24 | 119.40 | 0.00 |
| Down-regulator of transcription 1 | NM_026106 | Dr1 | 0.00 | 104.82 | 0.00 |
| Endothelin converting enzyme 1 | NM_199307 | Ece1 | −111.42 | −30.36 | −24.05 |
| HIF-1α | NM_010431 | Hif1a | −188.92 | 48.65 | 105.36 |
| IGF-II | NM_010514 | Igf2 | 0.00 | 0.00 | 119.20 |
| IL-6 | NM_031168 | Il6 | −198.98 | −101.71 | 159.58 |
| Protein phosphatase 2a, catalytic subunit, beta | NM_017374 | Ppp2cb | −167.21 | 4.83 | 159.59 |
| Proteasome (prosome, macropain) 28 subunit, beta | NM_011190 | Psme2 | 0.00 | −120.18 | 0.00 |
| Ribosomal protein L37a | NM_009084 | Rpl37a | 0.00 | 0.00 | 118.94 |
| Ribosomal protein S2 | NM_008503 | Rps2 | 0.00 | −36.51 | 109.70 |
| U1 small nuclear ribonucleoprotein polypeptide A | NM_009224 | Snrp70 | 0.00 | −121.53 | 107.78 |
| Superoxide dismutase 2, mitochondrial | NM_013671 | Sod2 | 0.00 | −125.43 | 0.00 |
| Spectrin β2 | NM_009260 | Spnb2 | 0.00 | −4.86 | 109.26 |
| Uncoupling protein 2 (mitochondrial, proton carrier) | NM_011671 | Ucp2 | 0.00 | −114.84 | 145.72 |
This table lists the 18 hypoxia inducible genes that were identified as being differentially regulated between the fracture calluses of HIF-1α+/+ and HIF-1α+/− mice. The gene names, accession numbers, symbols, and percent differences as given. Percent difference values of 0.00 indicate no difference, values > 0.00 indicate increased expression in HIF-1α+/+ samples, and values < 0.00 indicate increased expression in HIF-1α+/− samples.
The qPCR analysis of fracture callus PP2A expression relative to intact bone showed significant PP2A elevation for both genotypes at all time-points except for HIF-1α+/−at PFD 14 (Fig. 5A). Expression of PP2A in HIF-1α+/+ calluses rose to a PFD 14 peak of >2-fold above intact levels, whereas HIF-1α+/− callus expression peaked at ~1.75-fold on PFD 7 (Fig. 5A). When expression levels were compared between genotypes, no differences were apparent at PFDs 7 and 10, but significantly reduced expression (~60%) in HIF-1α+/− calluses was seen at PFD 14 (Fig. 5A). In contrast, BCL2 expression was significantly higher in HIF-1α+/− calluses on PFDs 10 (~80%) and 14 (~50%; Fig. 5B). Additionally, comparison of callus and intact BCL2 expression showed no apparent upregulation of BCL2 in HIF-1α+/+ calluses, but a steady rise in HIF-1α+/− calluses, reaching an ~2-fold peak on PFD 14 (Fig. 5B). These data suggest that elevated pro-apoptotic signaling in HIF-1α+/+ calluses, coupled with elevated anti-apoptotic signaling in HIF-1α+/− calluses, may result in decreased apoptosis in HIF-1α+/− calluses, explaining the observation of enhanced bone regeneration.
FIG. 5.
