

Abstract
Objective
Right ventricular contractile failure from acute RV pressure overload is an important cause of morbidity and mortality, but the mechanism of RV failure in this setting is incompletely defined. We hypothesized that RV dysfunction from acute RV pressure overload is in part due to activation of calpain, and that calpain inhibition would therefore attenuate RV dysfunction.
Methods
Anesthetized, open chest pigs were treated with the calpain inhibitor MDL-28170 or with inactive vehicle, and then subjected to acute RV pressure overload for 90 min. RV contractile function was assessed by the regional Frank-Starling relation. RV myocardial tissue was analyzed for evidence of calpain activation and calpain-mediated proteolysis.
Results
RV pressure overload caused severe contractile dysfunction, along with significant alterations in the endogenous calpain inhibitor calpastatin typical of calpain activation. MDL-28170 attenuated RV free wall dysfunction by more than 50%. However, there were no differences in degradation of spectrin, desmin, troponin-I or SERCA2 between SHAM operated pigs and pigs subjected to acute RV pressure overload, or between vehicle and MDL-28170 treated pigs.
Conclusions
Acute RV pressure overload causes calpain activation, and RV contractile dysfunction from acute RV pressure overload is attenuated by the calpain inhibitor MDL-28170; however, the effect is not explained by inhibition of calpain-mediated degradation of spectrin, desmin, troponin-I or SERCA2. Since this is the first report of any agent that can directly attenuate RV contractile dysfunction in acute RV pressure overload, further investigation of the mechanism of action of MDL-28170 in this setting is warranted.
Keywords: hypertension, pulmonary; ventricular dysfunction, right; myocardial contraction; calpain; protease inhibitors; calpastatin
Introduction
Right ventricular (RV) contractile failure from acute RV pressure overload is an important cause of morbidity and mortality in conditions such as massive pulmonary embolism, hypoxic pulmonary vasoconstriction, and following cardiopulmonary bypass and cardiac transplantation. More than 600,000 cases of pulmonary embolism occur in the United States each year; mortality remains high, and is strongly related to the development of RV dysfunction [1] [2] [3]. Acute RV contractile failure accounts for 50% of all cardiac complications and 19% of all early deaths in cardiac transplant patients [4], and acute RV contractile failure continues to be a major cause of morbidity and mortality in several other important classes of patients as well [4, 5].
It was long assumed that RV failure during acute pressure overload was simply a load-dependent effect, i.e., stroke volume diminished due to increased afterload, rather than an alteration of intrinsic RV contractility. Thus, therapeutic strategies for management of RV contractile failure have concentrated on maneuvers to reduce RV afterload, such as pulmonary thromboendarterectomy or fibrinolysis in pulmonary embolism [6, 7], inhaled nitric oxide following cardiopulmonary bypass or cardiac transplant [4], and on use of positive inotropic agents such as dobutamine. While there is no question that global RV ejection fraction sometimes improves with such therapeutic interventions, response to these interventions is highly variable.
Recently, we [8, 9] and others [10] have shown that intrinsic RV contractile dysfunction occurs in the setting of acute RV pressure overload. Using a porcine model of pulmonary artery constriction and release, we showed that RV contractile function remains significantly depressed after a brief period of pressure overload, despite restoration of normal loading conditions. The severity of RV dysfunction following pressure overload is directly related to the level of RV free wall stress during pressure overload [9]. Moreover, the RV contractile dysfunction observed after pressure overload occurs in the absence of detectable RV ischemia during or following pressure overload [8]. This persistent alteration in RV contractile function following brief pressure overload indicates that intrinsic alterations in RV contractile function may contribute to RV failure during RV pressure overload.
Obviously, clinical recovery from acute RV pressure overload is strongly influenced by success in alleviating whatever condition is responsible for pressure overload in the first place; but it is the RV contractile dysfunction, rather than the pressure overload per se, that directly contributes to morbidity and mortality. Therefore, any factors that affect the development of or recovery from RV contractile dysfunction must necessarily play direct and important roles in ultimate clinical outcome. To date, there has been little investigative activity directed toward this phenomenon, and there are currently no therapeutic interventions available to directly attenuate the development of RV contractile dysfunction from acute pressure overload.
Some investigators have shown that skeletal muscle injury and dysfunction following prolonged or strenuous exercise is associated with activation of the calcium-sensitive cysteine protease calpain, and that such activation is associated with degradation of several myofibrillar and cytoskeletal proteins [11]. Myocardial ischemia-reperfusion is also associated with calpain activation; the resulting “stunning” is attenuated by broad-spectrum cysteine protease inhibitors such as leupeptin [12], as well as by more specific calpain inhibitors [13, 14]. By analogy, we hypothesized that RV pressure overload leads to activation of calpain, and that RV dysfunction would therefore be attenuated by administration of a calpain inhibitor.
To test this hypothesis, we studied the effects of the narrow-spectrum dipeptidyl aldehyde calpain inhibitor MDL-28170 (calpain inhibitor III, Z-Val-Phe-CHO) on RV contractile function in our porcine model of acute RV pressure overload, and probed for alterations in substrates sometimes found to be degraded by calpain in other models of contractile dysfunction. We found evidence of calpain activation in acute RV pressure overload, and attenuation of RV dysfunction by a calpain inhibitor.
Materials and Methods
This investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the National Academy Press (Institute for Laboratory Animal Research, revised 1996). It was approved by the Animal Studies Subcommittees of the University of Colorado Health Sciences Center and the Denver Department of Veterans Affairs Medical Center.
Instrumentation
The experimental preparation was similar to that previously described by this laboratory [9]. Forty adolescent domestic farm pigs of either sex (Landrace strain, 25-30 kg) were sedated with ketamine HCl (25 mg/kg IM), followed by α-chloralose (100 mg/kg IV via an ear vein); deep anesthesia was then maintained throughout the remainder of the experiment with α-chloralose (30-50 mg/kg-hr IV). The pigs were wrapped in a recirculating warm water blanket to prevent hypothermia. Normal saline solution was infused continuously at 150-250 ml/hr IV. Pigs were intubated via tracheotomy and ventilated with an air-O2 mixture using a pressure-cycled ventilator adjusted to maintain pCO2 between 35 and 45 mmHg, pH between 7.35 and 7.45, and pO2 greater than 100 mmHg.
After exposure via median sternotomy, the pericardium was opened and the heart was instrumented as previously described [9]. A solid-state micromanometer catheter (Millar Instruments, Houston, TX) was introduced into the RV via an internal jugular vein. A transit-time ultrasonic flow probe (Transonic Systems, Ithaca, NY) was fitted around the main pulmonary artery for measurement of cardiac output. An umbilical tape snare was placed around the main pulmonary artery to produce RV pressure overload, and a hydraulic occluder was placed around the inferior vena cava to dynamically alter preload. An array of four piezoelectric crystals was implanted in the central anterior RV free wall for determination of segment shortening using a multichannel digital sonomicrometer (Sonometrics, London, Ontario, Canada). A second micromanometer catheter was inserted into the left ventricle via apical puncture, pacing wires were affixed to the left atrial appendage, and a bipolar pacing lead was affixed to the RV endocardium adjacent to the crystal array for measurement of the intramyocardial electrogram.
