

Abstract
Recent studies have suggested that abnormal regulation of protein phosphatase 2A (PP2A) is associated with Type 2 diabetes in rodent and human tissues. Results with cultured mouse myotubes support a mechanism for palmitate activation of PP2A, leading to activation of glycogen synthase kinase 3. Phosphorylation and inactivation of glycogen synthase by glycogen synthase kinase 3 could be the mechanism for long-chain fatty acid inhibition of insulin-mediated carbohydrate storage in insulin-resistant subjects. Here, we test the effects of palmitic acid on cultured muscle glycogen synthase and PP2A activities. Palmitate inhibition of glycogen synthase fractional activity is increased in subjects with high body mass index compared with subjects with lower body mass index (r = −0.43, P = 0.03). Palmitate action on PP2A varies from inhibition in subjects with decreased 2-h plasma glucose concentration to activation in subjects with increased 2-h plasma glucose concentration (r = 0.45, P < 0.03) during oral glucose tolerance tests. The results do not show an association between palmitate effects on PP2A and glycogen synthase fractional activity. We conclude that subjects at risk for Type 2 diabetes have intrinsic differences in palmitate regulation of at least two enzymes (PP2A and glycogen synthase), contributing to abnormal insulin regulation of glucose metabolism.
Keywords: skeletal muscle, cell culture, obesity, insulin resistance
Both obesity and insulin resistance have been reported as risk factors for development of Type 2 diabetes (7, 25). Resistance to insulin-mediated glucose disposal primarily involves reduced glucose storage in skeletal muscle (28). Glycogen synthase is a rate-determining enzyme for glucose storage (45). Skeletal muscle biopsies obtained during a hyperinsulinemic-euglycemic clamp show reduced glycogen synthase activity in insulin-resistant subjects with reduced glucose storage rates (8). Reduced glucose storage rates and glycogen synthase activity have also been observed during insulin infusion after 2 wk of overnutrition with an average weight gain of 3 kg for nondiabetic subjects with a mean body mass index (BMI) of 25 kg/m2 (32). These results indicate that increased insulin resistance and a significant reduction in glycogen synthase activity do not require long-term exposure to increased body mass and could apparently occur secondary to the short-term effects of increased nutrients. Elevated concentrations of plasma lipid and nonesterified fatty acids have been observed to reduce insulin-mediated glucose storage rates (20, 24). Lipid infusion for 2–4 h during a euglycemic-hyperinsulinemic clamp produces insulin resistance, which correlates with reduced skeletal muscle glycogen synthase activity (5). These observations may indicate that lipids or nonesterified fatty acids are functional components of the overnutrition-induced insulin resistance noted above (32).
Cultured human myotubes from individuals with Type 2 diabetes have reduced basal and insulin-stimulated glycogen synthase activity (14). Our group (33) has reported reduced glycogen synthase activity in the presence or absence of insulin using cultured myoblasts from subjects with reduced in vivo insulin-stimulated carbohydrate storage rates. These observations in insulin-resistant subjects indicate that muscle cells isolated in culture contain the same mechanisms for reduced insulin action that produce abnormal glycogen synthase activity in noncultured skeletal muscle biopsies. The positive correlation between in vivo insulin-stimulated carbohydrate storage rates and glycogen synthase activity in myoblasts incubated in the absence of mitogens, including insulin for 42 h (33), suggest that abnormally low glycogen synthase fractional activity (GSFA) could be produced without modulation of the insulin signal transduction pathway. On the basis of the response of skeletal muscle glycogen synthase to lipid infusion in vivo (5), our group (31) has tested the effects of an 18-h incubation of cultured myoblasts with palmitic acid. This study demonstrated a dose-dependent palmitate inhibition of glycogen synthase activity in cultured muscle cells tested in the absence of insulin.
The activity and phosphorylation state of glycogen synthase are regulated in part by glycogen synthase kinase 3 (GSK3) (39). Studies in mouse C2C12 muscle cells suggest that palmitate conversion to ceramide activates protein phosphatase 2A (PP2A), leading to the activation of GSK3 and an inhibition of glycogen synthesis (10, 38). In this study, we compare the effects of palmitate on the activity of glycogen synthase and PP2A in cultured muscle cells from subjects over a range of BMI levels and glucose tolerance.
METHODS
Subjects
Thirty-three Pima Indians and four Caucasians were admitted to the metabolic ward of the Obesity and Diabetes Clinical Research Section of the National Institutes of Health in Phoenix, Arizona. These individuals gave written informed consent for the studies, which were approved by the Institutional Review Board of the National Institute of Diabetes and Digestive and Kidney Diseases and the Tribal Council of the Gila River Indian Community. Fitness for the study was determined by medical history, physical examination, electrocardiography, and routine biochemical, hematological, and urine testing. None of the subjects was taking any medication, and no subject had any clinically significant abnormalities on these examinations. The subjects’ characteristics are shown in Table 1. Group 1 consists of all 26 of the 37 subjects with data on palmitate inhibition of myoblast glycogen synthase activity. Group 2 consists of all 25 of the 37 subjects with data on palmitate effects on myoblast PP2A activity. Group 3 consists of 14 of the 37 subjects with data on both enzymes; this group was used to test the relative effects of palmitate on glycogen synthase activity vs. PP2A activity. Percent body fat was calculated from body composition determined by dual-energy X-ray absorptiometry (42). Individuals with diabetes were excluded according to criteria of the World Health Organization after a 75-g oral glucose tolerance test conducted after 3 days on a weight-maintaining diet (20% protein, 50% carbohydrate, and 30% fat).
Table 1.
