

Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative movement disorder and in most patients its aetiology remains unknown. Molecular genetic studies in familial forms of the disease identified key proteins involved in PD pathogenesis, and support a major role for mitochondrial dysfunction, which is also of significant importance to the common sporadic forms of PD. While current treatments temporarily alleviate symptoms, they do not halt disease progression. Drugs that target the underlying pathways to PD pathogenesis, including mitochondrial dysfunction, therefore hold great promise for neuroprotection in PD. Here we summarize how the proteins identified through genetic research (α-synuclein, parkin, PINK1, DJ-1, LRRK2 and HTRA2) fit into and add to our current understanding of the role of mitochondrial dysfunction in PD. We highlight how these genetic findings provided us with suitable animal models and critically review how the gained insights will contribute to better therapies for PD.
Keywords: Genetics, Mitochondria, Neuroprotection, Nuclear-encoded proteins, Oxidative stress, Parkinson's disease, Treatment
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder with a prevalence of 1.8% in individuals of 65 years and older (de Rijk et al. 2000). Resting tremor, bradykinesia and rigidity are the cardinal clinical characteristics of PD. Neuropathological examination shows several affected brain regions, but the loss of dopaminergic (DAergic) neurons in the substantia nigra pars compacta (SNpc) is believed to be the most crucial (Braak et al. 2004). At the time of clinical presentation approximately 50–70% of DAergic neurons in the nigrostriatal system have been lost (Orth & Schapira 2002). Surviving neurons may contain Lewy bodies, intracytoplasmic protein aggregates mainly composed of α-synuclein (SNCA) (Spillantini et al. 1997). And these Lewy bodies are a second neuropathological feature of PD. Current treatments for PD, with levodopa as the most commonly used drug, are focused on the symptomatic improvement of motor features related to the above mentioned loss of DAergic neurons (Schapira 2005). More importantly levodopa, notwithstanding its symptomatic benefits, does not cure PD, nor does it halt the development of additional features during the course of PD, such as autonomic dysfunction, gait disturbance, freezing and dementia (Olanow et al. 2004).
Mitochondrial dysfunction has long been implicated in PD pathogenesis; this hypothesis arose with the discovery that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) produced PD-like symptoms in designer drug abusers (Langston et al. 1983). Its metabolite, 1-methyl-4-phenylpyridinium (MPP+), is actively transported into DAergic neurons by the dopamine transporter. Within these neurons MPP+ enters mitochondria, and selectively inhibits mitochondrial respiration at complex I of the electron transport chain (Krueger et al. 1990). Chronic infusion of rotenone, a highly selective complex I inhibitor, also reproduced behavioural (e.g. hypokinesia and rigidity) and neuropathological features of PD in rats (Betarbet et al. 2000; Sherer et al. 2003). These neurotoxins and neurotoxic animal models of PD renewed interest in possible environmental causes of PD, as similar compounds in the environment might play a causative role in the disease. In addition, genetic defects causing familial forms of PD have been identified in the last decade. Despite the rarity of these familial forms of PD (5–10% of the PD population) the identification of PD-linked genes has fuelled our understanding of possible pathogenic mechanisms of PD, and placed ubiquitin-proteasome system (UPS) dysfunction, oxidative stress and mitochondrial dysfunction at centre stage. Mutations or polymorphisms in both mitochondrial DNA (mtDNA) and nuclear DNA were implicated in causing PD or in affecting PD risk. Of the nuclear genes, mutations in SNCA, PARK2 (also known and hereafter referred to as parkin), PINK1 (PTEN induced putative kinase 1), PARK7 (also known and hereafter referred to as DJ-1), LRRK2 (leucine-rich repeat kinase 2) and HTRA2 (high temperature requirement A2) provide direct or indirect evidence for a major role of mitochondrial dysfunction in PD.
In this review we aim to highlight the recent genetic data on mtDNA polymorphisms and the nuclear-encoded SNCA, parkin, PINK1, DJ-1, LRRK2 and HTRA2, and focus on the relevance of these genes for mitochondrial function. Pathogenic theories focus on a combination of genetic and environmental risk factors, and thus we will also briefly consider environmental exposures relevant to mitochondrial dysfunction in PD. Our current understanding of the mitochondrial pathway to PD provided us with tools to create better animal models, as well as interesting entry points for therapy. These entry points will be discussed along with therapeutic drugs acting on them. Some of the drugs are already in clinical use; others are still in the primary stages of evaluation, but show great promise for modifying PD progression.
Mitochondria
Function and structure
The primary function of mitochondria is the generation of cellular energy in the form of adenosine 5′-triphosphate (ATP) by oxidative phosphorylation. In addition to energy production mitochondria play a role in the metabolism of e.g. amino acids and lipids, as well as intermediate metabolic pathways, calcium homeostasis and free radical scavenging.
Mitochondria are intracellular double membrane-bound structures (Fig. 1), and are partitioned in four main compartments: outer mitochondrial membrane (OMM), intermembrane space (IMS), inner mitochondrial membrane (IMM) and matrix. The five complexes – I [NADH (nicotinamide adenine dinucleotide, reduced form) ubiquinone oxidoreductase], II (succinate ubiquinone oxidoreductase), III (ubiquinone-cytochrome c reductase), IV (cytochrome oxidase) and V (ATP synthase) – of the mitochondrial respiratory chain, also called the electron transport chain, are all located in the IMM. Two mobile electron carriers, coenzyme Q (ubiquinone) and cytochrome c, are located in the IMM and IMS, respectively. The transport of electrons down the respiratory chain is energetically favourable. The released energy is used by complexes I, III and IV to transport protons from the matrix to the IMS, thus creating a proton and electrochemical gradient across the IMM. This gradient forms the basis of the inner mitochondrial transmembrane potential (ΔΨm) and is exploited by complex V of the respiratory chain to drive ATP synthesis.
Figure 1.
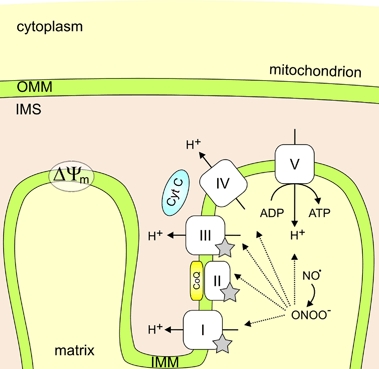
Mitochondrial structure and composition of the mitochondrial respiratory chain. The mitochondrial respiratory chain is a sequence of complexes found in the IMM that accepts electrons from electron donors such as NADH or succinate, shuttles these electrons across the IMM and creates a proton and electrochemical gradient. This gradient forms the basis of the inner mitochondrial transmembrane potential (ΔΨm) and is used to drive ATP synthesis by complex V of the respiratory chain. Complexes I, II and III generate ROS (indicated by grey stars). NO inhibits respiration by reversible binding to the oxygen binding site of complex IV (not shown) and is likely to be a physiological regulator of respiration. When cells are under oxidative stress, ROS will accumulate, react with NO, and form peroxynitrite (ONOO−); a strong oxidant thought to be responsible for the ‘pathological actions’ of NO. ONOO− inactivates the respiratory complexes (dotted lines), stimulates proton leakage through the IMM and might inhibit complex I by tyrosine nitration (for review see Brown & Borutaite 2002). Abbreviations: ΔΨm, inner mitochondrial transmembrane potential; ADP, adenosine 5′-diphosphate; CoQ, coenzyme Q; Cyt C, cytochrome c.
The mitochondria contain their own genome. mtDNA is a multicopy, maternally inherited, 16.5-kb circular double stranded DNA molecule without any histone coating. mtDNA is extremely compact (93% coding sequence) and the majority of proteins required to build and maintain functional mitochondria is therefore encoded by nuclear DNA, synthesized in the cytosol and imported into mitochondria, where they are targeted to one of the four mitochondrial compartments.
Oxidative stress
Oxidative stress is the result of an imbalance between excessive production of reactive oxygen species (ROS) and limited antioxidant defences. Mitochondria generate most of the ROS as a byproduct of oxidative phosphorylation. An estimated 2% of the oxygen (O2) consumed by mitochondria is converted to superoxide anion (O2−·) (Beal 2003), which is a precursor of most other ROS (Turrens 2003) (Fig. 2). These ROS can lead to oxidative damage of proteins, DNA and lipids (Raha & Robinson 2000).
Figure 2.
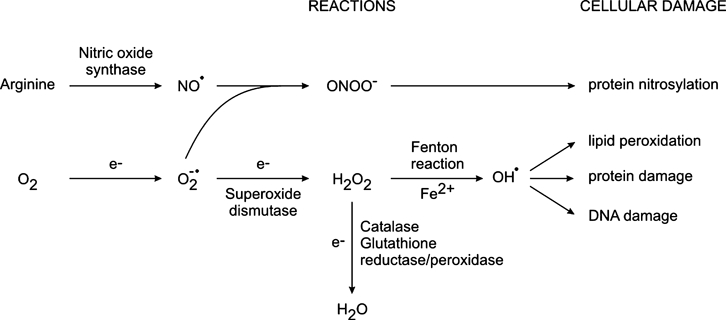
ROS. These include (1) free radicals (containing highly reactive unpaired electrons), such as superoxide (O2−·), nitric oxide (NO·) and hydroxyl radical (OH·); and (2) other molecular species, such as hydrogen peroxide (H2O2) and peroxynitrite (ONOO−). O2−· is converted to H2O2, either spontaneously or through a reaction catalysed by SOD (Fridovich 1995). H2O2 may in turn be fully reduced to water, by catalase or glutathione reductase/peroxidase, or partially reduced to hydroxyl radicals (OH·). The latter reaction (Fenton reaction) occurs in the presence of reduced transition metals (e.g. Fe2+), which may again be re-reduced by O2−·, propagating the process (Liochev & Fridovich 1994). Alternatively O2−· can also react with NO radicals (NO·; produced by nitric oxide synthase during conversion of arginine to citrulline) to form peroxynitrite (ONOO−) (Beckman & Koppenol 1996).
