

Abstract
Vitamin A (Retinol), a fat soluble vitamin, is an essential nutrient for normal functioning of the visual system, epithelial cell integrity and growth, immunity, and reproduction. Our group has investigated the effect of high doses of oral vitamin A on early childhood diarrhea in our prospective community-based studies from Northeast-Brazil and found a benefit role in reducing the mean duration but not incidence of diarrheal episodes. In this study, we explored the role of retinol supplementation in intestinal cell lines following Clostridium difficile toxin A (TxA) challenge. C. difficile is the most common anaerobic pathogen associated with antibiotic- diarrhea and pseudomembranous colitis. Since retinol is critical for the integrity of tight junctions and to modulate cell cycle, we have focused on changes in transepithelial electrical resistance (TEER) in Caco-2, a more differentiated intestinal cell line, and on models of cell proliferation, migration and viability in IEC-6 cells, an undifferentiated crypt cell line, following TxA injury. In this model, retinol therapy reduced apoptosis, improved cell migration, proliferation and prevented the reduction in TEER, following C. difficile TxA challenge in glutamine-free medium. These results suggest the role of retinol in protecting the intestinal epithelial barrier function from C. difficile TxA enterotoxic damage.
Keywords: Retinol, vitamin A, Clostridium difficile toxin A, transepithelial resistance, proliferation, migration, apoptosis
INTRODUCTION
Vitamin A (Retinol), a fat soluble vitamin, is an essential nutrient needed in small amounts for normal functioning of the visual system, growth and maintenance of epithelial cell integrity, immune function, and reproduction (Quadro et al., 2000; Ziegler et al., 2003; Zile et al., 1977). Vitamin A deficiency is a public health issue in many developing countries, where diarreal and respiratory illnesses are common, with consequences ranging from potentially blinding xerophthalmia to increased risks of infection and mortality (Grotto et al., 2003). In a study conducted with Bangladeshi children afflicted with acute Shigella spp. infection, vitamin A supplementation was associated with significant improvement in clinical recovery (Mitra et al., 1998). Additional studies in developing countries suggest that repletion of vitamin A in vitamin A-deficient individuals decreased the risk of diarrhea and gut barrier dysfunction, suggesting a critical role of this nutrient in the gut mucosal repair during infection (Barreto et al., 1994; Wang et al., 1997).
Retinol is the alcohol form of vitamin A and, within the enterocyte, is esterified with long-chain fatty acids for storage or conversion to active metabolites (Lissoos et al., 1995). The importance of vitamin A in maintenance of the gastrointestinal tract structure and function has been demonstrated in several animal studies, by the enhancement of the intestinal epithelial barrier with supplementation and the worsening with deprivation (Li and Tso, 2003; Zile et al., 1977). Vitamin A-deficient rats have significantly reduced villus height, sucrase and maltase activities (Li and Tso, 2003; Warden et al., 1997), impaired cell migration, and impaired mucosal protein synthesis at the translational level (Zaiger et al., 2004). Nonetheless, the beneficial role of vitamin A supplementation has been also demonstrated in animal models of small intestinal enteropathies (Beyzadeoglu et al., 1997).
Our group has investigated the effect of high-doses of oral vitamin A on early childhood diarrhea in our prospective community-based studies in high endemic areas of the Northeast Brazil and found a significant decrease in the mean duration but not in the incidence of diarrheal episodes (Walser et al., 1996). Furthermore, we have found significantly impaired mannitol absorption, increased lactulose:mannitol ratio, and reduced serum retinol concentrations associated with diarrheal illnesses and significant improvements in the intestinal barrier function (shown by L/M permeability ratio) and growth (seen by the high-per-age Z scores), following vitamin A and zinc supplementation (Quadro et al., 2000).
Clostridium difficile is the most common anaerobic pathogen that causes antibiotic-associated diarrhea and pseudomembranous colitis following antibiotic therapy (Bartlett et al., 1980; Guerrant et al., 1990). The estimated number of cases of C. difficile-associated disease exceeds 250,000 per year in the United States (Wilkins and Lyerly, 2003), with a total additional health care costs approaching US$1 billion annually (Kyne et al., 2002). Pathogenic strains of C. difficile produce two high molecular weight protein exotoxins, toxin A (TxA) and toxin B (TxB). Toxin A is a 308-kDa cytotoxin and enterotoxin that induces marked intestinal inflammation, fluid secretion, disruption of the intestinal epithelium and mucosal injury (Johal et al., 2004; Lima et al., 1988; Pothoulakis, 1996). Recently, our laboratory has studied the TxA-induced apoptotic pathway in T84 intestinal cells via caspase and Bid activation and further demonstrated the disruption of mitochondrial membrane potential and release of cytochrome c contributing to the epithelial disruption seen in the TxA challenge (Brito et al, 2002).
Although disruption of the epithelial intestinal barrier due to TxA enterotoxic injury has been extensively studied (Nusrat et al., 2001; Pothoulakis, 1996), to our knowledge, up to date no studies have addressed the role of retinol in protecting the C. difficile TxA-induced cell damage in intestinal cell lines. Since retinol is critical for establishing the integrity of tight junctions (Baltes et al., 2004) and to modulate the cell cycle (Hilakivi-Clarke et al., 2004), we have focused on changes in transepithelial resistance in Caco-2, a more differentiated intestinal cell line, and on models of proliferation and migration in IEC-6 cells, an undifferentiated crypt cell line, following TxA-induced cell injury.
MATERIALS AND METHODS
Reagents, drugs and toxin
Trypsin, Dulbecco's Modified Eagle Media, fetal bovine serum and antibiotic antimycotic solution were obtained from either Gibco BRL (Grand Island, NY) or Invitrogen (Carlsbad, CA). Water soluble-retinol (vitamin A) was obtained from Sigma (St. Louis, MO). A stock solution of retinol (5 mM) was prepared in deionized and sterile water immediately before use, handled in the dark, and kept at -20 °C in order to avoid isomerization and oxidation. Annexin V ApopAlert kit was obtained from Clontech (Palo Alto, CA). Tetrazolium salt WST-1 reagent was obtained from Roche (Mannheim, Germany). Mitomycin C was obtained from Roche applied Science (Roche, Indianapolis, IN). Purified toxin A from Clostridium difficile (strain #10463; molecular weight, 308 kDa) was kindly provided by Dr. Lyerly (Tech Laboratory, Blackburg, VA).
