

Abstract
A complex of high molecular mass proteins (PfRhopH) of the human malaria parasite Plasmodium falciparum induces host protective immunity and therefore is a candidate for vaccine development. Understanding the level of polymorphism and the evolutionary processes is important for advancements in both vaccine design and knowledge of the evolution of cell invasion in this parasite. In the present study, we sequenced the entire open reading frames of seven genes encoding the proteins of the PfRhopH complex (rhoph2, rhoph3, and five rhoph1/clag gene paralogs). We found that four rhoph1/clag genes (clag2, 3.1, 3.2, and 8) were highly polymorphic. Amino acid substitutions and indels are predominantly clustered around amino acid positions 1000-1200 of these four rhoph1/clag genes. An excess of nonsynonymous substitutions over synonymous substitutions was detected for clag8 and 9, indicating positive selection. The McDonald-Kreitman test with a Plasmodium reichenowi orthologous sequence also supports positive selection on clag8. Based on the ratio of interspecific genetic distance to intraspecific distance, the time to the most recent common ancestor of the clag2 and 8 polymorphisms was estimated to be 1.89 and 0.87 million years ago, respectively, assuming divergence of P. falciparum and P. reichenowi 6 million years ago. In addition to a copy number polymorphism, gene conversion events were detected for the rhoph1/clag genes on chromosome 3, which likely play a role in increasing the diversity of each locus. Our results indicate that a high diversity of the PfRhopH1/Clag multigene family is maintained by diversifying selection forces over a considerably long period.
Keywords: Gene conversion, Malaria, Polymorphism, Rhoptry, Selection
1. Introduction
Malaria infects more than 300 million people and kills 1-2 million each year. Efforts have been made to develop effective malaria vaccines, but none is available so far. Malaria is caused by the obligate intracellular protozoan Plasmodium parasites; and entry into erythrocytes is prerequisite for the growth in the mammalian host. After contact with the erythrocyte surface, parasite discharge the content of the microorganelles called the micronemes to establish a tight junction with the erythrocyte surface molecules; parasite then invaginates into a nascent parasitophorous vacuole (PV) [1, 2]. During formation of the PV, the parasite discharges the contents of another pair of microorganelles, the rhoptries [3]. The molecules located within these organelles play a key role in erythrocyte invasion and have been studied as vaccine targets, with the aim to induce antibodies to block invasion. One erythrocyte-binding molecule in the rhoptry is a complex of high-molecular-mass proteins called the RhopH complex [4, 5]. The RhopH complex is distributed throughout the erythrocyte and PV membrane (PVM) and has been detected in ring-stage parasites [6], suggesting an important role during PV establishment. The importance of the complex has further been emphasized by the failure of attempts to disrupt the pfrhoph3 gene locus, suggesting its necessity for parasite survival [7].
The RhopH complex comprises three distinct components: RhopH1, RhopH2, and RhopH3 [8-12]. The genes encoding RhopH1 are members of the rhoph1/clag gene family, which was originally defined by the cytoadherence linked asexual gene (clag) on chromosome 9 in P. falciparum (clag9) and consists of at least three members; clag2, 3.1, and 9 [13-15]. Although not yet determined experimentally, molecules encoded by clag3.2 and 8 are likely parts of the RhopH complex as judged by their similarity in amino acid sequence and transcription pattern with other members [15]. Because only one RhopH1/Clag participates to form a single RhopH complex [15, 16], five types of PfRhopH complex are expected to exist, each of which contains one rhoph1/clag gene product. In this report we employ ‘RhopH1/Clag’ (protein) and ‘rhoph1/clag’ (gene) as the family name, and ‘Clag’ (protein) and ‘clag’ (gene) for each member.
Erythrocyte-binding proteins discharged from P. falciparum merozoites are considered to be targets of host immune responses. Strong diversifying selections on microneme proteins have been detected (e.g., AMA-1 and EBA-175), suggesting that polymorphism of these proteins has been maintained to evade host immunity in parasite populations [17, 18]. Antibodies against the PfRhopH complex partially inhibit the growth of P. falciparum in vitro and in vivo, consistent with its potential as a vaccine target [19-21]. Although the RhopH complex has been shown to induce host protective immunity and is likely to be under host immune pressure, the genetic diversity and immunologic characteristics of this complex are not fully understood. Here, we analyzed sequence polymorphism in five rhoph1/clag members, rhoph2, and rhoph3 and show that some of the rhoph genes are under positive/diversifying selection. In addition, we assessed a population genetic mechanism that might drive the evolution of the rhoph1/clag multigene family.
2. Materials and methods
2.1. Malaria parasites
All cloned lines of P. falciparum were maintained in vitro, essentially as described previously [22]. The parasite lines examined originated from Southeast Asia (Dd2, FVO, Camp, T9/96, T9/102, K1, and Thai838), Papua New Guinea (MAD20), Central and South America (HB3, 7G8, DIV17, DIV29, DIV30, PC49, PC54, Santa Lucia, and Haiti), and Africa (RO33, 123/5, 128/4, SL/D6, LF4/1, 102/1, M2, M5, Fab9, 713, P13, and KMWII) and have been previously described [23-25]. Their geographic origins have also been previously described [26].
2.2. DNA and RNA isolation
Genomic DNA was obtained as described previously [24]. Total RNA was isolated from schizont stage-enriched HB3 and Dd2 parasite lines using the RNeasy mini kit (Qiagen, Valencia, CA). Complementary DNA was synthesized using random hexamers and an Omniscript reverse transcription kit (Qiagen) after DNase treatment.
2.3. Polymerase chain reaction (PCR) amplification and sequencing
Nucleotide sequences corresponding to open reading frame (ORF) were determined for five pfrhoph1/clag genes, rhoph2, and rhoph3 in four parasite lines: Dd2, HB3, 7G8, and FVO. DNA fragments were PCR amplified with KOD-Plus DNA polymerase (Toyobo, Japan) using a panel of oligonucleotides specific for the genes (Supplemental Table 1) and sequenced directly using an ABI PRISM® 310 genetic analyzer (Applied Biosystems, Foster City, CA) or sequenced after cloning into pGEM-T Easy® plasmid (multiple plasmid clones sequenced for each DNA fragment; Promega, Madison, WI). To PCR amplify DNA fragments including the entire ORF of clag3.1 or 3.2, LA Taq DNA polymerase (TaKaRa, Japan) was used with oligonucleotide primers 3.1F (5′-TGTGCAATATATCAAAGTGTACATGC-3′) and 3.1R (5′-TAGAAAATATTAGAATTGCTATTATGTAC-3′) or 3.2F (5′-AATAGTTGAGTACGCACTAATATGTC-3′) and 3.2R (5′-ACACAAATTCTTAATAATTATATAAAACC-3′), respectively. A highly polymorphic region identified in clag2, 3.1, 3.2, and 8 in this study was further analyzed by increasing the number of parasite lines (n = 25) from different geographic areas.
