

Abstract
We present here an optimized and cost-effective approach to saturation fluorescence labeling of protein thiols for proteomic analysis. We investigated a number of conditions and reagent concentrations including a disulfide reducing agent (TCEP), pH, incubation time, linearity of labeling, and saturating dye: protein thiol ratio with protein standards to gauge specific and non-specific labeling. Efficacy of labeling under these conditions was quantified using specific fluorescence estimation, defined as the ratio of fluorescence pixel intensities and Coomassie-stained pixel intensities of bands after digital imaging. Factors leading to specific vs. non-specific labeling in the presence of thiourea are also discussed.
We have found that reproducible saturation of available Cys residues of the proteins used as labeling standards (human carbonic anhydrase I, enolase, α-lactalbumin) is achieved at 50-100-fold excess of the uncharged maleimide-functionalized BODIPY™ dyes over Cys. We confirm our previous findings and those of others that the maleimide dyes are not impacted by the presence of 2M thiourea. Moreover, we establish that 2 mM TCEP used as reductant is optimal. We also establish further that labeling is optimal at pH 7.5 and complete after 30 min. Low non-specific labeling was gauged by the inclusion of non-Cys containing proteins (horse myoglobin, bovine carbonic anhydrase) to the labeling mixture. We also show that the dye exhibits little to no effect on the two dimensional mobilities of labeled proteins derived from cells.
Keywords: saturation fluorescence labeling, cysteine-labeling, proteomics, two-dimensional electrophoresis
1 Introduction
One of the many criticisms leveled against two dimensional gel electrophoresis (2DGE) is that the staining techniques used to visualize separated proteins are semi-quantitative, exhibit a small dynamic range and low sensitivity, and are not very reproducible (for recent reviews, see [References 1; 2; 3]. This may be due in part to the variation in protein reactivity with non-covalent dyes, protein precipitation at their isoelectric points (pI), incomplete solubilization by SDS, variability of gel composition and quality, and other factors. Quantitative standards are of little use because of the uniqueness of individual proteins with respect to dye reactivity and other, difficult to control factors. With the introduction of fluorescent dyes and high quality imagers, post-separation staining has improved in terms of sensitivity and dynamic range, but high coefficients of variation in abundance estimates remain a problem, even with these dyes [4].
Nevertheless, 2DGE remains the “gold standard” for differential proteomic analyses primarily due to its exquisite resolving power and its ability to efficiently compare protein abundances from multiple sources. It has been our long-term goal to address some of the difficulties in 2DGE, and accurate quantification is, in our view, one of the most important.
One promising approach applies fundamental biochemical principles, namely, pre-separation covalent labeling of proteins for proteomic analysis. This approach has garnered attention from the proteomics community because it promises accurate quantification, multiplexed separation and detection, and high sensitivity. Covalent labeling techniques generally fall into two categories: isotope (stable and radio) and fluorescence labeling. Stable Isotope labeling schemes (e.g., Stable Isotope Labeling of Amino Acids in Culture; SILAC [5; 6]; iTRAQ® [7]) exploit the exquisite resolution obtainable by even comparatively low-cost mass spectrometers (MS) to the degree that labels differing by one atomic mass unit (amu) are easily discriminated. Thus to compare two protein populations, each mixture can be covalently labeled with a reagent that differs by one amu from the label on the other mixture. The two populations can then be mixed and their detection multiplexed in order to obtain relative quantification of parental proteins. The advantage to this approach is that the stable isotopes permit ratiometric normalization of protein (peptide) recoveries to minimize the impact of artifactual loss. The difficulty in these approaches is the requirement of most mass spectrometers for low mass: charge ratios of analytes, hence the need to digest proteins to their constituent peptides. Moreover the increased complexity this imparts to the protein extracts dictates pre-MS separation strategies, leading to potential loss of sample. This is troublesome with respect to proteins that are in low abundance: artifactual loss may decrease their signals below the detection limits, and the stable isotopes do not, in and of themselves, increase MS sensitivity. In addition, relative quantification is performed at the analytical stage (MS), rather than the separation stage, thereby dictating that every peptide that is generated from the extract must be analyzed, regardless of its constitutive or inducible status. Thus there is no opportunity for a differential cut and even uninformative proteins must be analyzed. Finally, for quantification to work, the cognate peptides from the different sources must co-elute from the fractionation step and appear in the same MS scan; otherwise the opportunity for quantification is lost. Failure of cognate peptides to co-elute might occur, for example, when proteins undergo post-translational modification(s) as a result of the differential treatment of the cell that alters chromatographic or MS behavior. In this case, cognate peptides will not appear in the same MS scan, and therefore, will not be quantified.
The alternative strategy, covalent derivatization of protein samples with fluorescent dyes prior to separation by two dimensional electrophoresis (2DGE) is a technique finding increasing utility in proteomics (e.g. Differential In-Gel Electrophoresis; DIGE [8-10], or saturation labeling [8; 11]) due to its potential for quantitative accuracy and sensitivity. The advantages of this approach include the opportunity for multiplexing samples and inclusion of internal standards, the potential for detecting low abundance proteins, the ability to realize relative and absolute linear quantification (once the protein is identified), and the potential for real-time monitoring of electrophoresis. In contrast to the stable isotope strategies, quantification is performed after separation, but before MS identification. This means that the sample complexity is reduced before the identification phase, since only those proteins whose abundance levels are of interest need be analyzed by the MS instruments for the purpose of identification.
These advantages come with a price, however. Quantification requires reproducible saturation of targeted sites on any protein as well as absence of non-specific modifications. Detection of low abundance proteins requires instrumentation that exhibits low noise and low background, as well as the ability to prolong exposure times. In our view, the advantages outweigh the disadvantages, and with attention to the chemical as well as instrument demands, pre-separation labeling with fluorescence can contribute greatly to the needs of the proteomics community, and most importantly, permit accurate and reproducible detection of low-abundance proteins.
We and others [11-13] have previously examined conditions for labeling proteins with thiol-reactive dyes, and defined the basic properties these dyes must exhibit in order to function as viable candidates; these include high extinction coefficients, good quantum yields, pH insensitivity of fluorescence, low photo-bleaching, minimal effects on protein pIs, a high degree of chemical specificity, and the ability to achieve reproducible saturation of protein residues. In our study [11], we noted that thiourea, an important component of many protein solubilization cocktails, severely inhibited labeling of protein thiols by iodoacetamide dyes, but not by a maleimide dye, in agreement with others who observed similar reactivities [14]. For pre-separation fluorescence labeling to find widespread use in proteomics as a quantitative tool, its reactivity in varied chemical environments must be systematically studied. For this reason, and in view of the widespread use of thiourea in proteomic sample preparation, we continued our investigations into the use of fluorescence dyes for proteomic applications and present here optimal conditions for labeling protein thiol groups with a maleimide dye in the presence of thiourea, such that saturation labeling of thiols and minimal non-specific labeling occur.