Subject characteristics
| Group 1
|
Group 2
|
Group 3
|
||||
|---|---|---|---|---|---|---|
| Mean ± SD | Range | Mean ± SD | Range | Mean ± SD | Range | |
| Men/women | 15/11 | 17/8 | 8/6 | |||
| Age, yr | 29 ± 8 | 18–42 | 30 ± 9 | 18–45 | 30 ± 8 | 19–42 |
| Fasting insulin, pM | 220 ± 86 | 139–432 | 239 ± 78 | 131–432 | 238 ± 95 | 159–432 |
| Fasting glucose, mM | 5.0 ± 0.5 | 4.1–5.9 | 4.9 ± 0.6 | 3.8–5.9 | 4.9 ± 0.6 | 4.1–5.9 |
| 2 h glucose, mM | 7.1 ± 2.0 | 3.6–10.4 | 7.4 ± 1.7 | 4.1–10.4 | 7.8 ± 1.8 | 4.9–10.4 |
| Body fat, % | 31 ± 9 | 12–47 | 31 ± 9 | 14–47 | 30 ± 10 | 18–47 |
| Body mass index, kg/m2 | 32 ± 7 | 20–46 | 33 ± 7 | 22–46 | 32 ± 9 | 22–46 |
Group 3 consists of subjects common to groups 1 and 2.
Myoblast cultures
After an overnight fast, percutaneous muscle biopsies were obtained from the vastus lateralis muscle after local anesthesia of skin and fascia with 10 ml of bicarbonate-buffered 1% lidocaine (22). Primary myoblast cultures were established from human muscle with the use of a previously reported method from this laboratory (44). After the removal of fibroblasts from the primary culture, all subsequent cultureware pieces were collagen coated (Bio-coat; Becton Dickinson, Bedford, MA). Primary cultures were allowed to grow until 100% confluent in DMEM (GIBCO BRL 11885, Gaithersburg, MD), supplemented with 25 mM HEPES, 17% heat-treated FCS, 2 mM glutamine, 0.5% chick embryo extract, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.2% Fungizone (GIBCO BRL). These myoblasts were trypsinized and subcultured at 7.5 × 105 cells per 75-cm2 flask in the above growth medium with 10% FCS. Cells between three- and four-cell population doublings (primary cell monolayers are designated as 1-cell population doubling) were plated at 1.0 × 105 cells per 60-mm dish and were allowed to reach 80–90% confluence during 4 days in the above growth medium with 5% FCS. These cells were not tested for mycoplasmal contamination before measurement of the effects of palmitate on glycogen synthase and phosphatase activity.
Monolayers were washed twice with Dulbecco’s PBS and incubated at 37°C in DMEM (GIBCO BRL 11966) supplemented with 0.5 mM glucose, 25 mM HEPES, 100 U/ml penicillin, 10 μg/ml streptomycin, 0.2% Fungizone, and 0.5% BSA (fatty acid free; Sigma A7511, St. Louis, MO), plus or minus the indicated concentrations of sodium palmitate (Sigma P9767). The 0.5% BSA and indicated concentrations of palmitate were established by adding the required volumes of a 10% BSA, 25 mM HEPES solution in DMEM with or without 8 mM palmitate to the 18-h incubation medium. Sodium palmitate (8 mM) was prepared in DMEM, 25 mM HEPES, and 10% BSA by gentle stirring for 3.5 h in a 37–38°C water bath. Stock palmitate concentrations were verified by enzymatic analysis (Wako Chemicals, Richmond, VA). At the end of an 18-h incubation period, the cells remained preconfluent and contained <2.0% multinucleated myocytes. Cell monolayers were washed at 4°C once each in Dulbecco’s PBS with and without 0.2 mM phloridzin (Sigma P3449).
For measurements of glycogen synthase activity, cell monolayers were covered with 200 μl/dish of 30% glycerol, 10 mM EDTA, and 50 mM KF, pH 7.0. For measurements of phosphatase activity, cell monolayers were covered with 200 μl of 50 mM Tris·HCl, 1 mM EDTA, and 50 mM 2-mercaptoethanol, pH 7.0. The cell monolayers were frozen by floating the 60-mm dish on liquid N2. Frozen mono-layers were stored at −70°C.
Enzyme assays
Frozen cell monolayers for analysis of glycogen synthase or phosphatase activity were collected by scraping the dish at 4°C followed by homogenization (Omni International, Waterbury, CT) and centrifugation at 4°C, 10,000 g for 10 min. Protein was assayed (Bio-Rad Laboratories, Richmond, VA) on 40-μl aliquots of undiluted supernatant. The supernatant for assay of glycogen synthase was diluted 2.3-fold with 50 mM Tris, 130 mM KF, and 20 mM EDTA (pH 7.8), as previously described (22). The active forms (13) of glycogen synthase (GSA) were assayed at a physiological level of 0.17 mM glucose-6-phosphate (low G-6-P), and maximum glycogen synthase activity (MGS) was assayed at 7.2 mM G-6-P (high G-6-P). Activity units are in nanomole per minute per milligram of protein. GSFA is expressed as the activity ratio of GSA to MGS measured at 0.13 mM UDP-glucose. The inter- and intra-assay coefficients of variation for this glycogen synthase assay have been previously described (33).
Phosphatase activity was determined using [33P]phosphorylase(a) as described by Nimmo and Cohen (34). For phosphatase activity, 30 μl of supernatant were aliquoted for enzyme assay, with 40 μl frozen for protein assay. After preincubation of each supernatant aliquot with 1 μM or 3 nM okadaic acid (Sigma 07760) for 5 min at 30°C, 70 μl of preincubated [33P]phosphorylase(a) were added to each assay tube. After 6 min at 30°C, the reaction was stopped with 100 μl each of 17.5% TCA and 6 mg/ml BSA at 4°C. After 10 min at 4°C, reaction tubes were centrifuged at 10,000 g for 4 min, and unbound 33P-labeled inorganic phosphate was measured in 200-μl aliquots by liquid scintillation counting. Phosphatase activity was expressed as nano-moles per minute based on the specific activity of [33P]phosphorylase(a). [33P]phosphorylase(a) was prepared as previously described (23). In preliminary experiments using phosphorylase as a substrate, total phosphatase activity in the divalent cation-free myoblast extract described above was completely inhibited by 1 μM okadaic acid (data not shown). Type 2a activity was calculated as the amount of total activity inhibited by 3 nM okadaic acid (12). The interassay coefficient of variation for the PP2A assay was 5.3%. Approximately 70% of the total phosphatase activity was PP2A.