Intestinal epithelial cells culture
Rat intestinal jejunal crypt cells (IEC-6, passages 6-12) or human intestinal epithelial cell line (Caco-2, passages 40-51) were purchased from American Type Culture Collection (Rockville, MD) and were cultured at 37°C in a 5% CO2 incubator. For IEC-6 cells, the maintenance cell media was Dulbecco's Modified Eagle Media (DMEM; Gibco BRL, Grand Island, NY) supplemented with 5% fetal calf serum (FCS), 5mg bovine insulin, 50ug/ml of penicillin/streptomycin (DMEM; Gibco BRL, Grand Island, NY) and a final concentration of 1mM of sodium pyruvate. The media was changed thrice a week, according to standard culture protocols. For Caco-2 cells, the maintenance cell media was Dulbecco's Modified Eagle Media (DMEM; Gibco BRL, Grand Island, NY) supplemented with 10% fetal calf serum (FCS) and 100ug/ml of penicillin/streptomycin (DMEM; Gibco BRL, Grand Island, NY). The cultured cells were trypsinized with 0.25% EDTA trypsin when confluence was achieved.
WST-1 cell proliferation assay
Cell proliferation was measured indirectly using the tetrazolium salt WST-1 (4-[3-(4-iodophenyl)-2H-5-tetrazolio]-1-3-benzene disulfonate), according to the manufacturer recommendations. A 96-well plate was seeded with IEC-6 cells in a total concentration of 4 ×104 cells/ml in 100 μL of standard DMEM media. Cells were allowed to attach for 48 hours, when the media volume was removed and it was changed to standard medium or medium without glutamine with supplementation of vitamin A (0.01-100nM) or not, and then either incubated or not with toxin A (0.1μg/ml). After 24 and 48 hours, wells were incubated for 4 hours with 10μL of the tetrazolium salt and the absorbance was measured using an ELISA microplate reader at 450nm (reference range 420-480 nm). Tetrazolium salts are cleaved to formazan by mitochondrial enzymes in viable cells. Enhancement of the number of viable cells will result in an increase of the amount of the formazan dye, which is detectable by the ELISA reader. Therefore, this model indirectly measures the cell proliferation rate in a time manner.
Migration assay in intestinal cells
This assay was performed as previous described with modifications (Brito et al., 2005). IEC-6 cells were seeded in 6-well plates in a concentration of 5×104 cells/ml and cultivated in DMEM media with 5% FCS (Gibco BRL, Grand Island, NY), until confluence. Wells were then scratched along their diameter and extending 30mm in length to the right center corner, using a sterile razor blade. Prior to scratching, 50% of the media volume was removed from each study well. After scratching, the medium was changed to medium without glutamine with or without supplementation of vitamin A (0.01-100 nM), and then toxin A (0.01 μg/ml) was added. This dose was chosen based on our previous studies. In order to rule-out a proliferation component on cell migration, in some experiments 5 μg/ml of mitomycin C was added 15-20 min prior to the scraping to inhibit DNA synthesis and cell mitosis (Roche, Indianapolis, IN). In order to verify the effect of vitamin A on the migration following mechanical damage, the replacement of medium after scratching was made with standard medium or medium without glutamine (WG) with supplementation of vitamin A (0.01-100 nM) in absence of toxin A. The wells were then returned to the incubator, and after 24h, a column with the highest migration rate was selected and the cells were counted using an eye piece grid, according to Brito et al (2005).
Measurements of the transepithelial electrical resistance (TEER)
Electrical resistance across the Caco-2 cells was measured as described by Hidalgo et al (Hidalgo et al., 1989). Caco-2 monolayers, derived from a human colon adenocarcinoma, exhibit a phenotype that mimics those that occur in normal colonic cells in vivo. Twelve-cluster plate transwells (6.5mm insert diameter and a growth area of 0.33cm2) (Costar, Corning, NY) were used to address the monolayer permeability, which rises when electrical resistance drops. The Caco-2 cells were seeded at 5 × 105cells/ml in the upper compartment, on collagen-coated filters, and incubated at 37°C in a 5% CO2. The medium volumes in the apical and basal compartments were 0.4ml and 1.0 ml, respectively. In order to assay the role of retinol (0.01-100 nM) in reaching cell confluence, after trypsinization, detached Caco-2 cells (passage 40-45) were transferred to transwells and were seeded in glutamine-free medium with or without retinol supplementation. Changes in TEER were tracked daily until reaching >95% confluence, during 120 hours following cell seeding. In order to evaluate C. difficile toxin A challenge, we assayed TEER changes in Caco-2 cells following cell confluence. The standard medium was replaced with glutamine-free medium, supplemented with vitamin A (0.1-100 nM) or not (controls) and then toxin A was added (0.1 μg/ml). Transepithelial resistance of the Caco-2 monolayers was measured using a Millicell Electrical Resistance System with a dual electrode (Millipore Corp., Bedford, MA), by placing separate electrodes in the upper and lower wells, according to the manufacturer's instructions. The resistance from each well was subtracted from a blank value (obtained by inserting the electrodes in a transwell harboring a cell-fee medium) to ultimately calculate the monolayer resistance, which was multiplied by the area of the membrane to obtain TEER (Ω . cm2). Results from the toxin A assay were given as percentage of the initial measurement.
Flow cytometry for apoptosis and necrosis
Caco-2 cells were seeded onto 12-well plates in a concentration of 5×105 cells/ml. These cells were allowed to attach on the plate surface for 24 hours. After 24h, the medium was replaced by a novel medium either supplemented with vitamin A (0.01-100 nM) or not, and then incubated or not with toxin A (0.1μg/ml). Cells were trypsinized, centrifuged, and washed with medium, before incubation with Annexin V. Then, the cells were counted and diluted to 105 cells and rinsed with 1x binding buffer, and re-suspended in 200 μl of binding buffer. 5 μl of Annexin V and 10 μl of PI were added and incubated for 5-15 min in the dark. Apoptosis and necrosis were measured by flow cytometry analyses, on an EPICS XL-MCL platform, using a XL System II (Beckman Coulter, Fullerton, CA), using the ApoAlert Annexin V kit. Annexin V is a molecule that binds to phosphatidylserine (PS) and when conjugated to a fluorochrome detects apoptotic cells expressing PS on the reversed membrane surface. For this protocol, propidium iodide (PI) was also used to detect necrotic and late apoptotic cells, which express PI inside the membrane.
Data entrance and statistical analyses
The data were entered in a computer data set and validated by two different persons. Results are expressed as mean ± standard error (SEM), as generated by GraphPad Prism (GraphPad Software, San Diego, CA), using duplicate measurements for at least three independent experiments for each dose and time point. The differences between the experimental groups were compared by one way ANOVA, corrected by Bonferroni's multiple comparison test. Dose-response curve analyses were generated using the best to fit analyses by the GraphPad Prism software. Statistical significance was accepted at the level of p < 0.05.