2.4. P. reichenowi sequences
A TBLASTN search was performed against the P. reichenowi preliminary genome shotgun database (Dennis strain; Sanger Centre, UK) using Clag2, 3.1, 3.2, and 8 amino acid sequences as queries. For prclag2 and prclag8, sequences were assembled using SeqMan II accompanied with Lasergene software (DNASTAR Inc., Madison, WI) with manual corrections. Regions covered by at least two independent reads and showing identical sequences were selected and used for analysis (Supplemental Figs. S1 and S2). The generated sequences were 3273 bp long for prclag2, corresponding to nucleotide (nt) positions 193-870, 1021-1902, 2458-3432, and 3448-4185 of pfclag2 (3D7), and 2175 bp long for prclag8, corresponding to nt positions 1459-4173 of pfclag8 (3D7). For Clag3 orthologs in the P. reichenowi genome, only sequences possessing homology with the 5′ untranslated region (UTR) (reich908g11.plk) or 3′ UTR (reich1194c08.plk and reich289f06.qlk) were used.
2.5. Sequence alignment and analysis
The entire ORFs for the 7 PfRhopH complex-related genes (5 rhoph1/clag genes, rhoph2, and rhoph3) in four culture-adapted P. falciparum lines—Dd2 (Southeast Asia), 7G8 (Brazil), HB3 (Honduras), and FVO (Vietnam)—were aligned with those retrieved from a genome database (3D7 line, presumably African in origin) using a CLUSTAL W program [27] with manual corrections; nucleotide diversity (π) and its standard error (SE) were computed with the Jukes and Cantor method using MEGA 3.1 software [28] after excluding insertions/deletions (indels) and highly polymorphic regions. The mean numbers of synonymous substitutions per synonymous site (dS) and nonsynonymous substitutions per nonsynonymous site (dN) and their standard errors were computed using the Nei and Gojobori method [29] with the Jukes and Cantor correction, implemented in MEGA 3.1. The statistical difference between dS and dN was tested using a one-tailed Z-test with 500 bootstrap pseudosamples using MEGA 3.1. A value of dN significantly higher or lower than dS at the 95% confidence level was taken as evidence for positive or purifying selection, respectively. The dN:dS ratio was evaluated using a sliding window method (50 bases with a step size of 10 bases) in DnaSP 4.0 [30]. Positive selection was also evaluated using the McDonald-Kreitman test [31]. Before estimating the time to the most recent common ancestor (TMRCA) for P. falciparum clag2 and 8 polymorphism, the evolutionary rate constancy of clag2 and 8 between P. falciparum and P. reichenowi was validated using a Plasmodium yoelii ortholog PyRhopH1A (accession number AB060734) as an outgroup using Tajima’s relative rate test [32] implemented in MEGA 3.1. Mean and 95% confidence intervals (CI) for estimated TMRCA were computed based on the model assuming the distribution of the distance and the substitution rate were Gamma-distributed [33]. Gene conversion was evaluated for each exon using an algorithm by Betrán et al. [34] implemented in DnaSP 4.0.
Unrooted dendrograms of the pfrhoph1/clag members were constructed using the neighbor-joining and maximum parsimony methods in MEGA 3.1, and Tajima’s relative rate test was used to evaluate the evolutionary rate among members. Indels and highly polymorphic regions could not be satisfactorily aligned and were therefore excluded from the analysis. The sequences (3D7 parasite line) used to construct trees and the evolutionary rate were as follows: nt positions 154-312, 331-573, 727-1122, 1207-1266, 1324-1560, 1609-2988, 3004-3288, and 3382-3924 for clag3.2; nt positions 160-318, 337-579, 733-1128, 1213-1272, 1330-1566, 1615-2994, 3010-3294, and 3388-3930 for clag3.1; nt positions 223-381, 400-642, 799-1194, 1279-1338, 1390-1626, 1696-3075, 3091-3375, and 3553-4095 for clag2; nt positions 130-288, 307-549, 706-1101, 1186-1245, 1300-1536, 1606-2985, 3001-3285, and 3415-3957 for clag8; and nt positions 82-240, 265-507, 652-1047, 1132-1191, 1276-1512, 1582-2961, 2977-3261, and 3394-3936 for clag9.
3. Results
3.1. Polymorphism of the PfRhopH complex-related genes
All seven PfRhopH complex-related genes showed greater nucleotide diversity levels than the average (+2 SE) of 204 ORFs on P. falciparum chromosome (chr) 3 [35] (Table 1). Among the seven genes, clag2, 3.1, 3.2, and 8 are highly polymorphic with nucleotide diversity (π = 0.0053-0.0164) comparable to malaria vaccine candidate antigen protein genes such as eba-175 (π = 0.0030) and ama-1 (π = 0.0166) [17, 18]. The observed nucleotide diversity levels of clag2, 3.1, and 3.2 should be taken as minimum estimates, because indels and highly polymorphic regions were excluded from this analysis to obtain reliable alignments. The highly polymorphic nature of four rhoph1/clag genes at the nucleotide level extends to the amino acid level, which is represented by high dN values (Table 1). Thus, the genes encoding RhopH1/Clag are more polymorphic than RhopH2 and RhopH3.
Table 1.
Nucleotide diversity of the PfRhopH complex genes a
| Gene | n | indel | Sitesd | π | π (SE) | dN | dN (SE) | dS | dS (SE) | dN/dS | Pe |
|---|---|---|---|---|---|---|---|---|---|---|---|
| clag2 b | 5 | (+) | 4,317 | 0.0053 | (0.0008) | 0.0032 | (0.0007) | 0.0133 | (0.0028) | 0.24 | (0.0003) |
| clag3.1 b | 5 | (+) | 4,140 | 0.0164 | (0.0015) | 0.0062 | (0.0011) | 0.0582 | (0.0058) | 0.11 | (< 10-10) |
| clag3.2 b | 5 | (+) | 4,134 | 0.0138 | (0.0011) | 0.0063 | (0.0011) | 0.0445 | (0.0050) | 0.14 | (< 10-10) |
| clag8 | 5 | (-) | 4,182 | 0.0066 | (0.0007) | 0.0065 | (0.0011) | 0.0069 | (0.0020) | 0.94 | ns |
| clag9 | 5 | (-) | 4,020 | 0.0009 | (0.0003) | 0.0011) | (0.0004 | 0.0000 | (0.0000) | ∞ | 0.002 |
| rhoph2 | 5 | (-) | 4,134 | 0.0009 | (0.0003) | 0.0010 | (0.0004) | 0.0005 | (0.0005) | 2.00 | ns |
| rhoph3 | 5 | (-) | 2,691 | 0.0013 | (0.0004) | 0.0012 | (0.0005) | 0.0015 | (0.0010) | 0.80 | ns |
| clag2 c | 24 | (+) | 522 | 0.0131 | (0.0032) | 0.0114 | (0.0042) | 0.0192 | (0.0074) | 0.60 | ns |
| clag8 c | 26 | (-) | 585 | 0.0267 | (0.0042) | 0.0305 | (0.0060) | 0.0132 | (0.0061) | 2.31 | 0.020 |
| Chr 3 d | 5 | 202,069 | 0.00044 | (0.00006) | 0.00039 | (0.0060) | 0.00068 | (0.00010) | 0.57 |
n, number of sequences sampled; Sites, sites analyzed excluding noncoding sequences and alignment gaps; π, pairwise nucleotide diversity; dN, number of nonsynonymous substitutions over numbers of nonsynonymous sites; dS, number of synonymous substitutions over numbers of synonymous sites; SE, standard error computed using the Nei-Gojobori method with Jukes-Cantor correction. SE was estimated using the bootstrap method with 500 replication
For optimal sequence alignment, nt 3433-3435 was excluded from clag2, nt 3337-3447 from clag3.1, and nt 88-99 and 3343-3444 from clag3.2 for the analysis. Nucleotide numbering are after the 3D7 line sequences.