Experimental Protocols
Autonomic blockade was produced with atropine (0.2 mg/kg IV) and propranolol (1.0 mg/kg IV), and the heart was paced approximately 10 bpm faster than the spontaneous heart rate, to prevent potential alterations in indices of contractile function due solely to changes in sympathetic tone and heart rate [15].
Pigs were studied in two non-concurrent series
In Series 1, pigs subjected to acute RV pressure overload (RVPO, n=9) were compared with pigs not subjected to RV pressure overload (SHAM, n=7) to identify biochemical alterations associated with RV pressure overload. Following instrumentation, the preparation was monitored for 30 min to verify stability. In RVPO pigs, the pulmonary artery occluder was constricted over 5 to 10 min to obtain the highest RV pressure achievable without producing progressive systemic hypotension. During the next 90 min, the pulmonary artery constrictor was periodically readjusted as necessary to maintain the highest sustainable RV systolic pressure. After 90 min RV pressure overload, the constrictor was fully released. In our previous investigations, we used measurements of RV perfusion and transmyocardial lactate metabolism to establish that this protocol does not result in detectable RV ischemia [8]. Hemodynamic measurements were obtained under baseline conditions, during acute RV pressure overload, immediately after release of RV pressure overload, and 30 min later. SHAM pigs were instrumented identically to RVPO pigs except that the pulmonary artery was not instrumented, and underwent hemodynamic measurements at the same time intervals.
In Series 2, 24 pigs were subjected to acute RV pressure overload to explore the role of calpain in the development of RV contractile dysfunction. In these pigs, the proximal portion of the right coronary artery was dissected free and cannulated with a 23 gauge steel needle, and a transit-time ultrasonic flow probe was fitted around the right coronary artery just distal to the infusion needle for measurement of coronary blood flow. Half were pretreated with a calpain inhibitor (RVPO+INH, n=12) and half with inactive vehicle (RVPO+VEH, n=12) before being subjected to acute RV pressure overload. Heparin sodium 5000 u IV was administered to prevent thrombosis around the coronary infusion needle. MDL-28170 (Z-Val-Phe-CHO, CalBiochem) is a membrane-permeable cysteine protease inhibitor with a Ki for calpain of 8 nM [16]. After baseline measurements of hemodynamics and RV function were obtained, infusion of either MDL-28170 (10 μM per liter in 1% DMSO/phosphate buffered saline vehicle, pH 7.40) or inactive vehicle (1% DMSO/phosphate buffered saline) was begun, with the infusion rate adjusted to 10% of the measured coronary flow, as measured with the ultrasonic flow probe. The target coronary arterial concentration of 1 μM per liter was approximately 100-fold higher than the inhibitor’s Ki for calpain. The infusion was continued throughout the remainder of the experiment, with the rate adjusted periodically according to the measurement of RCA flow to maintain a consistent intracoronary concentration of inhibitor or vehicle.
Thirty min after beginning inhibitor or vehicle infusion, a second set of measurements of hemodynamics and RV function was repeated to determine the effects of treatment in the absence of RV pressure overload. The remainder of the protocol was identical to that described under Series 1, above.
In all pigs, euthanasia was performed at the conclusion of each experiment under deep general anesthesia with IV infusion of 10% KCl to induce ventricular fibrillation. Myocardial tissue was then immediately harvested from the region adjacent to the sonomicrometry crystals. Specimens were freeze-clamped on a liquid-nitrogen cooled mortar within 15 s of collection, and then stored under liquid nitrogen until analysis.
Hemodynamic Measurements
Hemodynamic data were digitized at 200 Hz and recorded during 10 s suspensions of mechanical ventilation. End-diastole was defined as the point corresponding to the peak of the R-wave of the intramyocardial electrogram. End-systole was defined as the point of maximum negative RV dP/dt.
The instantaneous RV free wall area was continuously calculated from the product of the longitudinal and transverse chord lengths between crystals, determined by the digital sonomicrometer. Regional external work was defined as the area of an RV pressure-wall area loop (i.e., the two-dimensional plot of RV pressure vs. RV free wall area) obtained during a single cardiac cycle normalized to the baseline end-diastolic regional RV free wall area.
To assess regional RV free wall contractile function independent of loading conditions, we used the regional Frank-Starling relation. The regional Frank-Starling relation (sometimes referred to as the “preload-recruitable stroke work relation”) has been extensively validated as a loading condition-insensitive index of contractility in both the LV [17] and the RV [18]. It is reliable in the RV free wall over large variations in LV pressure [19], and is superior to the end-systolic pressure dimension relation for in vivo assessment of RV contractile function under diverse conditions [20]. As previously described [9], data were recorded during 3-6 repetitions of 10 s occlusion of the inferior vena cava, spaced at 1-2 min intervals. The area of each RV free wall area-pressure loop obtained during a dynamic inferior vena cava occlusion (i.e., regional external work) was plotted against its corresponding end-diastolic RV free wall area. The slope and intercept of the resulting Frank-Starling relation were determined by linear regression. Regional RV Frank-Starling relations were determined at baseline, during Inhibitor or Vehicle treatment immediately before beginning RV pressure overload in Series 2 pigs, immediately after relief of RV pressure overload, and at the end of the experiment. Frank-Starling relations were not determined during RV pressure overload because the dynamic IVC occlusions result in unacceptable hemodynamic instability during severe RV pressure overload. We have found that interventions that alter contractility, result in changes in both slope and intercept of the regional Frank-Starling relation [21]. Therefore, to compare RV regional external work at recovery with the value determined at baseline under conditions of matched preload, we calculated preload-adjusted regional external work in each pig from the regional Frank-Starling relation slope and intercept with preload (end-diastolic RV free wall area) set at its baseline, steady state value. This index of contractile function integrates changes in both the slope and intercept of the regional Frank-Starling relation into a single estimate of loading condition-independent regional external work capacity [9].