2 Materials and Methods
2.1 Materials
Yeast enolase I (enol), bovine carbonic anhydrase II pI 5.4 (bCA), human carbonic anhydrase I (hCA), bovine alpha-lactalbumin A (α-lac), horse myoglobin (myo), Trishydroxymethyl aminomethane (Tris), sodium bicarbonate, sodium dodecyl sulfate (SDS), 2-mercaptoethanol (2-ME), dimethylsulfoxide (DMSO), thiourea, individual fluorescent molecular weight markers, Triton X-100, and Brilliant Blue G Colloidal Concentrate (colloidal Coomassie Blue) were from Sigma-Aldrich (St. Louis, MO). Urea was from Amersham Biotech (Piscataway, NJ). BODIPY® FL-N-(2-aminoethyl) maleimide (BODIPY FL-Mal) and BODIPY® FL C1-Iodoacetamide (BODIPY FL-Iaa) were from Molecular Probes (Eugene, OR). 3[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) was from Calbiochem (San Diego, CA) and Anatrace (Maumee, OH). Tris(2-carboxyethyl)phosphine (TCEP) was from Pierce (Rockford, IL). PD-10 columns (pre-packed Sephadex G 25) were from Amersham (Uppsala, Sweden; Piscataway, NJ). Precast NOVEX Tris-Gly and bis-Tris (NuPAGE) gels as well as NuPAGE MES running buffer concentrate and antioxidant were from Invitrogen (Carlsbad, CA). Perfect Protein Markers 10-225 KD was from Novagen (San Diego, CA). Fluorescence images were obtained on an AlphaImager (Alpha Innotech, San Leandro, CA) or ProXpress 2D (Perkin-Elmer, Waltham, MA). Stained gels were scanned on a Molecular Dynamics Personal Densitometer (Amersham, Piscataway, NJ). Labeling reactions were incubated on a Thermolyne Labquake rotator (Barnstead International, Dubuque, IA).
2.2 E. Coli Extraction
BL21 E. coli was put through a French press with 40mM Tris and centrifuged at 25,000 x g for 1 hr. The supernatant was collected and treated with 150U/ml Benzonase for 30 min at room temperature. The protein was precipitated overnight with acetone and pelleted at 25,000 x g for 1 hr. Five micrograms were removed and analyzed for protein and cysteine (Cys) content by amino acid analysis (Hitachi Model L-8800, Hitachi High Technologies America, Inc., San Jose, CA). The pellet was resuspended in 8M Urea + 1% CHAPS.
2.3 Protein Labeling
In general, proteins (12.5-50 μM) were denatured with 7M urea/2M thiourea or with 8M urea alone, along with 0.1% Triton X-100 or reduced Triton X-100, with 2% CHAPS, in 50 mM Tris-HCl, and reduced with TCEP for 20-30 min. In some cases 0.5 mM EDTA was added during the last 10 min of reduction. Dye (50 mM in DMSO) was added to the desired concentration, and the mixtures were incubated for up to 1.5 h. Unlabeled controls received DMSO in place of dye. The reactions were stopped with a 150 to 300-fold molar excess of 2-ME over dye and acidified with 3 μl 1N HCl per 100 μl reaction mix to further prevent undesired alkylation. All incubations were carried out at room temperature, in the dark. Typical reaction volume was 100 μl.
2.3 PD-10 Chromatography
In some cases the crude labeling mixes were applied to PD-10 columns and eluted with 0.1M sodium bicarbonate, pH 8.3. Following 2.0 ml of void volume, typically 0.5 ml or smaller fractions were collected. Peaks were located by fluorescence intensity and Coomassie Blue stain intensity on SDS NuPAGE gels, and the peak fractions were pooled.
2.4 Electrophoresis
Reduced SDS-PAGE in 1.5 mm, 14 or 16% Tris-Gly gels, or in bis-Tris NuPAGE gels with MES running buffer, was carried out according to the manufacturer’s instructions. Samples were generally diluted in half with sample buffer containing SDS, glycerol, and Tris. Because crude samples contained large amounts of 2-ME, additional 2-ME was not usually added to the SDS-PAGE sample buffer, except when running PD-10 eluates. Fronts were sometimes run off the gels in order to remove unreacted dye and obtain cleaner fluorescence images. Fluorescence images were obtained on an AlphaImager or Perkin-Elmer ProXpress 2D. Gels were then stained with colloidal Coomassie Blue, destained, and imaged on the Personal Densitometer.
Two dimensional electrophoresis was performed employing GE Healthcare IPGphor multiple sample IEF device for the first dimension, and Bio-Rad’s multiple gel SDS-PAGE systems (Protean Plus and Criterion Dodeca cells) for the second. Isoelectric focusing (IEF) was performed with 11 cm precast IPG strips (Bio-Rad). Sample aliquots were loaded onto dehydrated immobilized pH gradient (IPG) strips, and rehydrated overnight. IEF was performed at 20°C with the following parameters: 50 Volts (V), 11 h; 250 V, 1 h; 500 V, 1 h; 1000 V, 1 h; 8000 V, 2 h; 8000 V, 6 h. The IPG strips were then incubated in 4 ml of Equilibration Buffer (6 M urea, 2% SDS, 50 mM Tris-HCl, pH 8.8, 20% glycerol) containing 10 μl/ml tri-2 (2-carboxyethyl) phosphine (Geno Technology, Inc., St. Louis, MO) for 15 min at 22°C with shaking. The unlabeled samples were incubated in another 4 ml of Equilibration Buffer with 25 mg/ml iodoacetamide for 15 min at 22°C with shaking in order to ensure protein S-alkylation. Electrophoresis was performed at 150 V for 2.25 h, 4°C with precast 8-16% polyacrylamide gels in Tris-glycine buffer (25 mM Tris-HCl, 192 mM glycine, 0.1% SDS, pH 8.3).
After electrophoresis, the gels were fixed in Fix Buffer (10% methanol, 7% acetic acid in ddH20), stained with SYPRO-Ruby (Bio-Rad, Hercules, CA), and destained in Fix Buffer. The gels were scanned at a 100 μm resolution with the Perkin-Elmer ProXPRESS 2D™ Proteomic Imaging System (Boston, MA) using 460 nm excitation and 620 nm emission filters for SYPRO Ruby stained gels, or 480 nm excitation and 530 emission filters for BODIPY-labeled protein gels. The exposure time is adjusted to achieve a value of ∼55,000-63,000 pixel intensity (16-bit) on the most intense protein spots on the gel.