nt 3022-3606 of clag8 and nt 3106-3420 and 3436-3642 of clag2 were used.
Data from 204 ORF on P. falciparum chr 3 using five parasite lines [35].
P value indicates that dN is significantly greater than dS. Those shown in parenthesis indicate that dS are significantly greater than dN. The statistical difference between dS and dN was tested using an one-tail Z-test with 500 bootstrap pseudosamples implemented in MEGA 3.1. ns indicate not significant (P > 0.05)
Among the four RhopH1/Clag showing high polymorphism (Clag2, 3.1, 3.2, and 8), the majority of polymorphic sites are clustered in a region at amino acid (aa) positions 1000-1200 (Fig. 1). In addition, numerous polymorphic sites in this region have more than one amino acid substitution, whereas most polymorphisms in the other regions are dimorphic (at both nucleotide and amino acid levels). Most indels are also located in this region (Fig. 1, asterisks). Thus, the region at aa positions 1000-1200 of RhopH1/Clag is the most highly polymorphic region of the PfRhopH complex.
Fig. 1.
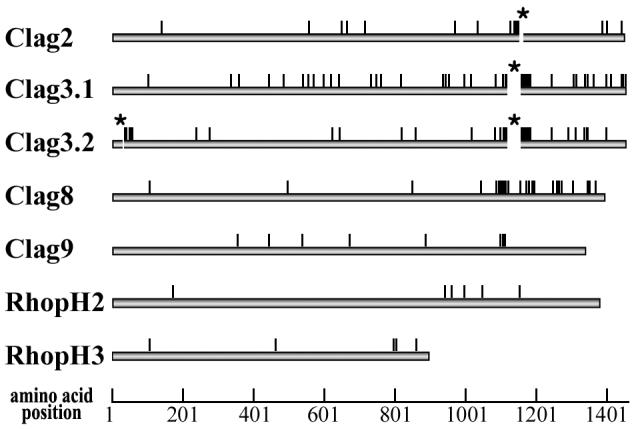
Locations of amino acid polymorphism of seven components of the PfRhopH complex among five P. falciparum parasite lines (3D7, HB3, Dd2, FVO, and 7G8). Indels are shown as gaps with asterisks (aa position 1145 for Clag2, aa positions 1113-1149 for Clag3.1, and aa positions 30-33 and 1115-1148 for Clag3.2). Numbers are those of 3D7 line sequences.
3.2. Gene conversion between clag3.1 and 3.2
Of interest, clag3.1 and 3.2 share some polymorphic sites. Because clag3.1 and 3.2 have 96.7% nucleotide identity (3D7 parasite line) and are located on chr 3 and separated by only 10 kb harboring one putative ORF (PFC0115c) (Fig. 2A), we assessed gene conversion between these two loci. Using an algorithm by Betrán et al. [34], we identified multiple gene conversion tracts located at nt positions 1314-1353, 1447-1452, 1612-1659, 1702-1785, 1852-1983, and 2148-2208 in 3D7 clag3.1; nt positions 3824-4240 in HB3 clag3.1; nt positions 189-247 in 7G8 clag3.1; nt positions 813-817 and 3821-4182 in 3D7 clag3.2; nt positions 88-151 in HB3 clag3.2; and nt positions 3320-3755 in 7G8 clag3.2 (Fig. 3). The detected conversion tracts had less than 5% informative nucleotides showing a mosaic origin, indicating that the probability of these tracts being involved in a recombination event more than once is negligible [34]. No gene conversion was detected between the other rhoph1/clag genes.
Fig. 2.
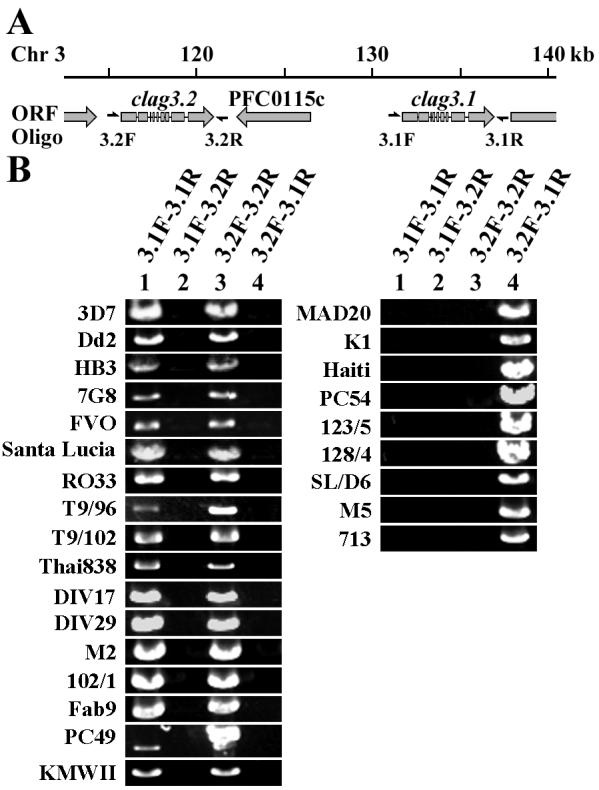
Copy number polymorphism of rhoph1/clag genes on chr 3. A) Genome organization around clag3.2 and 3.1 gene loci on chr 3. The locations of the oligonucleotide primers are indicated. Oligonucleotide 3.2F and 3.1F were designed on the 5′ UTR of clag3.2 and 3.1, respectively. Oligonucleotide 3.2R and 3.1R were designed on 3′ UTR of clag3.2 and 3.1, respectively. B) PCR-amplified DNA fragments of 26 P. falciparum lines with different combinations of oligonucleotides.
Fig. 3.