RESULTS
Effects of Retinol alone and following C difficile Toxin A on Transepithelial electrical Resistence (TEER)
In order to determine the time and dose effect of toxin A (TxA) on barrier cell function in human colonic Caco-2 cells, we measured TEER over 24h following TxA exposure, as previously described elsewhere (Liu et al., 2003). Exposure of Caco-2 monolayer to TxA led to a toxin dose-dependent significant decrease in the monolayer electrical resistance (Fig. 1A). TxA led to rapid and irreversible loss of electrical resistance, seen 24 hours following the challenge in all doses tested (0.1, 0.3, 1 and 3 μg/ml). We found a significant reduction (p< 0.05) of TEER in relation to the control as fast as 0.5 hour after the challenge in the dose of 3μg/mL; After 2h, TxA in the dose of 0.3μg/mL and 1μg/mL determined significant resistance decline, as well. After 3 hours of TxA incubation, significant reductions were seen for all the doses tested. The maximum effect of the highest and lowest dose occurred, respectively, after 2h and 6h, with no significant difference between them in this period. C. difficile TxA challenge determined an acute resistance drop seen in a dose and time dependent fashion. The beginning of the decline and the maximum effect were observed for the dose of 3μg/mL at 30 minutes and 2 hours, respectively. The dose of 0.1μg/mL TxA was chosen onwards to study the role of retinol in proliferation, TEER, apoptosis and necrosis, since this was the lowest TxA concentration, still with biologic activity, which enabled a conspicuous reduction of the transepithelial resistance, also demonstrated in our early studies (Brito et al., 2002; Brito et al., 2005).
FIGURE 1.


(A) C. difficile toxin A (TxA) (0.1-3μg/mL) effect on transepithelial electrical resistance (TEER) response after Caco-2 confluence. Caco-2 cells were grown on transwell-COL inserts in standard medium and were exposed to varying concentrations of TxA or sterile PBS vehicle (control). TEER was measured before, 30 minutes and until 24 hours after the addition of TxA. Values are means ± SEM. Statistical significance (P < 0.05) departure from the control is indicated by an asterisk, analyzed by ANOVA and Bonferroni's test. (B) Effect of retinol (0.01-100 nM) on Caco-2 monolayers' transepithelial electrical resistance (TEER) after C. difficile toxin A cell injury (0.1μg/mL). Transepithelial electrical resistance (TEER) was measured using Millicel Electrical Resistance System, by inserting electrodes into the apical and basal side of the well, according to the manufacturer's instructions. After cell confluence, TxA was added in the apical part, in a concentration of 0.1μg/mL. These cells were treated with different concentrations of retinol (0.01-100nM) and the control group received only deionized and sterile water as vehicle. Values are mean ± SEM. Statistical significance (P < 0.05) from the control is indicated by an asterisk, determined by Student's T test. Black bar represents TEER mean value from non-exposed Caco-2 cells seeded in a medium without glutamine (WG).
To study the retinol effect on the TxA-induced electrical resistance drop, cells were exposed during 24h to 0.1μg/mL TxA. Retinol at concentrations of 0.1nM (59.3 ± 1.3Ωcm2;p<0.05), 0.3nM (69.86 ±0.6 Ωcm2;p<0.05), 1 nM (66.4 ± 2.3Ωcm2;p<0.05), 3nM (42.5 ±3.05 Ωcm2;p<0.05) and 30nM (65.03 ± 0.625Ωcm2;p<0.05) prevented the reduction of TEER (Ω) (% of initial value), 3 hours following the TxA exposure, in relation to the untreated control challenged with TxA (59.3± 1.3Ωcm2). The doses of 0.1, 0.3 and 10nM (36.17± 0.02; 33.5± 1.8; 50.63±2.33 vs. 27.31±0.23Ωcm2; p<0.05, respectively) at 4h prevented the reduction in TEER in relation to the control with TxA. After six hours following C. difficile TxA exposure, the severity of TxA cytotoxicity outweighed the early observed retinol protection on TEER (Fig. 1B).
In order to evaluate the role of retinol alone, we also tracked cell confluence in transwells, by measuring non-TxA exposed Caco-2 short-time ongoing confluence (gain of TEER over time) in a medium without glutamine and enriched with retinol (0.01-100 nM). The rate of resistance gain was sustained significantly in all doses after 72 hours up to 120 hours, following initial measurement (data not shown).
Effect of C. difficile Toxin A and Retinol on cell proliferation
C. difficile toxin A (TxA) reduced cell proliferation in a dose-dependent fashion at 24 hours (Fig. 2A). The lowest dose of TxA to reduce proliferation was 0.01 μg/ml at 24h (reduction of 23.54% vs. control (standard media), p<0.001). The highest dose used (3.0μg/ml) reduced proliferation by 86.96% and 91.52% vs control at 24h and 48hours, respectively. Supplementation with retinol in standard medium (SM) did not significantly increased cell proliferation at 24 and 48 hours. Retinol supplementation was beneficial on cell proliferation in unchallenged IEC-6 seeded only in glutamine-free medium (WG) at a low dose o dose 0.75nM (10.53% vs. control–WG, P<0.024) following 24 hours of cell incubation (data not shown). The dose of 0.1μg/ml TxA was chosen to study the effect of retinol on cell proliferation, since this concentration induced significant reduction of cell proliferation at 24 hours (80.72% decline, p<0.001. Retinol supplementation increased cell proliferation after TxA-induced cell damage (0.1μg/mL) at a rate of 14.23%, 23.81%, 59.81%; 38.46%; 30.28%; 44.18% (doses of 0.01; 0.03; 0.1; 1.0; 10; 100nM of retinol, respectively), compared to controls with the presence of C. difficile TxA (Fig. 2B).
FIGURE 2.

(A) Dose and time response of C. difficile toxin A (0.001-3μg/mL) exposure on IEC-6 cell proliferation following 24 hours, showing inhibition of cell proliferation in vitro. Cell proliferation assay was assessed by reading the absorbance using an ELISA microplate reader at 450nm in 96-well, following 24 hour of C. difficile toxin A exposure. After 24 hours, wells were incubated for 2 hours with 10μL of tetrazolium salt and the absorbance was measured. *P<0.05 compared with the control containing non-exposed IEC-6 cells seeded in a standard medium (C), by ANOVA and Bonferroni's test. (B) Effect of retinol (vit A) on cell proliferation assay by detected absorbance using an ELISA microplate reader at 450nm in 96-well seeded IEC-6 cells. Retinol (0.01-100nM) was diluted in medium without glutamine. After 24hours, wells were incubated for 2 hours with 10uL of the tetrazolium salt and the absorbance was measure at 24 hours. *P<0.05 compared to the non-treated and toxin A exposed control (TxA), by ANOVA and Bonferroni's test.
Effects of C. difficile Toxin A and Retinol on IEC-6 migration
After 24 hours of TxA exposure (0.01μg/ml) following plate scraping, there was a significant reduction in cell migration, p<0.0001 (reaching a decline of 31%), as opposed to the unchallenged control. Following mechanical damage, retinol supplementation improved significantly (p<0.05) IEC-6 cell migration at all retinol concentrations tested (0.01-100 nM) following C. difficile-TxA exposure at 12 hours (4-8 fold increase) and 24 hours (5-9 fold increase), with mitomycin C pretreatment.