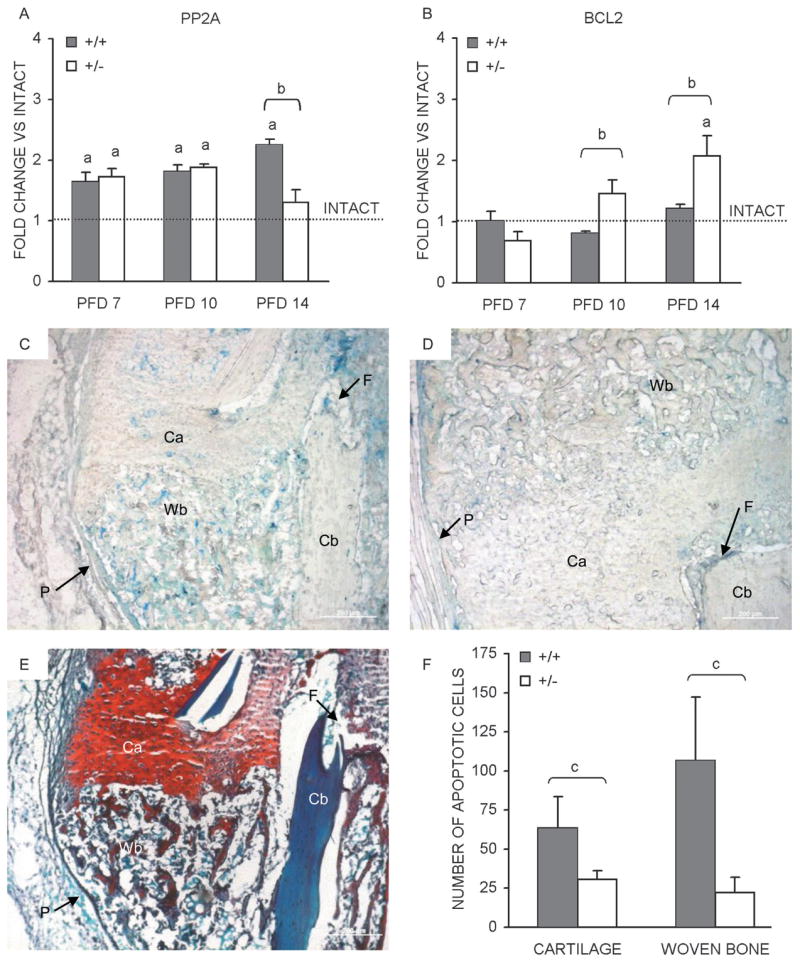
Fracture callus apoptosis. Temporal mRNA expression patterns, as determined by qPCR, for (A) PP2A and (B) BCL2, in the course of bone regeneration in HIF-1α+/+ and HIF-1α+/− mice are presented as bar graphs (average of three runs ± SD) indicating fold change vs. intact bone (dashed line). Photomicrographs of PFD 14 fracture calluses sections from (C) HIF-1α+/+ and (D) HIF-1α+/− mice after TUNEL staining was performed to identify apoptotic cells. Apoptotic cells (stained blue) are seen in regions of cartilage (Ca) and woven bone (Wb). Additionally labeled are regions of cortical bone (Cb), the fracture site (F), and periosteum (P). (E) An adjacent section stained with Safranin O/fast green to identify regions of cartilage (Ca, red) and woven bone (Wb, blue) is also presented. Scale bars represent 200 μm in all images. (F) Bar graph depicting the average number of apoptotic cells (N = 2 animals per time-point, two sections per animal) ± SD counted in regions of cartilage and woven bone from HIF-1α+/+ and HIF-1α+/− calluses. ap < 0.01, significant difference between intact and callus (Mann Whitney); bp < 0.05, significant difference between genotypes (Mann Whitney); cp < 0.03, significant difference between genotypes (Mann Whitney).