2.5 Image Processing and Analysis
Images acquired from the AlphaIMager were stored as “tif” files with no compression. The Coomassie stain and corresponding fluorescence images were first cropped to remove extraneous information and their image sizes (pixel dimensions) equalized with Photoshop (Adobe, San Jose CA). No other manipulations of the images were performed with Photoshop software. The files were then imported into IPLab (Scanalytics, Tysons Corner VA) where a region of interest (ROI) of equal size was defined for each band. Identical ROIs immediately in the vicinity of each band (containing no protein) were also defined and used for subsequent background correction. All ROIs were quantified for intensity and backgrounds corrected by subtraction. Specific fluorescence (SF) is defined as the ratio of background (Bf) -corrected pixel intensity sum across the ROI of the fluorescence band (Pf) and the background (Bc) -corrected pixel intensity sum across the corresponding ROI of the Coomassie stained band (Pc) according to the following formula:
In some cases, as noted in the figure legends, SF of the Cys-containing protein is calculated by subtraction of the SF of the non-Cys containing protein calculated in the same way.
2.6 Linearity of Labeling
Equimolar mixtures of enolase and myoglobin, ranging from 1.0 to 10,000 fmol (40 pM to 400 nM) were reduced and denatured with 8M urea, 0.1% TX-100, 50mM Tris pH 7.5, and 2 mM TCEP for 30 min in 25 μl. The proteins were labeled in duplicate with 750 pmol (30 μM; 75-fold excess over 10 pmol protein thiol) of BODIPY FL-Mal for 2 hr followed by quenching with 5 mM 2-ME for 30 min. All incubations were done at room temp. Reactions were frozen at -80°C overnight, and the proteins were run on duplicate 4-20% SDS gels. The gels were imaged on a Perkin-Elmer ProXpress 2D with 480/30 nm excitation and 535/50 nm emission filters for 60 sec. The fluorescent bands were quantified as described above for Image Processing and Analysis, and linear regression was performed on the duplicate intensities using Prism 4.0c (GraphPad, San Diego, CA).
3 Results
3.1 Labeling in thiourea with iodoacetamides vs. maleimides
This study focuses on pre-separation labeling with the dye, BODIPY FL-Mal. We selected it because we had previously shown that the BODIPY family of dyes meets our criteria for a good proteomics fluorescence dye [11]. The structures of BODIPY FL-Iaa and BODIPY FL-Mal are shown in Figure 1.
Figure 1. Dye Structures.

Upper: N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza--s-indacene-3-yl)methyl)iodoacetamide (BODIPY FL-Iaa). Lower: BODIPY®FL-N-(2-aminoethyl)maleimide (BODIPY FL-Mal). Structures courtesy of Molecular Probes, Eugene OR.
We previously showed strong inhibition of labeling with BODIPY TMR cadaverine Iaa in the presence of thiourea [11], corroborating the findings of Galvani et al. [14], that thiourea inhibits alkylation of thiol groups with iodoacetamide, and we found that Rhodamine Red C2 maleimide, as well as BODIPY FL-Mal, labeled efficiently in the presence of thiourea. To rule out the possibility that those differences were dye-specific, in this study we compared the maleimide and iodoacetamide forms of a single dye. Labeling was done at two pHs, to address the possible different pH requirements for optimal labeling with iodoacetamides vs. maleimides. Throughout this work, we include an internal protein standard containing no Cys to gauge the levels of non-specific labeling, and we show the Coomassie-stained protein as well as fluorescence images. In some instances, where disulfide reducing power is of issue, we use α-lac (containing 8 thiols). The advantage of this protein is that its incompletely labeled forms are observable as bands migrating between unlabeled and fully labeled species, thereby permitting direct visualization of reaction completeness.
We first determined the impact of labeling a mixture of enol (containing 1 thiol) and α-lac with BODIPY FL-Iaa or with BODIPY FL-Mal in 8M urea or in 7M urea/2M thiourea at pH 7.2 or 8.0 (Figure 2). The dye: thiol ratio in this experiment was 10:1 (2.25 mM dye: 225 μM thiol). Reduction was with 2 mM TCEP, giving a 0.88:1 reducer: dye ratio. The maleimide dye labeled efficiently in both chaotrope systems and at both pHs, as shown by the bright fluorescence of the bands and by the apparent molecular size shift of α-lac. In contrast, the iodoacetamide dye labeled well in urea, but was severely inhibited in thiourea, with virtually no labeling at pH 7.2 and very little at pH 8. These results confirm that the iodoacetamide but not the maleimide sulfhydryl reactivity is negatively affected by the presence of thiourea, and that this effect is not due to the structure of the dye molecule.
Figure 2. BODIPY FL-Iaa and -Mal Labeling In The Presence of Thiourea and/or Urea.
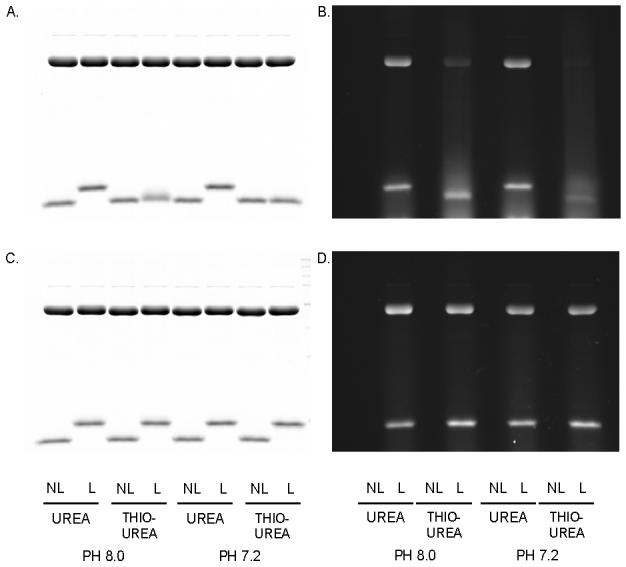
Enolase and α-lactalbumin (upper and lower protein bands, respectively) were labeled at two different pHs. A. BODIPY FL-Iaa: Coomassie; B. BODIPY FL-Iaa: fluorescence; C. BODIPY FL-Mal: Coomassie; D. BODIPY FL-Mal: fluorescence. Details are described in Materials and Methods. NL: proteins Not Labeled; L: proteins Labeled
3.2 Effect of TCEP concentration on completeness and specificity of labeling
Clearly, maximum reduction of protein thiols is critical to achieve reproducible labeling. The concentration of reducing agent must therefore be optimized without using an excessive amount that might lead to inhibition of labeling. Our previous results suggested interaction of the reducing agent, TCEP, with iodoacetamide and maleimide dyes, and the importance of the TCEP: dye ratio for complete and specific labeling [11]. To more systematically gauge this effect, we used α-lac, for its eight reducible Cys. Using this protein and bCA (MW 29,000, 0 Cys) as an internal non-specific labeling control, we examined the effect of TCEP concentration on labeling with a constant dye concentration in the presence of thiourea.