Retinol supplementation at concentrations of 0.1, 0.3, 1, 10 and 100 nM improved significantly IEC-6 cell migration at rates of 32, 49, 47, 88 and 62% at 12 hours and 66, 72, 81, 92 and 112%, at 24 hours respectively, following C. difficile-TxA exposure, without previous mitomycin C treatment (Fig. 3A). Migration rates from retinol supplemented and non-supplemented controls are depicted by representative microphotographs 24 hours following mechanical injury (Fig 3B).
FIGURE 3.
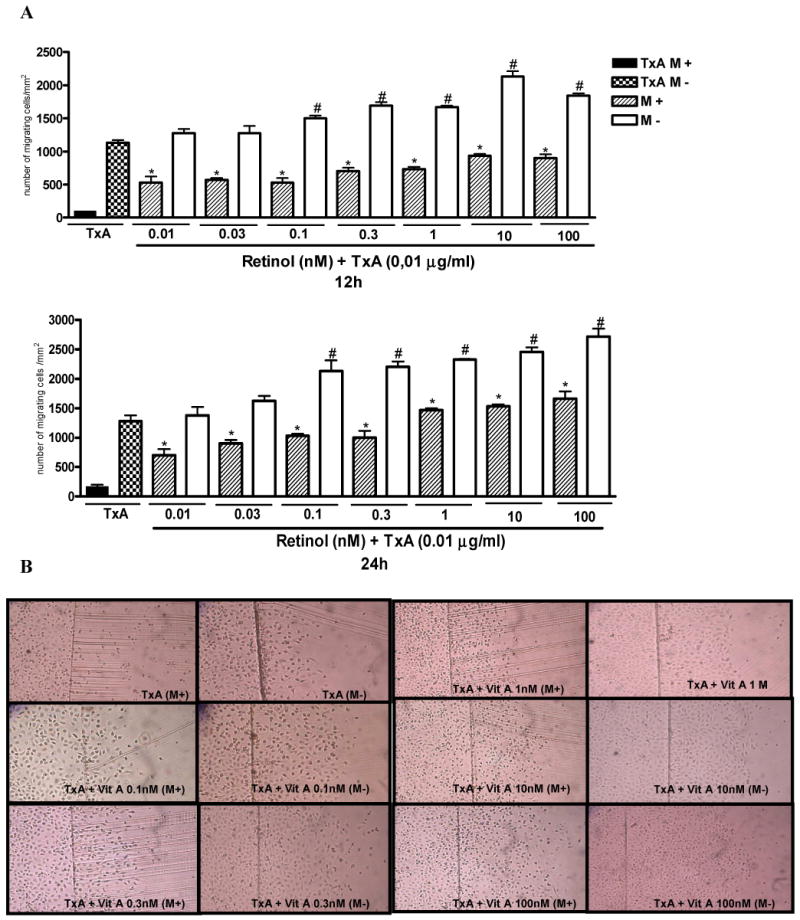
(A). Protective effect of retinol in IEC-6 cell migration following 12 and 24 hours of C. difficile toxin A (TxA) exposure (0.01μg/mL) with (M+) or without (M-) mitomycin C pretreatment (5μg/ml, 15-20 min prior to the scraping). IEC-6 cell monolayers were scraped and incubated with retinol (0.01-100nM) diluted in a glutamine-free medium, immediately after standard medium replacement. (B) Representative images of IEC-6 cell migration (X100) at 24 hours following TxA exposure (0.01μg/mL) and retinol supplementation (0.01-100nM) from 6-well plates with (M+) or without (M-) mitomycin C pretreatment. The bars represent mean ± SEM for the number of migrating cells per square millimeter of the scraped area. *P<0.05 compared to in cells incubated with TxA at 12 hours, #P<0.05, compared to in cells incubated with TxA at 24 hours, by ANOVA and Bonferroni's test.
Effects of C. difficile Toxin A and Retinol on cell viability, measured by flow cytometry analyses
Following 24h of TxA incubation (0.001-3μg/ml), the number of apoptotic cells was significantly increased in a dose-dependent way, starting from dose 0.1μg/ml, when compared with the glutamine-free non-exposed control (p<0.001, respectively). Likewise, we found a significant increase in the rate of necrotic cell death at the same starting dose of 0.1μg/ml (p<0.05), however reaching a plateau thereafter with increasing concentrations (Fig. 4A). Apoptotic cells were positive for annexin V-FITC only (lower right quadrant) and cells that were both necrotic and apoptotic were both positive for annexin V-FITC and propidium iodide (upper right quadrant). The viable cells did not stain for annexin V-FITC or with propidium iodide. Following 24h of TxA exposure in an enriched retinol medium, retinol was able to reduce TxA-induced apoptosis at almost all doses tested (0.03, 0.1, 1, 10 nM), p<0.05, but not at the lowest (0.01 nM) and the greatest doses (100 nM) tested. Similarly, retinol supplementation reduced necrosis signficantly, p<0.05, except at the concentration of 0.01 nM, in comparison to retinol-free medium (Fig. 4B and C, respectively).
FIGURE 4.