Biochemical Analyses
For polyacrylamide gel electrophoresis and Western immunoblotting, tissue was homogenized at 4°C using a glass-glass homogenizer in 10 vol of buffer (20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM DTT, 0.1 mM leupeptin, 1 mM AEBSF, and 10 μg/ml aprotinin). Homogenates were centrifuged at 4000 X g force for 5 min to pellet debris and the supernatant protein extracts were collected. Bradford protein assay was performed and protein concentrations equalized. Protein extracts were mixed with LDS sample buffer and sample reducing agent (Invitrogen), heated (70°C, 10 min), loaded at 20 μg total protein per sample well into a 4-12% gradient bis-tris or 3-8% tris-acetate mini gels (Invitrogen), and run using MES or TA buffer according to manufacturer recommendations. Proteins were transferred to PVDF membranes using NuPAGE transfer buffer (Invitrogen, 30 V, 60 min). Membranes were blocked in 5% nonfat milk/Tris buffered saline (50 mM Tris-HCl, 0.15 M NaCl, TBS; 2 hrs, RT) incubated with monoclonal antibodies (calpain 1 domain III, clone 9A4H8D3, Calbiochem, 1:5000; spectrin, clone AA6, Research Diagnostics, Concord, MA, 1:1000; desmin, clone DEU-10, Sigma, St. Louis, MO, 1:7500; troponin-I, clone MAB I7, Research Diagnostics, 1:50000; SERCA2, clone IID8, CalBiochem, 1:7500; calpastatin domain IV, clone 1F7E3D10, Calbiochem, 1:5000) diluted in Tris-Tween buffer (TBS pH 7.4 plus 0.1% Tween-20, TBST; 2 hrs, RT) and rinsed 3 times for 10 min in TBST. Membranes were simultaneously probed for tubulin (cat# ab7291, Abcam, Cambridge, MA, 1:10000) or β-actin (Abcam cat# 8226, 1:500) as a control for loading. Membranes were then incubated in HRP-conjugated anti-mouse Fab antibody diluted in TBST (1:7500, 1 hr, RT), rinsed 4 times in TBST for 10 min each, then visualized on x-ray film using either an enhanced chemiluminescence kit or using a TMB chromogenic assay (Pierce, Woburn, MA). Quantitation was performed by scanning and densitometric analysis (Phoretix, Nonlinear Dynamics, Durham, NC).
Six pigs from each group were randomly selected for the biochemical analyses so that the key comparisons (SHAM vs RVPO in Series 1; and RVPO+VEH vs RVPO+INH in Series 2) could be performed within a single gel, facilitating the quantitative analysis by eliminating inter-gel variability. To ensure that calpain-specific degradation bands could be identified in our system, gels loaded with myocardial homogenate digested in vitro by addition of calpain according to the protocol of Barta [22] were probed for spectrin, desmin and troponin-I.
Statistical Analysis
All results are reported as mean±SEM. Hemodynamic variables under each experimental condition and Western immunoblotting densitometry data were compared between groups (SHAM vs RVPO and RVPO+VEH vs RVPO+INH) using unpaired Student’s t-tests. Repeated measures ANOVA was performed to identify changes from baseline conditions to RVPO or recovery conditions within groups, and were thus used to determine whether MDL-28170 or Vehicle had independent hemodynamic effects under baseline conditions.
Results
All Series 1 pigs completed the experimental protocol. Two RVPO+INH and two RVPO+VEH pigs in Series 2 did not survive to complete the experimental protocol due to hemodynamic collapse during or following RV pressure overload; these pigs were excluded from the analysis. Table 1 shows hemodynamic data for the four groups of pigs: Series 1 SHAM operated pigs and RVPO pigs; and Series 2 pigs subjected to RV pressure overload with pretreatment with vehicle (RVPO+VEH) or intracoronary cysteine protease inhibitor MDL-28170 (RVPO+INH). Data are shown under baseline conditions, during RV pressure overload (or at the equivalent time in SHAM operated pigs not subjected to RV pressure overload), and 30 min following release of RV pressure overload (or equivalent time in SHAM).
Table 1. Hemodynamic Measurements.
All values are mean±SEM; p values are for comparison with Baseline within the same group using repeated measures ANOVA, with post-hoc testing by the Bonferroni/Dunn method
| Series 1 | Series 2 | ||||
|---|---|---|---|---|---|
| SHAM (n=7) | RVPO (n=9) | RVPO+VEH (n=10) | RVPO+INH (n=10) | ||
| Heart Rate (bpm) | |||||
| Baseline | 137±1 | 139±1 | 137±1 | 135±1 | |
| RV Pressure Overload | 138±2 | 142±2 | 136±1 | 136±1 | |
| Recovery | 138±1 | 140±1 | 137±1 | 137±1 | |
| RV Systolic Pressure (mmHg) | |||||
| Baseline | 29±2 | 30±1 | 34±1 | 31±1 | |
| RV Pressure Overload | 27±1 | 59±3 p<0.001 | 60±2 p<0.001 | 61±1 p<0.001 | |
| Recovery | 26±1 | 31±2 | 31±1 | 30±1 | |
| RV End-Diastolic Pressure (mmHg) | |||||
| Baseline | 5±2 | 3±1 | 5±1 | 3±1 | |
| RV Pressure Overload | 4±2 | 6±1 p<0.01 | 7±1 p<0.01 | 5±1 p=0.01 | |
| Recovery | 3±2 | 5±1 p<0.05 | 5±1 | 5±1 | |
| LV Systolic Pressure (mmHg) | |||||
| Baseline | 106±6 | 100±5 | 106±3 | 109±5 | |
| RV Pressure Overload | 110±6 | 91±6 p<0.05 | 90±4 p<0.01 | 101±5 | |
| Recovery | 110±7 | 107±5 | 100±3 | 102±4 | |
| Cardiac Output (l/min) | |||||
| Baseline | Not Obtained | 3.3±0.2 | 3.2±0.2 | 3.2±0.2 | |
| RV Pressure Overload | 2.8±0.2 | 2.2±0.3 | 2.6±0.3 | ||
| Recovery | 2.9±0.3 | 2.6±0.3 | 3.1±0.2 | ||
| RV Free Wall End-Diastolic Area (Recovery/Baseline) | |||||
| RV Pressure Overload | 0.97±0.01 | 1.21±0.04 p<0.01 | 1.22±0.05 p<0.01 | 1.14±0.03 p<0.01 | |
| Recovery | 0.96±0.02 | 1.16±0.06 p<0.05 | 1.12±0.03 p<0.01 | 1.02±0.02 | |
| Regional Frank-Starling Relation (Recovery/Baseline) | |||||
| Slope | 0.94±0.04 p=0.14 | 0.70±0.07 p<0.01 | 0.69±0.05 p<0.01 | 0.79±0.10 p=0.07 | |
| Intercept | 1.06±0.02 p<0.05 | 1.12±0.05 p<0.05 | 1.16±0.05 p<0.01 | 1.00±0.03 | |
Ninety minutes of acute RV pressure overload causes severe RV contractile dysfunction
There were no significant differences between groups with respect to baseline heart rate, systemic or RV pressure, or cardiac output. In SHAM pigs, there was a small decline (15±5%, p=0.02) in preload adjusted regional external work over the course of the experiment (SHAM example in Figure 1 and summary data in Figure 2). In pigs subjected to RV pressure overload, pulmonary artery constriction resulted in an RV systolic pressure of 59±3 mmHg, compared with baseline30±1 mmHg. RV systolic pressure returned to its baseline value after release of pulmonary artery constriction. In pigs subjected to RV pressure overload, we observed a 53±14% decline in our primary index of RV contractile function, preload-adjusted regional external work, similar to what we and others have reported previously (RVPO example in Figure 1, p<0.01 for summary data in Figure 2) [8-10]. The reduction in preload-adjusted regional external work was due to both a significant decline in slope and a significant increase in the x-axis intercept of the regional Frank-Starling relation (Table 1). In SHAM pigs, the minimal decline in preload adjusted regional external work over the course of the experiment was primarily due to a small increase in the x-axis intercept of the regional Frank-Starling relation.