Partial HIF-1α deficiency leads to reduced apoptosis
Because the largest differences in apoptotic signaling were observed on PFD 14, TUNEL staining was performed on callus sections from this time-point to assess differential apoptosis (Figs. 5C and 5D). Photomicrographs of TUNEL staining performed on HIF-1α+/+ (Fig. 5C) and HIF-1α+/−(Fig. 5D) callus sections showed apoptotic cells (labeled blue) present throughout the fracture calluses and most apparent within regions of cartilage (Ca) and woven bone (Wb). Additional staining was visible in the periosteum (P, arrow) and marrow near the fracture site (F, arrow); however, it was diffuse and cannot be resolved to any cellular phenotype. The cortical bone (Cb) showed no positive staining. Safranin O/fast green staining was performed on an adjacent section (Fig. 5E) to clearly distinguish between regions of cartilage (Ca, red) and woven bone (Wb, blue). Quantification of the number of positively stained chondrocytes from regions of cartilage and osteoblasts from regions of woven bone revealed significantly higher numbers of apoptotic cells in the fracture calluses of HIF-1α+/+ mice compared with those from HIF-1α+/− mice (Fig. 5F). In addition, the increase in apoptosis was much larger for osteoblasts (~375%) than for chondrocytes (~100%; Fig. 5F), indicating that inhibited osteoblast apoptosis contributed more to the observed enhancement in bone regeneration in HIF-1α+/− mice than inhibited chondrocyte apoptosis.
DISCUSSION
Ideally, examination of the function of HIF-1α in fracture repair would be carried out in homozygous HIF-1α–null mice rather than the heterozygotes used in these experiments. However, complete HIF-1α deficiency results in embryonic lethality,(23,24) and even conditional HIF-1α knockout limited to chondrocytes results in perinatal lethality,(13) thus necessitating the use of heterozygous mice. Previous experiments conducted with these mice have shown no phenotype under conditions of normoxia but significantly impaired responses to hypoxia.(25–28) The results of our experiments show that partial HIF-1α deficiency is also sufficient to alter the normal process of bone regeneration.
One of the most thoroughly studied hypoxia-regulated processes is angiogenesis, and a broad consensus has been reached establishing HIF-1α activation as a vital component of this process.(29–35) Furthermore, proper angiogenesis is imperative for bone regeneration,(8,9,36–39) with inhibition of fracture callus angiogenesis leading to abrogation of the reparative process.(40,41) Based on these data, we originally hypothesized that partial HIF-1α deficiency would result in impaired bone regeneration caused by decreased angiogenesis. To our surprise, the results of the morphometric and biomechanical analyses revealed the exact opposite: larger, more mineralized, stiffer and stronger calluses developed in the HIF-1α+/− mice. These results led to the surprising conclusion that partial HIF-1α deficiency enhances bone regeneration.
Although analyses of HIF-1α mRNA and protein levels revealed expected decreases in HIF-1α+/− calluses, we analyzed several angiogenesis markers to ascertain that no compensatory mechanism was promoting angiogenesis in an HIF-1α–independent fashion in these samples. As expected, global angiogenic signaling, as determined by microarray analysis (data not shown), and qPCR analysis of VEGF and platelet endothelial cell adhesion molecule (PECAM; data not shown) all indicate that decreased angiogenic signaling is present in HIF-1α+/− calluses. Because it is improbable that decreased angiogenesis could explain the unexpected finding of enhanced bone regeneration in HIF-1α+/− mice, we closely examined the hypoxia-inducible microarray data for molecular clues to the mechanism behind this response. Within this dataset, we observed that several genes belonging to the apoptosis pathway were differentially expressed, and the more stringent qPCR analyses established that an increase in proapoptotic signaling, coupled with a decrease in anti-apoptotic signaling, is present in HIF-1α+/+ calluses. Subsequent TUNEL staining showing reduced numbers of apoptotic chondrocytes and osteoblasts in HIF-1α+/− fracture calluses confirmed that differential signaling is correlated to differential apoptosis in these mice.
The relationship between hypoxia, HIF-1α, and apoptosis has been the subject of numerous studies that have documented both pro- and anti-apoptotic effects of HIF-1α activation, depending on the biological system.(42–44) The majority of these studies have used various tumor and cancerous cell models; however, a few neuronal models have also been examined. Of particular relevance to our findings is a study by Helton et al.(45) that showed a significant decrease in hippocampal apoptosis in partially HIF-1α–deficient mice after exposure to hypoxia. Additional experiments conducted on mice with complete HIF-1α deletion targeted to the brain also showed decreased neuronal death after vascular occlusion.(45) These results complement our findings and together offer compelling evidence that HIF-1α deficiency can protect cells from hypoxia-related apoptosis.