Gene conversion tracts in clag3.1 and clag3.2. Polymorphic codons (circles) in the coding sequences of Clag3.1 and 3.2 were compared in five P. falciparum lines. Clag3.1, black bar; Clag3.2, gray bar. Polymorphisms matching the paralogous sequence are shown in gray or black circles, respectively, and rare polymorphisms by an open circle. Exons are separated by vertical bar with the intron number at the bottom. Polymorphic sites that differ between consensus sequences are shown below the line classified as nonsynonymous (n), synonymous (s), and deletion (d). Gene conversion tracts identified using algorithm by Betrán et al. [34], wide gray bars.
Because gene conversion potentially accelerates nucleotide diversity, we evaluated the evolutionary rates of clag3.1 and 3.2. Results showed that clag2, 3.1, and 3.2 form a single clade and clag8 another (Fig. 4); thus we performed Tajima’s relative rate test using clag8 as an outgroup and found that the evolutionary rates between clag3.1 and 2 and between clag3.2 and 2 were significantly different for all combinations of the sequences from five parasite lines. Because clag3.1 and 3.2 were more diverse than clag2, clag3.1 and 3.2 appear to have evolved more rapidly than clag2.
Fig. 4.

Unrooted dendrograms of pfrhoph1/clag genes using nucleotide sequences from the 3D7 parasite line. The trees were constructed by the neighbor-joining and maximum parsimony methods using MEGA 3.1. Numbers on branches indicate bootstrap values (500 pseudoreplicates).
3.3. Amino acid polymorphism of the region around aa positions 1000-1200 of Clag2, 3.1, 3.2, and 8
Because extensive polymorphisms were observed around aa positions 1000-1200 in Clag2, 3.1, 3.2, and 8, we further analyzed polymorphism in this region with additional sequences from parasite lines originating worldwide. Alignment of Clag2 sequences showed multiple amino acid substitutions per site at multiple sites, e.g., five amino acids at aa position 1139 (K, R, S, G, and I). Indels were also observed (Supplemental Fig. S3). Clag8 has even higher levels of amino acid substitutions at between 1077 and 1136; five different amino acids (I, S, R, G, and N) at 1100, seven at 1101 (D, S, T, E, N, I, and K), six at 1104 (S, N, I, K, R, and T), and five at 1105 (G, D, T, S, and N) (Supplemental Fig. S4). Clag3.1 and 3.2 are also highly polymorphic (Fig. 5), which will be discussed later.
Fig. 5.

Polymorphism of Clag3. An amino acid region 1112-1150 (after 3D7 line Clag3.1 sequence) of P. falciparum (27 lines) was aligned. Geographic origins are shown at left: SE Asia or S, Southeast Asia; Am, America; P, Papua New Guinea; Santa, Santa Lucia cloned line. Identical, conserved, or semiconserved residues in the alignment are indicated with asterisk, colon, or period, respectively. The number of amino acid replacements at each position and the region with indels are shown at the bottom. Cys residue at aa position 1113 and Asn and Ala residues at aa position 1116 are masked.
3.4. Copy number polymorphism of rhoph1/clag genes on chr 3
Notably, when PCR amplification was performed to obtain DNA fragments of the entire ORFs of clag3.1 or 3.2, 17 parasite lines showed the 2 expected positive bands with the primer sets 3.1F-3.1R and 3.2F-3.2R, whereas 9 parasite lines showed a positive band only with the primer set 3.2F-3.1R, which suggests that these 9 parasite lines possessed a hybrid gene with clag3.2 sequence at the 5′ UTR and clag3.1 sequence at the 3′ UTR (Fig. 2B). DNA fragments were not amplified with other primer combinations, indicating that artificial amplification due to primer mispairing was negligible. This is consistent with a recent report by Chung et al. [36], who found that some parasite lines possess only a single rhoph1/clag on chr 3 by Southern blot hybridization. We here designate this clag3 gene as clag3h (clag3 hybrid; Clag3H for protein). In addition, we obtained two distinct sequences for clag3.1 from the KMWII parasite line (Fig. 5), using several cloned plasmids after experiencing difficulty in direct sequencing of PCR products. Sequences for clag2, 3.2, and 8 were easily obtained from the KMWII line by direct sequencing of the PCR products, supporting the assumption that this line was a clone. Thus, the KMWII line appears to possess at least three clag3-related sequences in the genome. This data suggests that the number of clag3-related sequences in P. falciparum varies from one to at least three.
To deduce the direction of the one-gene to two-gene (or vice versa) change, we searched P. reichenowi orthologs in the genome database and found one sequence read (reich908g11.plk) showing high similarity with the sequence around the start codon of clag3.1 and 3.2. We also found two reads (reich289f06.qlk and reich1194c08.plk) showing strong homology with the sequence around the stop codons of clag3.1 and 3.2. Comparison of the nucleotide sequences at the UTR revealed that reich908g11.plk and reich289f06.qlk were similar to the pfclag3.2 sequence and that reich1194c08.plk was similar to the pfclag3.1 sequence (Fig. 6). Thus, duplication of clag3.1 and 3.2 gene loci appears to predate the divergence of P. falciparum and P. reichenowi, suggesting that a single rhoph1/clag (clag3h) found in some P. falciparum lines is likely a result of an unequal crossover between two closely related genes. Notably, Clag3H had characteristic amino acids that were not observed in Clag3.1 and 3.2. For example, Ala at 1116 was found in three of nine Clag3H (30%). If Clag3H originated recently, for example during culture, the amino acid allele observed in Clag3H would also exist in Clag3.1 or 3.2; however, Ala at 1116 was not found in a total of 36 sequences of non-Clag3H protein sequences. Three in nine Clag3Hs is a significant excess compared to zero Ala at 1116 in 36 non-Clag3H sequences by Fisher’s exact test (P = 0.013). This suggests that at least some Clag3H have accumulated some unique amino acid substitutions since their creation.
Fig. 6.

Nucleotide sequence alignment of the 5′ and 3′ UTRs for P. falciparum clag3.1 and 3.2 genes and P. reichenowi orthologous sequences. A P. reichenowi sequence possessing homology with pfclag3.1 was not found in the current database. Nucleotide sequences corresponding to the ORF and the UTR are shown with upper case and lower case letters, respectively. Putative start and stop codons are boxed. Characteristic nucleotides are displayed in reverse (clag3.1) or masked with gray (clag3.2).
3.5. Selection on the PfRhopH complex
Positive selection was evaluated by comparing synonymous and nonsynonymous substitutions (Table 1). A significant excess of dN over dS was observed for clag9 (entire ORF of five parasite lines) and for clag8 (highly polymorphic region at nt positions 3022-3606 of 26 parasite lines), suggesting positive selection acting on these genes. A sliding window plot of dN:dS ratios revealed that clag2 and 8 had the highest peaks, around nt positions 3000-3600 (Fig. 7). It should be noted that the corresponding regions of clag3.1 and clag3.2 are the regions showing highly extensive polymorphism with indels (asterisks in Fig. 7), thereby preventing evaluation of dN:dS ratios in this region. The peak at the N-terminus of clag3.2 is due to introduction of part of the clag3.1 sequence into the HB3 line clag3.2 by gene conversion (see Fig. 3).