Figure 1.
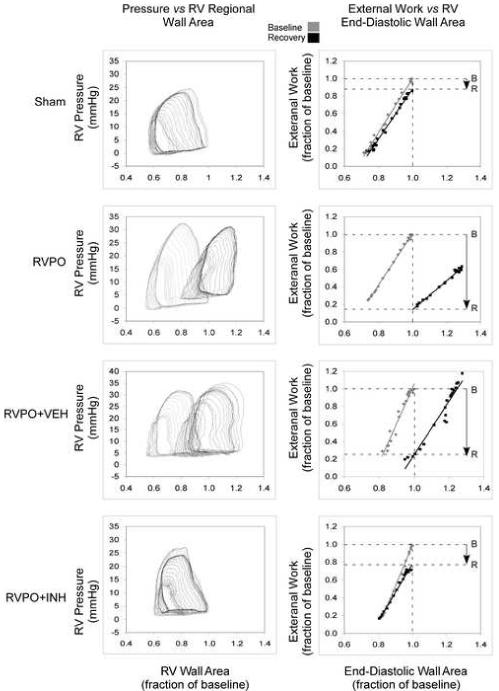
Examples of pressure versus RV free wall area (left column) and regional Frank-Starling relations (right column) obtained from a sham-operated pig (Sham, top), a pig subjected to RV pressure overload (RVPO, middle), a pig treated with inactive vehicle prior to being subjected to RV pressure overload (RVPO+VEH, next), and a pig treated with MDL-28170 prior to being subjected to RV pressure overload (RVPO+INH, bottom). Examples were chosen to accentuate the differences observed. Loops and relations in gray were obtained under baseline conditions; loops and relations in black were obtained following release of RV pressure overload. The horizontal dotted lines indicate baseline (B) and recovery (R) regional external work at baseline end-diastolic wall area (vertical dotted line). In these examples, there was a slight rightward shift in the regional Frank-Starling relation in the SHAM example, resulting in an 15% reduction in preload-adjusted regional external work; in the RVPO example, there was an 85% decline in preload-adjusted regional external work from baseline; in the RVPO+VEH example, the decline in preload adjusted regional external work was 75%; in the RVPO+INH example, the decline in preload-adjusted regional external work was attenuated, with only a 22% decline in preload-adjusted regional external work from baseline.
Figure 2. RV Free Wall Preload-Adjusted Regional External Work.

Average preload-adjusted regional external work in the RV free wall at the end of the experiment, normalized to baseline values, in SHAM pigs (n=7), untreated RV pressure overload pigs (RVPO, n=19), and RV pressure overload pigs treated with either vehicle (RVPO+VEH, n=10) or MDL-28170 (RVPO+INH, n=10). RV pressure overload resulted in a large and significant reduction in RV free wall contractile function; MDL-28170 treatment significantly attenuated the RV contractile dysfunction due to pressure overload (p < 0.05 for RVPO vs SHAM and p<0.01 for RVPO+INH vs RVPO+VEH treated pigs).
MDL-28170 attenuates RV contractile dysfunction due to acute RV pressure overload
Neither infusion of MDL-28170 nor of the inactive Vehicle had any significant effects on any hemodynamic measurement under baseline conditions, and did not alter the regional Frank-Starling relation prior to RV pressure overload (data not shown). However, MDL-28170 significantly attenuated RV dysfunction resulting from acute RV pressure overload: 30 min after release of pulmonary artery constriction, preload-adjusted regional external work of the RV free wall had declined by only 21±10% from baseline in RVPO+INH treated pigs, compared with the 68±10% decline from baseline in RVPO+VEH pigs (p=0.004 for difference, RVPO+INH example in Figure 1 and summary data in Figure 2). In RVPO+VEH pigs, RV contractile dysfunction after RV pressure overload was due to both a reduction in slope and a rightward shift in the regional Frank-Starling relation, as we have previously reported. In RVPO+INH pigs, there was a similar reduction in slope but no shift in x-axis intercept of the regional Frank-Starling relation (Table 1). The abolition of a rightward shift in the regional Frank-Starling relation by MDL-28170 treatment accounted for most of the relative preservation of contractile function in RVPO+INH compared with RVPO+VEH pigs.
There was a trend toward higher cardiac output in the RVPO+INH group compared with the RVPO+VEH group during RV pressure overload and recovery, although this difference did not reach statistical significance by two-way repeated measures ANOVA. End-diastolic wall area increased in RVPO+VEH vs RVPO+INH pigs, both during RVPO and 30 min after release of RVPO, with slightly higher RVEDP in RVPO+VEH vs RVPO+INH. The increase in end-diastolic wall area was comparable to what we have reported previously in open-chest, open pericardium pigs [9], and limited the decline in cardiac output that would otherwise be expected due to RV contractile dysfunction because of recruitment via the Frank-Starling mechanism.
Acute RV pressure overload caused alterations in the endogenous calpain inhibitor calpastatin without altering calpain-1 protein level
Because of the relatively short period of the experiment, we expected that calpain abundance would be unchanged in RVPO but that calpain would be activated by acute RV pressure overload, as occurs in several other pathological states, and then subsequently degrade structural or regulatory proteins. Calpain-1 is the calpain isoform most sensitive to calcium activation, and therefore most likely to be activated by calcium fluctuations associated with an increase in contractile activity [23]. As expected, we found no difference in calpain-1 abundance among any of the groups (Western immunoblotting data not shown).
Calpastatin is a potent and specific endogenous calpain inhibitor with no other known antiprotease activity. Calpastatin is thought to play an important role in calpain regulation, and alteration of calpastatin isoform abundance alters calpain activity in vivo [24]. Figure 3 shows that several isoforms of calpastatin were significantly altered during RV pressure overload (p<0.01 for overall effect). There was a trend toward a decrease in the high molecular 110 kDa calpastatin band (p=0.3), a significant decrease in a lower-molecular weight 41 kDa band (p=0.04), and a significant increase (p=0.02) in a low MW band.
Figure 3. RV calpastatin.

Western immunoblots of myocardium from six RVPO vs six SHAM pigs (Series 1) probed for calpastatin; *: p < 0.05 RVPO vs SHAM. There is a trend toward reduction of the 110 kDa calpastatin band by 20% (p=0.3), a significant reduction in the 41 kDa calpastatin band of 23% (p=0.04), and a significant increase in the low molecular weight band of 60% (p=0.02) resulting from RV pressure overload.