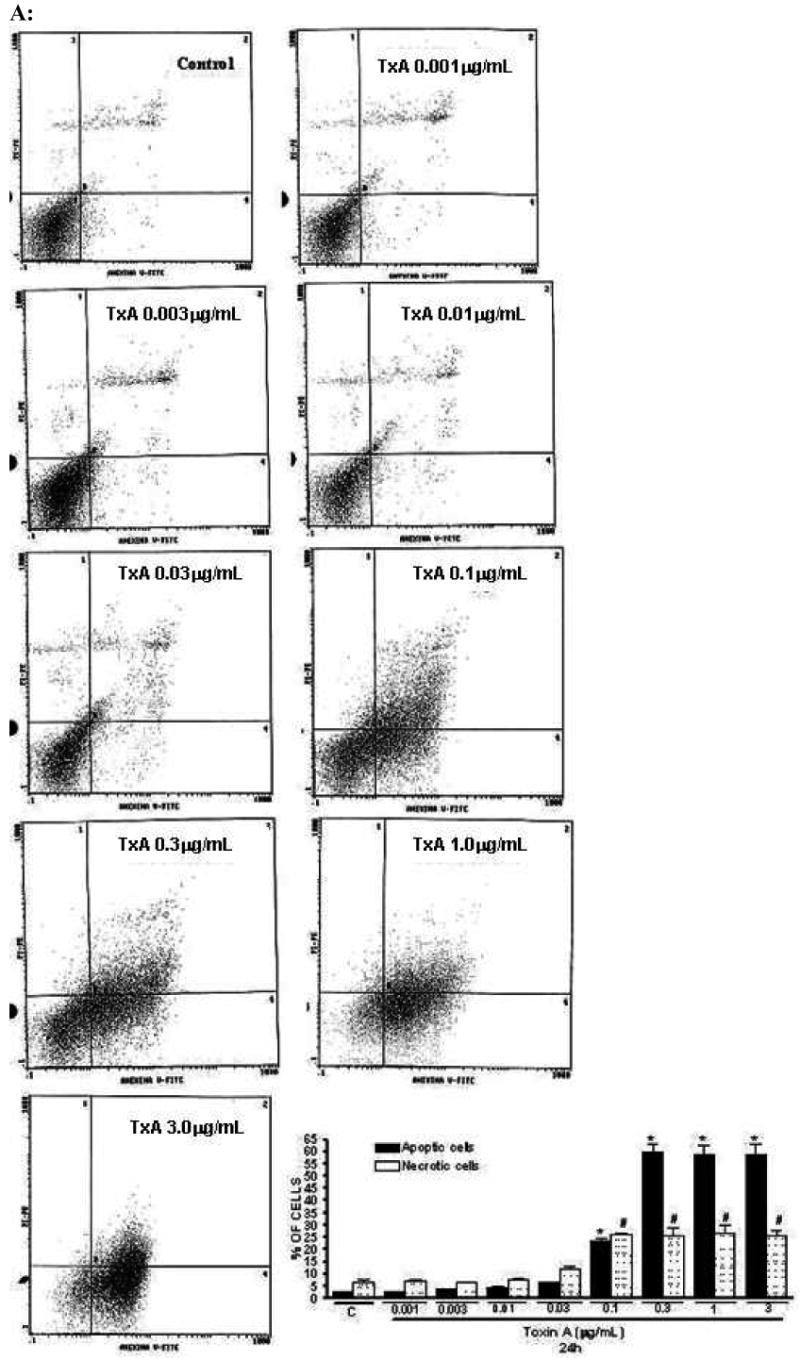
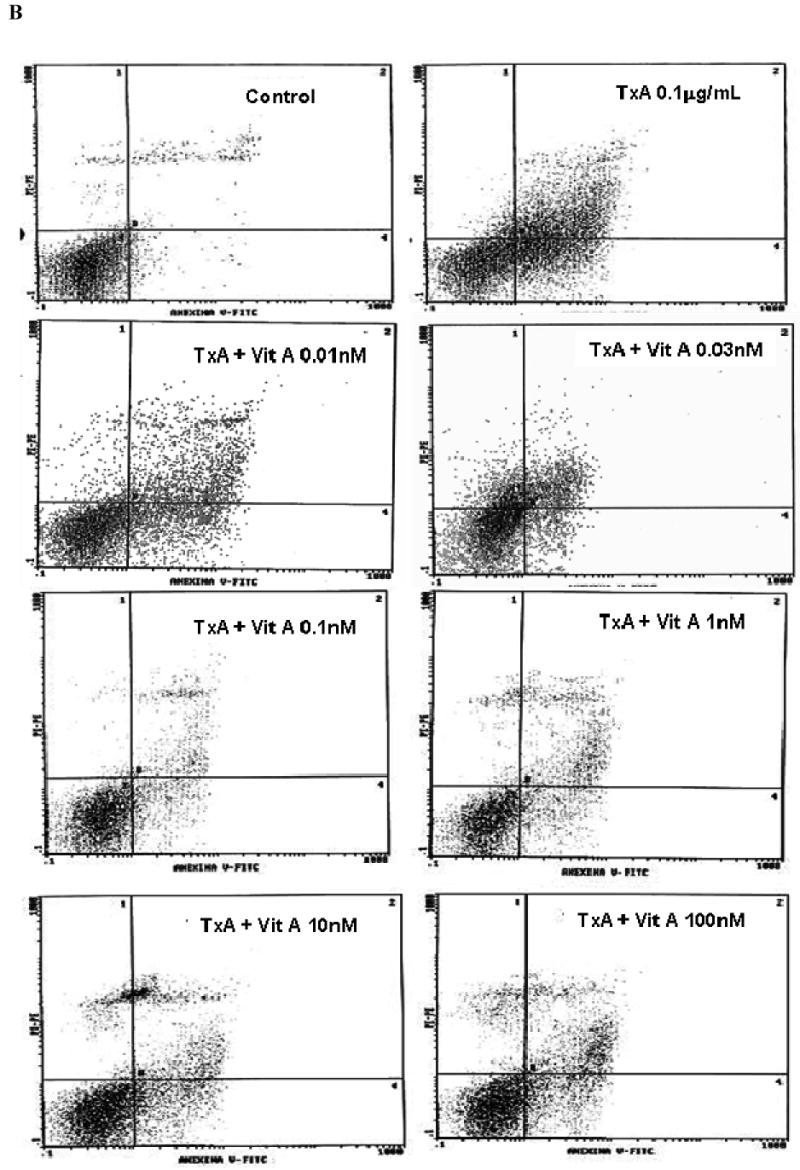

(A) Apoptosis and necrosis induced by C. difficile toxin A (TxA) (0.001-3μg/mL) in a dose-dependent fashion at 24 hours. IEC-6 cells were incubated for 2 hours with TxA and then harvest. Cells were stained with FITC-conjugated annexin V and propidium iodide and analyzed by flow cytometry. Results are shown as density plots with propidium iodide vs. annexin V-FITC. Viable cells have low annexin-V-FITC and low propidium iodide staining (lower-left quadrant), apoptotic cells have high annexin V-FITC and low propidium iodide staining (lower-right quadrant), and necrotic cells have high propidium iodide and annexin V-FITC staining (upper-right quadrant). Bars on the graph (bottom right) represent the percentage of apoptotic and necrotic cells. (B) C. difficile toxin A-induced apoptosis and necrosis are inhibited by retinol (Vit A). IEC-6 cells were incubated for 24 hours with TxA (0.1μg/mL) in culture medium containing Vit A (0.01-100nM) or medium deprived of Vit A. Cells were stained with FITC-conjugated annexin V and propidium iodide and analyzed by flow cytometry. Results are shown as density plots with propidium iodide vs. annexin V-FITC. Viable cells have low annexin-V-FITC and low propidium iodide staining (lower-left quadrant), apoptotic cells have high annexin V-FITC and low propidium iodide staining (lower-right quadrant), and necrotic cells have high propidium iodide and annexin V-FITC staining (upper-right quadrant).
(C) C. difficile toxin A-induced apoptosis and necrosis are inhibited by retinol (Vit A). IEC-6 cells were incubated for 24 hours with TxA (0.1μg/mL) in culture medium containing Vit A (0.01-100nM) or medium deprived of Vit A. Bars on the graph represent the percentage of apoptotic and necrotic cells (C). *P<0.05 compared to media with TxA by ANOVA and Bonferroni's test.