Data analyses
Data are expressed as means ± SE unless otherwise indicated. Statistical analyses were calculated with the Statistical Analysis System (SAS Institute, Cary, NC). P values were analyzed with Student’s paired and nonpaired t-tests. Simple correlations are Pearson product-moment correlations adjusted for age of the muscle donor where appropriate.
RESULTS
Glycogen synthase
GSFA in the absence of palmitate (control) is not significantly related to the BMI of the muscle donor (Fig. 1A). In the presence of 240 μM palmitate, GSFA is inversely correlated with BMI (r = −0.43, P = 0.03; Fig. 1B). The fraction of control GSFA remaining after cells are treated for 18 h with 240 μM palmitate decreases with increasing BMI (r = −0.50, P = 0.01; Fig. 1C). After palmitate treatment, glycogen synthase activity per unit protein measured in low G-6-P (GSA) and high G-6-P (MGS) are negatively, but not significantly, related to BMI (data not shown).
Fig. 1.
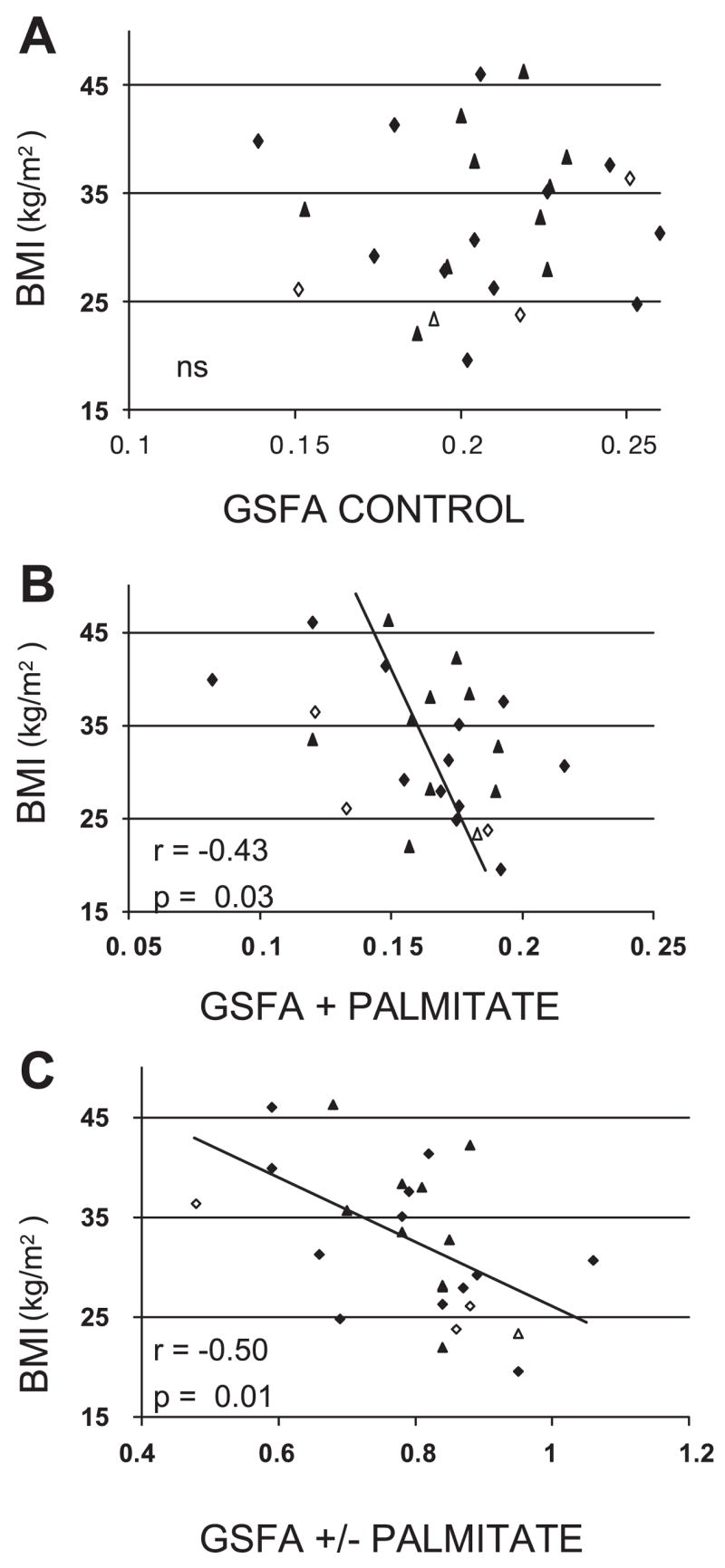
Relationship in 26 group 1 subjects between glycogen synthase fractional activity (GSFA) in control myoblasts (A) and 240 μM palmitate-treated myoblasts (B) and fraction of control GSFA remaining after myoblasts are treated with 240 μM palmitate vs. the body mass index (BMI) of the muscle donor (C). Diamonds signify men, and triangles signify women, with open symbols representing Caucasian subjects.