Despite evidence of altered calpain activity reflected by altered calpastatin, acute RV pressure overload did not cause detectable degradation of desmin, troponin, spectrin, or SERCA2
Calpain activation may cause degradation of structural proteins such as spectrin [25] and desmin [12], and regulatory proteins such as troponin-I [26] and SERCA2 [27]. Degradation of one or more of these proteins has been associated with development of contractile dysfunction in ischemia-reperfusion, cardiomyopathy, and skeletal muscle stress [26, 28]. Nevertheless, we saw no significant increase in degradation of spectrin, desmin, or troponin-I in RVPO compared with SHAM, and no attenuation of degradation of these proteins in RVPO+INH compared with RVPO+VEH (Figures 4 and 5). Likewise, there was no difference in SERCA2A degradation with RV pressure overload (data not shown).
Figure 4. Western immunoblots of RV myocardium.
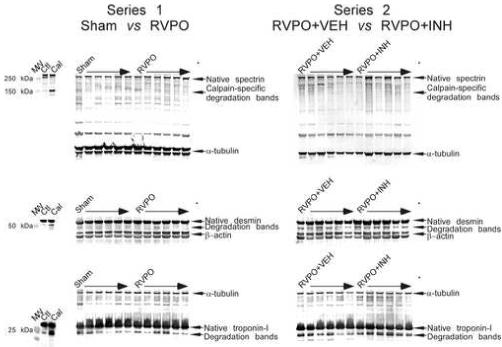
Panels on the left show myocardium from six SHAM operated pigs and six pigs subjected to RV pressure overload (RVPO) from Series 1. Panels on the right show myocardium from six pigs subjected to RV pressure overload pretreated with either inactive Vehicle (RVPO+VEH) or MDL-28170 (RVPO+INH) from Series 2. Blots were probed with antibodies to non-erythroid spectrin (clone AA6), desmin (clone DEU-10) and troponin-I (clone 8-I7), with either tubulin or β-actin used as controls for loading. Along the left side of the blots are additional lanes showing the effects of in vitro calpain digestion on spectrin, troponin-I and desmin. Molecular weight markers are designated MW. Ctl designates fresh RV myocardium obtained from a pig not subjected to RV pressure overload; Cal designates fresh RV myocardium incubated in vitro with purified human calpain I according to the protocol of Barta [15]. Calpain-specific degradation bands (identified by arrows at the right) could be detected easily in myocardium incubated in vitro with purified human calpain, but there were no detectable differences between the groups, indicating that there was no significant increase in calpain-mediated proteolysis of these proteins due to RV pressure overload, and no significant attenuation of calpain-mediated proteolysis of these proteins by MDL-28170. Both tubulin and β actin appear as doublets when run to for prolonged periods on electrophoresis gels; quantitation was performed by summing the bands.
Figure 5.
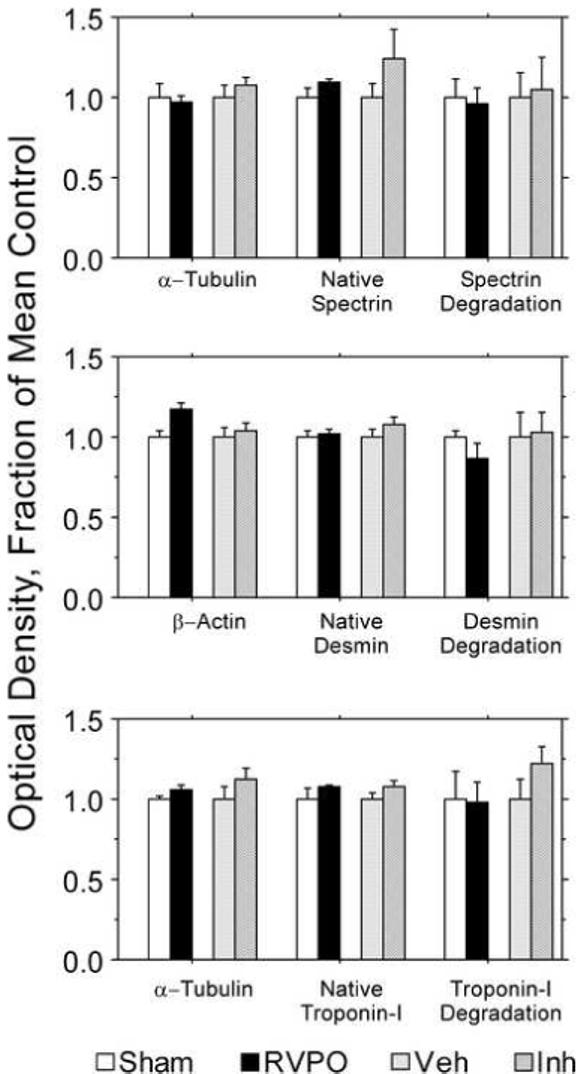
Densitometric analysis of the Western immunoblots shown in Figure 5 (see Figure 5 for description of immunoblots). There were no significant differences between groups for absolute values of degradation products, ratio of degradation products to native protein, or native protein normalized to the loading controls.
To ensure that our methods were capable of detecting calpain-mediated degradation of spectrin, desmin and troponin-I, samples of tissue homogenate digested in vitro with calpain were analyzed using the same system of antibodies. Spectrin, desmin and troponin-I degradation bands due to calpain digestion of tissue homogenate were easily detected in our system (leftmost bands of Figure 4). Therefore, it is unlikely that significant degradation of spectrin, desmin or troponin-I occurred during RV pressure overload or were attenuated by MDL-28170. Of note, the absence of an increase in desmin or troponin-I degradation in pigs subjected to RV pressure overload provides additional evidence that the dysfunction we observe following acute pressure overload is not the result of myocardial ischemia, since desmin and/or troponin-I degradation are commonly observed in that setting. Similarly, Muhlfeld et al did not detect evidence of ischemic myofibrillar damage in RVPO myocardium on transmission electron microscopy in a model of more severe and prolonged RVPO [29].
Discussion
Our results show that acute RV pressure overload alters calpain activity as reflected by altered calpastatin isoforms, and that RV contractile dysfunction from acute RV pressure overload is attenuated by a calpain inhibitor (MDL-28170). This is consistent with the hypothesis that RV contractile dysfunction after acute RV pressure overload is, at least in part, due to calpain activation. However, we have not yet identified the mechanism by which calpain causes RV contractile dysfunction in this setting. While it is possible our methods were insensitive to changes in spectrin, desmin or troponin-I degradation, tissue homogenate digested in vitro with calpain showed obvious degradation bands of native spectrin, desmin and troponin-I, typical of those that have been reported by other investigators in other models of contractile dysfunction [12, 26]. We did in fact detect low levels of spectrin, desmin and troponin-I degradation in all our samples, as has previously been reported in normal pig myocardium [30], but there were no differences between groups. Of note, the mouse monoclonal anti-troponin-I antibody we used (clone 8-I7) is reportedly more sensitive for the calpain-derived troponin-I degradation product than the mouse C5 clone or the rabbit polyclonal antibody used in the study reported by Thomas et al [30, 31]; therefore, it seems unlikely we would have missed a significant increase in troponin-I degradation.