Because apoptosis is responsible for removal of the large number of chondrocytes and osteoblasts that are generated during the proliferative phase of fracture repair,(46–48) decreased apoptosis in the calluses of HIF-1α+/− mice is therefore consistent with enhanced bone regeneration. A thorough analysis of the spatiotemporal progression of apoptosis within the fracture calluses of mice performed by Li et al.(47) identified PFD 16 as the point of maximal apoptosis, which is in agreement with our finding of a PFD 14 peak in pro-apoptotic signaling in HIF-1α+/+ calluses. Moreover, at PFD 14, we also observed the largest differences in apoptotic signaling between HIF-1α+/+ and HIF-1α+/− mice, indicating that partial HIF-1α deficiency affects apoptosis in the fracture callus at a pivotal time during bone regeneration.
Similar to our results, studies of fracture healing in TNF-α receptor knockout mice show an increase in fracture callus morphometry.(49) This difference is attributed to increased cartilage formation attendant with lower rates of chondrocyte apoptosis. Whereas the effects of TNF-α signaling abrogation during fracture repair are manifested in several other ways,(49) the effects on chondrocytes support our findings and aid in discerning the possible mechanism by which decreased chondrocyte apoptosis can enhance fracture repair. Specifically, lower rates of chondrocyte apoptosis in HIF-1α+/− calluses result in prolonged matrix secretion, leading to increases in ensuing endochondral ossification and contributing to the observed increases in the morphometric and mechanical properties of the calluses.
In the case of osteoblasts, inhibited apoptosis is surmised to enhance bone regeneration through increased intramembranous ossification resulting from prolonged activity. In a cortical defect model, Olmedo et al.(50) found that administration of IL-1β resulted in increased callus formation that was attributed to greater numbers of osteoblasts within the defect and lower rates of osteoblastic apoptosis. Whereas these results support our finding of decreased osteoblast apoptosis correlating with larger callus development, IL-1β administration also increased the rate of osteoblast proliferation.(50) This additional effect makes it difficult to distinguish between the relative contributions of proliferation and apoptosis to callus formation. However, it is clear that increased numbers of callus osteoblasts indeed correlate with larger callus development. Furthermore, because our results revealed higher rates of apoptosis in osteoblasts than chondrocytes, they indicate that increased intramembranous ossification contributes to the gains in mechanical properties seen in HIF-1α+/− fracture calluses to a greater degree than increased endochondral ossification.
The results of this series of experiments have shown a significant and unexpected phenotype of enhanced bone regeneration in HIF-1α+/− mice compared with wildtype littermates. The morphometric and biomechanical analyses provide strong evidence for this conclusion with subsequent molecular analyses identifying differential regulation of apoptosis as the likely basis of these differences. Although differential transcription of PP2A and BCL2, as well as differential rates of osteoblast and chondrocyte apoptosis, has been shown, the exact mechanism by which partial HIF-1α deficiency decreases the rate of apoptosis remains to be elucidated. In light of this limitation, future in vitro mechanistic experiments are being planned to address this critical issue.
The broader long-term goal underlying this research is to identify therapeutic targets for clinically accelerating bone regeneration. The results of these experiments show that partial HIF-1α deficiency enhances the process of bone regeneration by decreasing chondrocyte and osteoblast apoptosis. This presents the opportunity for a unique therapeutic approach to accelerate bone regeneration, namely apoptosis inhibition. This approach may be more efficacious than broad-based HIF-1α inhibition because it should not adversely affect angiogenesis, which is clearly required for expedient bone regeneration. Taken together, the results presented herein have not only increased our understanding of the role of HIF-1α in fracture repair, but have also led to the identification of a novel line of research to develop clinically useful therapeutics for the enhancement of skeletal regeneration.
