

Abstract
We previously reported that BMRF-2, an Epstein-Barr virus (EBV) glycoprotein, binds to β1 family integrins and is important for EBV infection of polarized oral epithelial cells. To further study the functions of BMRF-2, we constructed a recombinant EBV that lacks BMRF-2 expression by homologous recombination in B95-8 cells. We found that lack of BMRF-2 resulted in about 50% reduction of EBV attachment to oral epithelial cells, but not to B lymphocytes, suggesting that BMRF-2 is critical for EBV infection in oral epithelial cells, but not in B lymphocytes. In polarized oral epithelial cells, infection rate of the recombinant EBV virus was about 4- to 8-fold lower than the wild-type B95-8 virus. Cell adhesion assays using the BMRF-2 RGD peptide and its RGE and AAA mutants showed that the RGD motif is critical for BMRF-2 binding to integrins. These data are consistent with our previous observation that interactions between EBV BMRF-2 and integrins are critical for infection of oral epithelial cells with EBV.
Keywords: Epstein-Barr virus, BMRF-2, integrin, virion attachment, oral epithelial cells
Introduction
Epstein-Barr virus (EBV) is a human herpesvirus that infects about 90% of the human population and it is associated with lymphoid and epithelial neoplasia (Kieff and Rickinson A, 2001). EBV mainly infects B lymphocytes and epithelial cells; however, accumulating evidence indicates that it also may infect monocytes, macrophages, dendritic cells, and T cells (Guerreiro-Cacais et al., 2004; Kashiwagi et al., 2007; Knol et al., 2005; Masy et al., 2002; Savard et al., 2000; Schlitt et al., 2005; Shimakage et al., 1999; Tugizov et al., 2007).
Initiation of EBV infection requires attachment of virions to the host cell, followed by fusion of virus and cell membranes and the release of viral capsids into the cytoplasm (Hutt-Fletcher, 2007; Spear and Longnecker, 2003). It is well known that in B lymphocytes, EBV glycoprotein gp350/220 plays an important role in virus attachment by binding to cell surface receptor CD21. After attachment, fusion of the virus-cell membrane is triggered by interactions of HLA class II molecules with EBV glycoprotein gp42, which forms a complex with gHgL (Li et al., 1997; Li, Turk, and Hutt-Fletcher, 1995). EBV gB also is known to be important in the virus-B lymphocyte cell membrane fusion (Haan, Lee, and Longnecker, 2001). However, the mechanisms involved in EBV attachment to and entry into epithelial cells are less clear. Neither gp350/220 nor gp42 is required for EBV infection of epithelial cells, in contrast to gHgL and gB, which are essential (Hutt-Fletcher, 2007; Spear, 1993): gHgL is critical to virus attachment (Molesworth et al., 2000), and its coordinated functions with gB mediate fusion of the virus-cell membrane in epithelial cells (Borza et al., 2004; McShane and Longnecker, 2004; Omerovic, Lev, and Longnecker, 2005). Interactions between BMRF-2 and the β1 family of integrins also are critical for infection of polarized oropharyngeal epithelial cells with EBV (Tugizov, Berline, and Palefsky, 2003).
BMRF-2 is an EBV glycoprotein with highly hydrophobic, multi-span transmembrane domains and an arginine-glycine-aspartate (RGD) motif in its major extracellular domain (Tugizov, Berline, and Palefsky, 2003; Xiao et al., 2007). The RGD motif is present in extracellular matrix proteins, which bind to integrins and play a vital role in cell adhesion and cell-to-cell communication (Friedl, Brocker, and Zanker, 1998; Giancotti and Ruoslahti, 1999; Gilcrease, 2007; Ruoslahti, 1996). Viral proteins containing the RGD motif may serve as ligands for integrins leading to attachment of virions to host cells (Triantafilou, Takada, and Triantafilou, 2001). BMRF-2 is highly expressed in the upper differentiated layer of oral hairy leukoplakia (HL) lesions, where the virus actively replicates (Lagenaur and Palefsky, 1999; Palefsky et al., 1997; Xiao et al., 2007). The promoter of BMRF-2 is up-regulated by mechanisms known to induce differentiation of epithelial cells (Lagenaur and Palefsky, 1999). BMRF-2 is associated with the viral envelope and binds to α3, α5, αv, and β1 integrins through its RGD domain (Tugizov, Berline, and Palefsky, 2003; Xiao et al., 2007). Antibodies to β1 family integrins and to the BMRF-2 protein significantly block infection of EBV in polarized oral epithelial cells, which express high levels of integrins on the basolateral membranes (Tugizov, Berline, and Palefsky, 2003). Treatment with the same antibodies does not affect EBV infection in B lymphocytes, which express lower levels of integrins (Tugizov, Berline, and Palefsky, 2003). These findings indicate that interactions between EBV BMRF-2 and β1 family integrins may be critical for efficient EBV infection in oral epithelial cells but not B lymphocytes.
Binding of viral proteins with cell surface integrins plays an important role in initiating virus infection and dissemination (Triantafilou, Takada, and Triantafilou, 2001). Integrins are a large family of herterodimeric cell surface receptors consisting of multiple α and β subunits, most of which bind extracellular matrix proteins through their RGD motif (Friedl, Brocker, and Zanker, 1998; Giancotti and Ruoslahti, 1999; Gilcrease, 2007; Ruoslahti, 1996). Binding of viral proteins to integrins is mostly RGD-dependent, as seen in human herpesvirus 8 (α3β1) (Akula et al., 2002), EBV (α3, α5, αv, and β1) (Tugizov, Berline, and Palefsky, 2003), hantavirus (β3) (Gavrilovskaya et al., 1999), rotavirus (α2β1) (Ciarlet et al., 2002), foot-and-mouth disease virus (αvβ3) (Jackson et al., 1997), West Nile virus (αvβ3) (Chu and Ng, 2004), echovirus (αvβ6) (Triantafilou and Triantafilou, 2001) and coxsackie virus (αvβ6) (Williams et al., 2004). However, interactions between viral glycoproteins and integrins also can be RGD-independent, as shown in human cytomegalovirus (HCMV), which attaches to α2β1 and αvβ3 integrins through gB and gH, respectively (Feire, Koss, and Compton, 2004; Wang et al., 2005). Upon binding to viral attachment proteins, integrins may activate downstream signaling pathways leading to internalization of the virus into host cells (Krishnan et al., 2006; Medina-Kauwe, 2003; Pietiainen et al., 2004; Sharma-Walia et al., 2004; Wang et al., 2005).
In the present study, we further examined the role of BMRF-2 in the infection of oral epithelial cells using a recombinant EBV that lacks expression of BMRF-2. We found that lack of BMRF-2 in the virion envelope leads to a substantial reduction of EBV attachment and infection in oral epithelial cells, but not B lymphocytes.
Results
Selection of a marmoset B lymphoblastoid cell line producing recombinant EBV that lacks BMRF-2 expression
To construct the recombinant virus, EBV/ΔBMRF-2, B95-8 cells were transfected with linearized shuttle vector DNA, pB-GFP-NK (Fig. 1A), and neomycin-resistant cells were selected by G418. Because the poly-A signal for BMRF-1 mRNA is located within the BMRF-2 coding sequence, it is possible that knockout of the BMRF-2 gene also prevents BMRF-1 expression in EBV/ΔBMRF-2. To prevent this disruption, HSV-TK poly-A was inserted in the 3’ untranslated sequence of BMRF-1 just downstream of the upstream homologous sequence to provide a poly-A signal for BMRF-1 expression (Fig. 1A). We used a PCR method to screen neo-resistant clones that carried EBV/ΔBMRF-2 using primer sets A and B (Fig. 1A). Because each pair of primers consisted of one primer specific for the sequence present and one for the part of EBV genome not present in the shuttle vector, only recombinant viruses should be tested positive by PCR. Twenty-two of the 150 neomycin-resistant clones tested positive by both pairs of primers for recombination (data not shown). One of the positive clones, designated B27-BMRF-2low, showed the strongest PCR signal for both primer sets (Fig. 1B) and was chosen for further expansion and Southern blot analysis. Fig. 1C shows the results of a Southern blot test using both a BMRF-2 probe and an NK probe. The BMRF-2 probe detected WT EBV DNA as a 6.4 kb fragment and recombinant DNA as a 1.6 kb fragment, as expected. Only the WT EBV DNA was detected in B95-8 cells. Although both the WT and the recombinant EBV genome were detected in B27-BMRF-2low using the BMRF-2 probe, the lower band (recombinant) was much stronger than the upper band (WT), indicating that the cells contained mainly the recombinant EBV/ΔBMRF-2. The NK probe detects only EBV/ΔBMRF-2 DNA, which was expected in B27-BMRF-2low but not in B95-8 (Fig. 1C).
Fig. 1.