α-Lac was labeled at a concentration of 25 μM in the presence of 12.5 μM bCA in 7M urea/2M thiourea, at pH 7.2. The difference in protein amounts reflects the difference in the number of potentially reactive amines in each protein that might engage in side-reactions with maleimides [15]. Reduction was carried out for 40 min using 0, 0.5, 1, 2, 3, 4, 5, or 10 mM TCEP, followed by labeling with 2 mM BODIPY FL-Mal for 1.5 hour (dye: thiol ratio 10:1). The reducing agent: thiol ratio varied from 2.5: 1 to 50: 1, and the reducing agent: dye ratio varied from 0.25: 1 to 5: 1. The results are shown in Figure 3.
Figure 3. Effect of TCEP on BODIPY-FL-Mal Labeling.

A. Coomassie stain. Bovine carbonic anhydrase (upper band) and α-lactalbumin (lower band) simultaneously labeled with BODIPY-FL-Mal, each at a dye: thiol ratio of 10:1, in the presence of increasing concentrations of TCEP. B. Fluorescence image of A. C. Ratio (Specific Fluorescence) of pixel sum of α-lactalbumin fluorescence and Coomassie intensity (from A and B) with respect to TCEP concentration. Both fluorescence and Coomassie signals were corrected for background by subtraction. Details are described in Materials and Methods.
At 0.5 and 1 mM TCEP (TCEP: dye ratios of 0.25 and 0.5), α-lac displays multiple bands at intermediate positions between the unlabeled and the fully labeled bands on the SDS gel, suggesting incomplete modification. Maximum labeling appears at 2-3 mM TCEP (Figure 3A-C). Very long exposures reveal low labeling (<5%) of bovine carbonic anhydrase regardless of TCEP concentration (quantitative data not shown). Interestingly labeling of α-lac at 10 mM TCEP (TCEP: dye 5:1) seems to result in an electrophoretic behavior similar to that observed at 1 mM TCEP (Figure 3A), suggesting that interference with labeling occurs at a five-fold or greater excess of TCEP over dye.
3.3 Effect of dye concentration on completeness and specificity of labeling
Having found the maleimide-compatible range of TCEP to dye ratios, we investigated the optimal ratio of dye to protein thiol. HCA (MW 29,000), containing a single Cys residue, was chosen for this purpose. Equine myo (MW 17,000, no Cys) was included as a negative control to detect non-specific labeling.
Figure 4 shows labeling of hCA at 25 μM in the presence of 43 μM myo and 2.5 mM TCEP, with the dye concentration varying from 0.125 - 5 mM (dye: thiol 0-200:1). From the summary graph (Figure 4C), hCA appears to saturate readily at dye: thiol of 20:1 (0.5 mM dye) and remain at this level through dye: thiol ratio of 200:1 (5 mM dye) at 2.5 mM TCEP. Non-specific labeling appears in significant levels at dye: thiol ratios greater than 100:1 (2.5 mM dye).
Figure 4. BODIPIY FL-Mal Saturation.

Increasing concentrations of dye were incubated with a mixture of human carbonic anhydrase (hCA; upper band) and myoglobin (myo; lower bands) in A and B in 2.5 mM TCEP. A. Coomassie stained; B. Fluorescence. C. Specific Fluorescence (SF). SF is expressed as the ratio of the sum of pixel intensities of each hCA fluorescence band and the corresponding hCA Coomassie band minus the SF of myo. Both intensities were background corrected. Abscissa represents dye: protein molar ratio for myo (0 Cys) and dye: protein thiol for hCA (1 Cys). Further details are described in Materials and Methods.
Comparing these results with Figure 3, where the dye: thiol ratio was 10: 1 and the best results were obtained with 2 mM TCEP concentration (dye concentration was also 2 mM), the results in Figure 4 indicate saturation at 10-fold or greater excess dye when TCEP concentration was 2.5 mM (Figure 4C). Higher dye: thiol ratios are well tolerated and may approach 100-fold with little to no non-specific labeling. Both figures suggest a fine degree of interplay between TCEP, dye, and protein thiols, with a window of optimal concentrations. In light of these data, we routinely use 2 mM TCEP for reduction and 50- 100-fold dye: thiol ratios for labeling complex mixtures of proteins.
3.5 Effect of pH and reaction time on completeness and specificity of labeling
To measure completeness of alkylation, α-lac was labeled at 26.8 μM in the presence of 13.4 μM bCA as negative control. TCEP and dye were each 2 mM, while the dye: thiol ratio was 9.3: 1. The reactions were buffered at pH 6.5, 7.0, 7.2, 7.5, or 8.0 (Figure 5).
Figure 5. pH Dependence of BODIPY FL-Mal Labeling.
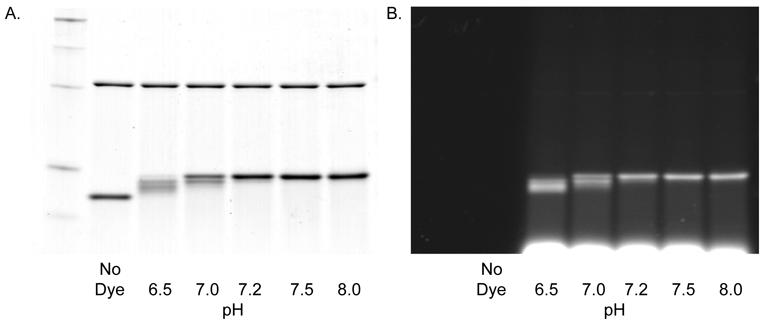
Bovine carbonic anhydrase (bCA, upper bands) and α-lactalbumin (lower bands) were mixed and labeled at the indicated pHs with 2mM TCEP and 1mM dye (dye:thiol ratio was 9:1). A. Coomassie stain; B. Fluorescence image. Details are described in Materials and Methods.