Acknowledgments
The authors thank Dr William Ahrens for help with the mouse fracture protocol, Dr Mark Horowitz for the fracture device, Dr Dominador Manalo for assistance in genotyping, Robert Gersch for assisting in tissue harvesting, Drs Clinton Rubin, Yi-Xian Qin, and Stefan Judex for helpful discussions and data analyses relating to μCT and mechanical testing, and Colleen Michaels for administrative assistance. This work was supported by a grant from the National Institutes of Health RO3AR476032 (MH).
Footnotes
The authors state that they have no conflicts of interest.
References
- 1.Ferguson CM, Miclau T, Hu D, Alpern E, Helms JA. Common molecular pathways in skeletal morphogenesis and repair. Ann NY Acad Sci. 1998;857:33–42. doi: 10.1111/j.1749-6632.1998.tb10105.x. [DOI] [PubMed] [Google Scholar]
- 2.Hadjiargyrou M, Lombardo F, Zhao SC, Ahrens W, Joo J, Ahn H, Jurman M, White DW, Rubin CT. Transcriptional profiling of bone regeneration—Insight into the molecular complexity of wound repair. J Biol Chem. 2002;277:30177–30182. doi: 10.1074/jbc.M203171200. [DOI] [PubMed] [Google Scholar]
- 3.Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA. Fracture healing as a post-natal developmental process: Molecular, spatial, and temporal aspects of its regulation. J Cell Biochem. 2003;88:873–884. doi: 10.1002/jcb.10435. [DOI] [PubMed] [Google Scholar]
- 4.Gersch RP, Lombardo F, McGovern SC, Hadjiargyrou M. Reactivation of Hox gene expression during bone regeneration. J Orthop Res. 2005;23:882–890. doi: 10.1016/j.orthres.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Zhong N, Gersch RP, Hadjiargyrou M. Wnt signaling activation during bone regeneration and the role of Dishevelled in chondrocyte proliferation and differentiation. Bone. 2006;39:5–16. doi: 10.1016/j.bone.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 6.Probst A, Spiegel HU. Cellular mechanisms of bone repair. J Invest Surg. 1997;10:77–86. doi: 10.3109/08941939709032137. [DOI] [PubMed] [Google Scholar]
- 7.Brighton CT, Krebs AG. Oxygen-tension of healing fractures in rabbit. J Bone Joint Surg Am. 1972;A54:323–331. [PubMed] [Google Scholar]
- 8.Brighton CT, Hunt RM. Early histological and ultrastructural-changes in medullary fracture callus. J Bone Joint Surg Am. 1991;73A:832–847. [PubMed] [Google Scholar]
- 9.Einhorn TA. The cell and molecular biology of fracture healing. Clin Orthop. 1998;355:S7–S21. doi: 10.1097/00003086-199810001-00003. [DOI] [PubMed] [Google Scholar]
- 10.Gerber HP, Ferrara N. Angiogenesis and bone growth. Trends Cardiovasc Med. 2000;10:223–228. doi: 10.1016/s1050-1738(00)00074-8. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL. Hypoxia-inducible factor 1: Master regulator of O-2 homeostasis. Curr Opin Genet Dev. 1998;8:588–594. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- 12.Steinbrech DS, Mehrara BJ, Saadeh PB, Chin G, Dudziak ME, Gerrets RP, Gittes GK, Longaker MT. Hypoxia regulates VEGF expression and cellular proliferation by osteoblasts in vitro. Plast Reconstr Surg. 1999;104:738–747. doi: 10.1097/00006534-199909030-00019. [DOI] [PubMed] [Google Scholar]
- 13.Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1 alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfander D, Cramer T, Schipani E, Johnson RS. HIF-1 alpha controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci. 2003;116:1819–1826. doi: 10.1242/jcs.00385. [DOI] [PubMed] [Google Scholar]