Numerous studies indicated the involvement of ROS and oxidative stress in PD pathogenesis, including reduced amounts of the thiol-reducing agent glutathione (Pearce et al. 1997) and elevated concentrations of iron (Fe) (Kienzl et al. 1995) in SN of PD patients. Loss of neuromelanin-containing DAergic cells is characteristic for PD and the dark brown pigment neuromelanin attracted attention to the auto-oxidation of dopamine, as it consists primarily of products of dopamine redox chemistry (Wakamatsu et al. 2003). Normal metabolism of dopamine, partly accomplished by monoamine oxidases (MAO), produces hydrogen peroxide (H2O2) (Maker et al. 1981). From this reaction alone, DAergic neurons are exposed to oxidative stress. In addition, dopamine can be oxidized to a dopamine quinone. This oxidation occurs spontaneously, is accelerated by the presence of transition metal ions, or can be enzyme-catalysed. The resulting dopamine quinone covalently modifies cellular macromolecules, which may serve as a mechanism for dopamine induced neurotoxicity (Stokes et al. 1999).
Apoptosis
Apoptotic cell death is characterized by marked nuclear and cellular shrinkage, membrane blebbing, chromatin condensation, nuclear fragmentation and the budding off of apoptotic bodies (Kerr et al. 1972). Apoptosis is triggered by a number of insults including e.g. misfolded proteins, ROS and mitochondrial complex inhibition (Bredesen et al. 2006), and is executed via two main pathways (Fig. 3), which eventually converge at the level of effector caspases activation and the subsequent cleavage of apoptotic substrates. Firstly, the death receptor (or extrinsic) pathway, which is initiated by activation of cell-surface death receptors (e.g. Fas), and secondly, the mitochondrial (or intrinsic) pathway, characterized by the release of mitochondrial pro-apoptotic proteins (e.g. cytochrome c) (Hengartner 2000).
Figure 3.
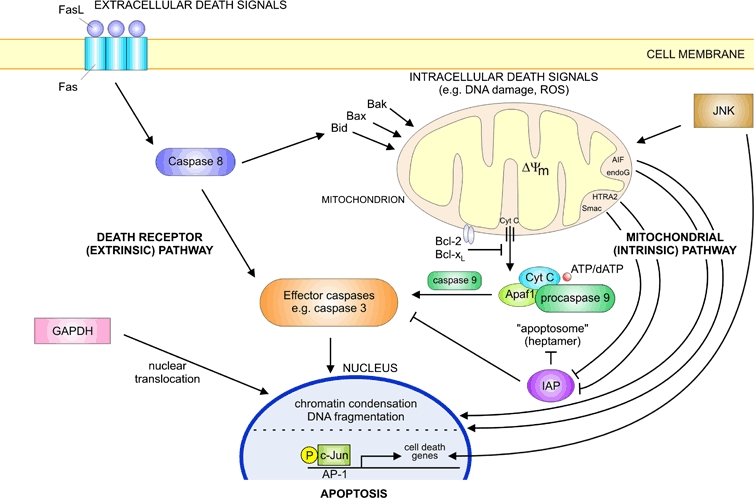
Schematic illustration of the major pathways leading to apoptosis. Apoptosis occurs through two main pathways. These are the death receptor (extrinsic) pathway which originates through the activation of cell-surface death receptors, for example Fas, and the mitochondrial (intrinsic) pathway which originates from mitochondrial release of cytochrome c. A distinct nuclear pathway of apoptosis arises through increased expression of GAPDH and its translocation from the cytoplasm to the nucleus, whereas the JNK signal-transduction pathway is activated in response to different stress stimuli. Bid, Bax and Bak represent pro-apoptotic Bcl-2 family members; Bcl-2 and Bcl-xL are antiapoptotic. Abbreviations: ΔΨm, inner mitochondrial transmembrane potential; AP-1, activating protein-1; Apaf1, apoptotic protease-activating factor 1; dATP, deoxyadenosine 5′-triphosphate; Cyt C, cytochrome c; endoG, endonuclease G; FasL, Fas ligand.
The pivotal event in the mitochondrial pathway is mitochondrial outer membrane permeabilization (MOMP) that leads to the release of several IMS proteins, such as cytochrome c, apoptosis inducing factor (AIF), endonuclease G, second mitochondria-derived activator of caspases (Smac) and HTRA2 (van Loo et al. 2002). MOMP can occur via two mechanisms: the first one involves the opening of the permeability transition (PT) pore, a protein complex at the contact site between OMM and IMM (Zamzami & Kroemer 2001), whereas the second mechanism appears to be mediated by direct action of Bcl-2 family members on the OMM (Green & Kroemer 2004). Cytochrome c, released from mitochondria following MOMP, assists in the formation of the apoptosome (a complex consisting of cytochrome c, apoptosis protease-activating factor 1, procaspase 9 and ATP/deoxyATP). Apoptosome oligomerization activates caspase 9, which then triggers activation of caspase 3 and other caspases in an amplication cascade ultimately causing cell death (Li et al. 1997).
Inhibitors of apoptosis (IAPs) can still inhibit active caspases (Holley et al. 2002), but the IAP-mediated block may in turn be released by proteins as Smac (Du et al. 2000) or HTRA2 (Martins et al. 2002). Two other proteins released from mitochondrial IMS during MOMP (AIF and endonuclease G) translocate to the nucleus and induce chromatin condensation and DNA fragmentation, independent of caspase activation (Li et al. 2001; Susin et al. 1999).
The c-Jun N-terminal kinase (JNK) signal-transduction pathway is also important for the execution of apoptosis in response to different stress stimuli (Davis 2000). JNK is part of a sequential kinase-signalling cascade involving three kinases. Mitogen-activated protein kinase (MAPK) kinase activates JNK, whereas MAPK kinase activation is mediated by MAPK kinase kinases, including the mixed lineage kinase (MLK) family in neurons. The activation of the JNK pathway rapidly induces its downstream target activating protein-1 transcription factor c-Jun, which plays a major role in the transcription of several pro-apoptotic genes (Ham et al. 2000). JNK acts as an effector of apoptosis through mitochondrial-dependent processes, as it catalyses the phosphorylation of antiapoptotic (Schroeter et al. 2003) and pro-apoptotic (Bhakar et al. 2003; Papadakis et al. 2006) Bcl-2 family members, induces release of cytochrome c (Schroeter et al. 2003) and Smac (Chauhan et al. 2003) and mediates a partial collapse of the mitochondrial membrane potential (Schroeter et al. 2003). Another distinct pathway of apoptosis arises through increased expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and its translocation from the cytoplasm to the nucleus (Ishitani et al. 1996; Sawa et al. 1997). In this pathway nitric oxide (NO) is the initial trigger to cell death (Hara et al. 2005).
Although an increased immunoreactivity for effectors of the mitochondrial apoptotic pathway (e.g. caspase 3) (Tatton 2000), an increased activation of JNK downstream targets (Hunot et al. 2004), as well as nuclear accumulation of GAPDH (Tatton 2000) have been detected in post-mortem SN of PD patients, there is still no clear consensus concerning the contribution of apoptosis to loss of DAergic neurons in PD (Tatton et al. 2003b). Necrosis (or passive cell death) represents a form of non-apoptotic cell death, but is not likely to play a major role in PD (Kostrzewa 2000) because necrosis is associated with energy failure, cell swelling and rupture followed by an inflammatory response; features that are largely absent in PD (Dickson 2007). Another form of non-apoptotic cell death that has gained interest in recent years is autophagic cell death, resulting from excessive levels of cellular autophagy. Autophagy complements the UPS pathway as it degrades long-lived proteins, protein aggregates and organelles (e.g. damaged mitochondria) through a lysosomal degradation pathway (Rubinsztein 2006). Whereas the survival-promoting role of autophagy in nutrient starvation is well accepted, its role in cell death is more controversial (Eskelinen 2005). Cross-talk between apoptotic and autophagic pathways has been reported, but their molecular interdependence is not yet clear (Ferraro & Cecconi 2007). However, mitochondria might represent the link at which apoptosis and autophagy interact, because mitochondria generate apoptotic signals, but are in turn removed by autophagy when damaged.
Environmental exposures and PD
The finding that humans intoxicated with MPTP develop a syndrome nearly identical to PD (Langston et al. 1983) lended support to the hypothesis that substances in the environment might contribute to PD aetiology. Indeed, epidemiological studies have implicated environmental risk factors such as rural living, consumption of well water and pesticide exposure in increased risk of PD (Priyadarshi et al. 2001). Conversely, cigarette smoking and coffee drinking are inversely associated with the risk for developing PD (Hancock et al. 2007; Hernan et al. 2002).
Cigarette smoking represents a good example of the relevance of an environmental toxin to mitochondrial function. Cigarette smoke is composed of more than 4000 compounds, but nicotine, the main alkaloid, seems to be particularly important. Nicotine reduces ROS generation from mitochondria (Cormier et al. 2003), and prevented neurotoxin-induced mitochondrial swelling and cytochrome c release in vitro through inhibition of the mitochondrial PT pore (Xie et al. 2005). The antioxidant properties of nicotine are likely important as well; e.g. nicotine may be protective against PD through complex formation with Fe2+, thus yielding Fenton-inactive Fe2+ (Fig. 2) and less oxidative stress (Linert et al. 1999). Chronic nicotine treatment was already shown to reduce paraquat-mediated nigrostriatal damage in a rodent model (Khwaja et al. 2007).
Genetics of PD
mtDNA genetics and PD
Studies using PD cybrids (cytoplasmic hybrids, through transfer of mtDNA of PD patients into mtDNA depleted cells) showed loss of complex I activity, increased O2 radical production, and increased susceptibility to MPTP-induced cell death (Swerdlow et al. 1996), suggesting that mtDNA abnormalities may be crucial in the pathogenesis of sporadic PD. Several studies reported inherited mtDNA microdeletions or single nucleotide mutations resulting in parkinsonism, typically as one feature of a multisystemic disorder, e.g. the prominent parkinsonism associated with Leber’s hereditary optic neuropathy caused by a single nucleotide mutation in the complex I ND4 gene (Simon et al. 1999). There is, however, less evidence for mtDNA involvement in non-syndromic PD. Nevertheless, specific mtDNA haplotypes were proposed to be implicated in PD risk (Autere et al. 2004; Ghezzi et al. 2005; Pyle et al. 2005; van der Walt et al. 2003). Interestingly, recent research into the extent of mtDNA deletions in SN DAergic neurons showed increased levels of clonally expanded somatic mtDNA deletions with ageing (Bender et al. 2006b; Kraytsberg et al. 2006), resulting in loss of mitochondrial function and cell death. The accumulation of mtDNA deletions was higher in PD patients as compared with age matched control individuals (Bender et al. 2006b).