Labeling appeared complete at pH 7.2 and above. There was a trace of fluorescence in the bCA band at pH 8.0 (very long exposure on the AlphaImager showed trace labeling at pH 8, and possibly at pH 7.5). Thus higher pH seems more efficient, but also resulted in some non-specific labeling. These results show that pH greater than 7.2 but less than 8.0 is best for efficient labeling, with little to no non-specific labeling.
Since non-specific reactivity is a concern with all of the maleimide dyes we have examined, we investigated the possibility that a shorter reaction time would minimize side reactions while still allowing complete labeling of thiols. The 1.5 hour reaction time used in experiments up to this point was based on our experience with iodoacetamide dyes.
α-lac was labeled (dye: thiol ratio 9.5:1) at 25 μM in the presence of 12 μM bovine carbonic anhydrase as negative control for 30, 60, and 90 min in 2 mM TCEP. We performed the labeling at pH 8.0 since we established in Figure 5 that only at this pH was non-specific labeling detectable.
Figure 6 shows minimal non-specific labeling at all three time points, even after gross overexposure (data not shown). Moreover, the figure shows that complete labeling under these conditions is achieved within 30 min, with no further increase in specific fluorescence, greatly improving the throughput of the procedure.
Figure 6. Time Course Labeling with BODIPY FL-Mal.
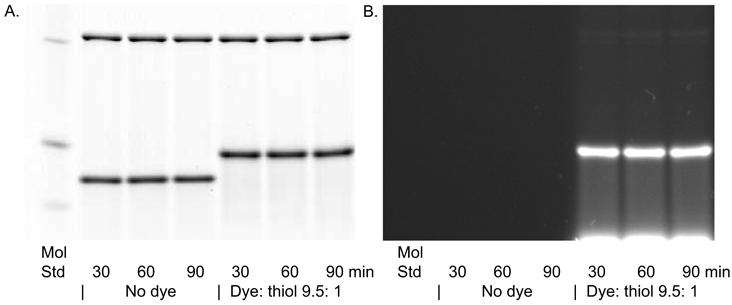
Bovine carbonic anhydrase (upper bands) and α-lactalbumin (lower bands) were mixed and labeled for the indicated times with 2 mM TCEP, dye:thiol ratio of 9.5:1, and at pH 8.0. A. Coomassie stain; B. Fluorescence image. Details are described in Materials and Methods.
3.6 Linearity of Labeling
Finally, using the optimized conditions described above, we labeled increasing amounts of equimolar enol and myo, from 1 fmol to 10 pmol (40 pM to 400 nM, respectively), with a constant 30 μM BODIPY FL-Mal (75-fold excess over 400 nM enol) in duplicate. Labeled mixtures were run on duplicate SDS gels without further processing, imaged, and quantified as described in the Methods. Figure 7 demonstrates linear labeling over at least 3-orders of magnitude of concentration. Linear regression of duplicates yielded a correlation coefficient of 0.9952, with a p-value of 1.000 for the residual Runs test (maximum significance for linear fit). The coefficient of variation of the replicates ranged from 1.9 to 9.4% ([Labeled Enol] = 25 to 10000 fmol), and 34% for 10 fmol. No significant labeling of myo was observed at any protein level (<1%).
Figure 7. Linearity of Labeling with BODIPIY FL-Mal and Enolase.

Increasing amounts of equimolar enolase and myoglobin (1-10,000 fmol) were incubated with 30 μM BODIPY FL-Mal (75-fold excess over 10,000 fmol enolase) in duplicate under the conditions described in Materials and Methods. Fluorescence intensities of duplicate SDS gels were measured in a Perkin-Elmer ProXpress 2D imager and intensities were background corrected. Linear regression was calculated with Prism 4.0c for Mac OS X. The correlation coefficient for the fitted line was 0.9951, and the p-value of the Runs Test of residuals was 1.000, indicating maximum linearity. Error bars represent SEM.
3.7 Impact on 2DGE Protein Mobility
To examine the impact of BODIPY FL-Mal modification of a complex protein extract, we performed the 2DGE separation of an E. coli extract with and without labeling (Figure 8). The gel containing unlabeled proteins was stained post-separation with Sypro Ruby and the protein spots matched against the BODIPY-labeled protein spots using Nonlinear Dynamics Progenesis software. The vectors defining the matched protein spots were obtained from the spot coordinates. Figure 8 (Upper) was generated by superimposition of the vectors on the gel containing the BODIPY-labeled proteins. The length and angle of the vectors summarize the relative difference in mobility between the BODIPY and the unlabeled proteins. Of the 1263 vectors describing proteins matched by the software, the average length was 6.6 ± 4.1 pixels. To place this in perspective, Figure 8 inset indicates the diameter of a circle containing the average vector length (Inset A), the 90% confidence diameter (Inset B.— 1 SD), and the 95% confidence diameter (Inset C. — 2 SD). The regions containing the greatest protein spot densities from both gels were cropped and only contrast and brightness adjusted with Photoshop to match and included in Figure 8 (Lower, Left and Right). These are included to demonstrate the similarities in spot numbers and migration. Protein spot intensities, however, do not necessarily match since the BODIPY labels reflect the number of cysteines per protein, whereas the Sypro Ruby intensities reflect efficiency of dye to protein binding.
Figure 8. Impact of BODIPY FL-Mal Labeling on Protein 2DGE Mobility.

UPPER: E.coli extracts were split into two fractions: one was labeled with BODIPY FL, and the other alkylated with iodoacetamide. Both were run on separate 2D gels as described in the text. The BODIPY-labeled proteins were imaged on a ProExpress 2D imager, and the unlabeled protein gel stained with Sypro Ruby and likewise imaged. The unmodified tif images were matched using Nonlinear Dynamics Progenesis with no operator modification. The match vectors (white segments) were obtained from the software and the image containing the match vectors overlaid on the BODIPY-labeled protein gel. Inset: A. Circle with average vector diameter (6.6 ± 4.1 pixels in original image); B. 90% confidence interval (10.7 pixels; 1 SD); C. 95% confidence interval (14.8 pixels; 2 SD). LOWER: Unprocessed images of gels used in UPPER. (Left) BODIPY FL-Mal labeled proteins; (Right) Sypro Ruby stained gel. Contrast, brightness, and pixel density of all gel images were adjusted with Photoshop only for publication, after image analysis.