GSA can be calculated as the product of GSFA and MGS. To investigate the contribution of these activity parameters to the negative relationship between BMI and glycogen synthase activity, group 1 was subdivided into subgroups below and above the median BMI of 33 kg/m2, removing the youngest subject and the oldest subject from the low and high BMI subgroups, respectively, to match the two subgroups for age. The clinical characteristics for these two subgroups are shown in Table 2. Myoblasts from these BMI subgroups (Fig. 2) have the same GSFA levels in the absence of exogenous palmitate. Palmitate produces a larger decrease in the GSFA of the high BMI subgroup (P < 0.03). In the presence and absence of exogenous palmitate, the high BMI subgroup tended to have lower MGS activities that were not statistically different from results for the low BMI subgroup. Within each subgroup, palmitate produced a significant decrease (P < 0.006 and P < 0.02) in MGS. This decrease in MGS, however, was not significantly different between the two subgroups. The product of MGS and GSFA in the presence of palmitate resulted in a GSA for the high BMI subgroup that was 73% of the GSA in the low BMI subgroup (P = 0.05). None of the measures of glycogen synthase activity in the presence or absence of palmitate correlated with 2-h plasma glucose concentrations (2HPG; data not shown).
Table 2.
Clinical characteristics of group 1 subgroups
| Body Mass Index
<33 kg/m2* |
Body Mass Index
>33 kg/m2 |
P | |
|---|---|---|---|
| Men/women | 7/5 | 6/5 | |
| Age, yr | 28 ± 8 | 29 ± 7 | NS |
| Fasting insulin, pM | 175 ± 25 | 270 ± 97 | <0.009 |
| Fasting glucose, mM | 4.7 ± 0.5 | 5.2 ± 0.4 | <0.02 |
| 2 h glucose, mM | 7.2 ± 1.8 | 7.0 ± 2.1 | NS |
| Body mass index, kg/m2 | 27 ± 3 | 39 ± 4 | <0.0001 |
| Body fat, % | 28 ± 7 | 37 ± 6 | <0.003 |
Values are means ± SD. NS, not significant.
One subject was omitted because no protein data were available.
Fig. 2.

Effect of 240 μM palmitate on group 1 subgroups with BMI less than (open bars, n = 12) or greater than (solid bars, n = 11) 33 kg/m2. Shown are GSFA results (top), maximum glycogen synthase activity results measured at high glucose-6-phosphate (MGS; middle), and glycogen synthase activity results measured at physiological glucose-6-phosphate (GSA; bottom). Activity is expressed as nmol·min−1·mg protein−1. #P < 0.02 for palmitate action within the BMI subgroup; *P < 0.03 comparing palmitate effects (0–240 μM) between BMI subgroups; &P = 0.05 vs. same variable for BMI <33 kg/m2.
Protein phosphatase
Myoblast PP2A activity in the presence or absence of palmitate did not show any relationship with BMI of the group 2 muscle donors (data not shown). In the absence of palmitate, no relationship was observed between myoblast PP2A activity and 2HPG (data not shown). Myoblast PP2A in the presence of palmitate was significantly correlated with 2HPG (r = 0.44, P = 0.03; Fig. 3A). When 2HPG increases for the myoblast donors, palmitate action on myoblast PP2A activity changed from inhibition to stimulation (r = 0.45, P < 0.03; Fig. 3B).
Fig. 3.

A: relationship in 25 group 2 subjects between 2-h plasma glucose concentration (2HPG) in muscle donor and 240 μM palmitate-treated myoblast protein phosphatase 2A (PP2A) activity. B: ratio of myoblast PP2A activity in the presence of 240 μM palmitate to PP2A activity under control conditions. Dashed line indicates the transition from inhibition to activation of PP2A. Diamonds signify men, and triangles signify women, with open symbols representing Caucasian subjects.
Group 3
All myoblasts from subjects in groups 1 and 2 with assays of both glycogen synthase and protein phosphatase were used for group 3 to compare palmitate action on the activities of glycogen synthase and type 2A protein phosphatase. The relationships of palmitate action on GSFA (Fig. 4A) or on PP2A (Fig. 4B) with BMI and 2HPG, respectively, were shown to be similar to those observed in the larger groups but were not significant in the 14 group 3 subjects. No correlation was observed for palmitate action on GSFA compared with palmitate action on PP2A (Fig. 4C). Palmitate action on GSA or MGS was also not correlated with palmitate effects on PP2A (data not shown).
Fig. 4.

A: relationship in the 14 group 3 subjects between BMI and the fraction of control GSFA remaining after myoblasts were treated with 240 μM palmitate. B: 2-h plasma glucose and the ratio of PP2A activity in the presence of 240 μM palmitate to control PP2A activity. C: effect of 240 μM palmitate on GSFA and PP2A. Dashed line indicates the transition from inhibition to activation of PP2A. Diamonds signify men, and triangles signify women, with open symbols representing Caucasian subjects.
DISCUSSION
Reduced insulin-mediated glucose disposal measured during a euglycemic-hyperinsulinemic clamp has been identified as a risk factor for Type 2 diabetes mellitus (7). Glycogen synthase regulates a rate-limiting step for glucose storage in skeletal muscle. Glucose storage has been identified as a principal site in vivo for reduced insulin action on glucose disposal (28). In insulin-resistant subjects, reduced glycogen synthase activity has been observed in muscle biopsies obtained during a euglycemic-hyperinsulinemic clamp (8). In cultured muscle cells from obese, insulin-resistant subjects, glycogen synthase activity is reduced in the presence or absence of insulin (14, 33). The mechanism for reduced glycogen storage and glycogen synthase activity in these subjects has not been identified; however, several studies have demonstrated a potential role for PP2A activity (2, 10, 17).