DISCUSSION
Retinol (and the oxidized form of retinoic acid) has been shown, at the transcriptional level, to activate and deactivate a myriad of genes coding for hormones, growth factors, structural proteins and cell death pathways (Blomhoff and Blomhoff, 2006). The transcriptional modulation activated by retinol involves members of the intracellular retinoic acid and retinoid X receptors (RXR), although these signaling pathways are not well understood. In this current study, we have focused on two intestinal cell lines (Caco-2 and IEC-6) to address the benefit of retinol in an in vitro model of epithelial cell damage due to C. difficile toxin A, yet not explored. Enterocytes are involved in vitamin A metabolism, since these cells deliver highly hydrophobic retinyl esters to chylomicrons to be released into the lymphatic system and are critically affected by vitamin A deficiency when mucosal damage occurs (Thomas et al., 2005).
We have explored changes in the epithelial monolayer integrity following C. difficile-induced cell damage, monitoring the transepithelial electrical resistance (TEER), which is the result of the physical sealing of cell-to-cell contacts by apical tight junctions (Musch et al., 2006), playing a role in cell differentiation. In this regard, Caco-2 cells are eligible intestinal cell lines, since Caco-2 cell sheets form polarized monolayers, developing a similar differentiated pattern seen in human enterocytes in vivo (Baltes et al., 2004). These cells embedded in an enriched medium can absorb retinol and are able to generate retinyl esters. Additionally, they can express cellular retinol binding protein-II (CRBP-II), which is critical for retinol solubilization within the cytoplasm (Lissoos et al., 1995) and might have a role during small bowel adaptive responses against mucosal injury (Wang et al., 1997). Our findings regarding the benefit of retinol in increasing TEER, as early as three hours, following TxA challenge, is unlikely to be because of only cell proliferation, since we could not find significant Caco-2 proliferation at these time points for all doses testes (unpublished data). However, we cannot rule out that cell proliferation might have role in the gain of TEER afterwards.
We also have explored the role of retinol in protecting cell proliferation and migration following the extensive cell damage induced by C. difficile TxA in IEC-6, a rat undifferentiated crypt cell line.
Cell proliferation and migration are tied kinetic phenomena acting together to switch cell function toward renewal and repair (Dignass, 2001). These changes seen along intestinal crypts after significant mucosal injury, can lead to compensatory crypt hyperplasia (Carneiro-Filho et al., 2004), required for epithelium restoration. In adult tissues, retinoids show different effects in regulating cellular proliferation, depending on cell type, by involving specific regulatory proteins of the cell cycle, including cyclins and cyclin-dependent kinases (Bohnsack and Hirschi, 2004). The exact meaning of these cell cycle mediators on vitamin A-induced cell healing warrants additional studies. Our findings with mitomycin C have shown that proliferation is an important component to cell migration. In this regard, Bulut et al have shown a significant stimulation of glucagons-like peptide 2 inducing migration in IEC-6 and IEC-18 monolayers but not in Caco-2 cells, effect which was mediated by tumor growth factor-β (Bulut et al, 2004), suggesting a synergistic role of retinol and growth factors in mediating wound healing upon changes of cell turn over involving the intestinal crypt.
Additionally, we have addressed the role of retinol in cell viability after C. difficile toxin A injury, by studying necrotic and apoptotic death in C. difficile toxin A-exposed IEC-6 cells, process that ultimately counterbalance cell renewal in vivo. The role of retinol in preventing cell apoptosis due to C. difficile toxin A might involve mitochondrial and cell death receptor pathways altogether, since the mechanism of the toxin A-induced apoptosis enrolls intrinsic and extrinsic caspase-activating pathways, by a cross-talk through Bid activation, as shown in our previous studies with T-84 cells (Brito et al., 2002; Carneiro et al., 2006). In non-intestinal tissue, retinoic acid was shown to be anti-apoptotic by reducing the oxidative mediated apoptosis via activator protein-1 (AP-1) pathway (Kitamura et al., 2002). On the other hand, retinoids might exert a pro-apoptotic effect by favoring differentiation rather than proliferation in tumor cells, a role that has been extensively studied in the recent development of anti-cancer therapy (Simoni and Tolomeo, 2001; Zamora et al., 2006). However, the mechanisms of the retinol anti-apoptotic effects following toxin A enterotoxic damage remains to be explored.
Our findings have highlighted the importance of retinol supplementation to enhance the intestinal barrier following a stressful condition, when other gut-trophic nutrients are deprived or when increase of their requirements are likely to occur. In favor of this is the observation that vitamin A supplementation was more beneficial in hastening the confluence of toxin A in non-exposed Caco-2 cells seeded in a glutamine-free medium than in standard medium. Furthermore, a recent study using cell monolayers has found a benefit of all-trans retinoic acid supplementation in enhancing Caco-2 differentiation and preventing apoptosis only under serum-free conditions (Baltes et al., 2004).
In a model of small bowel resection, vitamin A partial replenishment (50% of restored vitamin A serum levels) before surgery was able to reverse the effects of deficiency on enterocyte's apoptosis, although the regain of cell proliferation and migration was not fully reached, suggesting that higher availability of vitamin A is required for renewal kinetics (Swartz-Basile et al., 2000; Swartz-Basile et al., 2003).
These observations also give an explanation for some findings showing the greater benefit of vitamin A supplementation in children under heavy burdens of diarrhea when other gut-trophic micronutrients, such as glutamine, are usually depleted (Chen et al., 2003; Walser et al., 1996). The broad retinol protective action following toxin A exposure in intestinal cell lines, named enhanced cell proliferation and migration and anti-apoptotic action, may also explain the rapid recovery of the intestinal barrier function following other pathogen-born diarrheal illnesses in poor settings of the developing world (Walser et al., 1996). Recently, Wang et al have shown that pellet releasing retinoic acid was anti-apoptotic and stimulate crypt proliferation and enterocyte migration post-intestinal resection in vitamin A deficient rats, which was found to be mediated via changes in the extracellular matrix (Wang et al,. 2007).
In summary, we have confirmed the heavy cytotoxic effect of C. difficile toxin A in a dose-dependent fashion, as previously shown by our group, and found a potential benefit of retinol supplementation to enhance intestinal cell adaptative responses against C. difficile toxin A challenge. The medical attention to the physiopathology of C. difficile induced nosocomial infections has tremendously grown since the emerging of a more virulent and resistant strain and additional support nutritional therapies are important (Musher et al., 2006; Voth and Ballard, 2005). Based on our findings, this study shed light to the role of this nutritional therapy to restore the intestinal barrier function following C. difficile toxin A diarrhea, although molecular mechanisms and possible synergistic effects with other gut-trophic nutrients yet warrant further investigation. In this regard, specific mechanisms and pharmacological interactions of retinol alone or in combination with other micronutrients to enhance the intestinal barrier healing following C. difficile toxin A challenge are under way in our laboratory, especially addressing cyclin and Wnt-pathways in regulating cell proliferation.