- 15.Cramer T, Schipani E, Johnson RS, Swoboda B, Pfander D. Expression of VEGF isoforms by epiphyseal chondrocytes during low-oxygen tension is HIF-1 alpha dependent. Osteoarthritis Cartilage. 2004;12:433–439. doi: 10.1016/j.joca.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Komatsu DE, Hadjiargyrou M. Activation of the transcription factor HIF-1 and its target genes, VEGF, HO-1, iNOS, during fracture repair. Bone. 2004;34:680–688. doi: 10.1016/j.bone.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 17.Bonnarens F, Einhorn TA. Production of a standard closed fracture in laboratory animal bone. J Orthop Res. 1984;2:97–101. doi: 10.1002/jor.1100020115. [DOI] [PubMed] [Google Scholar]
- 18.Lombardo F, Komatsu D, Hadjiargyrou M. Molecular cloning and characterization of Mustang, a novel nuclear protein expressed during skeletal development and regeneration. FASEB J. 2004;18:52–61. doi: 10.1096/fj.03-0521com. [DOI] [PubMed] [Google Scholar]
- 19.Garcia A, Cayla X, Guergnon J, Dessauge F, Hospital V, Rebollo MP, Fleischer A, Rebollo A. Serine/threonine protein phosphatases PP1 and PP2A are key players in apoptosis. Biochimie. 2003;85:721–726. doi: 10.1016/j.biochi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Van Hoof C, Goris J. Phosphatases in apoptosis: To be or not to be, PP2A is in the heart of the question. Biochim Biophys Acta. 2003;1640:97–104. doi: 10.1016/s0167-4889(03)00029-6. [DOI] [PubMed] [Google Scholar]
- 21.Basu A, Haldar S. The relationship between BcI2, Bax and p53: Consequences for cell cycle progression and cell death. Mol Hum Reprod. 1998;4:1099–1109. doi: 10.1093/molehr/4.12.1099. [DOI] [PubMed] [Google Scholar]
- 22.Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, Andrews D, Knudson CM, Yang E. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003;22:5459–5470. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1α-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol. 1999;209:254–267. doi: 10.1006/dbio.1999.9253. [DOI] [PubMed] [Google Scholar]
- 25.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimoda LA, Manalo DJ, Sham JS, Semenza GL, Sylvester JT. Partial HIF-1alpha deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L202–L208. doi: 10.1152/ajplung.2001.281.1.L202. [DOI] [PubMed] [Google Scholar]
- 27.Kline DD, Peng YJ, Manalo DJ, Semenza GL, Prabhakar NR. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. Proc Natl Acad Sci USA. 2002;99:821–826. doi: 10.1073/pnas.022634199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation. 2003;108:79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- 29.Elson DA, Thurston G, Huang LE, Ginzinger DG, McDonald DM, Johnson RS, Arbeit JM. Induction of hypervascularity without leakage or inflammation in transgenic mice over-expressing hypoxia-inducible factor-1alpha. Genes Dev. 2001;15:2520–2532. doi: 10.1101/gad.914801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shyu KG, Wang MT, Wang BW, Chang CC, Leu JG, Kuan P, Chang H. Intramyocardial injection of naked DNA encoding HIF-1alpha/VP16 hybrid to enhance angiogenesis in an acute myocardial infarction model in the rat. Cardiovasc Res. 2002;54:576–583. doi: 10.1016/s0008-6363(02)00259-6. [DOI] [PubMed] [Google Scholar]
- 31.Mabjeesh J, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 32.Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, Rowan A, Yan Z, Campochiaro PA, Semenza GL. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- 33.Acker T, Plate KH. Hypoxia and hypoxia inducible factors (HIF) as important regulators of tumor physiology. Cancer Treat Res. 2004;117:219–248. doi: 10.1007/978-1-4419-8871-3_14. [DOI] [PubMed] [Google Scholar]