Free fatty acids have been described as an important link between obesity and Type 2 diabetes (3). They have been identified as agents causing reduced insulin secretion by the pancreas (49), elevated hepatic glucose production during a euglycemic-hyperinsulinemic clamp (6), and reduced insulin-mediated glucose disposal in peripheral tissues (4). The increased effects of free fatty acids in the tissues of obese and insulin-resistant subjects are generally assumed to result from increased cellular exposure to plasma free fatty acids secondary to the increased fat cell mass of obesity. Previous in vivo studies have demonstrated that lipid infusion produces inhibition of skeletal muscle glycogen synthase activity and reduced insulin-mediated glucose uptake (5, 20, 24).
We have previously reported palmitate inhibition of glycogen synthase in cultured human myoblasts (31). This observation is compatible with a report of palmitate or ceramide-mediated reduction in mouse C2C12 myotube glycogen synthesis (38). Here, we demonstrate increased palmitate inhibition of GSFA in cultured muscle cells from subjects over a range of BMI. Although the GSFA was decreased by palmitate in the myoblasts from the more obese subjects (Fig. 1), the control cell GSFA did not correlate with increasing BMI. The comparison of age-matched subgroups from group 1 subjects with BMI above or below 33 kg/m2 (Fig. 2) indicates that both GSFA and MGS are significantly decreased by palmitate in each subgroup. A decrease in MGS suggests a reduction in total available enzyme. A decrease in GSFA suggests increased phosphorylation and inactivation of available enzyme. These results indicate that palmitate can change both enzyme turnover rate and the phosphorylation state of glycogen synthase as mechanisms for reducing glucose storage rates. The significantly greater palmitate inhibition of GSFA in the high-BMI subgroup compared with the low-BMI subgroup suggests an abnormality in palmitate regulation of glycogen synthase phosphorylation state in the high-BMI subgroup. The results for MGS in Fig. 2 do not support an increased palmitate-mediated reduction in glycogen synthase protein in the high-BMI subgroup.
These observations indicate that equimolar concentrations of extracellular long-chain fatty acids could produce greater reduction of glucose storage in skeletal muscle of obese subjects in vivo because of an intrinsically greater sensitivity or response of GSFA to long-chain fatty acids. The increased palmitate action on glycogen synthase in myoblasts from obese subjects may also be the enzymatic explanation for the previously reported palmitate inhibition of insulin-stimulated glycogen synthesis in myotubes from insulin-resistant subjects (19). The latter study in myotubes and the results reported here are unique in describing increased palmitate action on the glucose storage pathway in cells from subjects at risk for developing Type 2 diabetes.
In an attempt to understand the mechanism for increased palmitate inhibition of GSFA in cultured muscle from obese subjects, we measured palmitate effects on PP2A activity under the same conditions used to measure glycogen synthase activity. Fourteen of these cultures with and without palmitate were simultaneously assayed for glycogen synthase activity and PP2A activity. This phosphatase is specifically identified by inhibition with nanomolar okadaic acid in the absence of divalent cations and represents a potentially large family of phosphatase enzymes that have been characterized as holoenzymes (reviewed in Ref. 18). Examples of this complex contain the catalytic subunit and a scaffolding subunit that interact with regulatory subunits. The number of different PP2A holoenzyme complexes involved in palmitate regulation of PP2A in myoblasts is not known. PP2A activity has been implicated in palmitate inhibition of glycogen synthesis (10). Palmitate is incorporated into ceramide (30), which can activate PP2A activity (11). In C2C12 mouse myotubes, palmitate activated PP2A, which apparently dephosphorylated and inactivated protein kinase B (10). Palmitate-initiated dephosphorylation of protein kinase B is associated with decreased phosphorylation of GSK3 and reduced glycogen synthesis after insulin stimulation (38). GSK3 phosphorylates several critical sites on glycogen synthase that, along with other kinase activities, progressively inhibit glycogen synthase activity (39, 40).
Abnormal regulation of PP2A activity in relation to diabetes has previously been reported in two different tissues. In the Goto-Kakizaki rat model for diabetes, adipocyte basal cytosolic PP2A activity was elevated compared with levels shown in controls and failed to suppress in the presence of insulin (2). In a study that used human skeletal muscle, PP2A catalytic α-subunit protein reduced insulin-mediated downregulation in individuals with Type 2 diabetes (17). This result indicates that the synthesis and degradation rates of a specific PP2A catalytic subunit isoform could describe a second mechanism that contributes to or explains elevated PP2A activity in diabetic subjects. In isolated β-cells, ceramide increases PP2A activity (26). In isolated rat islets (21), palmitate-induced increases in ceramide inhibit glucose-stimulated insulin gene expression. Long-term exposure to fatty acids inhibits glucose-induced insulin secretion in rats (49). Together, these studies of pancreatic cells indicate that impaired early insulin secretion, which is a principal risk factor for development of Type 2 diabetes in humans (48), could be secondary to fatty acid-mediated increases in PP2A activity.
In results reported here in human myoblasts from subjects over a range of 2HPG levels, palmitate action on PP2A activity changes from inhibition to activation as oral glucose tolerance deteriorates. Our group (31, 33) has previously rationalized that metabolic characteristics in the cultured myoblast, which relate to the clinical characteristics of the cell donor, are intrinsic to the cells from each research subject, as opposed to a response of each cell’s metabolism to the in vivo nutrient and hormonal differences at the time of tissue biopsy. Adipokines such as TNF-α, which can have large effects on muscle glucose metabolism (1), are therefore unlikely to be responsible for the alterations in palmitate action observed here in cultured muscle cells. The assumption that the stimulation of PP2A activity by palmitate is inherent in the myoblasts from subjects with worsening glucose tolerance suggests that a similar inherent increased palmitate response could exist in other tissues from the same subjects. It is possible, therefore, that a mechanism for increased palmitate stimulation of PP2A could be responsible for the abnormal regulation of glucose metabolism where PP2A has been suggested to regulate pathways associated with the development of diabetes. Examples include the adipocytes in Goto-Kakizaki diabetic rats (2), skeletal muscle glycogen synthesis in mouse and humans (10, 17, 19), and pancreatic cells in rats (21, 26).