PD genetics of nuclear-encoded proteins
Linkage analysis in extended families with highly penetrant Mendelian forms of PD has been successful in identifying specific disease-segregating mutations in previously unknown genes implicated in PD pathogenesis. Mutations in at least six genes were shown to cause familial parkinsonism: mutations in SNCA and LRRK2 account for autosomal dominant forms of PD, whereas mutations in parkin, PINK1, DJ-1 and ATP13A2 (Ramirez et al. 2006) show a recessive mode of inheritance (mutations in the latter are characteristic of Kufor-Rakeb syndrome, and will not be further discussed). In general autosomal dominant inherited mutations are gain-of-function mutations, while loss-of-function mutations are usually linked to recessive phenotypes. The use of animal models to evaluate gain- and loss-of-function mutations helped greatly in the illumination of the biological functions of the proteins encoded by these genes and yielded insights into their causal role in PD pathogenesis. Transgenic mice provide the ideal means to study possible disease-related gain-of-function mutations, knockout strategies in turn provide the opportunity to study diseases related to loss-of-function mutations.
The role of Mendelian genes in the common sporadic forms of PD is less known. On the other hand, association studies of candidate genes for PD try to define risk alleles that contribute to the sporadic forms of disease. Candidate gene studies evaluate genes based on their location in previously determined linkage regions (positional candidate genes), or based on a plausible biological function related to disease pathogenesis (functional candidate genes). Table 1 provides a general overview of the genetic causes of PD with mitochondrial involvement, whereas Fig. 4 depicts the proteins at their cellular action level.
Table 1.
Nuclear genes linked to autosomal inherited PD with mitochondrial involvement
| Protein | Function | Cellular localization | PD locus | Chromosomal location | Gene | Inheritance | Type of mutations | Key references |
|---|---|---|---|---|---|---|---|---|
| α-synuclein | Synaptic vesicle formation | Mainly cytoplasm | PARK1 | 4q21 | SNCA | Dominant | Missense mutations (p.Ala30Pro, p.Glu46Lys, p.Ala53Thr), duplication, triplication | Chartier-Harlin et al. 2004; Ibanez et al. 2004; Kruger et al. 1998; Polymeropoulos et al. 1997; Singleton et al. 2003; Zarranz et al. 2004 |
| Parkin | Ubiquitin E3 ligase | Mainly cytoplasm, OMM | PARK2 | 6q25.2–q27 | parkin | Recessive | Missense, nonsense and frameshift mutations, intraexonic deletions or insertions, promoter and exon deletions, exon multiplications | Hedrich et al. 2004b; Kitada et al. 1998; Mata et al. 2004 |
| Phosphatase and tensin homologue (PTEN)-induced kinase 1 | Mitochondrial kinase | Mainly IMM, occasionally OMM | PARK6 | 1p36 | PINK1 | Recessive | Missense, nonsense and frameshift mutations, exon deletions | Li et al. 2005; Marongiu et al. 2007; Rogaeva et al. 2004; Valente et al. 2004 |
| DJ-1 | Redox sensor, mitochondrial antioxidant | Cytoplasm, IMS and matrix | PARK7 | 1p36.33–p36.12 | DJ-1 | Recessive | Missense and frameshift mutations, insertions and exon deletions | Abou-Sleiman et al. 2003; Bonifati et al. 2003; Hague et al. 2003 |
| Leucine-rich repeat kinase 2 | Multifunctional kinase/GTPase | Cytoplasm, OMM | PARK8 | 12q12 | LRRK2 | Dominant | Missense mutations | Berg et al. 2005; Paisan-Ruiz et al. 2004; Zimprich et al. 2004 |
| High temperature requirement A2 | Pro-apoptotic protease, important for mitochondrial homeostasis | IMS | PARK13 | 2p12 | HTRA2 | Dominant* | Missense mutations | Strauss et al. 2005 |
In contrast with the other genes in the table, HTRA2 was not identified through linkage studies, but was associated with PD using a candidate gene approach.
Figure 4.
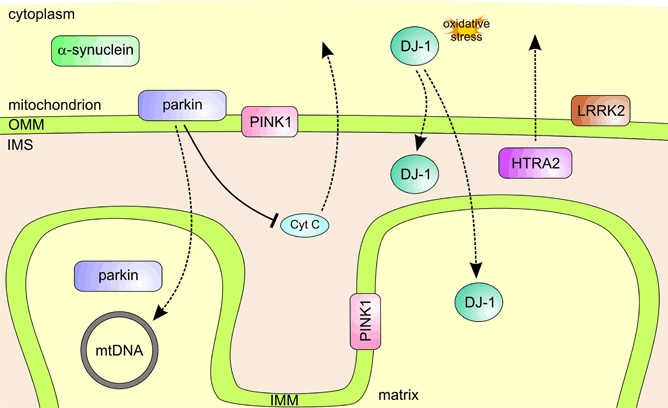
PD genes and their relation to mitochondria. Aggregation of mutant or overexpressed SNCA might be an upstream actor of mitochondrial alterations. Parkin associates with the OMM and was shown to play a role in mitochondrial biogenesis by regulating both transcription and replication of mtDNA. PINK1 has an N-terminal mitochondrial targeting motif and is localized to mitochondrial membranes, whereas oxidation of a key Cys-residue in DJ-1 leads to its relocalization to mitochondria. LRRK2 resides diffusely throughout the cytosol, but is partly associated with the OMM. HTRA2 resides in the IMS, wherefrom it is released upon apoptotic stimuli.
The inclusion protein SNCA
SNCA is the major constituent of Lewy bodies, the neuropathological hallmark of PD (Spillantini et al. 1997). SNCA is expressed throughout the brain (Solano et al. 2000) and is particularly enriched in presynaptic nerve terminals (Goedert 2001). It has potential roles in synaptic plasticity (George et al. 1995), regulation of dopamine neurotransmission (Abeliovich et al. 2000) and turnover of synaptic vesicles (Lotharius & Brundin 2002). In spite of extensive studies, the exact cellular function of SNCA remains unclear.
Missense mutations and multiplications of SNCA (4q21) cause autosomal dominant PD (Chartier-Harlin et al. 2004; Ibanez et al. 2004; Kruger et al. 1998; Polymeropoulos et al. 1997; Singleton et al. 2003; Zarranz et al. 2004), whereas association studies showed that increased SNCA expression because of variation in the SNCA promoter region conferred risk for sporadic PD (Maraganore et al. 2006; Pals et al. 2004).
Inducible expression of human p.Ala30Pro SNCA in PC12 cells (rat pheochromocytoma cells) decreased proteasome activity and increased sensitivity to mitochondria-dependent apoptosis (Tanaka et al. 2001). More recently human p.Ala53Thr SNCA overexpressing transgenic mice were reported to develop intraneuronal inclusions, as well as mtDNA damage and degeneration (Martin et al. 2006). Pathogenic p.Ala30Pro and p.Ala53Thr SNCA was also shown to be poorly degraded by chaperone-mediated autophagy (CMA) (Cuervo et al. 2004). Although these mutants bound with high affinity to the lysosomal membrane, they were not translocated into the lysosome. In addition to inhibiting their own degradation, they also blocked degradation of other CMA substrates; which is consistent with a toxic gain-of-function of these mutants.
On the other hand, loss of SNCA appears to have minimal effects because SNCA knockout mice were viable and fertile. Despite the lack of morphological abnormalities, these mice displayed a modest decrease in total striatal DA levels (Abeliovich et al. 2000), and a reduction in the reserve pool of synaptic vesicles in the hippocampus (Cabin et al. 2002).
The ubiquitin E3 ligase parkin
Ubiquitously expressed parkin encodes an ubiquitin E3 ligase (Shimura et al. 2000), which is a component of the UPS (Ciechanover 1998). Parkin was localized in mitochondria of proliferating cells and was also shown to play a role in mitochondrial biogenesis by regulating both transcription and replication of mtDNA (Kuroda et al. 2006).
Loss-of-function mutations in parkin cause autosomal recessive juvenile parkinsonism (Kitada et al. 1998). Parkin mutations may explain up to half of the patients with early onset PD and a family history compatible with recessive inheritance (Dekker et al. 2003). Mounting evidence suggests that heterozygous parkin mutations may increase susceptibility to late onset PD (Foroud et al. 2003; Oliveira et al. 2003; Sun et al. 2006; West et al. 2002). A promoter deletion in a family with a previously described heterozygous parkin exon 3-deletion recently extended the parkin mutation spectrum, as this compound heterozygous mutation was reported to result in complete absence of parkin expression (Lesage et al. 2007).
Drosophila parkin knockout mutants exhibited reduced lifespan, locomotor defects resulting from apoptotic muscle degeneration and male sterility. Mitochondrial structural alterations were prominent features of both muscle and germline pathology (Greene et al. 2003). Proteomics of midbrain from parkin knockout mice showed a decreased abundance of proteins involved in mitochondrial respiratory chain activity and protection against oxidative stress (Palacino et al. 2004).
Parkin overexpression models further support the role of parkin in mitochondria. Parkin overexpression in PC12 cells protected against ceramide-mediated cell death by delaying mitochondrial swelling, subsequent cytochrome c release and caspase 3 activation, and this protective effect was abrogated by parkin mutations (Darios et al. 2003). In human DAergic neuroblastoma cells (SH-SY5Y) parkin overexpression not only protected against apoptosis, but also decreased cellular ROS levels (Jiang et al. 2004).
On the other hand, mitochondrial dysfunction and oxidative stress can in turn affect parkin function, e.g. S-nitrosylation of parkin diminishes its ubiquitin E3 ligase activity and compromises its protective function (Chung et al. 2005). Such nitrosative stress combined with a single parkin mutation could lead to haploinsufficiency and might be particularly relevant for heterozygous parkin mutations associated with sporadic PD.
The mitochondrial kinase PINK1
The ubiquitously expressed PINK1 consists of an N-terminal mitochondrial targeting motif, a highly conserved serine–threonine kinase domain and a C-terminal autoregulatory domain (Silvestri et al. 2005). The PINK1 kinase is localized to the mitochondrial membranes (Gandhi et al. 2006).
Homozygous PINK1 mutations were first described in three consanguineous families (Valente et al. 2004). PINK1 loss-of-function mutations are estimated to account for 1–7% of early onset PD (Tan et al. 2006). Heterozygous PINK1 mutations were also reported, and were threefold enriched in sporadic PD patients compared with control individuals (Abou-Sleiman et al. 2006). It is hypothesized that heterozygous PINK1 mutations are a risk factor for the development of late onset PD (Khan et al. 2002).