Acknowledgments
This study was supported in part by two Brazilian funding agencies, CNPq and CAPES, and also international collaborative NIH Grants, including Forgarty and Ellison Grants # D43 TW006578-04S1, # HD053131-01, ICIDR program Grant # U01 AI026512-15S2. We would like to thank the support from the Grant # 55000645 from the HHMI International Infectious Disease Scholarship. A.A.F.L. Maciel is recipient of overseas fellowship program in Global Health and Clinical Research from the Forgarty Internacional Center and Ellison Medical Foundation. We would like to acknowledge the technical assistance of L.J. Barrett and M.C. Pinho on development of standard cell culture operation procedures, work instructions and laboratory maintenance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baltes S, Nau H, Lampen A. All-trans retinoic acid enhances differentiation and influences permeability of intestinal Caco-2 cells under serum-free conditions. Dev Growth Differ. 2004;46(6):503–514. doi: 10.1111/j.1440-169x.2004.00765.x. [DOI] [PubMed] [Google Scholar]
- 2.Barreto ML, Santos LM, Assis AM, Araujo MP, Farenzena GG, Santos PA, Fiaccone RL. Effect of vitamin A supplementation on diarrhoea and acute lower-respiratory-tract infections in young children in Brazil. Lancet. 1994;344(8917):228–231. doi: 10.1016/s0140-6736(94)92998-x. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett JG, Taylor NS, Chang T, Dzink J. Clinical and laboratory observations in Clostridium difficile colitis. Am J Clin Nutr. 1980;33 11:2521–2526. doi: 10.1093/ajcn/33.11.2521. [DOI] [PubMed] [Google Scholar]
- 4.Beyzadeoglu M, Balkan M, Demiriz M, Tibet H, Dirican B, Oner K, Pak Y. Protective effect of vitamin A on acute radiation injury in the small intestine. Radiat Med. 1997;15(1):1–5. [PubMed] [Google Scholar]
- 5.Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. J Neurobiol. 2006;66(7):606–630. doi: 10.1002/neu.20242. [DOI] [PubMed] [Google Scholar]
- 6.Bohnsack BL, Hirschi KK. Nutrient regulation of cell cycle progression. Annu Rev Nutr. 2004;24:433–453. doi: 10.1146/annurev.nutr.23.011702.073203. [DOI] [PubMed] [Google Scholar]
- 7.Brito GA, Carneiro-Filho B, Oria RB, Destura RV, Lima AA, Guerrant RL. Clostridium difficile toxin A induces intestinal epithelial cell apoptosis and damage: role of Gln and Ala-Gln in toxin A effects. Dig Dis Sci. 2005;50(7):1271–1278. doi: 10.1007/s10620-005-2771-x. [DOI] [PubMed] [Google Scholar]
- 8.Brito GA, Fujji J, Carneiro-Filho BA, Lima AA, Obrig T, Guerrant RL. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J Infect Dis. 2002;186(10):1438–1447. doi: 10.1086/344729. [DOI] [PubMed] [Google Scholar]
- 9.Bulut K, Meier JJ, Ansorge N, Felderbauer P, Schmitz F, Hoffmann P, Schmidt WE, Gallwitz B. Glucagon-like peptide 2 improves intestinal wound healing through induction of epithelial cell migration in vitro - evidence for TGF-β-mediated effect. Reg Pept. 2004;121:137–123. doi: 10.1016/j.regpep.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Carneiro BA, Fujii J, Brito GA, Alcantara C, Oria RB, Lima AA, Obrig T, Guerrant RL. Caspase and bid involvement in Clostridium difficile toxin A-induced apoptosis and modulation of toxin A effects by glutamine and alanyl-glutamine in vivo and in vitro. Infect Immun. 2006;74(1):81–87. doi: 10.1128/IAI.74.1.81-87.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carneiro-Filho BA, Oria RB, Wood RK, Brito GA, Fujii J, Obrig T, Lima AA, Guerrant RL. Alanyl-glutamine hastens morphologic recovery from 5-fluorouracil-induced mucositis in mice. Nutrition. 2004;20(10):934–941. doi: 10.1016/j.nut.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 12.Chen P, Soares AM, Lima AA, Gamble MV, Schorling JB, Conway M, Barrett LJ, Blaner WS, Guerrant RL. Association of vitamin A and zinc status with altered intestinal permeability: analyses of cohort data from northeastern Brazil. J Health Popul Nutr. 2003;21(4):309–315. [PubMed] [Google Scholar]
- 13.Dignass AU. Mechanisms and modulation of intestinal epithelial repair. Inflamm Bowel Dis. 2001;7(1):68–77. doi: 10.1097/00054725-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 14.Grotto I, Mimouni M, Gdalevich M, Mimouni D. Vitamin A supplementation and childhood morbidity from diarrhea and respiratory infections: a meta-analysis. J Pediatr. 2003;142(3):297–304. doi: 10.1067/mpd.2003.116. [DOI] [PubMed] [Google Scholar]
- 15.Guerrant RL, Hughes JM, Lima NL, Crane J. Diarrhea in developed and developing countries: magnitude, special settings, and etiologies. Rev Infect Dis. 1990;12 1:S41–S50. doi: 10.1093/clinids/12.Supplement_1.S41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96(3):736–749. [PubMed] [Google Scholar]
- 17.Hilakivi-Clarke L, Wang C, Kalil M, Riggins R, Pestell RG. Nutritional modulation of the cell cycle and breast cancer. Endocr Relat Cancer. 2004;11(4):603–622. doi: 10.1677/erc.1.00665. [DOI] [PubMed] [Google Scholar]
- 18.Johal SS, Solomon K, Dodson S, Borriello SP, Mahida YR. Differential effects of varying concentrations of clostridium difficile toxin A on epithelial barrier function and expression of cytokines. J Infect Dis. 2004;189(11):2110–2119. doi: 10.1086/386287. [DOI] [PubMed] [Google Scholar]
- 19.Kitamura M, Ishikawa Y, Moreno-Manzano V, Xu Q, Konta T, Lucio-Cazana J, Furusu A, Nakayama K. Intervention by retinoic acid in oxidative stress-induced apoptosis. Nephrol Dial Transplant. 2002;17 9:84–87. doi: 10.1093/ndt/17.suppl_9.84. [DOI] [PubMed] [Google Scholar]
- 20.Kyne L, Hamel MB, Polavaram R, Kelly CP. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin Infect Dis. 2002;34(3):346–353. doi: 10.1086/338260. [DOI] [PubMed] [Google Scholar]