Fig. 7.
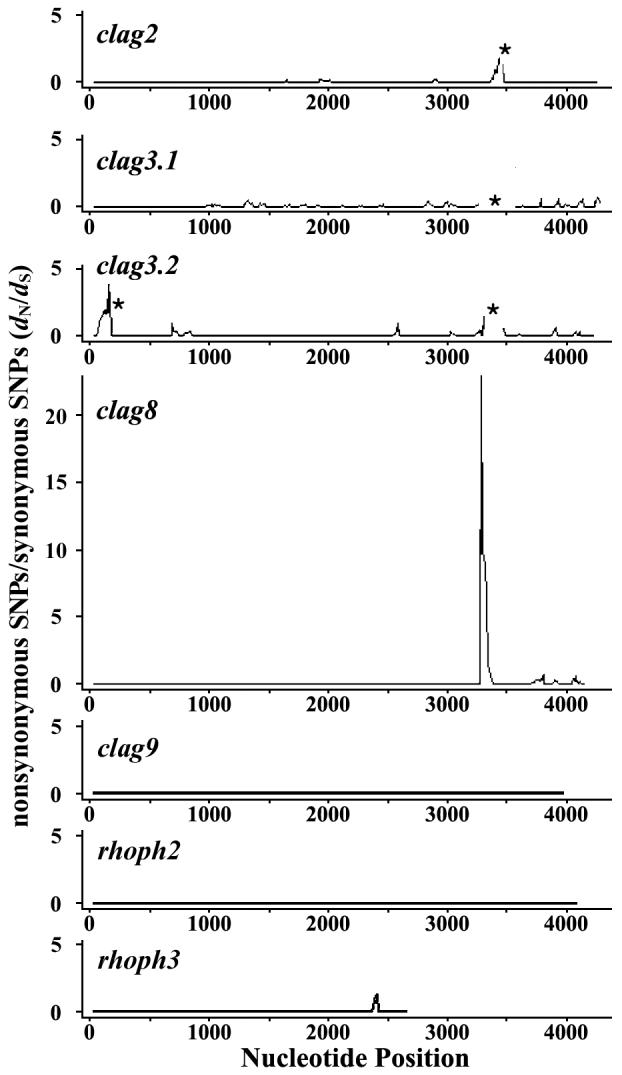
Sliding window plot of dN/dS ratio for seven genes of the PfRhopH complex. For optimum sequence alignment, nt positions 3433-3435 was excluded from clag2, nt positions 3337-3447 from clag3.1, and nt positions 88-99 and 3343-3444 from clag3.2 for the analysis (asterisks), because these regions were highly polymorphic with indels. Nucleotide numbers are those of 3D7 line sequences. Window length is 50 bp, and step size is 10 bp.
Positive selection was further evaluated by the McDonald-Kreitman test using P. reichenowi orthologs for clag2 and 8. Significant excess of intraspecific nonsynonymous substitutions over synonymous substitutions was observed in clag8 as compared with interspecies fixed differences of nonsynonymous and synonymous changes, suggesting positive selection (Table 2).
Table 2.
The McDonald-Kreitman test of selection for Plasmodium falciparum clag2 and 8
| Fixed differences between species |
Polymorphic sites within P. falciparum |
||||||
|---|---|---|---|---|---|---|---|
| Locus | na | No. sites | Syn | Nsynb | Syn | Nsyn | Pc |
| clag2 | 5 | 3273 | 23 | 47 | 18 | 12 | (0.011) |
| clag8 | 5 | 2715 | 36 | 55 | 11 | 38 | 0.030 |
n, number of P. falciparum lines used.
Syn, synonymous; Nsyn, nonsynonymous substitutions.
Fisher’s exact test (one-tailed) was used. P value indicates that Nsyn are significantly greater than Syn. Value in parenthesis indicates that Syn are significantly greater than Nsyn.
3.6. Early origin of the clag2 and 8 polymorphism
We estimated the TMRCA for clag2 and clag8 polymorphism using aligned regions. Distances of synonymous single-nt polymorphisms are 0.0139 ± 0.0031 for clag2 and 0.0106 ± 0.0030 for clag8. Distances between P. falciparum and P. reichenowi are 0.0455 ± 0.0082 and 0.0748 ± 0.0120 for clag2 and 8, respectively. Assuming that the divergence time of P. falciparum and P. reichenowi was 6 million years ago (mya) [37, 38], the estimated TMRCA of the polymorphism of clag2 and 8 are 1.89 (95% CI, 1.02-3.18) and 0.87 (95% CI, 0.42-1.54) mya, respectively.
4. Discussion
4.1. Diversifying selection on the rhoph1/clag gene loci
The present study revealed that the RhopH1/Clag-encoding genes clag2, 3.1, 3.2, and 8 contain a highly polymorphic region, particularly at nt positions 3000-3600. Diversifying selection increases nucleotide diversity (π), and an excess of dN to dS is indicative of positive selection favoring amino acid replacement [39]. Thus, the observed excess of dN to dS at nt positions 3000-3600 of clag8 suggests that the polymorphism in clag8 is positively maintained. An excess of dN to dS was also observed for clag9, indicating that this gene is also under positive selection. The most polymorphic region, in which positive selection was detected for clag8, was excluded from clag2, 3.1, and 3.2 due to extensive sequence variation that made sequence alignment unreliable. Further analysis is required to evaluate positive selection on these three rhoph1/clag genes.
To date, observation of such high levels of polymorphism for clag2, 3.1, 3.2, and 8 (π = 0.0053-0.0164; dN = 0.0032-0.0065) has not been reported for other known malaria rhoptry protein genes. The high polymorphism observed in clag2, 3.1, and 3.2 is consistent with the observation by Kidgell et al. [40] based on the hybridization of genomic DNA from a panel of parasite lines to an oligonucleotide array for the P. falciparum genome. In addition, the polymorphism levels are comparable to those of the microneme proteins such as eba-175 (π = 0.0030; dN = 0.0037) and ama-1 (π = 0.0166; dN = 0.0207), which are exposed to host immune responses [17, 18]. Rhoptry proteins are released into the PV and are considered to be minimally exposed to host immunity. If RhopH1/Clag polymorphism is generated by host immune pressure, the questions arises as to how RhopH1/Clag is exposed to host immunity. There are a few possible explanations. First, RhopH1/Clag may be released from the merozoites before attachment to the erythrocyte surface, thereby becoming a target of host immunity. Second, the RhopH complex, which is released into PVs, may be leaked to the surface of infected erythrocytes through the junction between invading parasite and the erythrocyte membrane. Leaked RhopH complex, and therefore parasite-infected erythrocytes, are then potential targets of host immunity. Indeed, the PfRhopH complex and rhoptry-associated protein 2 (RAP-2, RSP-2), another malaria rhoptry protein, have been detected on the erythrocyte surface upon parasite attachment to erythrocytes [41, 42]. The last possibility is that RhopH1/Clag, after release into the PV, may be distributed to the parasite-derived membranous network (i.e., Maurer’s clefts) in the erythrocyte cytosol, where it is exposed to host immunity. RhopH2 and RhopH3 have recently been observed in materials deriving from Maurer’s cleft by proteome analyses, consistent with this possibility [43, 44].