Downregulation of PINK1 expression by small interfering RNA (siRNA) decreased SH-SY5Y cell viability and increased apoptosis (Deng et al. 2005). RNA interference-mediated inactivation of the PINK1 homologue in Drosophila resulted in progressive loss of DAergic neurons. This neurodegeneration was suppressed by human antioxidant superoxide dismutase (SOD) 1, which suggests that PINK1 inactivation can induce neuronal death via an oxidative stress pathway (Wang et al. 2006). Notably, PINK1 inactivation in Drosophila leads to a phenotype that shares marked similarity with that of Drosophila parkin knockout mutants, including shortened lifespan, apoptotic muscle degeneration, male sterility and defects in mitochondrial morphology. Transgenic expression of parkin markedly ameliorated all PINK1 loss-of-function phenotypes, but not vice versa, suggesting that PINK1 and parkin function, at least in part, in the same pathway, with PINK1 functioning upstream of parkin (Clark et al. 2006; Park et al. 2006; Yang et al. 2006).
Overexpression of wild type PINK1 in SH-SY5Y cells attenuated neuronal apoptosis by reducing the release of cytochrome c and subsequent activation of caspases under basal and apoptotic stress conditions. However, PD-related mutations and an artificial kinase-dead mutant abolished this protective effect (Petit et al. 2005). Most likely, the PINK1 kinase exerts its neuroprotective effect by phosphorylating specific mitochondrial proteins and in turn modulating their functions. However, the physiological substrates of PINK1 remain unknown.
The antioxidative DJ-1
DJ-1 is expressed in a variety of tissues, including brain, and is partially localized to the mitochondrial matrix and IMS (Zhang et al. 2005a). DJ-1 is a H2O2 responsive protein, suggesting a function as antioxidant (Mitsumoto & Nakagawa 2001). DJ-1 acts as a redox sensor within cells and the acidification of a key Cys-residue (Cys106) seems to have an important signalling function. The same Cys-residue is also important for relocalization of DJ-1 to mitochondria (Canet-Aviles et al. 2004).
Recessively inherited DJ-1 missense and exonic deletion mutations were first identified in two European early onset PD families (Bonifati et al. 2003). Additional loss-of-function mutations have since been identified (Abou-Sleiman et al. 2003, 2004; Hague et al. 2003; Hedrich et al. 2004a). Loss-of-function mutations in DJ-1 are rare and are estimated to account for 1% of early onset PD (Lockhart et al. 2004). Single heterozygous DJ-1 mutations were also found (Clark et al. 2004; Hedrich et al. 2004a), but as for heterozygous mutations in parkin and PINK1, their effect remains elusive.
DJ-1 downregulation by siRNA in neuronal cell lines enhanced cell death by oxidative stress (Yokota et al. 2003). In Drosophila inhibition of DJ-1α, a Drosophila homologue of the human DJ-1, resulted in cellular accumulation of ROS, hypersensitivity to oxidative stress as well as dysfunction and degeneration of DAergic neurons (Yang et al. 2005). Knockout flies of DJ-1α and DJ-1β, the two DJ-1 homologues in Drosophila, displayed a selective sensitivity to environmental toxins such as paraquat and rotenone (Meulener et al. 2005). DJ-1α and DJ-1β are prominently localized to enlarged and swollen mitochondria, implicating that the localization of DJ-1 to mitochondria is a protection mechanism against oxidative stress (Park et al. 2005). DJ-1-deficient mice show no gross anatomical or neuronal abnormalities and have normal numbers of SN DAergic neurons (Chen et al. 2005; Goldberg et al. 2005; Kim et al. 2005), however, their nigrostriatal pathway is dysfunctional (Chen et al. 2005; Goldberg et al. 2005), leading to higher dopamine concentrations and possibly more cellular oxidative stress.
The multidomain and multifunctional LRRK2
LRRK2, also known as dardarin, is expressed at low levels in most tissues (Paisan-Ruiz et al. 2004) and shows an expression pattern in brain that directly relates to the nigrostriatal dopamine system. It is expressed in the dopamine target areas, striatum and frontal cortex, whereas the dopamine neurons themselves are devoid of LRRK2 messenger RNA (Galter et al. 2006). On a cellular level LRRK2 resides diffusely throughout the cytosol, but also associates with the OMM (West et al. 2005). Sequence analysis indicates that LRRK2 comprises several domains including a leucine-rich repeat domain, a GTPase (guanosine triphosphatase) domain Ras of complex proteins (Roc) followed by its associated C-terminal of Roc domain, a MLK-like domain and a C-terminal WD40 domain (Paisan-Ruiz et al. 2004; Zimprich et al. 2004). The kinase activity may be the link between LRRK2 and its role in PD pathogenesis.
Mutations in LRRK2 cause autosomal dominant late onset PD (Paisan-Ruiz et al. 2004; Zimprich et al. 2004). LRRK2 missense mutations reported so far are distributed along the protein (Berg et al. 2005; Khan et al. 2005; Paisan-Ruiz et al. 2004; Zimprich et al. 2004). p.Gly2019Ser is the most common pathogenic LRRK2 mutation in Caucasians and accounts for approximately 5% of familial PD and 1.5% of sporadic PD (Di Fonzo et al. 2005; Gilks et al. 2005; Nichols et al. 2005). We recently detected a founder effect of p.Arg1441Cys, the second most frequent pathogenic LRRK2 mutation, in the Belgian population (Nuytemans et al. in preparation).
p.Arg1441Cys in the GTPase domain and p.Gly2019Ser in the MLK-like domain were shown to result in increased kinase activity in vitro (West et al. 2005), a similar increase was seen for p.Ile2020Thr (Gloeckner et al. 2006). These findings are in favour of a gain-of-function mechanism for LRRK2-linked disease. Expression of p.Arg1441Cys and p.Gly2019Ser LRRK2 caused neuronal degeneration in both SH-SY5Y cells and mouse primary neurons (Smith et al. 2005). Overexpression of p.Gly2019Ser LRRK2 in rats did not alter normal appearance of DAergic neurons, but significantly increased apoptosis (MacLeod et al. 2006). Extensive in vivo validation of these data is, however, required.
The mitochondrial serine protease HTRA2
HTRA2 is ubiquitously expressed (Faccio et al. 2000) and nuclear encoded but carries a mitochondrial targeting sequence at its N-terminus. HTRA2 has an N-terminal IAP-binding motif, a serine protease domain and, at its C-terminus, a PDZ domain, which restricts access to the active site of the serine protease (Li et al. 2002; Verhagen et al. 2002). HTRA2 is localized to the IMS, but is released into the cytosol following apoptotic stimuli. In the cytosol it can induce apoptosis in a caspase-dependent manner by antagonizing caspase–IAP interaction, or in a caspase-independent manner relying on its serine protease activity (Hegde et al. 2002).
Transient HTRA2 overexpression in HEK293 cells (human embryonic kidney cells) induced apoptosis even in the absence of an apoptotic insult, whereas artificial protease-inactive mutant HTRA2 (Li et al. 2002) had lower death-inducing ability (Martins et al. 2002). In non-apoptotic conditions the proteolytic activity of HTRA2 is involved in maintaining mitochondrial homeostasis, as was first shown in the mnd2 mouse for motor neuron degeneration 2 (Jones et al. 2003) and later also in HTRA2 knockout mice (Martins et al. 2004). Both mnd2 and HTRA2 knockout mice died prematurely because of loss of striatal neurons. The identification of a p.Gly399Ser mutation in the HTRA2 PDZ domain in patients with sporadic PD (Strauss et al. 2005) was of direct relevance to PD. At the cellular level p.Gly399Ser HTRA2 impaired the regulation of proteolytic activity, caused mitochondrial swelling, decreased mitochondrial membrane potential and increased staurosporine-induced cell death. We recently identified another PD-specific HTRA2 mutation in the PDZ domain, p.Arg404Trp, in an extensive mutation analysis of over 250 Belgian PD patients (Bogaerts et al. unpublished data).
Other genes relevant to mitochondrial function and oxidative stress
Several polymorphisms in functional candidate genes related to mitochondrial function and oxidative stress have been studied for their possible association with PD. MAO, a principal enzyme of dopamine metabolism that has two isoforms: MAO-A and MAO-B, was tested for its supposed role in PD pathogenesis: a polymorphism in intron 13 of MAO-B was significantly associated with increased risk of PD (Kang et al. 2006; Tan et al. 2000b). Another polymorphism in exon 22 of inducible nitric oxide synthase (encoded by NOS2A), an enzyme that produces NO (Fig. 2), showed an inverse association with PD (Hague et al. 2004; Levecque et al. 2003). Although NO is a biological messenger, NO overproduction contributes to oxidative stress, impairment of mitochondria and overload of degradation pathways. Interestingly NOS2A polymorphisms were also reported to interact with smoking (Hancock et al. 2006). Glutathione S-transferase (GST) variants have been extensively studied in PD because of their ability to detoxify exogenous and endogenous toxic substances. Moreover, polymorphisms in GST Omega-1 and -2 were shown to modify age at onset of PD (Li et al. 2006). These genes represent only a handful of the list of functional candidate genes for PD, but their implication in PD at least supports the pathophysiological significance of mitochondrial dysfunction to PD.
The relevance of genetic findings in PD
The future of drug therapies depends on the development of animal models that strongly recapitulate the disease, not only to discover the mechanisms of disease but also to identify new neuroprotective drugs and to test their efficacy. The genetic findings described above provide a direct mechanistic link with PD and therefore animal models expressing the discovered PD-causing mutations probably mimic human PD better than neurotoxic animal models. The identified PD genes themselves represent direct targets for therapeutic application, e.g. by RNA interference. In addition, evidence from genetic studies indirectly suggests that mitochondrial dysfunction plays an integrative role in PD pathogenesis, and therefore mitochondrial function and mechanisms to cell death represent valid targets for potential neuroprotective therapies.
Direct relevance of genetic research to treatment
Transgenic mouse models of PD
The identification of causal PD genes in rare Mendelian forms of PD triggered the development of transgenic mouse models. The underlying hypothesis is that mutations causing Mendelian forms of PD highlight pathways, relevant to sporadic PD (Melrose et al. 2006). However all of these mouse models had shortcomings, of which the most notable is failure to exhibit degeneration of the nigrostriatal system, a key pathological feature of PD (Manning-Bog & Langston 2007). Efforts to create genetic models of PD in simple systems, e.g. Drosophila and yeast, have resulted in robust phenotypes. However, the principal value of these simple models of PD is perhaps that results in these systems might be used to improve current mouse models of PD (Whitworth et al. 2006).