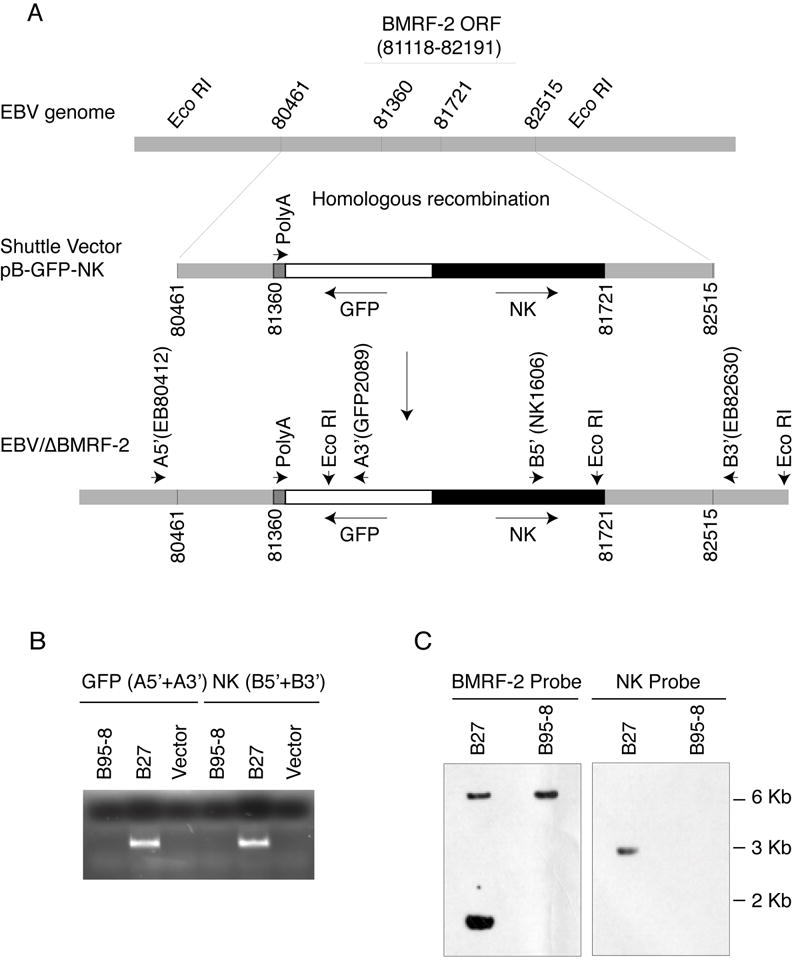
Construction of the recombinant EBV virus, EBV/ΔBMRF-2. A. Schematic view of the EBV genome and the shuttle vector for homologous recombination. Numbers represent the locations of the nucleotides in the EBV B95-8 genome (accession number NC_001345). B. Results of PCR using primer sets A (A5’+A3’, for GFP) and B (B5’+B3’, for NK) on genomic DNA from B95-8 and B27-BMRF-2low cells. C. Results of Southern blot analysis using a BMRF-2 probe and an NK probe and genomic DNA from B95-8 and B27-BMRF-2low cells digested with restriction enzyme Eco RI which gives a 6.4 kb (WT) band and a 1.6 kb (recombinant) band for the BMRF-2 probe, and a 3 kb (recombinant) band for the NK probe. A5’, A3’, B5’, and B3’ represents primers EB80412-31, GFP2089-2112, Nk1606-27, and EB82630-50, respectively.
To obtain cell lines producing only the recombinant EBV/ΔBMRF-2, we attempted to rescue the recombinant in two EBV-negative B lymphoblastoid cell lines, Akata 4E-3 and BJAB cells. At least 10 rescue experiments were performed in each line using 1-2 × 106 to 107 cells infected with virions at 2-10 MOI, and more than 3000 neo-resistant colonies were obtained initially. However, only a few colonies could be expanded and tested for the presence of the EBV/ΔBMRF-2 recombinant. Unfortunately, none of the surviving neo-resistant clones contained any EBV/ΔBMRF-2 genome, as it was either lost or had reverted to WT EBV.
Characterization of the B27-BMRF-2low virus
In the absence of pure recombinant EBV/ΔBMRF-2 virus, we characterized the B27-BMRF-2low cell line for the present study. To determine the ratio of WT EBV over EBV/ΔBMRF-2 in this cell line, we performed quantitative real-time PCR using a BZLF-1 probe for total EBV genome and an NK probe for the EBV/ΔBMRF-2 genome. We found that about 85% of genomic viral DNA copies in both intracellular and released virions of the B27-BMRF-2low cells were recombinant EBV DNA (Fig. 2A). To examine the stability of recombination in B27-BMRF-2low cells, we tested the ratio of recombinant/WT EBV in this cell line at different passages and found that the ratio is stable during the expansion of the B27-BMRF-2low cell line over more than 30 passages under G418 selection.
Fig. 2.
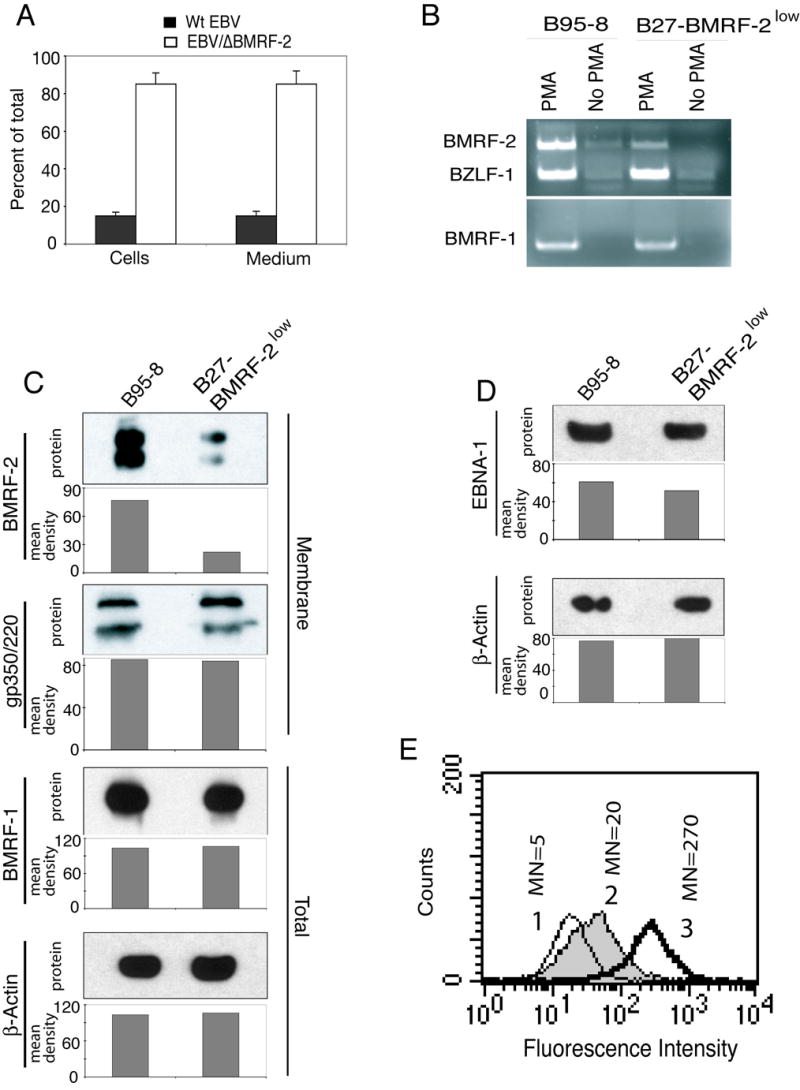
Viral gene expression in B95-8 and B27-BMRF-2low cells. A. The ratio of recombinant EBV to WT EBV in B27-BMRF-2low cells. Total EBV copy numbers were quantitated using the BZLF-1 probe and recombinant virions were quantitated by the NK probe, numbers represent the percentages of total viral genome copy numbers. B. RT-PCR detection of BMRF-1, BMRF-2, and BZLF-1 expression in B95-8 and B27-BMRF-2low cells. 5 × 106 each of B95-8 and B27-BMRF-2low cells were treated with or without PMA (30 ng/ml) for 5 days before total RNA was extracted. Five μl of cDNA (out of 20 μl reaction) were used as PCR templates. The top panel shows expression of both BMRF-2 and BZLF-1 in the same PCR reaction. The bottom panel shows expression of the BMRF-1 gene. C. Western blot analysis showing BMRF-2, gp350/220, and BMRF-1 expression in B95-8 and B27-BMRF-2low cells. For BMRF-2 and gp350 detection, 10 μg of the membrane fraction proteins were loaded onto each well of a 7 M urea SDS-PAGE gel. BMRF-1 was detected using total cell lysates separated on a NuPage 10% Bis-Tris gel. D. Western blot detection of EBNA-1 and β-actin expression in noninduced B95-8 and B27-BMRF-2low cells. 10 μg of total cell lysates were loaded onto each well in a NuPage 10% Bis-Tris gel. The mean density of pixels of the protein bands shown in panels C and D were measured by the NIH Image software, and the results of each gel are shown as a bar graph under each blot and background values were subtracted from each protein band. E. Cell surface detection of BMRF-2 by flow cytometry in B95-8 and B27-BMRF-2low cells using rat anti–BMRF-2 serum. Histograms are labeled as the following: 1, normal rat serum staining of these two cell lines (since the histograms were almost identical, only one is shown); 2, B27-BMRF-2low cells reacted with rat anti-BMRF-2 serum; 3, B95-8 cells reacted with rat anti-BMRF-2 serum. MN, mean fluorescence intensity.
To compare expression of BMRF-2, BMRF-1, EBNA-1, BZLF-1, and gp350/220 in B95-8 and B27-BMRF-2low cells, we performed RT-PCR, Western blot and flow cytometry assays. Small amounts of BMRF-2 RNA were detected by RT-PCR under noninducing conditions in B95-8 cells, but not in B27-BMRF-2low cells (Fig. 2 B). After PMA induction, expression of all three genes—BMRF-2, BZLF1, and BMRF-1—was greatly increased in B95-8 cells. In B27-BMRF-2low cells, however, comparable levels of expression were found only for BZLF-1 and BMRF-1. The amount of BMRF-2 RNA in B27-BMRF-2low cells was substantially lower than that in B95-8 cells (Fig. 2 B). These data indicate that knockout of BMRF-2 did not affect expression of BZLF-1 and BMRF-1, which are critical for EBV lytic infection.