Palmitate or ceramide have previously been described as metabolites capable of desensitizing insulin action in mouse myotubes (10) and in 3T3-L1 adipocytes (41). An increased palmitate-desensitizing action was described above for insulin-stimulated glycogen synthesis in insulin-resistant compared with normal subjects (19). This desensitization may involve ceramide activation of specific kinases, which compete for phosphorylation of insulin receptor substrate-1, reducing insulin action (15). The results presented here for increased palmitate inhibition of GSFA and stimulation of PP2A are unlikely to be secondary to desensitization of insulin receptor substrate-1 to insulin in muscle cells, which have been in serum-free and insulin-free conditions for 18 h. These observations support the concept for an abnormality in palmitate action that works independent of insulin signal transduction and may explain both alterations in basal metabolism and contribute to reduced insulin action in subjects at risk for Type 2 diabetes.
The absence of an inverse relationship between palmitate inhibition of GSFA and palmitate stimulation of PP2A activity suggests that the PP2A activity measured here in the postmitochondrial supernatant may not be the primary source of palmitate regulation of the synthase in the same subcellular fraction. Given the complex regulatory schemes and multiple isoforms available for PP2A regulation (reviewed in Ref. 18), the failure to observe a phosphatase-to-synthase relationship does not preclude the possibility for involvement of a PP2A enzyme in the abnormal regulation of GSFA. Further studies are needed to identify the specific isoforms of PP2A regulated by palmitate in human muscle and which complex can regulate glycogen synthase activity.
Additional mechanisms should be considered to explain palmitate action on glycogen synthase activity. Palmitate stimulates the activation of 5′-AMP-activated protein kinase (AMPK) by AMPK kinase. AMPK increases fatty acid oxidation in L6 myotubes (46) and phosphorylates a glycogen synthase peptide in rat skeletal muscle that can inhibit synthase activity (9). If AMPK activity were increased in skeletal muscle of obese subjects, it could explain both the previously reported increase in palmitate oxidation (31) and the increased inhibition of glycogen synthase reported here.
The previous study from this laboratory on palmitate inhibition of glycogen synthase activity in human myoblasts (31) failed to show any relationship between palmitate action on glycogen synthase activity and variations in muscle donor insulin resistance or BMI. To increase palmitate oxidation rates (27) and GSFA (16), the previous study omitted glucose during the 18-h palmitate treatment. The authors (31) speculated that the absence of glucose during palmitate treatment could have blocked an abnormal response of myoblasts from obese insulin-resistant subjects to palmitate effects on glycogen synthase activity. The different conclusion reached here using glucose in the study of the relationship between palmitate action on GSFA and the BMI of the myoblast donors could be based on one or a combination of potential glucose effects on palmitate inhibition of GSFA. The addition of glucose in the present study would 1) likely reduce palmitate oxidation (27), potentially providing more palmitate for increased ceramide production in the higher BMI subjects. The resulting increased inhibition of protein kinase B and activation of GSK3 could increase inhibition of GSFA as noted above and reduce GLUT4 translocation to the plasma membrane, reducing glucose uptake (43). The addition of glucose would also 2) lower control GSFA (16), which based on the reported hierarchical phosphorylation of the multiple phosphorylation sites on glycogen synthase (37) could provide different phosphorylation sites on the synthase with altered specificity for fatty acid stimulated kinase activities; 3) increase glucose available for formation of glucosamine-6-phosphate, leading to increased glycosylation and inhibition of glycogen synthase (35) by the hexosamine pathway (29). Here, palmitate could increase glucose flux through the hexosamine pathway by the inhibition of glycolysis described for the glucose-fatty acid cycle (36) or by increasing glutamine fructose-6-phosphate aminotransferase (47). The results presented here in myoblasts from human subjects demonstrate an increased response to palmitate for 1) inhibition of GSFA as subject BMI increases and 2) activation of PP2A in subjects with more impaired oral glucose tolerance. Both of these observations are made in the insulin-free state, but the underlying mechanism could cause or contribute to previously identified examples of reduced insulin action.
Acknowledgments
We thank the clinical staff for professional assistance. Most of all, we are grateful to the volunteers for cooperation during this study. We also thank Shannon Parrington and Tom Brookshire, PA-C, in our research unit for excellent technical support.
GRANTS
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases.