Given its clear link with PD, a variety of SNCA transgenic mice have been generated, varying in tissue specificity, mutation and expression level (for a review see Fernagut & Chesselet 2004). The developed SNCA transgenic models proved to be valuable in partially recapitulating the predisposition of certain neuronal populations to SNCA aggregation. Parkin knockout mice had normal numbers of DAergic neurons, but displayed subtle deficits in behaviour and DAergic neurotransmission (Goldberg et al. 2003; Itier et al. 2003). Similar to parkin knockouts, loss of DJ-1 did not result in loss of SN DAergic neurons (Chen et al. 2005; Goldberg et al. 2005; Kim et al. 2005), but again subtle behavioural deficits and increased striatal DA reuptake were reported. So far PINK1 knockout mice have not been reported, but silencing of PINK1 expression in mice by use of RNA interference did not cause a loss of DAergic neurons either (Zhou et al. 2007). LRRK2 transgenic mice have also not been reported. It is hoped that these transgenic mice display a more robust pathological phenotype (Melrose et al. 2006). However, considerable variability in pathological phenotype was already documented for several pathogenic LRRK2 mutations (Rajput et al. 2006; Zimprich et al. 2004).
Yet no perfect animal model for PD exists; most transgenic mouse lines do not show damage to the nigrostriatal system, but rather have subtle changes. Opposed to this, neurotoxic animal models do show degeneration of SN DAergic neurons. Combining these animal models might thus be very fruitful to develop new and better PD models. Such fused genetic and neurotoxic models might better capture the full spectrum of PD features, and provide excellent means to study gene–environment interactions (Manning-Bog & Langston 2007). Nevertheless, genetic mouse models have at least illustrated that neuronal dysfunction in PD most likely occurs long before cell loss. Current transgenic or knockout mouse models might indeed provide insights into the early stages of PD (Fleming et al. 2005), allowing us to decipher pathological processes that take place in brain exposed to a PD-causing insult before degeneration of the nigrostriatal system occurs, or before the disease begins elsewhere (Litvan et al. 2007).
Gene-based therapy
A straightforward approach, supported by genetic findings, is the possibility to lower SNCA levels. Indeed, given the gain-of-function mechanism of PD-linked SNCA mutations, silencing expression of mutant SNCA or reducing overexpression of wild-type SNCA is an attractive target for therapeutic RNA interference (Gonzalez-Alegre 2007). Allele-specific silencing of mutant SNCA was already shown in vitro, as well as suppression of overexpressed human SNCA in vivo in rat brain (Sapru et al. 2006). However, these studies need replication in different animal models of PD. A vaccination approach was also tested: immunization of human SNCA transgenic mice with recombinant human SNCA led to decreased SNCA accumulation in neuronal cell bodies and synapses, as well as reduced neurodegeneration (Masliah et al. 2005). Human trials on the proposed vaccine are currently underway (Singh et al. 2007). For parkin, PINK1 and DJ-1, where PD is caused by loss-of-function mechanisms, increasing expression is likely to ameliorate the phenotype (Farrer 2006). For example, parkin gene therapy was already shown to be effective against SNCA overexpression in rats (Mochizuki et al. 2006). Last but not least, the identification of LRRK2 mutations as a frequent cause of familial and sporadic PD was a landmark discovery in PD research. The increased kinase activity observed for mutant LRRK2 (West et al. 2005) may, if proved in vivo, translate relatively quickly into a novel treatment option involving kinase inhibition (Litvan et al. 2007). However, the physiological targets for LRRK2 activity are not yet known, hence uncertainty remains whether we can target this kinase activity without detrimental effects.
Drugs targeting mitochondrial pathways to PD
A scientific rationale, evidence of blood brain barrier (BBB) penetration, adequate safety data, efficacy in animal models and/or preliminary efficacy data in humans are requirements to identify promising neuroprotective drugs in PD (Ravina et al. 2003). In contrast with symptomatic treatments, neuroprotective drugs aim to slow or halt the progression of PD by a direct effect on the vulnerable cells (Olanow & Jankovic 2005).
The neuroprotective drugs include dopamine agonists and MAO-B inhibitors, which are already used for years to improve symptoms in the early stages of PD. Based on our current understanding of PD pathogenesis and mitochondria-related mechanisms of cell death, neuroprotective drugs can be divided into drugs that (1) attenuate mitochondrial apoptosis, (2) reduce oxidative stress in mitochondria or (3) directly target mitochondrial function (Fig. 5).
Figure 5.
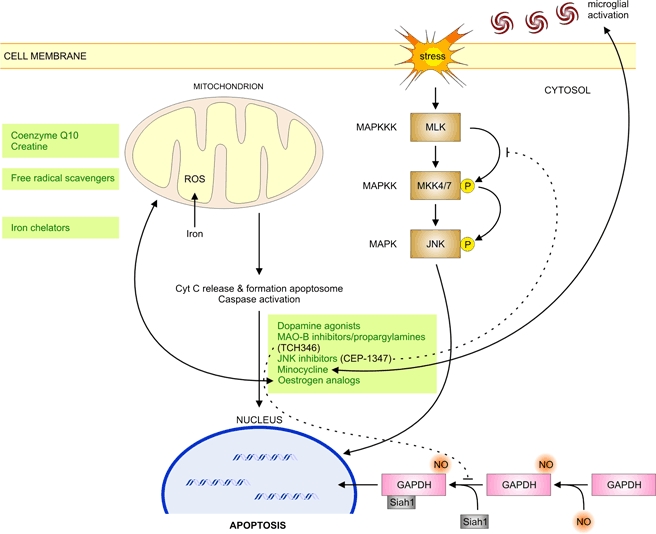
Entry points for PD therapy involving mitochondria. Green boxes highlight potential neuroprotective drugs at their respective action levels, and dotted lines indicate premature termination of clinical trials for promising neuroprotective drugs. Abbreviations: Cyt C, cytochrome c; MAPKK, MAPK kinase; MAPKKK, MAPKK kinase; MKK4/7, MAPK kinases 4/7; P, phosphorylated.
Antiapoptotic
Dopamine agonists
Dopamine agonists have proven efficacy in the early stages of PD. Dopamine agonists directly stimulate dopamine receptors and unlike levodopa their symptomatic effect does not depend on the presence of dopamine producing neurons (Junghanns et al. 2004). The neuroprotective effect of dopamine agonists may be the result of different mechanisms, including a ‘levodopa sparing’ effect (delaying the need for levodopa therapy) (Holloway et al. 2000; Rascol et al. 2000), antioxidative properties by scavenging free radicals (Iida et al. 1999), as well as inhibition of apoptosis (Nair et al. 2003).
Pramipexole is perhaps the best-studied dopamine agonist. Pramipexole decreased apoptotic cell death in SH-SY5Y cells exposed to MPP+ and rotenone (Kitamura et al. 1998; Schapira et al. 2002) and was shown to inhibit opening of the PT pore, reduce release of cytochrome c and decrease activation of caspase 3 (Cassarino et al. 1998; Gu et al. 2004; King et al. 2001). In vivo studies showed that marmosets (non-human primates) pretreated with pramipexole had significantly greater numbers of surviving SNpc neurons upon MPTP treatment (Iravani et al. 2006). The need for pretreatment, which is in agreement with earlier observations in SH-SY5Y cells (Gu et al. 2004; Kitamura et al. 1998), does not preclude clinical application of pramipexole, but rather emphasizes the importance of pramipexole administration early in the disease process to protect the remaining SN neurons from future dysfunction or degeneration.
Two prospective randomized double-blind clinical trials in early PD patients (diagnosed within 5 years) supported the neuroprotective effect of dopamine agonists (Schapira 2003), but further trials are needed to confirm their neuroprotective potential.
MAO-B inhibitors and propargylamines
MAO-A and MAO-B are tightly associated with the OMM and oxidatively deaminate monoamine neurotransmitters (e.g. dopamine), as well as exogenous monoamines (e.g. tyramine). MAO inhibitors were considered in PD therapy as dopamine sparing drugs, however, the first generation of non-selective MAO inhibitors had a major side-effect, known as the ‘cheese reaction’ (hypertensive crisis because of potentiation of the sympathomimetic activity of tyramine by peripheral inhibition of its MAO metabolism). Selective MAO-B inhibitors, such as the propargylamine-related l-deprenyl (Birkmayer et al. 1975) and the more potent rasagiline (Siderowf & Stern 2006), do not have such effect (Youdim & Weinstock 2004). Neuroprotection by l-deprenyl (Birkmayer et al. 1985) and other propargylamines is most likely because of interference with apoptotic signalling pathways rather than by MAO-B inhibition itself (Tatton et al. 2003a). In addition, l-deprenyl was shown to inhibit the formation of SNCA fibrils and to destabilize preformed SNCA fibrils (Ono et al. 2007).
In SH-SY5Y cells rasagiline suppressed apoptosis by preventing the fall in ΔΨm and opening of the mitochondrial PT pore, preventing nuclear translocation of GAPDH (Youdim & Weinstock 2001) and increasing expression of antiapoptotic Bcl-2 family members (Akao et al. 2002). In rats and marmosets l-deprenyl and rasagiline attenuated the neurotoxic effects of MPTP (Kupsch et al. 2001; Wu et al. 2000).
The neuroprotective effect of l-deprenyl and rasagiline was tested in prospective randomized multicentre placebo-controlled studies: the DATATOP (Deprenyl and Tocopherol Antioxidative Therapy for Parkinson’s disease) (Shoulson et al. 1993) and TEMPO study (TVP-1012 (rasagiline mesylate) in Early Monotherapy for Parkinson’s disease Outpatients) (Parkinson Study Group 2004a). The TEMPO study showed that PD patients treated with rasagiline for 12 months showed less functional decline than patients whose treatment was delayed for 6 months. Such difference cannot be simply explained by a symptomatic effect, which was a confounder in studies of l-deprenyl (Olanow et al. 1995; Shoulson et al. 2002), and is consistent with rasagiline having a neuroprotective effect. A larger double-blind delayed-start study is under way to confirm these data.
Another promising drug with potential neuroprotective effect is TCH346 (also known as CGP3466). TCH346 incorporates a propargyl ring within its molecular structure and resembles l-deprenyl, but does not inhibit MAO-B and was therefore not anticipated to have confounding symptomatic effects (Olanow et al. 2006).