Calpain-mediated degradation of several structural and regulatory proteins has been hypothesized to be mechanistically important in the development of contractile dysfunction in other models of myocardial contractile dysfunction. These include, among others, the structural proteins spectrin, desmin, and titin, and the regulatory proteins troponin-I, troponin-T, protein kinase c epsilon, and SERCA2. However, which proteins are degraded by calpain, and how endogenous and exogenous calpain inhibitors affect each of them, is highly model dependent. For example, no-flow ischemia-reperfusion appears to result in troponin-I degradation in perfused hearts [26], while low-flow ischemia-reperfusion in vivo may have a greater effect on troponin-T [32]. Anthracycline induced cardiac dysfunction is associated with titin degradation without any evidence of troponin degradation [33]. Desmin has been reported to be degraded by calpain in ischemic rat hearts [28], although in right heart failure from neonatal hypoxic pulmonary hypertension desmin organization changes without degradation [34]. Other regulatory proteins such as SERCA2 [27]and PKC epsilon [13] have been reported to be substrates for calpain in other settings.
Thus, the mechanism by which calpain affects contractile dysfunction appears to be highly variable. This may be due to compartmentalization of calpain activity [35], which restricts the substrates available to calpain depending on the setting. In our model, it seems likely that a substrate other than troponin-I, desmin, spectrin or SERCA2 is mechanistically important in the development of contractile dysfunction in our model [23], and/or that the important effects of calpain in acute RV pressure overload are compartmentalized. Emerging evidence suggests that focal adhesion complex structure is regulated in part by calpain [36], and is altered in numerous pathologic states including pressure overload [37]. We have begun studies to determine whether calpain and calpastatin play a role in altering focal adhesion complexes in acute RV pressure overload.
We interpret the changes in calpastatin we found as indicating altered calpain activity, but we did not measure calpain activity directly. Although methods for direct assessment of calpain activity have been reported, they may not reflect physiologically relevant in vivo calpain activity. For example, fluorometric assays of calpain activity measure total potential calpain activity at a specific point in time by adding calcium in excess to activate all the calpain present in a tissue homogenate, an approach that shows total potential calpain activity rather than the physiologic calpain activity present at relatively low intracellular calcium concentrations. Moreover, such methods cannot account for time varying changes in calpain activity in vivo due to time varying changes in calcium concentration. In our model, tissue was obtained from the RV free wall after restoration of control loading conditions, when intracellular calcium levels had presumably normalized, so it is likely that in vivo calpain activity at the end of the experiment was different from what it had been during pressure overload, and any direct measurement of calpain activity would not have correctly reflected the activity at the time of RV pressure overload. Moreover, while it was previously believed that calpain is distributed throughout the cytoplasm and regulated primarily by intracellular calcium concentration, more recent data suggest that calpain activity is tightly regulated via association with calpastatin and through intracellular redistribution and compartmentalization [23, 38]. Thus, measurement of calpain activity is not straightforward, and conventional enzymatic assay techniques may not be ideally suited to determining in vivo calpain activity.
Other investigators have also interpreted alterations in calpastatin as a reflection of altered calpain activity. Sorimachi et al and Enns et al both found that ischemia/reperfusion results in a decline in calpastatin inhibitory activity with no change in fluorometrically determined calpain activity [39, 40], but that calpain inhibition improves post-ischemic contractile function. Wei et al found altered calpain activity due to a change in calpastatin inhibitory activity in a model of sepsis-mediated muscle proteolysis [41]. Shi et al found down regulation of the 41 kDa isoform of calpastatin in kidney following renal ischemia/reperfusion similar to what we observed in the RV [42]. Alterations in calpastatin, which may be degraded by calpain or regulated through other mechanisms, are more likely to persist, and thus are likely more sensitive indicators of altered calpain activity.
Given that calpastatin has no known function other than regulation of calpain [23], it seems most likely that the alterations in calpastatin we and other investigators have identified indicate modulation of calpain activity. How calpastatin regulates calpain is incompletely understood, but it appears that different calpastatin domains can independently regulate calpain activity and subcellular location [43]. Thus, while alteration in calpastatin isoform abundance implies altered calpain activity, it is not possible at this time to predict exactly how a particular calpastatin isoform change will alter calpain activity.
Aside from the alteration in calpastatin isoforms we found, attenuation of RV dysfunction by MDL-28170 strongly implies a calpain-mediated effect, since that compound is relatively specific for calpain [44]. However, it remains possible that a cysteine protease other than calpain is mechanistically involved in the development of contractile dysfunction and was inhibited by MDL-28170 in this model. For example, although MDL-28170 is considered relatively specific for calpain, cathepsin B, a lysosomal cysteine protease, is also inhibited by MDL-28170 at a similar Ki of 12-24 nM [44]. We looked for evidence of cathepsin B activation by performing Western immunoblotting for myosin heavy chain [45] using the MF20 anti-myosin clone (Developmental Studies Hybridoma Bank, Iowa), but found no differences in myosin heavy chain degradation either (data not shown).
We have not yet determined the effects of calpain inhibition on calpastatin isoform abundance in our model of acute RV pressure overload. However, calpain inhibition would not necessarily be expected to affect calpastatin isoform abundance, since calpastatin is also regulated independently of calpain [42, 46]. For example, changes in calpastatin isoforms during ischemia/reperfusion in hearts may [40] or may not be [39] affected by calpain inhibition. Thus, in our model, administration of MDL-28170 could have attenuated an increase in calpain activity due to RV pressure overload induced release of calpastatin inhibitory activity without affecting calpastatin isoform content directly.
This study provides the initial observation that protease inhibition improves intrinsic contractile function in RV pressure overload. These observations not only provide an impetus to further exploration of the mechanism for these salutary effects, but may provide a rationale for investigation of the potential clinical utility of protease inhibition in acute RV pressure overload, particularly in cases where development of RV contractile dysfunction can be anticipated such as following complex cardiac surgery procedures.
Acknowledgments
Supported by the Department of Veterans Affairs Medical Research Service, NIH HL68606 (CRG), NIH HL49944 (GGS), and NIH F32HL076072 (HA).