Western blot analysis of BMRF-2 and gp350/220 in the membrane fraction of PMA-induced B95-8 and B27-BMRF-2low cells showed similar expression of gp350/220 in B95-8 and B27-BMRF-2low cells, but greatly reduced expression of BMRF-2 in B27-BMRF-2low cells (Fig. 2C). Under PMA induction, we found the rate of lytic induction is similar in both B95-8 and B27-BMRF-2low cells (data not shown). Analysis of total cell extracts showed that BMRF-1 expression was similar in both B95-8 and B27-BMRF-2low cells (Fig. 2C). These data indicate that expression of two lytic proteins of EBV (gp350/220 and BMRF-1) was not affected by knockout of BMRF-2. To determine whether knockout of BMRF-2 affected the expression of latent EBV genes, such as EBNA-1, we examined the protein expression of EBNA-1 in latent B95-8 and B27-BMRF-2low cells by Western blotting. Under noninduced conditions, the amount of EBNA-1 protein was also similar in both cell lines (Fig. 2D), suggesting that EBNA-1 expression also was not affected by disruption of the BMRF-2 gene.
To determine surface expression of the BMRF-2 protein in B95-8 and B27-BMRF-2low cells, PMA-induced cells were analyzed by flow cytometry assay using rat anti–BMRF-2 serum. We observed a substantial reduction of BMRF-2 on B27-BMRF-2low cells compared with B95-8 cells, with a mean fluorescence intensity of 17 and 270, respectively (Fig. 2E). This result correlated with Western blot analysis, which showed a low level of BMRF-2 protein in the membrane fraction of B27-BMRF-2low cells (Fig. 2C).
BMRF-2 may not be required for virion assembly and release
To determine whether knockout of BMRF-2 affects assembly of viral particles, we examined the morphology of virions in PMA-induced B95-8 and B27-BMRF-2low cells under electron microscopy, and we found no differences in the morphology of virions from these two cell lines (data not shown). To investigate whether BMRF-2 plays a role in the release of progeny virions, the B85-8 and B27-BMRF-2low cells were grown under the same conditions and induced by PMA and butyric acid for 5 days. Cells and media were then collected separately and examined for copies of the EBV genome by quantitative real-time PCR. We found no difference in the rate of release between B95-8 and B27-BMRF-2low viruses, indicating that virions from B27-BMRF-2low cells were released as efficiently as from B95-8 cells (Fig. 3A). To determine whether BMRF-2 was present in released WT and B27-BMRF-2low virions, double gradient-purified WT and B27-BMRF-2low viruses were examined for BMRF-2 and gp350/220 protein expression by Western blot assays (Fig. 3B). Analysis of BMRF-2 in released virions from B95-8 cells showed the presence of multiple smeary, but strong bands, which is typical for this protein due to its highly hydrophobic nature (Xiao et al., 2007). In contrast, virions from B27-BMRF-2low cells showed only weak bands, which represent BMRF-2 expressed from the WT virus that was present among the BMRF-2 knockout virus population (about 85%). Quantitative analysis of BMRF-2 protein bands showed that mean pixels were 158 and 29 for B95-8 and B27-BMRF-2low virions, respectively (Fig. 3B). Analysis of the same samples for gp350/220 showed an equal amount in both viruses with a mean pixel value of 160 each. These data clearly demonstrate that the amount of BMRF-2 protein in B27-BMRF-2low virions was about 80% less than the amount in B95-8 virions and that lack of BMRF-2 did not affect the egress of progeny virions.
Fig. 3.
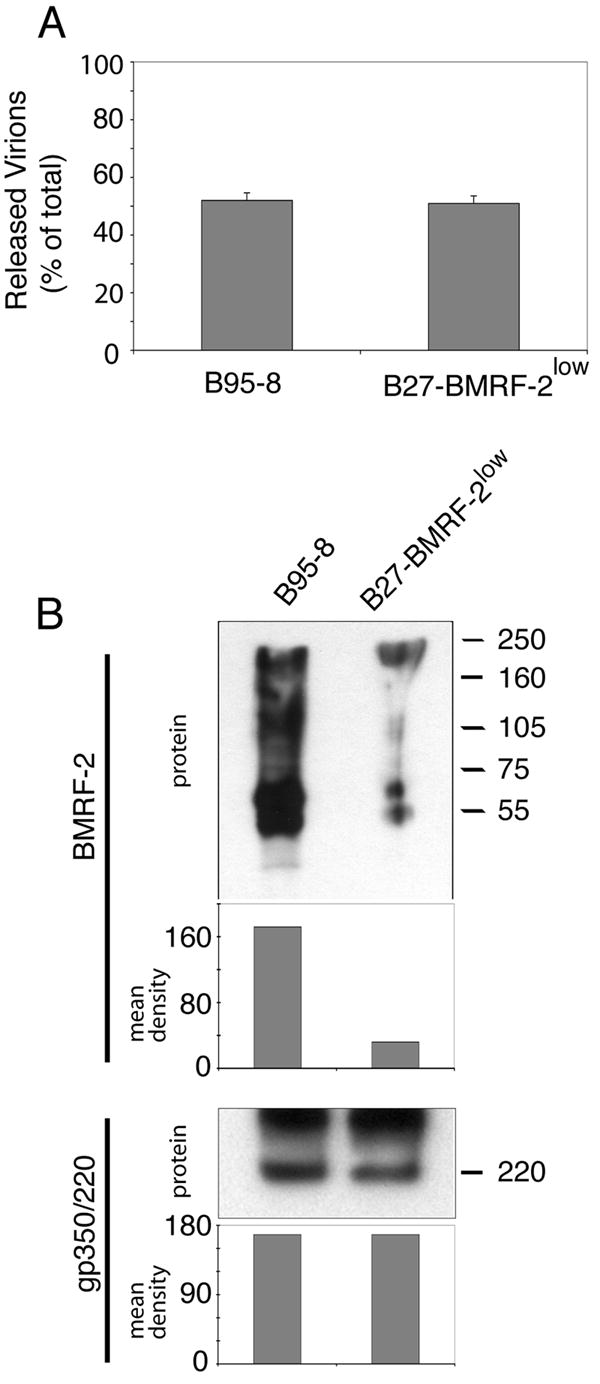
Virion release from B95-8 and B27-BMRF-2low cells. A. Analysis of released virions by quantitative real-time PCR. B95-8 and B27-BMRF-2low cells were cultured in the presence of PMA and butyric acid for 5 days, and total genomic DNA from cells or media was extracted separately. EBV genome copy numbers were quantitated by real-time PCR and number of released virions in medium from B95-8 or B27-BMRF-2low cells were shown as % of total, which equals to the number of virions from cells and medium together. Error bars represent standard deviations (SD) of three independent experiments. B. Western blot analysis of double gradient purified virions from B95-8 or B27-BMRF-2low cells using rat anti–BMRF-2 serum. 10 μg of total viral proteins were loaded onto each well of a 7 M urea SDS-PAGE gel.
BMRF-2 is required for EBV attachment to epithelial cells but not to B lymphocytes
In the above experiments, we confirmed that B27-BMRF-2low cells produce predominantly virions containing the EBV/ΔBMRF-2 genome (about 85%), and only about 15% of WT viral genome as demonstrated by real-time PCR quantification of WT and EBV/ΔBMRF-2 genome copy numbers (Fig. 2 A). Also, Western blot analysis of purified B27-BMRF-2low and wild-type B95-8 virions showed that B27-BMRF-2low virions contain substantially lower amount of BMRF-2 protein than in B95-8 virus (Fig. 3B). Therefore, we used B27-BMRF-2low virions to determine whether BMRF-2 is essential for EBV attachment to oral epithelial and B lymphocytes. For attachment assays, HSC-3sort, Detroitsort and Akata 4E-3 cells were incubated with B95-8 or B27-BMRF-2low virions at 5 MOI/cell and 25 MOI/cell on ice for 60 min, and the amounts of attached virions were quantitated by real-time DNA PCR. We found that binding of B27-BMRF-2low virions to HSC-3sort and Detroitsort cells was about 50% less than that of B85-8 virus (Fig. 4A). However, the amount of virions attached to EBV-negative Akata 4E-3 cells was similar for both B95-8 and B27-BMRF-2low virus (Fig. 4A).
Fig. 4.