TCH346 prevented nuclear translocation of GAPDH in a cellular system (Hara et al. 2006), and in bilaterally MPTP-treated monkeys TCH346 prevented death of SN DAergic neurons (Andringa et al. 2003). Preclinical safety studies and small trials in healthy volunteers showed that TCH346 was well tolerated and free of clinically significant adverse effects.
A highly anticipated first double-blind placebo-controlled clinical trial was already completed (Olanow et al. 2006). Unfortunately, no significant differences were found between any of the tested doses and placebo with respect to the primary outcome (time to disability requiring DAergic treatment) or secondary outcome measures [e.g. changes in Unified Parkinson’s Disease Rating Scale (UPDRS)] in patients with early untreated PD.
Nevertheless, the evidence for neuroprotection by l-deprenyl and nasagiline still deserves further study. Both propargylamines differ from TCH346 by their additional MAO-B inhibitory effects, which might not be redundant to explain the apparent differences in their potential neuroprotective effects.
JNK inhibitors
The first successful compound targeting the JNK signalling pathway was CEP-1347 (Maroney et al. 1998). CEP-1347 does not directly inhibit JNK, but reduces neuronal cell death by MLK inhibition (Maroney et al. 2001). JNK activation is relevant to PD pathogenesis, as activated JNK was associated with SNCA pathology in SNCA p.Ala30Pro transgenic mice (Frasier et al. 2005) and loss of parkin function was already shown to activate the JNK pathway in Drosophila (Cha et al. 2005).
CEP-1347 attenuated the neurotoxic effects of MPP+ in differentiated SH-SY5Y cells (Mathiasen et al. 2004), and reduced MPTP-mediated nigrostriatal DAergic neuron loss in vivo in mice (Saporito et al. 1999). A placebo-controlled pilot study showed that CEP-1347 was safe and well tolerated by PD patients (Parkinson Study Group 2004b), and this supported the initiation of a large trial of longer duration with CEP-1347: the PRECEPT study (Parkinson Research Examination of CEP-1347 Trial).
The PRECEPT study was, however, prematurely abrogated when an interim analysis showed that it would be futile to continue experimental treatment. In all tested CEP-1347 dose regimens more patients had reached the primary and secondary end-points compared with patients randomized to placebo (Shoulson et al. 2006). Again these findings contrasted with research in preclinical models that predicted favourable effects of CEP-1347 on the progression of PD.
Minocycline
Minocycline, a second-generation tetracycline, has a proven safety record in humans. Among several activities minocycline impairs microglial activation, neuroinflammation and apoptosis (Yong et al. 2004). In midbrain cultures of parkin knockout mice minocycline neuroprotection was related to inactivation of microglial cells and protection against neuronal apoptotic cell death (Casarejos et al. 2006). Increasing evidence suggests that its antiapoptotic effect is achieved through mechanisms acting at the level of mitochondria.
Minocycline inhibited mitochondrial PT mediated cytochrome c release in isolated brain mitochondria (Zhu et al. 2002), and in kidney cells minocycline not only induced antiapoptotic Bcl-2, but also antagonized pro-apoptotic Bcl-2 family members (Wang et al. 2004). Furthermore, minocycline was shown to prevent nigrostriatal DAergic neurodegeneration in the MPTP mouse model of PD (Du et al. 2001; Wu et al. 2002). In contrast with numerous reports of beneficial effects of minocycline, some studies reported significant exacerbation of MPTP-induced damage by minocycline in mice (Yang et al. 2003) and monkeys (Diguet et al. 2004). It remains, however, plausible that minocycline has different, sometimes even deleterious, effects according to its mode of administration and dose. A recent study suggested that the mitochondrial effects of minocycline contribute to toxicity rather than tissue protection at high dosing in animals and humans (Mansson et al. 2007). The possible harmful effects of minocycline must thus be ceaselessly kept in mind when considering minocycline use in clinical PD settings.
A randomized, double-blind, futility clinical trial of minocycline in early PD patients could not reject minocycline as futile (Ravina et al. 2006). Such futility trials are intended to determine if treatments are worthy of larger and longer term studies, or if they should be abandoned (Elm et al. 2005). Although the results of this futility study do not provide evidence for clinical use of minocycline in the treatment of PD, they indicate that minocycline at least merits further consideration for larger clinical trials.
Oestrogen analogues
Postmenopausal oestrogen replacement therapy in women with early PD was positively associated with lower symptom severity (Saunders-Pullman et al. 1999) and suggested a potential neuroprotective effect of oestrogens. Indeed, numerous in vitro and in vivo studies established that oestrogens act as neuroprotectants when challenged by various toxic stresses.
Several effects might explain the beneficial actions of oestrogens on brain; including anti-inflammatory activity and protection against apoptosis (Maggi et al. 2004). These effects appear to be primarily mediated through activation of intracellular oestrogen receptors (ER) and result in modulated transcription of oestrogen target genes, e.g. upregulated expression of antiapoptotic Bcl-2 (Garcia-Segura et al. 1998). Neuroprotective effects of oestrogens might also result from ER-independent mechanisms, including antioxidant effects (Moosmann & Behl 1999) related to their basic chemical properties as hydrophobic phenolic molecules. Mechanistic and structure–activity studies with oestrogens yielded a model in which they intercalate into cell membranes where they block lipid peroxidation reactions, and thereby stabilize membrane structure (Behl et al. 1997), which is especially critical to mitochondrial function.
Both the naturally occurring 17β-oestradiol (17β-E2) and its 17α-oestradiol (17α-E2) isomer, which is at least 200-fold less active as transactivating hormone, protected DAergic neurons from MPP+-induced neuronal death (Sawada et al. 2002). 17β-E2 showed beneficial effects in animal models of PD using MPTP as lesioning agent (Callier et al. 2000; Ramirez et al. 2003), and 17α-E2 treatment in 6-hydroxydopamine injected rats later showed significantly improved behavioural outcomes (Dykens et al. 2005b).
A first small double-blind placebo-controlled clinical trial of 17α-E2 was completed including eight healthy postmenopausal women; the tested doses of 17α-E2 were well tolerated and no adverse events were reported (Dykens et al. 2005a). However, the neuroprotective, or better mitoprotective, effects of 17α-E2 remain to be tested in PD patients.
Antioxidants
Iron chelators
Alterations in cellular iron metabolism and iron-induced oxidative stress are important factors in the pathogenesis of PD (Bharath et al. 2002; Riederer et al. 1989; Sofic et al. 1991; Youdim et al. 1993). Moreover, iron was shown to accelerate the aggregation of SNCA (Kalivendi et al. 2004) and to induce alterations of parkin solubility, with depletion of soluble functional parkin leading to reduced proteasomal activities and increased cell death (Wang et al. 2005). Reducing the SN iron content through iron chelation might thus be a straightforward therapeutic approach to the treatment of PD (Zhang et al. 2005b).
Desferrioxamine, the prototype of iron chelators (Jiang et al. 2006), blocked iron-induced oxidative damage in SK-N-SH neuroblastoma cells (DAergic origin) (Sangchot et al. 2002). In rat brain mitochondria desferrioxamine also protected mitochondrial complex I activity against inhibition by 6-hydroxydopamine, possibly through a combined neuroprotective effect of iron chelation and NADH dehydrogenase activation (Glinka et al. 1996, 1998). The neuroprotective effect of preinjected desferrioxamine was later confirmed in rat (Ben Shachar et al. 1991; Youdim et al. 2004) and mice models (Lan & Jiang 1997).
As desferrioxamine has poor BBB penetration, other lipophilic chelators with enhanced BBB penetration have been synthesized; e.g. VK-28 (Ben Shachar et al. 2004). Novel iron chelators were further developed based on VK-28, that combine its iron chelating potency with a MAO inhibitory propargylamine moiety (Youdim et al. 2005), e.g. the multifunctional M30. M30 exhibited neuroprotective activity upon in vitro and in vivo testing (Gal et al. 2005; Zheng et al. 2005), but its effect in PD patients remains to be evaluated. Nevertheless, as multifunctional compounds possess various targets within the central nervous system, they may provide better efficacy and utility as potential disease-modifying drugs (Youdim & Buccafusco 2005).
Free radical scavengers
Under physiological conditions several cellular mechanisms are able to neutralize a possible excess of free radicals and thereby prevent oxidative stress. These comprise free radical scavengers, which are either endogenously produced molecules or nutrients, and scavenger enzymes such as SOD, glutathione peroxidase and catalase (Fig. 2). The free radical scavengers include vitamins A, C and E, as well as glutathione in its reduced form, and melatonin (Contestabile 2001). RNA interference-mediated inactivation of the Drosophila PINK1 homologue already showed that addition of SOD1 and vitamin E to the flies’ diet inhibited degeneration of photoreceptor neurons (Wang et al. 2006).
The lipid-soluble vitamin E received much attention as a chain-breaking antioxidant that prevents propagation of the radical chain (Burton et al. 1983). In vitro studies indicated oxidation of vitamin E upon incubation with free radicals in rat brain mitochondria (Vatassery et al. 1995) and protection against H2O2-induced oxidative stress in a murine hippocampal-derived cell line (Behl 2000). However in vivo studies did not consistently find positive effects of vitamin E administration (Gong et al. 1991; Roghani & Behzadi 2001).
Already in 1987 α-tocopherol, the biologically most active form of vitamin E, was selected for the first clinical trial of neuroprotection in PD: the DATATOP study (Shoulson et al. 1993). This study did not show beneficial effects of supplemental α-tocopherol intake in PD patients, but a more recent meta-analysis suggested that diets rich in vitamine E might exert a protective effect against the development of PD (Etminan et al. 2005). There is, however, some concern that high-dosage vitamin E supplementation may be harmful (Miller et al. 2005).
Use of reduced gluthathione as a free radical scavenger is hampered by a poor BBB permeability (Zeevalk et al. 2007). On the other hand, the pineal hormone melatonin is a highly lipophilic free radical scavenger (Reiter et al. 1993) and antioxidant (Tan et al. 2000a). Some of the melatonin metabolites resulting from scavenging actions are also efficient free radical scavengers (Lopez-Burillo et al. 2003). The action of melatonin as a free radical scavenger is thus a sequence of scavenging reactions, resulting in a cascade of protective reactions.