3.8 Impact on Protein Solubility During 2DGE
An important question arises regarding the solubility of BODIPY-labeled proteins due to the increase in overall protein hydrophobicity by the modification. This is of particular concern when IEF is complete, when the local protein concentration is at its maximum and may result in precipitation and failure to transfer to the 2nd dimension gel. We exploited the protein fluorescence by performing IEF of a complex protein mixture (extracted from eol-1 cells) labeled with BODIPY FL-Mal and run in a standard IEF gel (not an IPG strip). The gel was imaged, a lane cut out, and the proteins run into a 2nd dimension SDS gel in typical fashion. After electrophoresis, the IEF region was re-imaged to look for retained proteins. No difference was observed between the BODIPY-labeled and the unlabeled extracts stained with Sypro Ruby in either the 1st dimension IEF gel or the 2nd dimension SDS gel (data not shown).
4 Discussion
Covalent labeling of protein thiols with fluorescent dyes is an attractive strategy for proteomic analyses for a number of reasons. If performed with saturating ratios of dye to thiols (>50 to 1), it is most likely to result in accurate quantification and reproducible separations. Because most proteomic procedures perform sulfhydryl alkylation to prevent artifactual oxidation, our approach accomplishes this as well, just with a fluorescent dye. This leads to a separation pattern that very closely (if not identically) matches the pattern obtained with the conventional sulfhydryl blocking agents, provided that the dyes are uncharged. Although saturation labeling with DIGE Cy3 and Cy5 dyes has proven itself to be more accurate than post-separation detection with adsorptive dyes for proteomic analyses by 2DGE, in our view its implementation suffers from several difficulties. For example, the labeling protocol by the manufacturer (GE Healthcare) for the Cy3- and Cy5-maleimide saturation dyes recommends a dye concentration that corresponds to a dye: thiol labeling ratio of between 1.7 and 15 to 1, depending upon assumptions regarding the average molecular weight of proteins. We have shown that saturation cannot be assured unless total cysteine content of a preparation is known and the dye to thiol ratio exceeds 50 to 1, at least with the BODIPY and other dyes. In our previous study [11], we investigated the reactivity of various uncharged and zwitterionic (neutral) cysteine-reactive dyes. In all cases, including zwitterionic dyes, saturation required at least 40- to 50-fold excess dye over protein thiols. Any differences observed among the dyes were largely in their reactivities with non-cysteine containing protein residues, and/or quenching at higher dye excess. Although we did not assay the maleimide Cy3 and Cy5 dyes, it is unlikely that their cysteine-specific reactivity will be greatly different than any of these other dyes. This may raise a concern that the DIGE saturation protocol is not truly saturating, thus impacting its accuracy and reproducibility. A second issue is that Cy3 and Cy5 are charged but neutral by virtue of their quaternary amine (pKa > 12) and sulfonic acid (pKa < 1) moieties [16]. Thus, their impact on the pI of modified proteins will depend on the degree of substitution but will at the least compress the IEF separation profiles on a 2D gel, making acidic proteins more basic and basic proteins more acidic. In general, it is more desirable to increase resolution rather than decrease it, and to modify the proteins uniformly and maintain them in a state that most closely resembles their unmodified, denatured state. We believe that the best way to accomplish this is to truly saturate proteins with uncharged fluorescent dyes.
To address the suitability for MS identification, the maleimide DIGE dyes have been shown to be compatible with MS analyses — with both modified and unmodified model peptides from a labeling mixture (dye: thiol 2:1) appearing in a MALDI scan [16] - and we, as well, have previously demonstrated the suitability of the BODIPY dyes for MALDI MS [11].
Our purpose in pursuing this labeling strategy is to push the limits of conventional 2D gel electrophoresis to the highest level of quantitative accuracy and sensitivity, and to do so in the most cost effective manner. Thus, we have optimized a labeling strategy with off-the-shelf dyes that match our criteria to meet these goals. Previously, we examined the advantages and considerations of pre-separation covalent saturation fluorescence labeling of protein thiol groups, with emphasis on iodoacetamide-derivatized dyes [11]. We noted that while labeling with iodoacetamide dyes was severely inhibited in the presence of thiourea, such was not the case with maleimide dyes. However, several maleimide dyes that we surveyed exhibited significant non-specific labeling in 8M urea. Saturation was readily achievable with both dye types, and optimization centered on identifying a window of conditions that eliminated non-specific labeling while still allowing for saturation labeling. In this study, we extended our survey and show, among other things, that specificity is not significantly affected by choice of chaotrope (8M urea or 7M urea/2M thiourea). Moreover, we show that with careful choice of dye, dye: thiol ratio, reducer concentrations, pH, and time, conditions can be selected to obtain linear, saturation labeling with minimal non-specific labeling to detect a broad concentration range of proteins.
An important point to emphasize is that the amount of dye necessary to accomplish saturation must be based on the accurate determination of total Cys content for the protein extract to be labeled. We rely on amino acid analysis (AAA) for this estimate, and it remains arguably the most accurate means for quantifying specific amino acid residues and total protein. Labeling with sufficient dye to achieve excess based on AAA estimation of Cys, and performing labeling under conditions that maximally reduce and denature proteins, eliminates the “mass influence” of abundant proteins because in the denatured state (a pre-requisite for saturation fluorescence labeling) all Cys should be equally reactive, regardless of whether they belong to a low-abundance or high-abundance protein; i.e., they are likely to be labeled and respond similarly to the excess of dye.
4.1 Optimization of labeling with BODIPY FL-Mal in the presence of thiourea
A direct comparison of labeling with the iodoacetamide and the maleimide versions of BODIPY FL in urea or urea/thiourea confirmed that the differences we had previously seen between other iodoacetamide and maleimide dyes in the two chaotrope systems were due to the sulfhydryl-selective reactive group. Consistent with our previous observations, labeling by BODIPY FL-Iaa, but not by BODIPY FL-Mal, was strongly inhibited in thiourea. We therefore concluded that the maleimide form of BODIPY might allow better labeling efficiency in thiourea with maximum fluorescence yield under optimal reducing and saturating conditions.
As with the iodoacetamide-derivatized dyes we previously examined, optimization of labeling with maleimide-derivatized dyes involves a matrix of interactive effects and conditions. Using BODIPY FL-Mal, we addressed the effects of the TCEP: dye and dye: thiol ratios, of pH, and of reaction time on completeness and specificity of labeling. The critical reactions are the reduction of disulfides by TCEP and the alkylation of free thiols by the maleimide moiety of the dye. Other important reactions that may occur are the binding of dye to TCEP [11; 17] and the binding of dye to non-thiol sites on proteins. To ensure quantitative accuracy, the latter reaction is to be avoided; and to ensure saturation, the former is to be minimized while still achieving sufficient reducing power for protein disulfide reduction. The apparent interaction of TCEP and dye may be minimized by using 2 mM TCEP to achieve complete protein disulfide reduction. A large excess of TCEP, however, may deplete the dye to levels below the concentration necessary to achieve saturation. In this study, the best results were obtained with a TCEP concentration at 2 mM, and dye: thiol ratio greater than 10:1 for highly purified proteins. For ill-defined complex extracts, we have found a more optimal ratio to be about 75:1.