References
- 1.Arner P. Insulin resistance in type 2 diabetes—role of the adipokines. Curr Mol Med. 2005;5:333–339. doi: 10.2174/1566524053766022. [DOI] [PubMed] [Google Scholar]
- 2.Begum N, Ragolia L. Altered regulation of insulin signaling components in adipocytes of insulin-resistant type II diabetic Goto-Kakizaki rats. Metabolism. 1998;47:54–62. doi: 10.1016/s0026-0495(98)90193-7. [DOI] [PubMed] [Google Scholar]
- 3.Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes. 1997;46:3–10. [PubMed] [Google Scholar]
- 4.Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest. 1994;93:2438–2446. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boden G, Jadali F, White J, Liang Y, Mozzoli M, Chen X, Coleman E, Smith C. Effects of fat on insulin-stimulated carbohydrate metabolism in normal men. J Clin Invest. 1991;88:960–966. doi: 10.1172/JCI115399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boden G, She P, Mozzoli M, Cheung P, Gumireddy K, Reddy P, Xiang X, Luo Z, Ruderman N. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-κB pathway in rat liver. Diabetes. 2005;54:3458–3465. doi: 10.2337/diabetes.54.12.3458. [DOI] [PubMed] [Google Scholar]
- 7.Bogardus C, Lillioja S, Bennett PH. Pathogenesis of NIDDM in Pima Indians. Diabetes Care. 1991;14:685–690. doi: 10.2337/diacare.14.7.685. [DOI] [PubMed] [Google Scholar]
- 8.Bogardus C, Lillioja S, Stone K, Mott D. Correlation between muscle glycogen synthase activity and in vivo insulin action in man. J Clin Invest. 1984;73:1185–1190. doi: 10.1172/JCI111304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carling D, Hardie DG. The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta. 1989;1012:81–86. doi: 10.1016/0167-4889(89)90014-1. [DOI] [PubMed] [Google Scholar]
- 10.Cazzolli R, Carpenter L, Biden TJ, Schmitz-Peiffer C. A role for protein phosphatase 2A-like activity, but not atypical protein kinase Cζ, in the inhibition of protein kinase B/Akt and glycogen synthesis by palmitate. Diabetes. 2001;50:2210–2218. doi: 10.2337/diabetes.50.10.2210. [DOI] [PubMed] [Google Scholar]
- 11.Chalfant CE, Kishikawa K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem. 1999;274:20313–20317. doi: 10.1074/jbc.274.29.20313. [DOI] [PubMed] [Google Scholar]
- 12.Cohen P, Klumpp S, Schelling DL. An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 1989;250:596–600. doi: 10.1016/0014-5793(89)80803-8. [DOI] [PubMed] [Google Scholar]
- 13.Guinovart J, Salavert A, Massague J, Ciudad C, Salsas E, Itarte E. Glycogen synthase: a new activity ratio assay expressing a high sensitivity to the phosphorylation state. FEBS Lett. 1979;106:284–288. doi: 10.1016/0014-5793(79)80515-3. [DOI] [PubMed] [Google Scholar]
- 14.Henry RR, Ciaraldi TP, Abrams-Carter L, Mudaliar S, Park KS, Nikoulina SE. Glycogen synthase activity is reduced in cultured skeletal muscle cells of non-insulin-dependent diabetes mellitus subjects. Biochemical and molecular mechanisms. J Clin Invest. 1996;98:1231–1236. doi: 10.1172/JCI118906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herschkovitz A, Liu YF, Ilan E, Ronen D, Boura-Halfon S, Zick Y. Common inhibitory serine sites phosphorylated by IRS-1 kinases, triggered by insulin and inducers of insulin resistance. J Biol Chem. 2007;282:18018–18027. doi: 10.1074/jbc.M610949200. [DOI] [PubMed] [Google Scholar]
- 16.Hidaka H, Howard BV, Kosmakos FC, Fields RM, Craig JW, Bennett PH, Larner J. Insulin stimulation of glycogen synthase in cultured human diploid fibroblasts. Diabetes. 1980;29:806–810. doi: 10.2337/diacare.20.10.806. [DOI] [PubMed] [Google Scholar]
- 17.Hojlund K, Poulsen M, Staehr P, Brusgaard K, Beck-Nielsen H. Effect of insulin on protein phosphatase 2A expression in muscle in type 2 diabetes. Eur J Clin Invest. 2002;32:918–923. doi: 10.1046/j.1365-2362.2002.01098.x. [DOI] [PubMed] [Google Scholar]
- 18.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kausch C, Staiger H, Staiger K, Krutzfeldt J, Matthaei S, Haring HU, Stumvoll M. Skeletal muscle cells from insulin-resistant (non-diabetic) individuals are susceptible to insulin desensitization by palmitate. Horm Metab Res. 2003;35:570–576. doi: 10.1055/s-2003-43501. [DOI] [PubMed] [Google Scholar]
- 20.Kelley DE, Mokan M, Simoneau JA, Mandarino LJ. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J Clin Invest. 1993;92:91–98. doi: 10.1172/JCI116603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem. 2003;278:30015–30021. doi: 10.1074/jbc.M302548200. [DOI] [PubMed] [Google Scholar]
- 22.Kida Y, Esposito-Del Puente A, Bogardus C, Mott DM. Insulin resistance is associated with reduced fasting and insulin-stimulated glycogen synthase phosphatase activity in human skeletal muscle. J Clin Invest. 1990;85:476–481. doi: 10.1172/JCI114462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kida Y, Raz I, Maeda R, Nyomba BL, Stone K, Bogardus C, Sommercorn J, Mott DM. Defective insulin response of phosphorylase phosphatase in insulin-resistant humans. J Clin Invest. 1992;89:610–617. doi: 10.1172/JCI115627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleiber H, Munger R, Jallut D, Tappy L, Felley C, Golay A, Frasca-rolo P, Jequier E, Felber JP. Interaction of lipid and carbohydrate metabolism after infusions of lipids or of lipid lowering agents: lack of a direct relationship between free fatty acid concentrations and glucose disposal. Diabetes Metab. 1992;18:84–90. [PubMed] [Google Scholar]