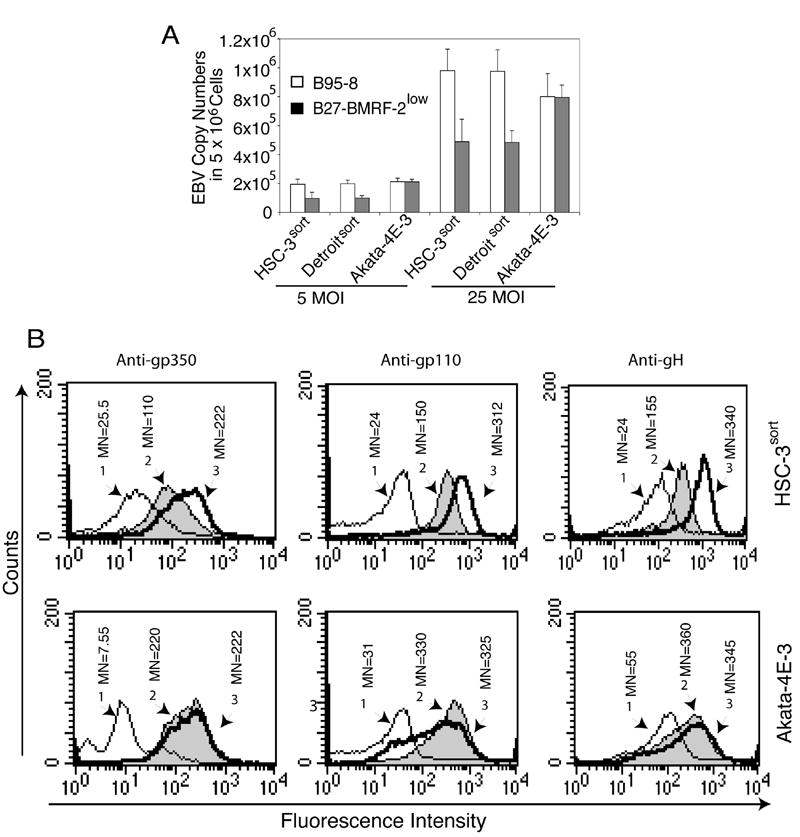
Analysis of attachment of B95-8 and B27-BMRF-2low virus to oral epithelial and Akata 4E-3 B lymphocytes. A. Detection of virus attachment by real-time PCR. The B95-8 and B27-BMRF-2low viruses were added independently to HSC-3sort, Detroitsort, and Akata 4E-3 cells and incubated at 4°C for 1 h. Cells were washed, and genomic DNA extracted and bound virions were examined by real-time PCR. B. Evaluation of virus binding by flow cytometry assay. HSC-3sort and Akata 4E-3 cells were incubated with B95-8 or B27-BMRF-2low viruses for 1 h at 4°C and washed, and attached virions were detected using mouse monoclonal anti-gp250/350/ (left panel), anti-gp110 (anti-gB, middle panel), or anti-gH (right panel). The amount of virions bound to the cell surface was examined by flow cytometry and the results are shown as histograms labeled as follows: 1, cells incubated with the B95-8 virus and reacted with mouse IgG1; 2 and 3, cells incubated with the B27-BMRF-2low and the B95-8 virus, respectively, and reacted with one of the above monoclonal antibodies. MN, mean fluorescence intensity.
To confirm the results of real-time PCR assays, we examined the attachment of B95-8 and B27-BMRF-2low viruses to HSC-3sort and Akata 4E-3 cells by flow cytometry using anti-gp350/220, anti-gp110 (gB), and anti-gH antibodies. We found a significant reduction in the binding of B27-BMRF-2low virus to HSC-3sort cells, compared with B95-8 virus, but a similar rate at which both viruses bind to the Akata 4E-3 cells (Fig. 4B). These data clearly indicate that the binding of the B27-BMRF-2low virus to oral epithelial cells is substantially reduced, but did not change in B lymphocytes. In addition, these data show that the amounts of glycoproteins gp350/220, gB and gH are also normal in B27-BMRF-2low virus.
BMRF-2 is important for EBV infection of polarized oral epithelial cells
Our previous work showed that EBV infection in polarized oral epithelial cells occurs from their basolateral membranes and that BMRF-2 may play a critical role in this infection (Tugizov, Berline, and Palefsky, 2003; Xiao et al., 2007). To confirm this observation, we examined the infectivity of both B95-8 and B27-BMRF-2low viruses in polarized HSC-3sort, Detroitsort, and OCO cells. Polarized oral epithelial cells were infected with either B95-8 virus or B27-BMRF-2low virus at 100 MOI from the basolateral surfaces. At 5 days after infection, EBV-infected cells were detected by immunofluorescence staining using an antibody specific for gp350/220. Immunostaining of gp350/220 in polarized HSC-3sort, Detroitsort (data not shown), and OCO cells (Fig. 5A) showed localization of this protein to the membrane and cytoplasm, as observed in our earlier work (Tugizov, Berline, and Palefsky, 2003; Xiao et al., 2007). Quantitative analysis of infected cells showed that infection rates of the B95-8 virus were 4- to 8-fold higher than those of the B27-BMRF-2low virus in polarized oral epithelial cells (Fig. 5B). The gp350/220-positive cells were 4.1%, 5.1%, and 13.5% in HSC-3sort, Detroitsort, and OCO cells, respectively, after B95-8 virus infection, but only 0.5%, 0.8%, and 3.8%, respectively, after B27-BMRF-2low virus infection. These data show that B27-BMRF-2low virus has substantially lower efficiency in infecting polarized oral epithelial cells from their basolateral membranes.
Fig. 5.
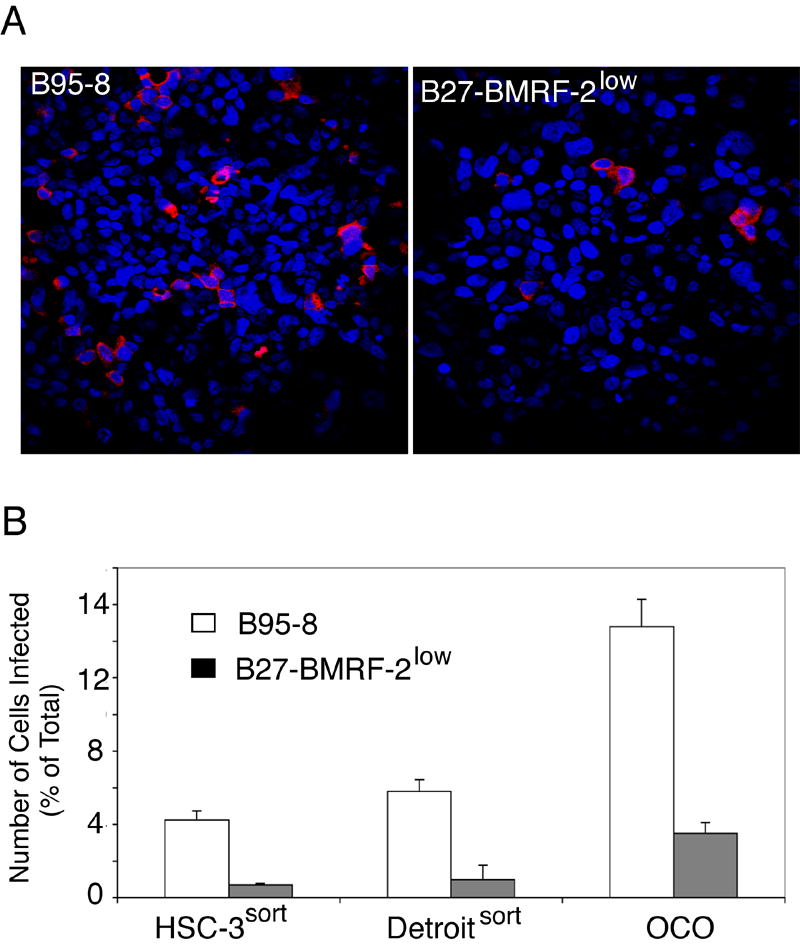
Infection of polarized oral epithelial cells with B95-8 or B27-BMRF-2low virus. A. Polarized OCO cells were infected with B95-8 or B27-BMRF-2low virus from their basolateral membranes at 100 MOI. At 5 days after infection, cells were fixed and stained with mouse anti-gp350/220 antibody (red). Cell nuclei were stained in blue. B. Polarized HSC-3sort, Detroitsort and OCO cells were infected with B95-8 or B27-BMRF-2low virus from their basolateral membranes at 100 MOI/cell. Cells were fixed at 5 days postinfection and immunostained with gp350/220 antibodies; positive cells were counted. Numbers shown are the average percentage of gp350/220–positive cells from individual filer membranes. Error bars show SD (n=3).
The extracellular RGD motif of BMRF-2 mediates HSC-3sort cell adhesion
The above experiments showed that attachment and entry of B27-BMRF-2low virus into oropharyngeal epithelial cells was significantly impaired, indicating that BMRF-2 is critical in this process. Our earlier findings showed that antibodies to BMRF-2, and β1 and/or α5β1 integrins inhibit EBV attachment to oral epithelial cells and that BMRF-2 RGD binds to β1 and αv integrins (Tugizov, Berline, and Palefsky, 2003; Xiao et al., 2007), suggesting that the extracellular domain containing the RGD motif may play a key role in BMRF-2’s function. To determine the role of the BMRF-2 RGD motif in attachment of the virus to oral epithelial cells, we examined adhesion of HSC-3sort cells to 96-well plates coated with BMRF-2 RGD peptide or its mutant derivatives.
The purified BMRF-2 RGD fragment consisting of 48 amino acids (Fig. 6A) was used to coat 96-well plates. These experiments showed that HSC-3sort cells attached to the BMRF-2 RGD peptide-coated plates in a dose-dependent manner (Fig. 6B), the efficiency of which was similar to fibronectin at higher concentrations (Fig. 6B). Binding of HSC-3sort cells to BMRF-2 RGD–coated plates was specifically blocked by rat anti-sera to BMRF-2 and β1 integrin, and mouse monoclonal anti-αv antibodies, indicating that adhesion is due to specific interaction between this peptide and integrins, such as β1 and αv. Two EBV-positive human sera previously shown to bind to BMRF-2 on the surface of 293 cells transfected with BMRF-2 (Xiao et al., 2007) also significantly reduced adhesion of HSC-3sort cells to BMRF-2 RGD–coated plates (Fig. 6C). To confirm that interactions between BMRF-2 and integrins were RGD dependent, we tested the efficiency of HSC-3sort cell adhesion to mutant BMRF-2 RGD peptides, BMRF-2 RGE, and BMRF-2 AAA. Substitution of RGD with RGE significantly reduced HSC-3sort cell adhesion, and replacement of the RGD sequence with AAA eliminated the ability of the RGD peptide to mediate adhesion of HSC-3sort cells to the plates (Fig. 6D). These results demonstrate that the RGD sequence is critical for BMRF-2 binding to integrins.
Fig. 6.