- 21.Li E, Tso P. Vitamin A uptake from foods. Curr Opin Lipidol. 2003;14(3):241–247. doi: 10.1097/00041433-200306000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Lima AA, Lyerly DM, Wilkins TD, Innes DJ, Guerrant RL. Effects of Clostridium difficile toxins A and B in rabbit small and large intestine in vivo and on cultured cells in vitro. Infect Immun. 1988;56(3):582–588. doi: 10.1128/iai.56.3.582-588.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lissoos TW, Davis AE, Levin MS. Vitamin A trafficking in Caco-2 cells stably transfected with cellular retinol binding proteins. Am J Physiol. 1995;268(2 Pt 1):G224–G231. doi: 10.1152/ajpgi.1995.268.2.G224. [DOI] [PubMed] [Google Scholar]
- 24.Liu TS, Musch MW, Sugi K, Walsh-Reitz MM, Ropeleski MJ, Hendrickson BA, Pothoulakis C, Lamont JT, Chang EB. Protective role of HSP72 against Clostridium difficile toxin A-induced intestinal epithelial cell dysfunction. Am J Physiol Cell Physiol. 2003;284(4):C1073–C1082. doi: 10.1152/ajpcell.00134.2002. [DOI] [PubMed] [Google Scholar]
- 25.Mitra AK, Alvarez JO, Wahed MA, Fuchs GJ, Stephensen CB. Predictors of serum retinol in children with shigellosis. Am J Clin Nutr. 1998;68(5):1088–1094. doi: 10.1093/ajcn/68.5.1088. [DOI] [PubMed] [Google Scholar]
- 26.Musch MW, Walsh-Reitz MM, Chang EB. Roles of ZO-1, occludin, and actin in oxidant-induced barrier disruption. Am J Physiol Gastrointest Liver Physiol. 2006;290(2):G222–G231. doi: 10.1152/ajpgi.00301.2005. [DOI] [PubMed] [Google Scholar]
- 27.Musher DM, Logan N, Mehendiratta V. Epidemic Clostridium difficile. N Engl J Med. 2006;354(11):1199–1203. doi: 10.1056/NEJMc053654. [DOI] [PubMed] [Google Scholar]
- 28.Nusrat A, von Eichel-Streiber C, Turner JR, Verkade P, Madara JL, Parkos CA. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect Immun. 2001;69(3):1329–1336. doi: 10.1128/IAI.69.3.1329-1336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pothoulakis C. Pathogenesis of Clostridium difficile-associated diarrhoea. Eur J Gastroenterol Hepatol. 1996;8(11):1041–1047. doi: 10.1097/00042737-199611000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Quadro L, Gamble MV, Vogel S, Lima AA, Piantedosi R, Moore SR, Colantuoni V, Gottesman ME, Guerrant RL, Blaner WS. Retinol and retinol-binding protein: gut integrity and circulating immunoglobulins. J Infect Dis. 2000;182 1:S97–S102. doi: 10.1086/315920. [DOI] [PubMed] [Google Scholar]
- 31.Simoni D, Tolomeo M. Retinoids, apoptosis and cancer. Curr Pharm Des. 2001;7(17):1823–1837. doi: 10.2174/1381612013397168. [DOI] [PubMed] [Google Scholar]
- 32.Swartz-Basile DA, Rubin DC, Levin MS. Vitamin A status modulates intestinal adaptation after partial small bowel resection. JPEN J Parenter Enteral Nutr. 2000;24(2):81–88. doi: 10.1177/014860710002400281. [DOI] [PubMed] [Google Scholar]
- 33.Swartz-Basile DA, Wang L, Tang Y, Pitt HA, Rubin DC, Levin MS. Vitamin A deficiency inhibits intestinal adaptation by modulating apoptosis, proliferation, and enterocyte migration. Am J Physiol Gastrointest Liver Physiol. 2003;285(2):G424–G432. doi: 10.1152/ajpgi.00524.2002. [DOI] [PubMed] [Google Scholar]
- 34.Thomas S, Prabhu R, Balasubramanian KA. Retinoid metabolism in the rat small intestine. Br J Nutr. 2005;93(1):59–63. doi: 10.1079/bjn20041306. [DOI] [PubMed] [Google Scholar]
- 35.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18(2):247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walser BL, Lima AA, Guerrant RL. Effects of high-dose oral vitamin A on diarrheal episodes among children with persistent diarrhea in a northeast Brazilian community. Am J Trop Med Hyg. 1996;54(6):582–585. doi: 10.4269/ajtmh.1996.54.582. [DOI] [PubMed] [Google Scholar]
- 37.Wang JL, Swartz-Basile DA, Rubin DC, Levin MS. Retinoic acid stimulates early cellular proliferation in the adapting remnant rat small intestine after partial resection. J Nutr. 1997;127(7):1297–1303. doi: 10.1093/jn/127.7.1297. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, Tang Y, Rubin DC, Levin MS. Chronically administered retinoic acid has trophic effects in the rat small intestine and promotes adaptation in a resection model of short bowel syndrome. Am J Physiol Gastrointest Liver Physiol. 2007 doi: 10.1152/ajpgi.00567.2006. [DOI] [PubMed] [Google Scholar]
- 39.Warden RA, Noltorp RS, Francis JL, Dunkley PR, O'Loughlin EV. Vitamin A deficiency exacerbates methotrexate-induced jejunal injury in rats. J Nutr. 1997;127(5):770–776. doi: 10.1093/jn/127.5.770. [DOI] [PubMed] [Google Scholar]
- 40.Wilkins TD, Lyerly DM. Clostridium difficile testing: after 20 years, still challenging. J Clin Microbiol. 2003;41(2):531–534. doi: 10.1128/JCM.41.2.531-534.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zaiger G, Nur T, Barshack I, Berkovich Z, Goldberg I, Reifen R. Vitamin A exerts its activity at the transcriptional level in the small intestine. Eur J Nutr. 2004;43(5):259–266. doi: 10.1007/s00394-004-0466-2. [DOI] [PubMed] [Google Scholar]
- 42.Zamora M, Ortega JA, Alana L, Vinas O, Mampel T. Apoptotic and anti-proliferative effects of all-trans retinoic acid. Adenine nucleotide translocase sensitizes HeLa cells to all-trans retinoic acid. Exp Cell Res. 2006;312(10):1813–1819. doi: 10.1016/j.yexcr.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 43.Ziegler TR, Evans ME, Fernandez-Estivariz C, Jones DP. Trophic and cytoprotective nutrition for intestinal adaptation, mucosal repair, and barrier function. Annu Rev Nutr. 2003;23:229–261. doi: 10.1146/annurev.nutr.23.011702.073036. [DOI] [PubMed] [Google Scholar]
- 44.Zile M, Bunge C, Deluca HF. Effect of vitamin A deficiency on intestinal cell proliferation in the rat. J Nutr. 1977;107(4):552–560. doi: 10.1093/jn/107.4.552. [DOI] [PubMed] [Google Scholar]