- 25.Knowler WC, Pettitt DJ, Savage PJ, Bennett PH. Diabetes incidence in Pima Indians: contributions of obesity and parental diabetes. Am J Epidemiol. 1981;113:144–156. doi: 10.1093/oxfordjournals.aje.a113079. [DOI] [PubMed] [Google Scholar]
- 26.Kowluru A, Metz SA. Ceramide-activated protein phosphatase-2A activity in insulin-secreting cells. FEBS Lett. 1997;418:179–182. doi: 10.1016/s0014-5793(97)01379-3. [DOI] [PubMed] [Google Scholar]
- 27.Krutzfeldt A, Spahr R, Mertens S, Siegmund B, Piper HM. Metabolism of exogenous substrates by coronary endothelial cells in culture. J Mol Cell Cardiol. 1990;22:1393–1404. doi: 10.1016/0022-2828(90)90984-a. [DOI] [PubMed] [Google Scholar]
- 28.Lillioja S, Bogardus C. Obesity and insulin resistance: lessons learned from the Pima Indians. Diabetes Metab Rev. 1988;4:517–540. doi: 10.1002/dmr.5610040508. [DOI] [PubMed] [Google Scholar]
- 29.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem. 1991;266:4706–4712. [PubMed] [Google Scholar]
- 30.Merrill AH, Jr, Jones DD. An update of the enzymology and regulation of sphingomyelin metabolism. Biochim Biophys Acta. 1990;1044:1–12. doi: 10.1016/0005-2760(90)90211-f. [DOI] [PubMed] [Google Scholar]
- 31.Mott DM, Hoyt C, Caspari R, Stone K, Pratley R, Bogardus C. Palmitate oxidation rate and action on glycogen synthase in myoblasts from insulin-resistant subjects. Am J Physiol Endocrinol Metab. 2000;279:E561–E569. doi: 10.1152/ajpendo.2000.279.3.E561. [DOI] [PubMed] [Google Scholar]
- 32.Mott DM, Lillioja S, Bogardus C. Overnutrition induced decrease in insulin action for glucose storage: in vivo and in vitro in man. Metabolism. 1986;35:160–165. doi: 10.1016/0026-0495(86)90118-6. [DOI] [PubMed] [Google Scholar]
- 33.Mott DM, Pratley RE, Bogardus C. Postabsorptive respiratory quotient and insulin-stimulated glucose storage rate in nondiabetic Pima Indians are related to glycogen synthase fractional activity in cultured myoblasts. J Clin Invest. 1998;101:2251–2256. doi: 10.1172/JCI1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nimmo GA, Cohen P. The regulation of glycogen metabolism. Purification and characterisation of protein phosphatase inhibitor-1 from rabbit skeletal muscle. Eur J Biochem. 1978;87:341–351. doi: 10.1111/j.1432-1033.1978.tb12383.x. [DOI] [PubMed] [Google Scholar]
- 35.Parker GJ, Lund KC, Taylor RP, McClain DA. Insulin resistance of glycogen synthase mediated by o-linked N-acetylglucosamine. J Biol Chem. 2003;278:10022–10027. doi: 10.1074/jbc.M207787200. [DOI] [PubMed] [Google Scholar]
- 36.Randle PJ, Kerbey AL, Espinal J. Mechanisms decreasing glucose oxidation in diabetes and starvation: role of lipid fuels and hormones. Diabetes Metab Rev. 1988;4:623–638. doi: 10.1002/dmr.5610040702. [DOI] [PubMed] [Google Scholar]
- 37.Roach PJ. Control of glycogen synthase by hierarchal protein phosphorylation. FASEB J. 1990;4:2961–2968. [PubMed] [Google Scholar]
- 38.Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem. 1999;274:24202–24210. doi: 10.1074/jbc.274.34.24202. [DOI] [PubMed] [Google Scholar]
- 39.Skurat AV, Roach PJ. Phosphorylation of sites 3a and 3b (Ser640 and Ser644) in the control of rabbit muscle glycogen synthase. J Biol Chem. 1995;270:12491–12497. doi: 10.1074/jbc.270.21.12491. [DOI] [PubMed] [Google Scholar]
- 40.Skurat AV, Wang Y, Roach PJ. Rabbit skeletal muscle glycogen synthase expressed in COS cells. Identification of regulatory phosphorylation sites. J Biol Chem. 1994;269:25534–25542. [PubMed] [Google Scholar]
- 41.Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol. 1998;18:5457–5464. doi: 10.1128/mcb.18.9.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tataranni PA, Ravussin E. Use of dual-energy X-ray absorptiometry in obese individuals. Am J Clin Nutr. 1995;62:730–734. doi: 10.1093/ajcn/62.4.730. [DOI] [PubMed] [Google Scholar]
- 43.Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001;50:2563–2571. doi: 10.2337/diabetes.50.11.2563. [DOI] [PubMed] [Google Scholar]
- 44.Thompson DB, Pratley R, Ossowski V. Human primary myoblast cell cultures from non-diabetic insulin resistant subjects retain defects in insulin action. J Clin Invest. 1996;98:2346–2350. doi: 10.1172/JCI119046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villar-Palasi C, Larner J. Insulin-mediated effect on the activity of UDPG-glycogen transglucosylase of muscle. Biochim Biophys Acta. 1960;39:171–173. doi: 10.1016/0006-3002(60)90142-6. [DOI] [PubMed] [Google Scholar]
- 46.Watt MJ, Steinberg GR, Chen ZP, Kemp BE, Febbraio MA. Fatty acids stimulate AMP-activated protein kinase and enhance fatty acid oxidation in L6 myotubes. J Physiol. 2006;574:139–147. doi: 10.1113/jphysiol.2006.107318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weigert C, Klopfer K, Kausch C, Brodbeck K, Stumvoll M, Haring HU, Schleicher ED. Palmitate-induced activation of the hexosamine pathway in human myotubes: increased expression of glutamine:fructose-6-phosphate aminotransferase. Diabetes. 2003;52:650–656. doi: 10.2337/diabetes.52.3.650. [DOI] [PubMed] [Google Scholar]
- 48.Weyer C, Pratley RE, Tataranni PA. Role of insulin resistance and insulin secretory dysfunction in the pathogenesis of type 2 diabetes mellitus: lessons from cross-sectional, prospective, and longitudinal studies in Pima Indians. Curr Opin Endocrinol Diabetes. 2002;9:130–138. [Google Scholar]
- 49.Zhou YP, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest. 1994;93:870–876. doi: 10.1172/JCI117042. [DOI] [PMC free article] [PubMed] [Google Scholar]