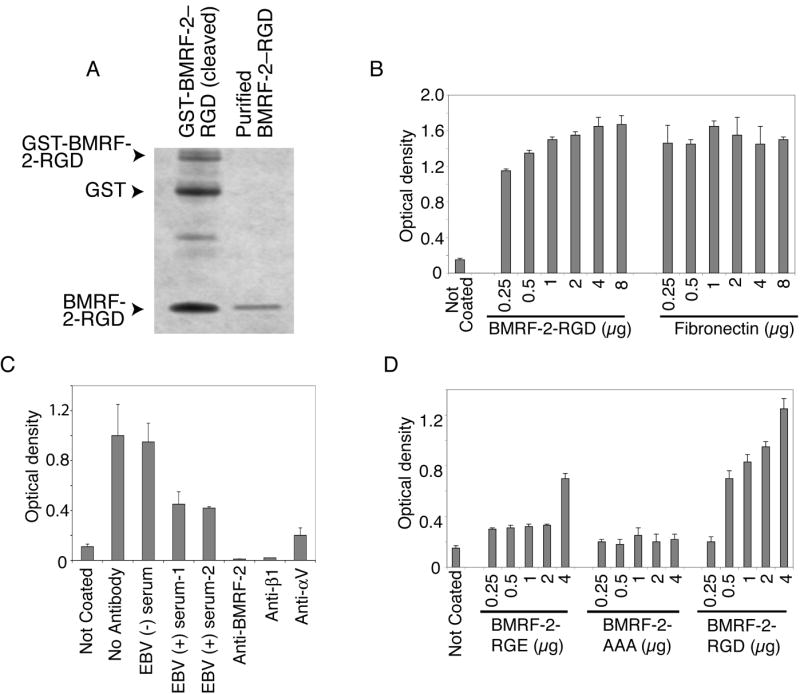
Adhesion of HSC-3sort cells to BMRF-2 RGD–coated plates. A. Purification of the BMRF-2 RGD peptide: BMRF-2 RGD fragment was cleaved by thrombin from the GST-BMRF-2 RGD fusion protein and purified by Centricon-10. Ten μg of GST-BMRF-2 RGD fusion protein (left lane) and 1 μg of purified BMRF-2 RGD peptide (right lane) were separated on an SDS PAGE gel and stained by simple blue (Invitrogen). B. Adhesion of HSC-3sort cells to a Maxisorp 96-well plate coated with the BMRF-2 RGD peptide or fibronectin at different concentrations. C. Adhesion of HSC-3sort cells was blocked by different antibodies: EBV-negative serum, EBV-positive serum, or rat anti-BMRF-2 were added to a Maxisorp 96-well plate coated with the BMRF-2 RGD peptide before HSC-3sort cells were applied to the plate; Anti-β1 and anti-αv antibodies were incubated with HSC-3sort cells on ice for 60 min before they were added to the BMRF-2 RGD–coated plate. D. Adhesion of HSC-3sort cells to the wells of a Maxisorp 96-well plate coated with PBS, BMRF-2 RGD peptide, BMRF-2 RGE peptide, or BMRF-2 AAA peptide. B-D, Cell adhesion was examined by measuring attached cells after crystal violet staining using an ELISA-plate reader with a 595 nm absorbance filter. Error bars represent SD of three independent experiments.
Discussion
Our previous work showed that BMRF-2 binds to β1 and αv family integrins through its RGD domain and that antibodies to BMRF-2, and β1 and/or α5β1 integrins reduce EBV attachment to polarized oral epithelial cells, suggesting that the interaction of BMRF-2 with integrins may be an important determinant for EBV infection in these cells (Tugizov, Berline, and Palefsky, 2003; Xiao et al., 2007). In the present study, our goal was to further investigate the role of BMRF-2 in the infection of oral epithelial cells with EBV by a constructing recombinant EBV that lacks BMRF-2 expression. In homologous recombination experiments using B95-8 cells, we selected the clonal cell line B27-BMRF-2low that produced predominantly BMRF-2 knockout virus. However, we were unable to rescue pure EBV recombinant that lacks BMRF-2. We cannot explain the difficulties encountered in rescuing pure BMRF-2 knockout EBV, as BMRF-2 has not been shown to be an essential protein for EBV latency. It is also unlikely that insertion of the GFP-NK cassette influences expression of other genes essential for EBV replication or latency. By RT-PCR and Western blot analysis we showed that BMRF-1, gp350/220, and BZLF-1 were expressed normally in B27-BMRF-2low cells (Fig. 2B and C). Using Western blot analysis, we found that the level of EBNA-1 expression in B27-BMRF-2low cells was similar to that in B95-8 cells (Fig. 2D), indicating that knockout of BMRF-2 does not affect expression of EBNA-1, a protein that is essential for maintaining EBV latency (Leight and Sugden, 2000). A great deal of our efforts have been put to use the EBV-bacterial artificial chromosome system (kindly provided by Drs. K Takada and T Kanda (Kanda et al., 2004) for constructing BMRF-2 knockout EBV recombinants, but this approach also has not been successful for us. Although it is not ideal to use a mixture of WT and recombinant virus in the current study, valuable information had been published by other investigators using nonrescued EBV recombinants, which were also created in B95-8 cells (Speck et al., 1999).
Detailed analysis of B27-BMRF-2low cells showed that about 85% of the EBV virions from these cells contain the EBV/ΔBMRF-2 genome (Fig. 2A) and the amount of the BMRF-2 protein is significantly lower in B27-BMRF-2low virus than in B95-8 virus (Fig. 3B), and therefore this model was suitable to study the function of BMRF-2. Virions lacking BMRF-2 protein were released normally from B27-BMRF-2low cells suggesting that BMRF-2 may not be critical for virus egress although the BMRF-2 protein can be partially complemented by the WT virus. Substantial reduction in attachment (about 50%) of B27-BMRF-2low virus to oral HSC-3sort and Detroitsort indicated that BMRF-2 was responsible for virion attachment to the surface of oral epithelial cells. However its ability to attach to and infect EBV-negative Akata 4E-3 cells is similar to that of B95-8 virus (Fig. 4). These data suggest that BMRF-2 function is restricted to oral epithelial cells, which express high levels of β1 family integrins (Tugizov, Berline, and Palefsky, 2003). This observation confirms our previous finding that antibodies against BMRF-2, β1 and/or α5β1 integrins reduced EBV infection in polarized oral epithelial cells but not in B lymphocytes (Tugizov, Berline, and Palefsky, 2003).
Infection of polarized oral epithelial cells by B27-BMRF-2low virus leads to significant reduction in infection (Fig. 5), which was consistent with our previous findings that antibodies to BMRF-2 and β1 family integrins also inhibit EBV infection in polarized oral epithelial cells (Tugizov et al., 1995). Proteomic analysis of purified EBV showed that the level of BMRF-2 in the virion envelope is very low (Johannsen et al., 2004). However, our data show that virions lacking BMRF-2 substantially lose their binding and infectious activity in oral epithelial cells (Fig. 4 and Fig. 5), indicating that the presence of BMRF-2 in the virion envelope is critical for EBV attachment to and infection of these cells. Previously, we showed that full separation of BMRF-2 in commonly used protein-gel systems is not always feasible due to its highly hydrophobic nature, which generates a multi-spanning glycoprotein with 10 potential transmembrane domains (Tugizov, Berline, and Palefsky, 2003; Xiao et al., 2007). Therefore, it may not be possible to determine the exact amount of this protein in the virion envelope by standard gel-separation systems.
We previously reported that BMRF-2 binds to integrins β1, α3, α5, and αv through its RGD-containing extracellular domain fused to the GST protein (Tugizov, Berline, and Palefsky, 2003; Xiao et al., 2007). In this report, we show that the BMRF-2 RGD peptide mediates adhesion of HSC-3sort cells in a dose-dependent manner. The efficiency of BMRF-2 RGD–mediated cell adhesion was comparable to cell adhesion to the fibronectin, which is a main ligand for α5β1 integrin (Larsen et al., 2006; Takagi, 2004). BMRF-2 RGD–dependent cell adhesion was specifically blocked by antibodies to BMRF-2, and β1 and αv integrins. Substitution of the RGD sequence with RGE or AAA also reduced or eliminated the ability of the BMRF-2 RGD peptide to mediate adhesion of HSC-3sort cells (Fig. 6). These findings clearly demonstrate that BMRF-2 is a virion attachment glycoprotein that mediates binding of EBV to oral epithelial cells through the interaction of its RGD-containing extracellular domain with integrins.
Accumulating evidence indicates that binding of viral proteins with integrins may establish initial virion attachment to the surface of the host cell (Adam, 2001; Akula et al., 2002; Jackson et al., 1997; Triantafilou and Triantafilou, 2001). The well-known glycoproteins of human cytomegalovirus, gB and gH, and human herpesvirus-8 gB bind cell surface integrins and facilitate virion binding (Akula et al., 2002; Feire, Koss, and Compton, 2004; Wang et al., 2005). Recent work by May et al., (May et al., 2005) showed that the BMRF-2 homologue of the gamma-herpesvirus-68, ORF-58, also may function as an attachment protein. Interestingly, the attachment function of ORF-58 is mediated by its extracellular loop (between aa 163 and aa 200), which is highly homologous with the extracellular loop of BMRF-2. However, ORF-58 does not have the RGD motif in its extracellular domain, suggesting that ORF-58 binding to host cells may be independent of integrins.
In summary, our results show that BMRF-2 serves as an attachment protein for EBV in oral epithelial cells and therefore may play an important role in early events of EBV infection within the oropharyngeal mucosal epithelium. Moreover, our data show that a minimal role for BMRF-2 in EBV infection of B lymphocytes, suggesting that BMRF-2 may have specific functions in the epithelium, as most β1 and αv integrins are highly functional and generate multiple signaling events in highly adhesive epithelial cells, in contrast to B lymphocytes (Friedl, Brocker, and Zanker, 1998; Friedl, Zanker, and Brocker, 1998; Giancotti and Ruoslahti, 1999; Gilcrease, 2007; Larsen et al., 2006; Takagi, 2004; Yamada, Pankov, and Cukierman, 2003). Further studies are needed to understand the downstream signaling events initiated by BMRF-2–integrin interactions and their role in the infection of oral epithelial cells with EBV.