- 34.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 35.Patel TH, Kimura H, Weiss CR, Semenza GL, Hofmann LV. Constitutively active HIF-1alpha improves perfusion and arterial remodeling in an endovascular model of limb ischemia. Cardiovasc Res. 2005;68:144–154. doi: 10.1016/j.cardiores.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Glowacki J. Angiogenesis in fracture repair. Clin Orthop. 1998;355:S82–S89. doi: 10.1097/00003086-199810001-00010. [DOI] [PubMed] [Google Scholar]
- 37.Street J, Bao M, deGuzman L, Bunting S, Peale FV, Jr, Ferrara N, Steinmetz H, Hoeffel J, Cleland JL, Daugherty A, van Bruggen N, Redmond HP, Carano RA, Filvaroff E. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc Natl Acad Sci USA. 2002;99:9656–9661. doi: 10.1073/pnas.152324099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarkka T, Sipola A, Jamsa T, Soini Y, Yla-Herttuala S, Tuukkanen J, Hautala T. Adenoviral VEGF-A gene transfer induces angiogenesis and promotes bone formation in healing osseous tissues. J Gene Med. 2003;5:560–566. doi: 10.1002/jgm.392. [DOI] [PubMed] [Google Scholar]
- 39.Carvalho RS, Einhorn TA, Lehmann W, Edgar C, Al-Yamani A, Apazidis A, Pacicca D, Clemens TL, Gerstenfeld LC. The role of angiogenesis in a murine tibial model of distraction osteogenesis. Bone. 2004;34:849–861. doi: 10.1016/j.bone.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 40.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 41.Hausman MR, Schaffler MB, Majeska RJ. Prevention of fracture healing in rats by an inhibitor of angiogenesis. Bone. 2001;29:560–564. doi: 10.1016/s8756-3282(01)00608-1. [DOI] [PubMed] [Google Scholar]
- 42.Piret JP, Mottet D, Raes M, Michiels C. Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem Pharmacol. 2002;64:889–892. doi: 10.1016/s0006-2952(02)01155-3. [DOI] [PubMed] [Google Scholar]
- 43.Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol. 2004;57:1009–1014. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett. 2006;237:10–21. doi: 10.1016/j.canlet.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 45.Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S, Johnson RS, Lipton SA, Barlow C. Brain-specific knock-out of hypoxia-inducible factor-1alpha reduces rather than increases hypoxic-ischemic damage. J Neurosci. 2005;25:4099–4107. doi: 10.1523/JNEUROSCI.4555-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landry P, Sadasivan K, Marino A, Albright J. Apoptosis is coordinately regulated with osteoblast formation during bone healing. Tissue Cell. 1997;29:413–419. doi: 10.1016/s0040-8166(97)80027-4. [DOI] [PubMed] [Google Scholar]
- 47.Li G, White G, Connolly C, Marsh D. Cell proliferation and apoptosis during fracture healing. J Bone Miner Res. 2002;17:791–799. doi: 10.1359/jbmr.2002.17.5.791. [DOI] [PubMed] [Google Scholar]
- 48.Ford JL, Robinson DE, Scammell BE. The fate of soft callus chondrocytes during long bone fracture repair. J Orthop Res. 2003;21:54–61. doi: 10.1016/S0736-0266(02)00087-6. [DOI] [PubMed] [Google Scholar]
- 49.Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Tsay A, Fitch J, Barnes GL, Graves DT, Einhorn TA. Impaired fracture healing in the absence of TNF-alpha signaling: The role of TNF-alpha in endochondral cartilage resorption. J Bone Miner Res. 2003;18:1584–1592. doi: 10.1359/jbmr.2003.18.9.1584. [DOI] [PubMed] [Google Scholar]
- 50.Olmedo ML, Landry PS, Sadasivan KK, Albright JA, Meek WD, Routh R, Marino AA. Regulation of osteoblast levels during bone healing. J Orthop Trauma. 1999;13:356–362. doi: 10.1097/00005131-199906000-00006. [DOI] [PubMed] [Google Scholar]