Pharmacological doses of melatonin, which greatly exceed endogenous levels, were shown to be neuroprotective in different neurotoxic models of PD (Joo et al. 1998; Thomas & Mohanakumar 2004), but to our knowledge melatonin has not yet been tested for the treatment of PD in clinical trials.
Targeting mitochondrial function
Coenzyme Q10
Coenzyme Q shuttles electrons from complexes I/II to complex III of the electron transport chain, and the predominant form of coenzyme Q in humans is coenzyme Q10. In addition, ubiquinol (its reduced form) functions as antioxidant, prevents lipid peroxidation in most subcellular membranes and also protects mitochondrial membrane proteins and DNA from ROS-induced oxidative damage (Ernster & Dallner 1995).
Coenzyme Q10 was shown to prevent in vitro mitochondrial PT pore opening upon apoptotic stimuli (Papucci et al. 2003). In aged mice coenzyme Q10 attenuated MPTP-induced loss of striatal dopamine and DAergic axons (Beal et al. 1998). In addition, PD patients showed lower levels of coenzyme Q10 in platelet mitochondria (Shults et al. 1997), as well as elevated levels of oxidized coenzyme Q10 (Sohmiya et al. 2004) compared with control individuals.
A pilot study in PD patients showed that oral administration of coenzyme Q10 was well tolerated and caused a trend towards increased complex I activity (Shults et al. 1998). A later study also showed reduced functional decline in PD patients taking coenzyme Q10 (Shults et al. 2002). More recently, a randomized, double-blind, futility clinical trial of coenzyme Q10 in early PD could not reject coenzyme Q10 as futile (The NINDS NET-PD Investigators 2007). However, large trials are still needed to evaluate the long term effects of coenzyme Q10 intake and to assess its disease-modifying potential in the treatment of PD patients.
Creatine
The endogenous guanidine-derived creatine is a substrate for cytosolic and mitochondrial creatine kinases (CK). Aerobic glycolysis, coupled to mitochondrial ATP synthesis via oxidative phosphorylation, is the primary pathway of ATP synthesis in brain, but in addition the CK/phosphocreatine system provides a rapid alternative source for ATP synthesis (Wallimann et al. 1992). Mitochondrial CK together with high cytoplasmic creatine levels also inhibits mitochondrial PT and the consequent triggering of apoptosis (Andres et al. 2005).
In primary neuronal cultures creatine exerted neuroprotective effects against neurotoxic insults as MPP+ exposure (Andres et al. 2005). Oral supplementation with creatine protected against MPTP-induced dopamine depletion in mice and reduced damage to the nigrostriatal DAergic system (Matthews et al. 1999).
A double-blind placebo-controlled pilot study on the effect of oral creatine supplementation on PD progression did not find a treatment effect on UPDRS scores, although a significantly smaller dose increase of DAergic therapy in creatine-treated patients indicated a potential neuroprotective effect (Bender et al. 2006a). A randomized, double-blind, futility clinical trial of creatine in early PD could not reject creatine as futile (Ravina et al. 2006). Creatine thus remains a promising neuroprotective drug for PD, but its neuroprotective potential may only be shown by large multicentre studies over an extended observation period.
Conclusion
Molecular genetic studies identified key proteins involved in PD pathogenesis. Mutations or polymorphisms in mtDNA, but especially mutations in the nuclear-encoded SNCA, parkin, PINK1, DJ-1, LRRK2 and HTRA2 provided direct or indirect links between mitochondria and PD pathogenesis. Other chromosomal loci still await refinement and characterization (Bogaerts et al. 2007; Gasser et al. 1998; Hicks et al. 2002; Pankratz et al. 2003) and will further increase our knowledge of the basic mechanisms underlying PD pathogenesis.
Perhaps the most relevant question for research into the causes of PD is: how can we exploit the gained knowledge on mitochondrial involvement in PD to develop better therapies? Neuroprotective therapies that slow or halt disease progression remain an unmet need. Our insights into the pathogenic mechanisms of cell death in PD led to several potential neuroprotective drugs, which have actions as attenuating mitochondrial apoptosis and reducing oxidative stress in mitochondria, or directly target mitochondrial function. The selection of drugs covered in this review all exert at least one of the three above mentioned actions, along with possible involvements in other non-mitochondrial pathways to PD.
However, no drug has yet been shown to be neuroprotective in PD patients, although several were tested in clinical trials. This discrepancy, reported for several potential neuroprotective drugs targeting different elements of the mitochondrial pathway, raises serious concerns as to the suitability of currently available models (Waldmeier et al. 2006). Particularly, the currently used animal models, typically acute or subacute neurotoxic models, might not adequately recapitulate the pathogenesis of the slower progressive neurodegenerative process in humans. Therefore, genetic models of PD are highly desirable because they produce a delayed onset and a neurodegenerative process that probably mimics human PD better than neurotoxic models.
On the other hand, the clinical end-points used to assess neuroprotective drugs in PD, such as need for symptomatic treatment or change in UPDRS score, may be confounded by symptomatic effects, or may simply be too insensitive. True biomarkers that accurately reflect disease progression remain an urgent priority. An additional concern is that DA neurons may already be beyond rescue when clinical features appear. This reinforces the need for biomarkers to identify patients who are in the presymptomatic or early stages of PD. Ideally, neuroprotective drugs should be administered during this early period (Halbig et al. 2004).
Last but not least a very important questions remains: do we target the right pathway? Moreover, how can we fit all the cellular processes uncovered by genes involved in familial forms of PD in the mitochondrial pathway? And is this mitochondrial pathway also relevant to sporadic forms of PD? Association studies in sporadic PD patients, showing modest effects for candidate genes involved in mitochondrial function and oxidative stress, support this contention. On the other hand, monogenic PD explains only 20% of early onset PD and less than 3% of late onset PD at best (Klein 2006). In most PD patients, the loss of DAergic neurons results from a combination of exogenous stresses and a genetic predisposition. Given this complexity, it is unlikely that any neuroprotective drug will be applicable to the full spectrum of PD patients. However, it is more plausible that mixtures of therapies targeting several different processes converging in a common pathway may prove useful in PD neuroprotection. The recent development of multifunctional compounds represents a move into that direction.
As the repeated failure of neuroprotective drugs in clinical trials has unfortunately shown, we must acknowledge that at present we merely have a rough sketch of the mitochondrial pathway to neurodegeneration in PD. This sketch turns out inadequate to predict fruitful neuroprotective approaches and further research is definitely needed. Nevertheless, the future looks bright. Our understanding of PD pathogenesis is rapidly evolving, boosted by a massive amount of research into the genetic causes of PD, the influence of genetic and environmental susceptibility factors on disease penetrance and manifestation, and the development of better models to test potential neuroprotective therapies. New insights from these future studies will (re)direct our efforts and eventually will provide us with neuroprotective therapies that might really make the change in the treatment of PD.
Glossary
Association study: Study of the correlation between a genetic variant in a population and a disease. If a correlation is observed, then there is said to be an association between the variant and the disease.
Candidate gene: Gene with evidence for a possible role in the disease under study.
DAergic: Related to or activated by dopamine.
Early PD: Early stages of PD with subtle impairment. In clinical trials, early PD patients have Hoehn and Yahr stage I (unilateral disease) or stage II (bilateral disease with preservation of postural reflexes) PD for less than 5 years. The Hoehn and Yahr rating scale is a simple and popular scale to establish the severity of PD with stages ranging from I to V (wheelchair-bound or walking only with assistance).
Early onset PD: Having an onset at or before 45 years of age.
Haploinsufficiency: Situation in which half of the normal level of gene expression is not sufficient to permit cells to function normally.
Haplotype: Sequential set of alleles present on the same chromosome.
Linkage analysis: Mapping loci by genotyping genetic markers in families to identify chromosome regions implicated in disease. Such linked regions are of great help in the identification of genetic variants causing disease.
Meta-analysis: A statistical analysis combining the results of several independent studies.
Mitochondrial respiratory chain (electron transport chain): Collective term used to describe the peptides, organized in five enzymatic mitochondrial complexes (I–V), and the electron shuttle molecules that are needed to produce ATP.
Oxidative phosphorylation: Phosphorylation of adenosine 5′-diphosphate to ATP driven by the transfer of electrons to oxygen.
SH-SY5Y: Human neuroblastoma subclonal cell line of the neuroepithelioma cell line SK-N-SH. These cells are widely used in studies addressing the molecular mechanisms of neurodegenerative disease.
UPS: Cellular system responsible for the degradation of damaged or misfolded proteins. Ubiquitin molecules are attached to lysine residues of a given protein and target the protein for destruction by the multienzyme proteasome.
UPDRS: Unified Parkinson’s Disease Rating Scale used to monitor the progression of PD. It is the most widely used clinical rating scale for PD and consists of a four-part evaluation: I non-motor experiences of daily living, II motor experiences of daily living, III motor examination and IV motor complications.