Material and Methods
Cell culture
The EBV-producing marmoset B lymphoblastoid cell line B95-8 EBV-negative BJAB and Akata 4E-3 cells (a gift from Dr. L Hutt-Fletcher at Louisiana State University, Health Sciences Center, Shreveport, Louisiana) were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum and antibiotics. Two EBV permissive cell lines, HSC-3sort tongue and Detroitsort pharyngeal, and the HSC-3sort/BMRF-2 cell line constitutively expressing BMRF-2 were previously established in our laboratory (Tugizov, Berline, and Palefsky, 2003; Xiao et al., 2007). The cells were maintained in Dulbecco’s Modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) (HyClone, Logan, Utah), 200 mM L-glutamine, 0.1 mg/ml streptomycin, and 100 U/ml penicillin. Primary tongue keratinocyte (OCO) cells were previously established from human tongue tissue (University of California, San Francisco, Committee on Human Research approval #RS00908) in our laboratory and were grown in keratinocyte growth medium (Cambrix Bio Science, Walkersville, Maryland) (Tugizov, Berline, and Palefsky, 2003).
Construction of recombinant EBV that lacks expression of BMRF-2
A recombinant virus, designated EBV/ΔBMRF-2, was constructed by a homologous recombination method (Fig. 1.) To disrupt expression of the BMRF-2 gene, we used a PCR-generated DNA fragment containing green fluorescence protein (GFP) and neomycin/kanamycin (NK) resistance (aminoglycoside phosphotransferase) expression cassettes to replace 360 nucleotides (81361-81720) of the BMRF-2 open reading frame (ORF) in EBV B95-8 strain (accession number NC_001345). The GFP is driven by the HCMV immediate early promoter and the NK protein is expressed under the SV40 promoter. After recombination, the BMRF-2 protein was truncated after 81 amino acids (81118-81361). The shuttle vector plasmid, designated pB-GFP-NK, containing the upstream (EBV genome 80461-81360) and downstream (EBV genome 81721-82515) homologous sequences, the GFP expression cassette, and the neomycin resistance expression cassette (Fig. 1) was constructed by PCR-cloning using the following primers: 1) Primers for amplification of the upstream homologous sequence: bmrf1-5, ggtgacactcaatccggatc; bmrf1-3bam, gtaggatccattcagcatgatggctggag. 2) Primers for amplification of the downstream homologous sequence: bmrf2-dn5pst, tacctgcagattcggtggcatccctgaag; bmrf2-dn3gccgacttctggaacatttg. 3) Primers for amplification of the GFP-expression cassette: cmv-p-gfp5, gtattaccgccatgcattag; cmv-p-gfp3, aaccacaactagaatgcagtg. 4) Primers for amplification of the NK resistance expression cassette: Nk-5, tcagttagggtgtggaaagtc; Nk-3, caaacgacccaacaccgtgcg. The upstream and downstream homologous sequences were first cloned into psp72 by PCR cloning. The HSV-thymidine kinase poly-adenylation signal (HSV-TK-pA: sense strand, gatcaataaaaagacagaataaag; antisense strand, gatcctttattctgtctttttatt) were then inserted into the BamH I site at the 3’end of the upstream homologous sequence. The PCR products of the GFP and NK expression cassettes were inserted into the BamH I and Pst I sites, respectively, by blunt-ended ligation after treating the vector and PCR products with T4 DNA polymerase. For homologous recombination, the plasmid DNA of pB-GFP-NK was linearized with restriction enzyme Pvu I. Twenty μg of the linearized DNA were transfected into 5 × 106 B95-8 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, California) according to the manufacturer’s protocol. Forty-eight h after transfection, cells were plated into 96-well plates with 104 cells per well in 200 μl of selection medium (RPMI 1640 with 10% FBS and 800 μg/ml G418), and half of the culture medium was replaced every 5 days with fresh selection medium. Three weeks after transfection, G418-resistant colonies were expanded and tested by PCR for recombination. Two pairs of primers were used to test recombination between the shuttle vector and the EBV genome: Primer set A (for GFP), A5’ (EB80412), 5’-gcttcctcgttgcagaagtg and A3’ (Gfp2089), 5’-tgttgttaacttgtttattgcagc; and primer set B (for NK), B5’ (Nk1606), 5’-taactgacacacattccacagc and B3’ (EB82630), 5’-tatcggatgcctcacgcgaag).
To rescue pure recombinant EBV/ΔBMRF-2, cells containing the recombinant viral genome were induced with phorbol 12-myristate-13-acetate (PMA) (30 ng/ml) and butyric acid (4 μM) for 5 days; supernatant was collected by centrifugation and filtered through 0.8μm filters. For each rescue experiment, 1.5-2 × 106 each of EBV-negative Akata 4E-3 or BJAB cells were incubated with 1 ml of growth medium containing 0.5, 0.25, or 0.05 ml of the above supernatant for 90 min with gentle shaking (160 rpm) at 37°C. Cells were then collected by centrifugation at 500 g for 5 min and cultured in growth medium for 36-48 h before the cells were plated into 96-well plates at 104 cells/well. G418-resistant colonies were selected as above.
Real-time DNA PCR and reverse transcription PCR (RT-PCR)
Real-time DNA PCR assays to quantitate total EBV genome copies were performed using a probe specific for the BZLF-1 gene, as previously described (Xiao et al., 2007). EBV/ΔBMRF-2 was quantitated using the primers and probe specific for the neomycin resistance gene: forward primer, 5’-GCATACGCTTGATCCGGCTA; reverse primer, TGATCGACAAGACCGGCTTC; probe: CTGCCCATTCGACCACCAAGCG. For RT-PCR, the total RNA of each sample was extracted from 4-5 × 106 cells using the RNeasy kit (Qiagen, Valencia, California) according to the protocols provided by the manufacturer. Reverse transcription reaction was performed using the iScript reverse transcriptase kit from Bio-Rad (Hercules, California) according to the manufacturer’s protocol. Regular RT-PCR was performed using gene-specific primers for BZLF-1, BMRF-1, and BMRF-2 and single-stranded cDNA as templates.
Flow cytometry assays
Expression of BMRF-2 on the cell surface was determined by flow cytometry using rat anti–BMRF-2 serum (1:50). Cells were washed once with cold phosphate buffered saline (PBS) and incubated with primary antibodies in PBS (pH 7.4) containing 1% bovine serum albumin (BSA) for 1 h on ice. Cells were then washed three times (5 min each) in PBS by centrifugation and reacted with phycoerythrin-labeled goat–anti-rat antibodies for 30 min at 4°C. Cells were analyzed in a fluorescence-activated cell sorting (FACS) cytometer (Becton-Dickinson and Company, San Jose, California).
Gradient purification of EBV viruses
Production of EBV was achieved by culturing cells with PMA (30 ng/ml) and butyric acid (4 μM) (Sigma) for 5 days, after which virion-containing media were clarified by centrifugation at 500 g for 5 min and filtered through a 0.8-μm membrane. Gradient purification of EBV was performed as previously described (Xiao et al., 2007). Briefly, virions were concentrated by centrifugation at 16000 × g for 90 min and resuspended in Tris-Sodium chloride-bacitracin (TNB) buffer (10 mM Tris HCl, pH 7.2, 150 mM NaCl, 100μg/ml bacitracin [Sigma]). Each aliquot of 0.5 ml of concentrated virions was then loaded onto 9 ml of 24-42% Histodenz gradient (Sigma) and centrifuged at 70000 × g for 2 h, and the upper three 0.5 ml fractions were collected and combined. Purified virions were then washed in TNB buffer three times using Amicon Ultra-100 centrifugal filter devices (Millipore, Billerica, Massachusetts) to remove Histodenz. The purification process was repeated once for double gradient purification. Titers of gradient purified EBV were then determined by real-time DNA PCR as described above.
Western blot assays
The BMRF-1 protein was detected using a Western blot assay with monoclonal antibody (mAb) to EBV p52/50 (Ea D, Advanced Biotechnologies, Columbia, Maryland) and NuPage 10% Bis-Tris gel (Invitrogen). The BMRF-2 protein was detected using rat anti–BMRF-2 serum (Tugizov, Berline, and Palefsky, 2003), and gp350/220 was detected using a mouse monoclonal antibody (ViroStat, Portland, Maine). To detect BMRF-2 and gp350/220 in cells producing the wild-type (WT) EBV or EBV/ΔBMRF-2, the membrane fractions of B95-8 (WT) and B27-BMRF-2low (EBV/ΔBMRF-2) cells were extracted and proteins were separated on SDS-polyacrylamide gel with 7 M urea, as previously described (Xiao et al., 2007). Antibody to EBNA-1 was purchased from Advanced Biotechnologies (Columbia, Maryland). Anti–β-actin was obtained from Ambio (Austin, Texas). All secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, Pennsylvania). The protein bands were visualized using the ECL Western blot assay and quantitated by measuring the intensity of pixels (mean density) of protein bands using NIH image software.
Quantitation of released virions
To measure the rate of virion release, B95-8 or B27-BMRF-2low cells were cultured for 5 days with PMA (30 ng/ml) and butyric acid (4 μM); cells and culture media were separated by centrifugation at 500 g for 5 min. Virions in the supernatants were concentrated by centrifugation at 16000 g for 90 min. Cellular and viral genomic DNA were then extracted using AquaPure genomic DNA kits (Bio-Rad). Copy numbers of the EBV genome in the cells or media were then quantitated by real-time DNA PCR as described above.