There are no obvious associations between particular haplotypes and their geographic origins, and most haplotypes co-exist in different geographic areas, similar to other known polymorphic antigens such as MSP-1 [45, 46]. RhopH1/Clag polymorphism might be maintained in natural parasite populations to evade host immunity. Using a T-cell epitope prediction algorithm (SYFPEITHI software) [47], we found that binding of a predicted T-cell epitope peptide to a particular HLA allotype was dramatically affected by the RhopH1/Clag polymorphism in silica. For example, aa positions 1094-1108 of 3D7 line Clag8 (KRISTSIDHISGGKW) was predicted as a T-cell epitope of HLA-DRB1*1101 with a score of 22, but the score for the corresponding region of Camp line Clag8 (MRISSTSTYISNNEW) was 0, emphasizing a potential involvement in immune evasion of RhopH1/Clag polymorphism. We consider the algorithm useful because the score of HLA-DRB1*0701 for the PfCSP Th2R domain, a well characterized malaria polymorphic T-cell epitope peptide, is 22 for the K1 parasite line (KIQYSLSTEWSPCSV) but only 12 for that of the 3D7 line (KIQNSLSTEWSPCSV).
4.2. Evolution of PfRhopH1/Clag polymorphism of the extant P. falciparum population
Gene conversion has been reported for other P. falciparum loci, such as falcipain 2 [48] and var [49], as a source of genetic diversity. In this study, we show that clag3.1/3.2, which interchange their sequences by gene conversion, evolved more rapidly than clag2. The precise function of the RhopH complex remains unknown, and thus whether the gene conversion observed was functionally advantageous or neutral is also unknown; however, gene conversion can be a mechanism for antigenic variation to evade host immunity. Some examples include the vsg gene of Trypanosoma brucei, causative agent of African sleeping sickness, the ves gene of a cattle parasite Babesia bovis [50], and var genes in P. falciparum [51].
Based on the shared hybridization pattern between clag3.1 and 3.2, Chung et al. [36] proposed that these genes are alleles of the same locus; however, because the origin of two rhoph1/clag loci on chr 3 appear to predate the P. falciparum-P. reichenowi divergence, these should be categorized as paralogous genes but not the same gene. Shared features between these loci detected by Southern blot hybridization by Chung et al. can be simply explained by the gene conversion identified in this study. clag3h could be generated by an unequal crossover between clag3.1 and 3.2 of a set of chromosomes during meiosis. If such crossover had occurred, parasite lines possessing three rhoph1/clag on chr 3 would be expected, with a molecule having its 5′ end derived from clag3.1 and its 3′ end from clag3.2 (Supplemental Fig. S5, model 1); however, this type of rhoph1/clag was not detected in this study. KMWII line appears to possess 3 rhoph1/clag genes on chr 3, but the third rhoph1/clag on chr 3 appears not to be generated by the mechanism described above, because this rhoph1/clag was obviously a duplicated clag3.1 gene amplified with the clag3.1-specific primer set. Thus, clag3h is more likely a product of a recombination event between clag3.1 and 3.2 on the same chromosome (Supplemental Fig. F5, model 2). Because a unique amino acid of Clag3H (e.g., Ala at aa position 1116) suggests a relatively old origin of clag3h, recombination events between clag3.1 and 3.2 might be rare in the natural population.
Four highly polymorphic rhoph1/clag genes contained unexpectedly large numbers of synonymous substitutions. Based on the ratio of interspecific distance to intraspecific distance, the TMRCA of the polymorphism of P. falciparum clag2 and 8 were estimated to be 1.89 (95% CI, 1.02-3.18) and 0.87 (95% CI, 0.42-1.54) mya, respectively. Although there is still controversy surrounding its accuracy, TMRCA of the extant P. falciparum population was estimated to be approximately 0.1-0.2 mya based on the genetic distance in nuclear genome housekeeping genes between P. falciparum and P. reichenowi [52, Tanabe, unpublished data]. Thus, polymorphism of clag2 and 8 appears to be generated between the divergence of P. falciparum and P. reichenowi and TMRCA of the extant P. falciparum populations. Early origins of the polymorphism have been suggested for merozoite surface proteins PfMSP-1 and PfMSP-2, for which the origin of the polymorphism was proposed to predate the P. falciparum-P. reichenowi divergence (thus termed ‘ancient origin’), or TMRCA of the extant P. falciparum population, respectively [53, 54]. Early origins of the polymorphism older than TMRCA of extant P. falciparum populations would suggest that rhoph1/clag polymorphisms confer an advantage to the parasite and were positively selected for during the recent evolution of P. falciparum.
In summary, four factors appear to affect current rhoph1/clag polymorphism; i) older origin than TMRCA of the extant P. falciparum population; ii) gene conversion and iii) copy number polymorphism for rhoph1/clag on chr 3; and iv) positive diversifying selection. Multigene families play important roles in many aspects of malaria biology, e.g., responsibility for redundancy of erythrocyte invasion or antigenic variation of parasite-infected erythrocytes. Given the abundance of multigene families in the P. falciparum genome [55], combination of the mechanisms described in this study can be a powerful driving force to generate high biologic redundancy for parasite survival.
Supplementary Material
Acknowledgements
We thank NIAID intramural editor Brenda Rae Marshall for assistance. Sequence data of P. reichenowi was obtained from The Sanger Centre website (http://www.sanger.ac.uk/Projects/P_reichenowi/). This work was supported in part by Grants-in-Aid for Scientific Research 15406015 (to MT), 18390131 (to KT), and 17590372 and 17406009 (to OK) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA (to XS).