Thanks to Jennifer Fox for technical assistance with the Western immunoblotting data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hirsh J, Hoak J, American Heart Association Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis (in consultation with the Council on Cardiovascular Radiology) Circulation. 1996;93(12):2212–45. doi: 10.1161/01.cir.93.12.2212. [DOI] [PubMed] [Google Scholar]
- [2].Goldhaber SZ, Visani L, De Rosa M. Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER) Lancet. 1999;353(9162):1386–9. doi: 10.1016/s0140-6736(98)07534-5. [DOI] [PubMed] [Google Scholar]
- [3].Goldhaber SZ. Echocardiography in the management of pulmonary embolism. Ann Intern Med. 2002;136(9):691–700. doi: 10.7326/0003-4819-136-9-200205070-00012. [DOI] [PubMed] [Google Scholar]
- [4].Stobierska-Dzierzek B, Awad H, Michler RE. The evolving management of acute right-sided heart failure in cardiac transplant recipients. J Am Coll Cardiol. 2001;38(4):923–31. doi: 10.1016/s0735-1097(01)01486-3. [DOI] [PubMed] [Google Scholar]
- [5].Dell’Italia LJ. The right ventricle: anatomy, physiology, and clinical importance. Curr Probl Cardiol. 1991;16(10):653–720. doi: 10.1016/0146-2806(91)90009-y. [DOI] [PubMed] [Google Scholar]
- [6].Menzel T, Wagner S, Kramm T, Mohr-Kahaly S, Mayer E, Braeuninger S, et al. Pathophysiology of impaired right and left ventricular function in chronic embolic pulmonary hypertension: changes after pulmonary thromboendarterectomy. Chest. 2000;118(4):897–903. doi: 10.1378/chest.118.4.897. [DOI] [PubMed] [Google Scholar]
- [7].D’Armini AM, Cattadori B, Monterosso C, Klersy C, Emmi V, Piovella F, et al. Pulmonary thromboendarterectomy in patients with chronic thromboembolic pulmonary hypertension: hemodynamic characteristics and changes. Eur J Cardiothorac Surg. 2000;18(6):696–701. doi: 10.1016/s1010-7940(00)00584-4. discussion -2. [DOI] [PubMed] [Google Scholar]
- [8].Greyson C, Xu Y, Cohen J, Schwartz GG. Right ventricular dysfunction persists following brief right ventricular pressure overload. Cardiovasc Res. 1997;34(2):281–8. doi: 10.1016/s0008-6363(97)00038-2. [DOI] [PubMed] [Google Scholar]
- [9].Greyson C, Xu Y, Lu L, Schwartz GG. Right ventricular pressure and dilation during pressure overload determine dysfunction after pressure overload. Am J Physiol Heart Circ Physiol. 2000;278(5):H1414–20. doi: 10.1152/ajpheart.2000.278.5.H1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kerbaul F, Rondelet B, Motte S, Fesler P, Hubloue I, Ewalenko P, et al. Effects of norepinephrine and dobutamine on pressure load-induced right ventricular failure. Crit Care Med. 2004 Apr;32(4):1035–40. doi: 10.1097/01.ccm.0000120052.77953.07. [DOI] [PubMed] [Google Scholar]
- [11].Belcastro AN, Shewchuk LD, Raj DA. Exercise-induced muscle injury: a calpain hypothesis. Mol Cell Biochem. 1998;179(12):135–45. doi: 10.1023/a:1006816123601. [DOI] [PubMed] [Google Scholar]
- [12].Matsumura Y, Saeki E, Inoue M, Hori M, Kamada T, Kusuoka H. Inhomogeneous disappearance of myofilament-related cytoskeletal proteins in stunned myocardium of guinea pig. Circ Res. 1996;79(3):447–54. doi: 10.1161/01.res.79.3.447. [DOI] [PubMed] [Google Scholar]
- [13].Urthaler F, Wolkowicz PE, Digerness SB, Harris KD, Walker AA. MDL-28170, a membrane-permeant calpain inhibitor, attenuates stunning and PKC epsilon proteolysis in reperfused ferret hearts. Cardiovasc Res. 1997;35(1):60–7. doi: 10.1016/s0008-6363(97)00099-0. [DOI] [PubMed] [Google Scholar]
- [14].Yoshida K, Inui M, Harada K, Saido TC, Sorimachi Y, Ishihara T, et al. Reperfusion of rat heart after brief ischemia induces proteolysis of calspectin (nonerythroid spectrin or fodrin) by calpain. Circ Res. 1995;77(3):603–10. doi: 10.1161/01.res.77.3.603. [DOI] [PubMed] [Google Scholar]
- [15].Spratt JA, Tyson GS, Glower DD, Davis JW, Muhlbaier LH, Olsen CO, et al. The end-systolic pressure-volume relationship in conscious dogs. Circulation. 1987;75(6):1295–309. doi: 10.1161/01.cir.75.6.1295. [DOI] [PubMed] [Google Scholar]
- [16].Mehdi S. Cell-penetrating inhibitors of calpain. Trends Biochem Sci. 1991 Apr;16(4):150–3. doi: 10.1016/0968-0004(91)90058-4. [DOI] [PubMed] [Google Scholar]
- [17].Glower DD, Spratt JA, Snow ND, Kabas JS, Davis JW, Olsen CO, et al. Linearity of the Frank-Starling relationship in the intact heart: the concept of preload recruitable stroke work. Circulation. 1985;71(5):994–1009. doi: 10.1161/01.cir.71.5.994. [DOI] [PubMed] [Google Scholar]
- [18].Morris JJ, 3rd, Pellom GL, Murphy CE, Salter DR, Goldstein JP, Wechsler AS. Quantification of the contractile response to injury: assessment of the work-length relationship in the intact heart. Circulation. 1987;76(3):717–27. doi: 10.1161/01.cir.76.3.717. [DOI] [PubMed] [Google Scholar]
- [19].Chow E, Farrar DJ. Effects of left ventricular pressure reductions on right ventricular systolic performance. Am J Physiol. 1989;257(6 Pt 2):H1878–85. doi: 10.1152/ajpheart.1989.257.6.H1878. [DOI] [PubMed] [Google Scholar]
- [20].Karunanithi MK, Michniewicz J, Copeland SE, Feneley MP. Right ventricular preload recruitable stroke work, end-systolic pressure-volume, and dP/dtmax-end-diastolic volume relations compared as indexes of right ventricular contractile performance in conscious dogs. Circ Res. 1992;70(6):1169–79. doi: 10.1161/01.res.70.6.1169. [DOI] [PubMed] [Google Scholar]
- [21].Lu L, Xu Y, Zhu P, Greyson C, Schwartz GG. A common mechanism for concurrent changes of diastolic muscle length and systolic function in intact hearts. Am J Physiol Heart Circ Physiol. 2001;280(4):H1513–8. doi: 10.1152/ajpheart.2001.280.4.H1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Barta J, Toth A, Edes I, Vaszily M, Papp JG, Varro A, et al. Calpain-1-sensitive myofibrillar proteins of the human myocardium. Mol Cell Biochem. 2005 Oct;278(12):1–8. doi: 10.1007/s11010-005-1370-7. [DOI] [PubMed] [Google Scholar]
- [23].Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003 Jul;83(3):731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- [24].Wendt A, Thompson VF, Goll DE. Interaction of calpastatin with calpain: a review. Biol Chem. 2004 Jun;385(6):465–72. doi: 10.1515/BC.2004.054. [DOI] [PubMed] [Google Scholar]