The pH and the dye: thiol ratio combine to impact completeness and specificity of labeling. Alkylation of a thiol with an iodoacetamide involves nucleophilic displacement of the iodine by the thiolate anion, producing a thioether [15]. With a pKa of about 8.3 for protein thiols, it is clear that the Cys thiol will be most reactive at high pH. Side reactions that may occur include alkylation of His, Lys, Met, or Tyr residues by the iodoacetamide [18]. In contrast, maleimides proceed via Michael addition where the nucleophile (thiolate) reacts with the olefin (maleimide). Maleimides are not known to react with Met or His, but at high pH, reactivity with Lys and Tyr are favored over thiols [19; 20]. Moreover, above pH 8, maleimides may be hydrolyzed to a mixture of non-reactive maleamic acid isomers. Since alkylation is favored for sulfhydryls and amines (average pKa for ε-Lys = 10.8) in the unprotonated state, increasing the pH between 7.0 and 8.0 increased both specific and non-specific labeling. At pH 7.5, sulfhydryls will be reactive (unprotonated) about 13%, and ε—amines about 0.05% of the time, while at pH 8.0, the reactivity of both groups increases to 68 and 0.2%, respectively. Clearly, maintaining the pH at 7.5 while increasing the dye: thiol ratio and TCEP: dye ratio will thereby yield greater sulfhydryl specificity and drive the reaction to completion. Stated in general terms, the pH range supporting complete and specific labeling for a given dye can be manipulated to some extent by increasing or decreasing the dye: thiol ratio, while keeping the TCEP: dye ratio constant. Accordingly, we investigated the optimum pH range that would allow complete labeling with the maleimide dye while minimizing side reactions, and found optimal labeling at pH 7.5.
Labeling with BODIPY FL-Mal in thiourea was rapid, and relatively insensitive to prolonged reaction times; no differences in completeness or specificity were observed at 30, 60, or 90 min of incubation. This result contrasts with the iodoacetamides in 8M urea, where we typically labeled for 90 min to ensure saturation.
4.1 Linearity of Labeling
Our measured limit of detection at a signal to noise of 2:1 in the Perkin-Elmer imager used in the linearity of labeling experiments was 10 fmol (unpublished observations), which was also the lowest signal measurable from the gels in this experiment (Figure 7). This amount corresponds to a protein copy number of 3000 molecules from 2 million cells, the number of cells that typically yields 200 μg of protein and the typical 2D gel load (13.3 × 8.7 cm) used in our facility. We have shown that our labeling approach can label this small amount of protein quantitatively and accurately and responds linearly through at least 3+ orders of magnitude of concentration. Moreover, this is accomplished all the while maintaining specificity for the Cys thiol, even in the presence of 75,000- fold excess of dye (10 fmol enol and 750 pmol BODIPY FL-Mal) as no labeling of the internal myo control was observed at any protein amount (<1%).
4.2 Impact of BODIPY Modification on Protein Mobility in 2DGE
We have shown, as expected, that BODIPY-labeled proteins exhibit little, if any, mobility difference with unlabeled, but sulfhydryl-blocked proteins. This is due to the uncharged nature of the dye that does not alter the protein pI upon modification. We might have expected a small impact on size-based mobility (the 2nd dimension), however, since this is dependent upon the net binding of SDS to the protein (typically 1.4 gm/gm protein), unless this ratio is changed by the conjugation of the dye, little if any change should be observed, and this is demonstrated in Figure 8. Moreover, most of the match vectors that define the relative positions of the BODIPY-labeled proteins and their unlabeled counterparts fall within the average diameter of spot itself. Clearly, the dye contributes marginally, if at all, to the two dimensional mobility of the labeled proteins.
It should be noted in comparison of the two gels shows unique protein spots in both gels: those that are unique to the Sypro-stained gel indicate proteins likely to contain no cysteine; those BODIPY-proteins that are unique indicate proteins that do not bind the Sypro Ruby dye. More detailed investigation is required, however, to establish this more definitively.
The coincidence of mobility confers considerable additional advantage when picking the spots for MS identification. Using our approach, one can be assured that the spot designated to be picked will contain the maximum amount of protein, as compared to the “minimal” Cy dyes (DIGE) or even the “saturation” Cy dyes (saturation DIGE), where significant mobility differences occur between labeled and unlabeled protein spots.
4.3 Removal of excess dye
In many cases it is desirable to perform the 2D or other analysis on a crude extract, to minimize protein loss and to ensure that the relative amounts of all the proteins remain unaltered. Several strategies were found to reduce extraneous fluorescence in crude preparations. After labeling is complete and the reaction has been stopped with 2-ME, any excess dye is present as the 2-ME conjugate and perhaps other components of the reaction mixture. Since the non-protein-associated dye intercalates into the SDS micelle sufficiently so that it migrates to the bottom of the gel, one strategy is to prolong the run time. While this strategy risks losing low molecular weight proteins, the use of high acrylamide concentrations (14% or more) can give a good margin of safety for all but very small proteins. Proteins as small as 10 KD were still resolved at some distance from the bottom of the gel (note the lowest molecular weight marker in Figs. 5A and 6A). Alternatively, free dye may be readily removed by washing the gel with 10% ethanol prior to imaging. The solution must be changed often, lest the free dye diffuse into the gel and impart a high background to the image. We have found that when this occurs, a useful method for reducing the diffuse fluorescence sometimes evident throughout the gel is acidification of the reaction mix prior to SDS-PAGE. Besides yielding lower background fluorescence in SDS gels, this treatment prevents further N-alkylation, and we routinely acidified our samples after stopping with 2-ME.
In some cases it is desirable to remove the excess dye before further analysis. Generally excess dye could be efficiently removed by gel filtration on PD-10 columns in either sodium bicarbonate at pH 8.3, or citrate at acidic pH (data not shown). Both the concentrated fluorescence at the bottom and the diffuse fluorescence sometimes observed in our gels were effectively removed.
Conventional dialysis of crude labeled preparations invariably resulted in precipitation of the sample. Dialysis against 8M urea in SpectraPor Float-A-Lyzer mini dialysis units avoided precipitation of the sample, but the lengthy exposure to urea at room temperature increases the likelihood of carbamylation, with resultant alteration of isoelectric points and horizontal displacement of spots on 2D gels.