Abbreviations
- aa
amino acid
- chr
chromosome
- clag
cytoadherence-linked asexual gene(s)
- CI
confidence interval
- mya
million years ago
- nt
nucleotide
- ORF
open reading frame(s)
- PCR
polymerase chain reaction
- PV
parasitophorous vacuole
- PVM
PV membrane
- SE
standard error
- TMRCA
time to the most recent common ancestor
- UTR
untranslated region(s)
Footnotes
Sequence data from this article have been deposited with the GenBank™/EMBL/DDBJ databases under accession numbers AB250801-AB250912.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124:755–66. doi: 10.1016/j.cell.2006.02.006. [DOI] [PubMed] [Google Scholar]
- [2].Kaneko O. Erythrocyte invasion: Vocabulary and grammar of the Plasmodium rhoptry. Parasitol Int. 2007;56:255–62. doi: 10.1016/j.parint.2007.05.003. [DOI] [PubMed] [Google Scholar]
- [3].Bannister LH, Mitchell GH, Butcher GA, Dennis ED. Lamellar membranes associated with rhoptries in erythrocytic merozoites of Plasmodium knowlesi: a clue to the mechanism of invasion. Parasitology. 1986;92:291–303. doi: 10.1017/s0031182000064064. [DOI] [PubMed] [Google Scholar]
- [4].Sam-Yellowe TY, Perkins ME. Interaction of the 140/130/110 kDa rhoptry protein complex of Plasmodium falciparum with the erythrocyte membrane and liposomes. Exp Parasitol. 1991;73:161–71. doi: 10.1016/0014-4894(91)90019-s. [DOI] [PubMed] [Google Scholar]
- [5].Rungruang T, Kaneko O, Murakami Y, et al. Erythrocyte surface glycosylphosphatidyl inositol anchored receptor for the malaria parasite. Mol Biochem Parasitol. 2005;140:13–21. doi: 10.1016/j.molbiopara.2004.11.017. [DOI] [PubMed] [Google Scholar]
- [6].Lustigman S, Anders RF, Brown GV, Coppel RL. A component of an antigenic rhoptry complex of Plasmodium falciparum is modified after merozoite invasion. Mol Biochem Parasitol. 1988;30:217–24. doi: 10.1016/0166-6851(88)90090-4. [DOI] [PubMed] [Google Scholar]
- [7].Cowman AF, Baldi DL, Duraisingh M, et al. Functional analysis of proteins involved in Plasmodium falciparum merozoite invasion of red blood cells. FEBS Lett. 2000;476:84–8. doi: 10.1016/s0014-5793(00)01703-8. [DOI] [PubMed] [Google Scholar]
- [8].Holder AA, Freeman RR, Uni S, Aikawa M. Isolation of a Plasmodium falciparum rhoptry protein. Mol Biochem Parasitol. 1985;14:293–303. doi: 10.1016/0166-6851(85)90057-x. [DOI] [PubMed] [Google Scholar]
- [9].Brown HJ, Coppel RL. Primary structure of a Plasmodium falciparum rhoptry antigen. Mol Biochem Parasitol. 1991;49:99–110. doi: 10.1016/0166-6851(91)90133-q. [DOI] [PubMed] [Google Scholar]
- [10].Shirano M, Tsuboi T, Kaneko O, Tachibana M, Adams JH, Torii M. Conserved regions of the Plasmodium yoelii rhoptry protein RhopH3 revealed by comparison with the P. falciparum homologue. Mol Biochem Parasitol. 2001;112:297–9. doi: 10.1016/s0166-6851(00)00366-2. [DOI] [PubMed] [Google Scholar]
- [11].Ling IT, Kaneko O, Narum DL, et al. Characterisation of the rhoph2 gene of Plasmodium falciparum and Plasmodium yoelii. Mol Biochem Parasitol. 2003;127:47–57. doi: 10.1016/s0166-6851(02)00302-x. [DOI] [PubMed] [Google Scholar]
- [12].Kaneko O, Tsuboi T, Ling IT, et al. The high molecular mass rhoptry protein, RhopH1, is encoded by members of the clag multigene family in Plasmodium falciparum and Plasmodium yoelii. Mol Biochem Parasitol. 2001;118:237–45. doi: 10.1016/s0166-6851(01)00391-7. [DOI] [PubMed] [Google Scholar]
- [13].Holt DC, Gardiner DL, Thomas EA, et al. The cytoadherence linked asexual gene family of Plasmodium falciparum: are there roles other than cytoadherence? Int J Parasitol. 1999;29:939–44. doi: 10.1016/s0020-7519(99)00046-6. [DOI] [PubMed] [Google Scholar]
- [14].Ling IT, Florens L, Dluzewski AR, et al. The Plasmodium falciparum clag9 gene encodes a rhoptry protein that is transferred to the host erythrocyte upon invasion. Mol Microbiol. 2004;52:107–18. doi: 10.1111/j.1365-2958.2003.03969.x. [DOI] [PubMed] [Google Scholar]
- [15].Kaneko O, Yim Lim BY, Iriko H, et al. Apical expression of three RhopH1/Clag proteins as components of the Plasmodium falciparum RhopH complex. Mol Biochem Parasitol. 2005;143:20–8. doi: 10.1016/j.molbiopara.2005.05.003. [DOI] [PubMed] [Google Scholar]
- [16].Ghoneim A, Kaneko O, Tsuboi T, Torii M. The Plasmodium falciparum RhopH2 promoter and first 24 amino acids are sufficient to target proteins to the rhoptries. Parasitol Int. 2007;56:31–43. doi: 10.1016/j.parint.2006.11.001. [DOI] [PubMed] [Google Scholar]
- [17].Polley SD, Conway DJ. Strong diversifying selection on domains of the Plasmodium falciparum apical membrane antigen 1 gene. Genetics. 2001;158:1505–12. doi: 10.1093/genetics/158.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Baum J, Thomas AW, Conway DJ. Evidence for diversifying selection on erythrocyte-binding antigens of Plasmodium falciparum and P. vivax. Genetics. 2003;163:1327–36. doi: 10.1093/genetics/163.4.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Siddiqui WA, Tam LQ, Kramer KJ, et al. Merozoite surface coat precursor protein completely protects Aotus monkeys against Plasmodium falciparum malaria. Proc Natl Acad Sci USA. 1987;84:3014–8. doi: 10.1073/pnas.84.9.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cooper JA, Ingram LT, Bushell GR, et al. The 140/130/105 kilodalton protein complex in the rhoptries of Plasmodium falciparum consists of discrete polypeptides. Mol Biochem Parasitol. 1988;29:251–60. doi: 10.1016/0166-6851(88)90080-1. [DOI] [PubMed] [Google Scholar]
- [21].Doury JC, Bonnefoy S, Roger N, Dubremetz JF, Mercereau-Puijalon O. Analysis of the high molecular weight rhoptry complex of Plasmodium falciparum using monoclonal antibodies. Parasitology. 1994;108:269–80. doi: 10.1017/s0031182000076113. [DOI] [PubMed] [Google Scholar]
- [22].Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–5. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- [23].Jongwutiwes S, Tanabe K, Nakazawa S, Uemura H, Kanbara H. Coexistence of GP195 alleles of Plasmodium falciparum in a small endemic area. Am J Trop Med Hyg. 1991;44:299–305. doi: 10.4269/ajtmh.1991.44.299. [DOI] [PubMed] [Google Scholar]
- [24].Kaneko O, Soubes SC, Miller LH. Plasmodium falciparum: invasion of Aotus monkey red blood cells and adaptation to Aotus monkeys. Exp Parasitol. 1999;93:116–9. doi: 10.1006/expr.1999.4441. [DOI] [PubMed] [Google Scholar]
- [25].Su X, Ferdig MT, Huang Y, et al. A genetic map and recombination parameters of the human malaria parasite Plasmodium falciparum. Science. 1999;286:1351–3. doi: 10.1126/science.286.5443.1351. [DOI] [PubMed] [Google Scholar]