Virus attachment assay
Virus was propagated by inducing B95-8 or B27-BMRF-2low cells with 30 ng/ml PMA and 4 μM butyric acid (both from Sigma) for 5 days. Virus was then concentrated using Amicon Ultra-100 centrifugal filter devices (Millipore) and viral titer was determined by quantitative real-time PCR. Virus attachment to cells was measured according to the method described by Shannon-Lowe et al. (Shannon-Lowe et al., 2005) with some modifications. HSC-3sort and Detroitsort cells were grown into 80% confluence, dissociated with enzyme-free cell dissociation buffer (Invitrogen), and washed once with ice-cold PBS. EBV-negative Akata 4E-3 cells were washed once with ice-cold PBS. About 5 × 105 cells were incubated with different amounts of EBV virions in PBS at 0°C for 60 min on a rotator. Cells were then washed three times with PBS to remove unbound virions, and genomic DNA was extracted using AquaPure genomic DNA kits (Bio-Rad). Virus copy numbers were quantitated by real-time PCR as described above. Virus binding to cells was also detected by flow cytometry using the following primary antibodies: monoclonal anti-gp250/350 (Virostat, Portland, Maine), anti-gHgL (E1D1) (a gift from Dr. L Hutt-Fletcher at Louisiana State University Health Sciences Center, Shreveport, Louisiana), and anti-gp110 (Sykesville, Maryland). After primary antibody reaction, flow cytometry analyses were then performed as described above.
EBV infection of polarized oral epithelial cells
Polarization of oral epithelial cells and their infection with EBV were as described previously (Tugizov, Berline, and Palefsky, 2003). Briefly, HSC-3sort tongue, Detroitsort, and primary tongue epithelial cells were cultured on 24-mm diameter Costar Transwell filters (Corning Life Sciences, Corning, New York) to form a polarized monolayer. Polarized HSC-3sort tongue (between passages 18-22) and Detroitsort pharyngeal (between passages 17-20) cells were infected with cell-free EBV at 1000 multiplicity of infection (MOI)/cell through their basolateral membranes. Since only 10% of virions may traverse filter pores, the actual MOI will be about 100 virions/cell (Tugizov, Berline, and Palefsky, 2003). Five days after infection, cells were fixed with 3% paraformaldehyde in PBS at 4°C for 30 min and immunostained with mouse monoclonal anti-gp350/220 antibody and Texas red–labeled goat anti-mouse secondary antibodies. Cells were analyzed using a krypton-argon laser coupled with a Bio-Rad MRC2400 confocal head (Bio-Rad). The data were analyzed using Laser Sharp software (Bio-Rad).
Site-directed mutagenesis of the BMRF-2 protein
The extracellular domain containing the RGD motif (BMRF-2 RGD, amino acids 171 to 218) was previously constructed as a glutathione-S-transferase (GST) –BMRF-2 RGD fusion protein. The QuikChange Site-Directed Mutagenesis Kit (Stratagene, San Diego, California) was used to mutate the RGD sequence in the BMRF-2 RGD peptide using the GST–BMRF-2 RGD fusion gene as a template and the following HPLC-purified primers (Invitrogen). Primers for the BMRF-2 RGE mutant are forward primer 5’-tttctgcgcccggggacaacattcggtggc-3’ and reverse primer 5’-gccaccgaatgttgtccccgggcgcagaaa. Primers for the BMRF-2 AAA mutant are forward primer 5’-cattttctgcgccgccgcagctcattcggtggcatc-3’ and reverse primer 5’-gatgccaccgaatgagctgcggcggcgcagaaaatg. Mutant plasmids were created according to the protocols provided by the manufacturer. Correct substitutions were confirmed by DNA sequencing.
GST-fusion proteins were propagated in the Escherichia coli Bl-21 strain (Xiao et al., 2007) and purified using Sepharose-4 beads. BMRF-2 peptides were cleaved by thrombin (Amersham, Piscataway, New Jersey) according to protocols provided by the manufacturer. After thrombin cleavage, peptide fragments were washed three times and concentrated in PBS using Amicon Ultra-10 centrifugal filter devices (Millipore). Endotoxin was measured using the Limulus Amebocyte Lysate assay kit E-TOXATE (Sigma), and the endotoxin level in purified proteins was less than 0.005 EU/mg.
Cell adhesion assay
Cell adhesion assays were carried out according to published protocols (Wang et al., 2003) with some modifications. Briefly, Maxisorp 96-well plates (Nalge Nunc International, Rochester, New York) were coated with 100 μl of BMRF-2 RGD, BMRF-2 RGE, BMRF-2 AAA peptide, or fibronectin at different concentrations overnight at 4°C in PBS, washed three times with cold PBS, and incubated with 1% BSA in PBS at 4°C for 2 h to block nonspecific protein binding. The plates were then washed three times with PBS before use. HSC-3sort cells grown to 80% confluence were dissociated with enzyme-free Cell Dissociation Buffer (Invitrogen) and washed once with serum-free DMEM. To determine the role of integrins in cell adhesion, cells were incubated with rat anti-β1 mAb (AIIB2, Hybridoma Bank, The University of Iowa, Iowa City, Iowa) or mouse anti-αv mAb (MAB 2021Z) (Chemicon, Temecula, California) for 1 h at 4°C. In parallel experiments the RGD-coated plates were incubated with rat anti-BMRF-2 serum, EBV-negative (Blackhawk BioSystem, Inc., San Ramon, California) or EBV-positive (gift from Evelyn Lennette, Virolab, Berkeley, California) human serum. Cells were resuspended in DMEM and added to the protein-coated plates at 2 × 104 cells/well in 100 μl DMEM. After 1 h incubation at 37°C in a 5% CO2 incubator, the plates were washed four times with DMEM to remove unbound cells. Attached cells were then fixed with 4% paraformaldehyde, stained with crystal violet, and quantitated by measuring the absorbance at 595 nm in an enzyme-linked immunosorbent assay plate-reader (Spectra Max340, Molecular Devices, Sunnyvale, California).
Acknowledgments
We thank Dr. L. Hutt-Fletcher (University of Louisiana, Shreveport) for providing Akata 4E-3 cells and antibodies to EBV gHgL, Dr. K. Copren for performing the real-time PCR assays, M. Turner for establishment of OCO primary tongue keratinocytes and D. Airo for editorial assistance. This project was supported by National Institute of Health grants R01 DE14894 and R21 DE016009 (to S.T).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam T. Exploitation of host factors for efficient infection by Shigella. Int J Med Microbiol. 2001;291(4):287–98. doi: 10.1078/1438-4221-00132. [DOI] [PubMed] [Google Scholar]
- Akula SM, Pramod NP, Wang FZ, Chandran B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell. 2002;108(3):407–19. doi: 10.1016/s0092-8674(02)00628-1. [DOI] [PubMed] [Google Scholar]
- Borza CM, Morgan AJ, Turk SM, Hutt-Fletcher LM. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J Virol. 2004;78(10):5007–14. doi: 10.1128/JVI.78.10.5007-5014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu JJ, Ng ML. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J Biol Chem. 2004;279(52):54533–41. doi: 10.1074/jbc.M410208200. [DOI] [PubMed] [Google Scholar]
- Ciarlet M, Crawford SE, Cheng E, Blutt SE, Rice DA, Bergelson JM, Estes MK. VLA-2 (alpha2beta1) integrin promotes rotavirus entry into cells but is not necessary for rotavirus attachment. J Virol. 2002;76(3):1109–23. doi: 10.1128/JVI.76.3.1109-1123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feire AL, Koss H, Compton T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc Natl Acad Sci U S A. 2004;101(43):15470–5. doi: 10.1073/pnas.0406821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Brocker EB, Zanker KS. Integrins, cell matrix interactions and cell migration strategies: fundamental differences in leukocytes and tumor cells. Cell Adhes Commun. 1998;6(23):225–36. doi: 10.3109/15419069809004478. [DOI] [PubMed] [Google Scholar]
- Friedl P, Zanker KS, Brocker EB. Cell migration strategies in 3-D extracellular matrix: differences in morphology, cell matrix interactions, and integrin function. Microsc Res Tech. 1998;43(5):369–78. doi: 10.1002/(SICI)1097-0029(19981201)43:5<369::AID-JEMT3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Gavrilovskaya IN, Brown EJ, Ginsberg MH, Mackow ER. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J Virol. 1999;73(5):3951–9. doi: 10.1128/jvi.73.5.3951-3959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285(5430):1028–32. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Gilcrease MZ. Integrin signaling in epithelial cells. Cancer Lett. 2007;247(1):1–25. doi: 10.1016/j.canlet.2006.03.031. [DOI] [PubMed] [Google Scholar]
- Guerreiro-Cacais AO, Li L, Donati D, Bejarano MT, Morgan A, Masucci MG, Hutt-Fletcher L, Levitsky V. Capacity of Epstein-Barr virus to infect monocytes and inhibit their development into dendritic cells is affected by the cell type supporting virus replication. J Gen Virol. 2004;85(Pt 10):2767–78. doi: 10.1099/vir.0.80140-0. [DOI] [PubMed] [Google Scholar]
- Haan KM, Lee SK, Longnecker R. Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein-Barr virus-induced membrane fusion. Virology. 2001;290(1):106–14. doi: 10.1006/viro.2001.1141. [DOI] [PubMed] [Google Scholar]