- [25].Newcomb JK, Pike BR, Zhao X, Hayes RL. Concurrent assessment of calpain and caspase-3 activity by means of western blots of protease-specific spectrin breakdown products. Methods Mol Biol. 2000;144:219–23. doi: 10.1385/1-59259-050-0:219. [DOI] [PubMed] [Google Scholar]
- [26].Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res. 1997;80(3):393–9. [PubMed] [Google Scholar]
- [27].French JP, Quindry JC, Falk DJ, Staib JL, Lee Y, Wang KK, et al. Ischemia-reperfusion-induced calpain activation and SERCA2a degradation are attenuated by exercise training and calpain inhibition. Am J Physiol Lung Cell Mol Physiol. 2006 Jan;290(1):H128–36. doi: 10.1152/ajpheart.00739.2005. [DOI] [PubMed] [Google Scholar]
- [28].Papp Z, van der Velden J, Stienen GJ. Calpain-I induced alterations in the cytoskeletal structure and impaired mechanical properties of single myocytes of rat heart [see comments] Cardiovasc Res. 2000;45(4):981–93. doi: 10.1016/s0008-6363(99)00374-0. [DOI] [PubMed] [Google Scholar]
- [29].Muhlfeld C, Coulibaly M, Dorge H, Sellin C, Liakopoulos O, Ballat C, et al. Ultrastructure of right ventricular myocardium subjected to acute pressure load. Thorac Cardiovasc Surg. 2004 Dec;52(6):328–33. doi: 10.1055/s-2004-821272. [DOI] [PubMed] [Google Scholar]
- [30].Thomas SA, Fallavollita JA, Lee TC, Feng J, Canty JM., Jr. Absence of troponin I degradation or altered sarcoplasmic reticulum uptake protein expression after reversible ischemia in swine [see comments] Circ Res. 1999;85(5):446–56. doi: 10.1161/01.res.85.5.446. [DOI] [PubMed] [Google Scholar]
- [31].Foster DB, Van Eyk JE. In search of the proteins that cause myocardial stunning [editorial; comment] Circ Res. 1999;85(5):470–2. doi: 10.1161/01.res.85.5.470. [DOI] [PubMed] [Google Scholar]
- [32].Colantonio DA, Van Eyk JE, Przyklenk K. Stunned peri-infarct canine myocardium is characterized by degradation of troponin T, not troponin I. Cardiovasc Res. 2004 Aug 1;63(2):217–25. doi: 10.1016/j.cardiores.2004.04.006. [DOI] [PubMed] [Google Scholar]
- [33].Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, et al. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J Biol Chem. 2004 Feb 27;279(9):8290–9. doi: 10.1074/jbc.M308033200. [DOI] [PubMed] [Google Scholar]
- [34].Lemler MS, Bies RD, Frid MG, Sastravaha A, Zisman LS, Bohlmeyer T, et al. Myocyte cytoskeletal disorganization and right heart failure in hypoxia- induced neonatal pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2000;279(3):H1365–76. doi: 10.1152/ajpheart.2000.279.3.H1365. [DOI] [PubMed] [Google Scholar]
- [35].Murphy RM, Verburg E, Lamb GD. Ca2+ activation of diffusible and bound pools of {micro}-calpain in rat skeletal muscle. J Physiol. 2006 Oct 15;576(Pt 2):595–612. doi: 10.1113/jphysiol.2006.114090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bhatt A, Kaverina I, Otey C, Huttenlocher A. Regulation of focal complex composition and disassembly by the calcium-dependent protease calpain. J Cell Sci. 2002 Sep 1;115(Pt 17):3415–25. doi: 10.1242/jcs.115.17.3415. [DOI] [PubMed] [Google Scholar]
- [37].Franchini KG, Torsoni AS, Soares PH, Saad MJ. Early activation of the multicomponent signaling complex associated with focal adhesion kinase induced by pressure overload in the rat heart [In Process Citation] Circ Res. 2000;87(7):558–65. doi: 10.1161/01.res.87.7.558. [DOI] [PubMed] [Google Scholar]
- [38].Melloni E, Averna M, Stifanese R, De Tullio R, Defranchi E, Salamino F, et al. Association of calpastatin with inactive calpain: a novel mechanism to control the activation of the protease? J Biol Chem. 2006 Aug 25;281(34):24945–54. doi: 10.1074/jbc.M601449200. [DOI] [PubMed] [Google Scholar]
- [39].Enns D, Karmazyn M, Mair J, Lercher A, Kountchev J, Belcastro A. Calpain, calpastatin activities and ratios during myocardial ischemia-reperfusion. Mol Cell Biochem. 2002 Dec;241(12):29–35. doi: 10.1023/a:1020861120368. [DOI] [PubMed] [Google Scholar]
- [40].Sorimachi Y, Harada K, Saido TC, Ono T, Kawashima S, Yoshida K. Downregulation of calpastatin in rat heart after brief ischemia and reperfusion. J Biochem (Tokyo) 1997 Oct;122(4):743–8. doi: 10.1093/oxfordjournals.jbchem.a021818. [DOI] [PubMed] [Google Scholar]
- [41].Wei W, Fareed MU, Evenson A, Menconi MJ, Yang H, Petkova V, et al. Sepsis stimulates calpain activity in skeletal muscle by decreasing calpastatin activity but does not activate caspase-3. Am J Physiol Regul Integr Comp Physiol. 2005 Mar;288(3):R580–90. doi: 10.1152/ajpregu.00341.2004. [DOI] [PubMed] [Google Scholar]
- [42].Shi Y, Melnikov VY, Schrier RW, Edelstein CL. Downregulation of the calpain inhibitor protein calpastatin by caspases during renal ischemia-reperfusion. Am J Physiol Lung Cell Mol Physiol. 2000 Sep;279(3):F509–17. doi: 10.1152/ajprenal.2000.279.3.F509. [DOI] [PubMed] [Google Scholar]
- [43].Kawasaki H, Emori Y, Suzuki K. Calpastatin has two distinct sites for interaction with calpain--effect of calpastatin fragments on the binding of calpain to membranes. Arch Biochem Biophys. 1993 Sep;305(2):467–72. doi: 10.1006/abbi.1993.1448. [DOI] [PubMed] [Google Scholar]
- [44].Chatterjee S, Gu ZQ, Dunn D, Tao M, Josef K, Tripathy R, et al. D-amino acid containing, high-affinity inhibitors of recombinant human calpain I. J Med Chem. 1998 Jul 16;41(15):2663–6. doi: 10.1021/jm980035y. [DOI] [PubMed] [Google Scholar]
- [45].Tsuchida K, Aihara H, Isogai K, Hanada K, Shibata N. Degradation of myocardial structural proteins in myocardial infarcted dogs is reduced by Ep459, a cysteine proteinase inhibitor. Biol Chem Hoppe Seyler. 1986 Jan;367(1):39–45. doi: 10.1515/bchm3.1986.367.1.39. [DOI] [PubMed] [Google Scholar]
- [46].Barnoy S, Glaser T, Kosower NS. Calpain and calpastatin in myoblast differentiation and fusion: effects of inhibitors. Biochimica et biophysica acta. 1997 Sep 11;1358(2):181–8. doi: 10.1016/s0167-4889(97)00068-2. [DOI] [PubMed] [Google Scholar]