This study is an extension of our earlier work that surveyed the optimal fluors and overall conditions for saturation covalent fluorescence labeling in 8M urea. Because of the importance of thiourea for increasing protein recovery in cell lysates and its widespread use, we undertook this study to counter its confounding influence and optimize the conditions that are compatible with pre-separation covalent saturation labeling in the 7M urea/2M thiourea chaotrope system. The information presented here provides optimized conditions for saturation labeling with maleimide-derivatized, uncharged fluorescence dyes in the presence of thiourea.
We have extrapolated this performance to a typical proteomics experiment, where the Cys content was estimated by amino acid analysis. Labeling was then accomplished by the optimized protocol presented here in the presence of a 75-fold excess of BODIPY FL-Mal dye. In our experience, this amount of dye is well below the maximum solubility of the dyes and permits a high degree of quantitative accuracy and dynamic range at a well-affordable cost. We continue this effort and our results with complex mixtures of proteins, multiple dyes, and internal protein standards and will be published elsewhere.
5.0 Acknowledgements
The authors would like to acknowledge the excellent technical assistance in generating the data for Figure 7 by Ms. Susan Stafford of the Biomolecular Resource Facility at the University of Texas Medical Branch. This work was supported in part by NIH/NHLBI contract N01-HV-28184 to The University of Texas Medical Branch (A. Kurosky, Principal Investigator), and NHLBI SCCOR P50HL083794-01 to The University of Texas Health Science Center at Houston (D. Milewicz, Principal Investigator). The authors would also like to acknowledge support from Lynx Therapeutics, Inc. where the bulk of this work was performed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6.0 References
- [1].Gygi SP, Corthals GL, Zhang Y, Rochon Y, Aebersold R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc Natl Acad Sci U S A. 2000;97:9390–5. doi: 10.1073/pnas.160270797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Monteoliva L, Albar JP. Differential proteomics: an overview of gel and non-gel based approaches. Brief Funct Genomic Proteomic. 2004;3:220–39. doi: 10.1093/bfgp/3.3.220. [DOI] [PubMed] [Google Scholar]
- [3].Spandidos A, Rabbitts TH. Sub-proteome Differential Display: Single Gel Comparison by 2D Electrophoresis and Mass Spectrometry. Journal of Molecular Biology. 2002;318:21–31. doi: 10.1016/S0022-2836(02)00052-9. [DOI] [PubMed] [Google Scholar]
- [4].Turck CW, Falick AM, Kowalek JA, Lane WS, Lilley KS, Phinney BS, Weintraub ST, Witkowska HE, Yates NA. ABRF-PRG06: Relative Protein Quantification. Association of Biomolecular Resource Facilities 2006; Long Beach, CA: 2006. [DOI] [PubMed] [Google Scholar]
- [5].Taruvinga M, Jackson AA, Golden MH. Comparison of 15N-labelled glycine, aspartate, valine and leucine for measurement of whole-body protein turnover. Clin Sci (Lond) 1979;57:281–3. doi: 10.1042/cs0570281. [DOI] [PubMed] [Google Scholar]
- [6].Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- [7].Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin D. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Prot. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- [8].Greengauz-Roberts O, Stoppler H, Nomura S, Yamaguchi H, Goldenring JR, Podolsky R, Lee JR, Dynan WS. Saturation labeling with cysteine-reactive cyanine fluorescent dyes provides increased sensitivity for protein expression profiling of laser-microdissected clinical specimens. Proteomics. 2005;5:1746–1757. doi: 10.1002/pmic.200401068. [DOI] [PubMed] [Google Scholar]
- [9].Tonge R, Shaw J, Middleton B, Rowlinson R, Rayner S, Young J, Pognan F, Hawkins E, Currie I, Davison M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001;1:377–96. doi: 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- [10].Ünlü M, Morgan ME, Minden JS. Difference gel electrophoresis: A single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- [11].Tyagarajan K, Pretzer EL, Wiktorowicz JE. Thiol-reactive dyes for fluorescence labeling of proteomic samples. Electrophoresis. 2003;24:2348–58. doi: 10.1002/elps.200305478. [DOI] [PubMed] [Google Scholar]
- [12].Ercal N, Yang P, Aykin N. Determination of biological thiols by high-performance liquid chromatography following derivatization by ThioGlo maleimide reagents. J. Chromatogr. B. 2001;752:287. doi: 10.1016/s0378-4347(00)00560-0. [DOI] [PubMed] [Google Scholar]
- [13].Urwin VE, Jackson P. Two-Dimensional Polyacrylamide Gel Electrophoresis of Proteins Labeled with the Fluorophore Monobromobimane Prior to First-Dimensional Isoelectric Focusing: Imaging of the Fluorescent Protein Spot Patterns Using a Cooled Charge-Coupled Device. Anal. Biochem. 1993;209:57–62. doi: 10.1006/abio.1993.1082. [DOI] [PubMed] [Google Scholar]
- [14].Galvani M, Rovatti L, Hamdan M, Herbert B, Righetti PG. Protein alkylation in the presence/absence of thiourea in proteome analysis: a matrix assisted laser desorption/ionization-time of flight-mass spectrometry investigation. Electrophoresis. 2001;22:2066–74. doi: 10.1002/1522-2683(200106)22:10<2066::AID-ELPS2066>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- [15].Lundblad RL. Chemical Reagents for Protein Modification. CRC Press; Boca Raton, FL: 2004. [Google Scholar]
- [16].Wheeler RWTSLWSLSFWJCJX. Mass spectrometric analysis of maleimide CyDye labelled model peptides. Rapid Communications in Mass Spectrometry. 2003;17:2563–2566. doi: 10.1002/rcm.1220. [DOI] [PubMed] [Google Scholar]
- [17].Shafer DE, Inman JK, Lees A. Reaction of Tris(2-carboxyethyl)phosphine (TCEP) with maleimide and alpha-haloacyl groups: anomalous elution of TCEP by gel filtration. Anal Biochem. 2000;282:161–4. doi: 10.1006/abio.2000.4609. [DOI] [PubMed] [Google Scholar]
- [18].Lapko VN, Smith DL, Smith JB. Identification of an artifact in the mass spectrometry of proteins derivatized with iodoacetamide. J Mass Spectrom. 2000;35:572–5. doi: 10.1002/(SICI)1096-9888(200004)35:4<572::AID-JMS971>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- [19].Smyth DG, Blumenfeld OO, Konigsberg W. Reactions of N-ethylmaleimide with peptides and amino acids. Biochem J. 1964;91:589–95. doi: 10.1042/bj0910589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brewer CF, Riehm JP. Evidence for possible nonspecific reactions between N-ethylmaleimide and proteins. Anal. Biochem. 1967;18:248–255. [Google Scholar]