- Hutt-Fletcher LM. Epstein-Barr virus entry. J Virol. 2007;81(15):7825–32. doi: 10.1128/JVI.00445-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson T, Sharma A, Ghazaleh RA, Blakemore WE, Ellard FM, Simmons DL, Newman JW, Stuart DI, King AM. Arginine-glycine-aspartic acid-specific binding by foot-and-mouth disease viruses to the purified integrin alpha(v)beta3 in vitro. Journal of Virology. 1997;71(11):8357–61. doi: 10.1128/jvi.71.11.8357-8361.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A. 2004;101(46):16286–91. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Yajima M, Ahsan N, Tanaka M, Takada K. Production of high-titer Epstein-Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J Virol. 2004;78(13):7004–15. doi: 10.1128/JVI.78.13.7004-7015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi Y, Kawashima H, Sato S, Ioi H, Amaha M, Takekuma K, Hoshika A, Oshiro H, Matsubayashi J, Mukai K. Virological and immunological characteristics of fatal virus-associated haemophagocytic syndrome (VAHS) Microbiol Immunol. 2007;51(1):53–62. doi: 10.1111/j.1348-0421.2007.tb03890.x. [DOI] [PubMed] [Google Scholar]
- Kieff E, Rickinson AB. Epstein-Barr virus and its replication. In Fields Virology. In: Knipe DM, Howley PM, editors. Epstein-Barr virus and its replication. 4. Lippincott Williams and Willkins; New York: 2001. [Google Scholar]
- Knol AC, Quereux G, Pandolfino MC, Khammari A, Dreno B. Presence of Epstein-Barr virus in Langerhans cells of CTCL lesions. J Invest Dermatol. 2005;124(1):280–2. doi: 10.1111/j.0022-202X.2004.23570.x. [DOI] [PubMed] [Google Scholar]
- Krishnan HH, Sharma-Walia N, Streblow DN, Naranatt PP, Chandran B. Focal adhesion kinase is critical for entry of Kaposi’s sarcoma-associated herpesvirus into target cells. J Virol. 2006;80(3):1167–80. doi: 10.1128/JVI.80.3.1167-1180.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagenaur LA, Palefsky JM. Regulation of Epstein-Barr virus promoters in oral epithelial cells and lymphocytes. J Virol. 1999;73(8):6566–72. doi: 10.1128/jvi.73.8.6566-6572.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M, Artym VV, Green JA, Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol. 2006;18(5):463–71. doi: 10.1016/j.ceb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Leight ER, Sugden B. EBNA-1: a protein pivotal to latent infection by Epstein-Barr virus. Rev Med Virol. 2000;10(2):83–100. doi: 10.1002/(sici)1099-1654(200003/04)10:2<83::aid-rmv262>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. Journal of Virology. 1997;71(6):4657–62. doi: 10.1128/jvi.71.6.4657-4662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Turk SM, Hutt-Fletcher LM. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J Virol. 1995;69(7):3987–94. doi: 10.1128/jvi.69.7.3987-3994.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masy E, Adriaenssens E, Montpellier C, Crepieux P, Mougel A, Quatannens B, Goormachtigh G, Faumont N, Meggetto F, Auriault C, Groux H, Coll J. Human monocytic cell lines transformed in vitro by Epstein-Barr virus display a type II latency and LMP-1-dependent proliferation. J Virol. 2002;76(13):6460–72. doi: 10.1128/JVI.76.13.6460-6472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May JS, de Lima BD, Colaco S, Stevenson PG. Intercellular gamma-herpesvirus dissemination involves co-ordinated intracellular membrane protein transport. Traffic. 2005;6(9):780–93. doi: 10.1111/j.1600-0854.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- McShane MP, Longnecker R. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc Natl Acad Sci U S A. 2004;101(50):17474–9. doi: 10.1073/pnas.0404535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Kauwe LK. Endocytosis of adenovirus and adenovirus capsid proteins. Adv Drug Deliv Rev. 2003;55(11):1485–96. doi: 10.1016/j.addr.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. Journal of Virology. 2000;74(14):6324–32. doi: 10.1128/jvi.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omerovic J, Lev L, Longnecker R. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with epithelial and B cells. J Virol. 2005;79(19):12408–15. doi: 10.1128/JVI.79.19.12408-12415.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palefsky JM, Peñaranda ME, Pierik LT, Lagenaur LA, MacPhail LA, Greenspan D, Greenspan JS. Epstein-Barr virus BMRF-2 and BDLF-3 expression in hairy leukoplakia. Oral Diseases. 1997;3 Suppl 1(8):S171–6. doi: 10.1111/j.1601-0825.1997.tb00353.x. [DOI] [PubMed] [Google Scholar]
- Pietiainen V, Marjomaki V, Upla P, Pelkmans L, Helenius A, Hyypia T. Echovirus 1 endocytosis into caveosomes requires lipid rafts, dynamin II, and signaling events. Mol Biol Cell. 2004;15(11):4911–25. doi: 10.1091/mbc.E04-01-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- Savard M, Belanger C, Tardif M, Gourde P, Flamand L, Gosselin J. Infection of primary human monocytes by Epstein-Barr virus. J Virol. 2000;74(6):2612–9. doi: 10.1128/jvi.74.6.2612-2619.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitt A, Blankenberg S, Weise K, Gartner BC, Mehrer T, Peetz D, Meyer J, Darius H, Rupprecht HJ. Herpesvirus DNA (Epstein-Barr virus, herpes simplex virus, cytomegalovirus) in circulating monocytes of patients with coronary artery disease. Acta Cardiol. 2005;60(6):605–10. doi: 10.2143/AC.60.6.2004932. [DOI] [PubMed] [Google Scholar]
- Shannon-Lowe C, Baldwin G, Feederle R, Bell A, Rickinson A, Delecluse HJ. Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J Gen Virol. 2005;86(Pt 11):3009–19. doi: 10.1099/vir.0.81153-0. [DOI] [PubMed] [Google Scholar]
- Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J Virol. 2004;78(8):4207–23. doi: 10.1128/JVI.78.8.4207-4223.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakage M, Kimura M, Yanoma S, Ibe M, Yokota S, Tsujino G, Kozuka T, Dezawa T, Tamura S, Ohshima A, Yutsudo M, Hakura A. Expression of latent and replicative-infection genes of Epstein-Barr virus in macrophage. Arch Virol. 1999;144(1):157–66. doi: 10.1007/s007050050492. [DOI] [PubMed] [Google Scholar]
- Spear PG. Membrane fusion induced by herpes simplex virus. In: Bentz J, editor. Viral fusion mechanisms. CRC Press; Boca Raton, FL: 1993. pp. 201–232. [Google Scholar]
- Spear PG, Longnecker R. Herpesvirus entry: an update. J Virol. 2003;77(19):10179–85. doi: 10.1128/JVI.77.19.10179-10185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck P, Kline KA, Cheresh P, Longnecker R. Epstein-Barr virus lacking latent membrane protein 2 immortalizes B cells with efficiency indistinguishable from that of wild-type virus. J Gen Virol. 1999;80(Pt 8):2193–203. doi: 10.1099/0022-1317-80-8-2193. [DOI] [PubMed] [Google Scholar]
- Takagi J. Structural basis for ligand recognition by RGD (Arg-Gly-Asp)-dependent integrins. Biochem Soc Trans. 2004;32(Pt3):403–6. doi: 10.1042/BST0320403. [DOI] [PubMed] [Google Scholar]
- Triantafilou K, Takada Y, Triantafilou M. Mechanisms of integrin-mediated virus attachment and internalization process. Crit Rev Immunol. 2001;21(4):311–22. [PubMed] [Google Scholar]
- Triantafilou K, Triantafilou M. A biochemical approach reveals cell-surface molecules utilised by Picornaviridae: Human Parechovirus 1 and Echovirus 1. J Cell Biochem. 2001;80(3):373–81. doi: 10.1002/1097-4644(20010301)80:3<373::aid-jcb110>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Tugizov S, Herrera R, Veluppillai P, Greenspan J, Greenspan D, Palefsky JM. Epstein-Barr Virus (EBV)-Infected Monocytes Facilitate Dissemination of EBV within the Oral Mucosal Epithelium. J Virol. 2007;81(11):5484–96. doi: 10.1128/JVI.00171-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugizov S, Wang Y, Qadri I, Navarro D, Maidji E, Pereira L. Mutated forms of human cytomegalovirus glycoprotein B are impaired in inducing syncytium formation. Virology. 1995;209:580–591. doi: 10.1006/viro.1995.1290. [DOI] [PubMed] [Google Scholar]
- Tugizov SM, Berline JW, Palefsky JM. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat Med. 2003;9(3):307–14. doi: 10.1038/nm830. [DOI] [PubMed] [Google Scholar]
- Wang FZ, Akula SM, Sharma-Walia N, Zeng L, Chandran B. Human herpesvirus 8 envelope glycoprotein B mediates cell adhesion via its RGD sequence. J Virol. 2003;77(5):3131–47. doi: 10.1128/JVI.77.5.3131-3147.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Huang DY, Huong SM, Huang ES. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med. 2005;11(5):515–21. doi: 10.1038/nm1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CH, Kajander T, Hyypia T, Jackson T, Sheppard D, Stanway G. Integrin alpha v beta 6 is an RGD-dependent receptor for coxsackievirus A9. J Virol. 2004;78(13):6967–73. doi: 10.1128/JVI.78.13.6967-6973.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Palefsky JM, Herrera R, Tugizov SM. Characterization of the Epstein-Barr virus glycoprotein BMRF-2. Virology. 2007;359(2):382–96. doi: 10.1016/j.virol.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Pankov R, Cukierman E. Dimensions and dynamics in integrin function. Braz J Med Biol Res. 2003;36(8):959–66. doi: 10.1590/s0100-879x2003000800001. [DOI] [PubMed] [Google Scholar]