- [26].Mu J, Ferdig MT, Feng X, et al. Multiple transporters associated with malaria parasite responses to chloroquine and quinine. Mol Microbiol. 2003;49:977–89. doi: 10.1046/j.1365-2958.2003.03627.x. [DOI] [PubMed] [Google Scholar]
- [27].Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kumar S, Tamura K, Nei M. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–63. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- [29].Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–26. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- [30].Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–7. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- [31].McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–4. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- [32].Tajima F. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics. 1993;135:599–607. doi: 10.1093/genetics/135.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Haubold B, Wiehe T. Statistics of divergence times. Mol Biol Evol. 2001;18:1157–60. doi: 10.1093/oxfordjournals.molbev.a003902. [DOI] [PubMed] [Google Scholar]
- [34].Betrán E, Rozas J, Navarro A, Barbadilla A. The estimation of the number and the length distribution of gene conversion tracts from population DNA sequence data. Genetics. 1997;146:89–99. doi: 10.1093/genetics/146.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mu J, Duan J, Makova KD, et al. Chromosome-wide SNPs reveal an ancient origin for Plasmodium falciparum. Nature. 2002;418:323–6. doi: 10.1038/nature00836. [DOI] [PubMed] [Google Scholar]
- [36].Chung WY, Gardiner DL, Anderson KA, Hyland CA, Kemp DJ, Trenholme KR. The CLAG/RhopH1 locus on chromosome 3 of Plasmodium falciparum: Two genes or two alleles of the same gene? Mol Biochem Parasitol. 2007;151:229–32. doi: 10.1016/j.molbiopara.2006.11.004. [DOI] [PubMed] [Google Scholar]
- [37].Escalante AA, Ayala FJ. Phylogeny of the malarial genus Plasmodium, derived from rRNA gene sequences. Proc Natl Acad Sci USA. 1994;91:11373–7. doi: 10.1073/pnas.91.24.11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Goodman M. The genomic record of Humankind’s evolutionary roots. Am J Hum Genet. 1999;64:31–9. doi: 10.1086/302218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hughes AL, Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;335:167–70. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- [40].Kidgell C, Volkman SK, Daily J, et al. A systematic map of genetic variation in Plasmodium falciparum. PLoS Pathog. 2006;2:e57. doi: 10.1371/journal.ppat.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sam-Yellowe TY, Shio H, Perkins ME. Secretion of Plasmodium falciparum rhoptry protein into the plasma membrane of host erythrocytes. J Cell Biol. 1988;106:1507–13. doi: 10.1083/jcb.106.5.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Douki JB, Sterkers Y, Lepolard C, et al. Adhesion of normal and Plasmodium falciparum ring-infected erythrocytes to endothelial cells and the placenta involves the rhoptry-derived ring surface protein-2. Blood. 2003;101:5025–32. doi: 10.1182/blood-2002-12-3710. [DOI] [PubMed] [Google Scholar]
- [43].Sam-Yellowe TY, Fujioka H, Aikawa M, Hall T, Drazba JA. A Plasmodium falciparum protein located in Maurer’s clefts underneath knobs and protein localization in association with Rhop-3 and SERA in the intracellular network of infected erythrocytes. Parasitol Res. 2001;87:173–85. doi: 10.1007/pl00008572. [DOI] [PubMed] [Google Scholar]
- [44].Vincensini L, Richert S, Blisnick T, et al. Proteomic analysis identifies novel proteins of the Maurer’s clefts, a secretory compartment delivering Plasmodium falciparum proteins to the surface of its host cell. Mol Cell Proteomics. 2005;4:582–93. doi: 10.1074/mcp.M400176-MCP200. [DOI] [PubMed] [Google Scholar]
- [45].Ferreira MU, Kaneko O, Kimura M, Liu Q, Kawamoto F, Tanabe K. Allelic diversity at the merozoite surface protein-1 (MSP-1) locus in natural Plasmodium falciparum populations: a brief overview. Mem Inst Oswaldo Cruz. 1998;93:631–8. doi: 10.1590/s0074-02761998000500013. [DOI] [PubMed] [Google Scholar]
- [46].Sakihama N, Ohmae H, Bakote B, Kawabata M, Hirayama K, Tanabe K. Limited allelic diversity of Plasmodium falciparum merozoite surface protein 1 gene from populations in the Solomon Islands. Am J Trop Med Hyg. 2006;74:31–40. [PubMed] [Google Scholar]
- [47].Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–9. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- [48].Nielsen KM, Kasper J, Choi M, et al. Gene conversion as a source of nucleotide diversity in Plasmodium falciparum. Mol Biol Evol. 2003;20:726–34. doi: 10.1093/molbev/msg076. [DOI] [PubMed] [Google Scholar]
- [49].Freitas-Junior LH, Bottius E, Pirrit LA, et al. Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature. 2000;407:1018–22. doi: 10.1038/35039531. [DOI] [PubMed] [Google Scholar]
- [50].Dzikowski R, Deitsch K. Antigenic variation by protozoan parasites: insights from Babesia bovis. Mol Microbiol. 2006;59:364–6. doi: 10.1111/j.1365-2958.2005.05007.x. [DOI] [PubMed] [Google Scholar]
- [51].Kraemer SM, Kyes SA, Aggarwal G, et al. Patterns of gene recombination shape var gene repertoires in Plasmodium falciparum: comparisons of geographically diverse isolates. BMC Genomics. 2007;8:45. doi: 10.1186/1471-2164-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tanabe K, Sakihama N, Hattori T, et al. Genetic distance in housekeeping genes between Plasmodium falciparum and Plasmodium reichenowi and within P. falciparum. J Mol Evol. 2004;59:687–94. doi: 10.1007/s00239-004-2662-3. [DOI] [PubMed] [Google Scholar]
- [53].Polley SD, Weedall GD, Thomas AW, Golightly LM, Conway DJ. Orthologous gene sequences of merozoite surface protein 1 (MSP-1) from Plasmodium reichenowi and P. gallinaceum confirm an ancient divergence of P. falciparum alleles. Mol Biochem Parasitol. 2005;142:25–31. doi: 10.1016/j.molbiopara.2005.02.012. [DOI] [PubMed] [Google Scholar]
- [54].Roy SW, Ferreira MU, Hartl DL. Evolution of allelic dimorphism in malarial surface antigens. Heredity. doi: 10.1038/sj.hdy.6800887. doi:10.1038/sj.hdy.6800887. [DOI] [PubMed] [Google Scholar]
- [55].Michon P, Stevens JR, Kaneko O, Adams JH. Evolutionary relationships of conserved cysteine-rich motifs in adhesive molecules of malaria parasites. Mol Biol Evol. 2002;19:1128–42. doi: 10.1093/oxfordjournals.molbev.a004171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.