Acknowledgments
The research performed by the Neurodegenerative Brain Diseases Group of the VIB Department of Molecular Genetics is in part supported by the Special Research Fund of the University of Antwerp, the Fund for Scientific Research Flanders (FWO-F), the Institute for Science and Technology – Flanders and the Interuniversity Attraction Poles program P6/43 of the Belgian Science Policy Office. V.B. holds a PhD fellowship and J.T. a postdoctoral fellowship of the FWO-F.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JMG, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW. The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann Neurol. 2003;54:283–286. doi: 10.1002/ana.10675. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Healy DG, Wood NW. Causes of Parkinson’s disease: genetics of DJ-1. Cell Tissue Res. 2004;318:185–188. doi: 10.1007/s00441-004-0922-6. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Muqit MMK, McDonald NQ, Yang YX, Gandhi S, Healy DG, Harvey K, Harvey RJ, Deas E, Hatia K, Quinn N, Lees A, Latchman DS, Wood NW. A heterozygous effect for PINK1 mutations in Parkinson’s disease? Ann Neurol. 2006;60:414–419. doi: 10.1002/ana.20960. [DOI] [PubMed] [Google Scholar]
- Akao Y, Maruyama W, Yi H, Shamoto-Nagai M, Youdim MBH, Naoi M. An anti-Parkinson’s disease drug, N-propargyl-1(R)-aminoindan (rasagiline), enhances expression of anti-apoptotic Bcl-2 in human dopaminergic SH-SY5Y cells. Neurosci Lett. 2002;326:105–108. doi: 10.1016/s0304-3940(02)00332-4. [DOI] [PubMed] [Google Scholar]
- Andres RH, Huber AW, Schlattner U, Perez-Bouza A, Krebs SH, Seiler RW, Wallimann T, Widmer HR. Effects of creatine treatment on the survival of dopaminergic neurons in cultured fetal ventral mesencephalic tissue. Neuroscience. 2005;133:701–713. doi: 10.1016/j.neuroscience.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Andringa G, Eshuis S, Perentes E, Maguire RP, Roth D, Ibrahim M, Leenders KL, Cools AR. TCH346 prevents motor symptoms and loss of striatal FDOPA uptake in bilaterally MPTP-treated primates. Neurobiol Dis. 2003;14:205–217. doi: 10.1016/s0969-9961(03)00125-6. [DOI] [PubMed] [Google Scholar]
- Autere J, Moilanen JS, Finnila S, Soininen H, Mannermaa A, Hartikainen P, Hallikainen M, Majamaa K. Mitochondrial DNA polymorphisms as risk factors for Parkinson’s disease and Parkinson’s disease dementia. Hum Genet. 2004;115:29–35. doi: 10.1007/s00439-004-1123-9. [DOI] [PubMed] [Google Scholar]
- Beal MF. Bioenergetic approaches for neuroprotection in Parkinson’s disease. Ann Neurol. 2003;53:S39–S47. doi: 10.1002/ana.10479. [DOI] [PubMed] [Google Scholar]
- Beal MF, Matthews RT, Tieleman A, Shults CW. Coenzyme Q(10) attenuates the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged. Brain Res. 1998;783:109–114. doi: 10.1016/s0006-8993(97)01192-x. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Am J Physiol Cell Physiol. 1996;40:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Behl C. Vitamin E protects neurons against oxidative cell death in vitro more effectively than 17-beta estradiol and induces the activity of the transcription factor NF-kappa B. J Neural Transm. 2000;107:393–407. doi: 10.1007/s007020070082. [DOI] [PubMed] [Google Scholar]
- Behl C, Skutella T, Lezoualch F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- Ben Shachar D, Eshel G, Finberg JP, Youdim MB. The iron chelator desferrioxamine (Desferal) retards 6-hydroxydopamine-induced degeneration of nigrostriatal dopamine neurons. J Neurochem. 1991;56:1441–1444. doi: 10.1111/j.1471-4159.1991.tb11444.x. [DOI] [PubMed] [Google Scholar]
- Ben Shachar D, Kahana N, Kampel V, Warshawsky A, Youdim MBH. Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lesion in rats. Neuropharmacology. 2004;46:254–263. doi: 10.1016/j.neuropharm.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Bender A, Koch W, Elstner M, Schombacher Y, Bender J, Moeschl M, Gekeler F, Muller-Myhsok B, Gasser T, Tatsch K, Klopstock T. Creatine supplementation in Parkinson disease: a placebo-controlled randomized pilot trial. Neurology. 2006a;67:1262–1264. doi: 10.1212/01.wnl.0000238518.34389.12. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006b;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Berg D, Schweitzer K, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease*. Brain. 2005;128:3000–3011. doi: 10.1093/brain/awh666. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Howell JL, Paul CE, Salehi AH, Becker EBE, Said F, Bonni A, Barker PA. Apoptosis induced by p75NTR overexpression requires jun kinase-dependent phosphorylation of bad. J Neurosci. 2003;23:11373–11381. doi: 10.1523/JNEUROSCI.23-36-11373.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharath S, Hsu M, Kaur D, Rajagopalan S, Andersen JK. Glutathione, iron and Parkinson’s disease. Biochem Pharmacol. 2002;64:1037–1048. doi: 10.1016/s0006-2952(02)01174-7. [DOI] [PubMed] [Google Scholar]
- Birkmayer W, Knoll J, Riederer P, Youdim MBH, Hars V, Marton J. Increased life expectancy resulting from addition of L-deprenyl to madopar treatment in Parkinson’s-disease – a longterm study. J Neural Transm. 1985;64:113–127. doi: 10.1007/BF01245973. [DOI] [PubMed] [Google Scholar]
- Birkmayer W, Riederer P, Youdim MBH, Linauer W. Potentiation of anti akinetic effect after L-DOPA treatment by an inhibitor of Mao-B, Deprenil. J Neural Transm. 1975;6:303–326. doi: 10.1007/BF01253131. [DOI] [PubMed] [Google Scholar]
- Bogaerts V, Engelborghs S, Kumar-Singh S, Goossens D, Pickut B, van der Zee J, Sleegers K, Peeters K, Martin JJ, Del-Favero J, Gasser T, Dickson D, Wszolek Z, De Deyn P, Theuns J, Van Broeckhoven C. A novel locus for dementia with Lewy bodies: a clinically and genetically heterogeneous disorder. Brain. 2007 doi: 10.1093/brain/awm167. doi: 10.1093. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Borutaite V. Nitric oxide inhibition of mitochondrial respiration and its role in cell death. Free Radic Biol Med. 2002;33:1440–1450. doi: 10.1016/s0891-5849(02)01112-7. [DOI] [PubMed] [Google Scholar]
- Burton GW, Joyce A, Ingold KU. Is vitamin-e the only lipid-soluble, chain-breaking antioxidant in human-blood plasma and erythrocyte-membranes. Arch Biochem Biophys. 1983;221:281–290. doi: 10.1016/0003-9861(83)90145-5. [DOI] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callier S, Morissette M, Grandbois M, Di Paolo T. Stereospecific prevention by 17 beta-estradiol of MPTP-induced dopamine depletion in mice. Synapse. 2000;37:245–251. doi: 10.1002/1098-2396(20000915)37:4<245::AID-SYN1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarejos MJ, Menendez J, Solano RM, Rodriguez-Navarro JA, Garcia de Yebenes J, Mena MA. Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J Neurochem. 2006;97:934–946. doi: 10.1111/j.1471-4159.2006.03777.x. [DOI] [PubMed] [Google Scholar]
- Cassarino DS, Fall CP, Smith TS, Bennett JP. Pramipexole reduces reactive oxygen species production in vivo and in vitro and inhibits the mitochondrial permeability transition produced by the Parkinsonian neurotoxin methylpyridinium ion. J Neurochem. 1998;71:295–301. doi: 10.1046/j.1471-4159.1998.71010295.x. [DOI] [PubMed] [Google Scholar]
- Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, Kim JM, Chung J, Cho KS. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci U S A. 2005;102:10345–10350. doi: 10.1073/pnas.0500346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. alpha-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Li GL, Hideshima T, Podar K, Mitsiades C, Mitsiades N, Munshi N, Kharbanda S, Anderson KC. JNK-dependent release of mitochondrial protein, Smac, during apoptosis in multiple myeloma (MM) cells. J Biol Chem. 2003;278:17593–17596. doi: 10.1074/jbc.C300076200. [DOI] [PubMed] [Google Scholar]
- Chen LN, Cagniard B, Mathews T, Jones S, Koh HC, Ding YM, Carvey PM, Ling ZD, Kang UJ, Zhuang XX. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280:21418–21426. doi: 10.1074/jbc.M413955200. [DOI] [PubMed] [Google Scholar]
- Chung KK, Dawson VL, Dawson TM. S-nitrosylation in Parkinson’s disease and related neurodegenerative disorders. Methods Enzymol. 2005;396:139–150. doi: 10.1016/S0076-6879(05)96014-X. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Clark LN, Afridi S, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Singleton A, De Vrieze FW, Hardy J, Mayeux R, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Analysis of an early-onset Parkinson’s disease cohort for DJ-1 mutations. Mov Disord. 2004;19:796–800. doi: 10.1002/mds.20131. [DOI] [PubMed] [Google Scholar]
- Contestabile A. Antioxidant strategies for neurodegenerative diseases. Expert Opin Ther Pat. 2001;11:573–585. [Google Scholar]
- Cormier A, Morin C, Zini R, Tillement JP, Lagrue G. Nicotine protects rat brain mitochondria against experimental injuries. Neuropharmacology. 2003;44:642–652. doi: 10.1016/s0028-3908(03)00041-8. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, Brice A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12:517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- de Rijk MC, Launer LJ, Berger K, Breteler MMB, Dartigues JF, Baldereschi M, Fratiglioni L, Lobo A, Martinez-Lage J, Trenkwalder C, Hofman A. Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurology. 2000;54:S21–S23. [PubMed] [Google Scholar]
- Dekker MCJ, Bonifati V, van Duijn CM. Parkinson’s disease: piecing together a genetic jigsaw. Brain. 2003;126:1722–1733. doi: 10.1093/brain/awg172. [DOI] [PubMed] [Google Scholar]
- Deng H, Jankovic J, Guo Y, Xie W, Le W. Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH–SY5Y. Biochem Biophys Res Commun. 2005;337:1133–1138. doi: 10.1016/j.bbrc.2005.09.178. [DOI] [PubMed] [Google Scholar]
- Di Fonzo A, Rohe CF, Ferreira RJ, Chien HF, Vacca L, Stocchi F, Guedes L, Fabrizio E, Manfredi M, Vanacore N, Goldwurm S, Breedveld G, Sampaio C, Meco G, Barbosa E, Oostra BA, Bonifati V. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet. 2005;365:412–415. doi: 10.1016/S0140-6736(05)17829-5. [DOI] [PubMed] [Google Scholar]
- Dickson DW. Linking selective vulnerability to cell death mechanisms in Parkinson’s disease. Am J Pathol. 2007;170:16–19. doi: 10.2353/ajpath.2007.061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diguet E, Fernagut PO, Wei X, Du YS, Rouland R, Gross C, Bezard E, Tison F. Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur J Neurosci. 2004;19:3266–3276. doi: 10.1111/j.0953-816X.2004.03372.x. [DOI] [PubMed] [Google Scholar]
- Du CY, Fang M, Li YC, Li L, Wang XD. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Du YS, Ma ZZ, Lin SZ, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DLG, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykens JA, Moos WH, Howell N. Development of 17 alpha-estradiol as a neuroprotective therapeutic agent – rationale and results from a phase I clinical study. Future Horm Ther What Basic Sci Clin Studies Teach Us. 2005a;1052:116–135. doi: 10.1196/annals.1347.008. [DOI] [PubMed] [Google Scholar]
- Dykens JA, Wersinger C, Sidhu A. 17 beta- and 17 alpha-Estradiol are non-competitive inhibitors of dopamine uptake: implications for Parkinson’s disease models and therapeutics. Drug Dev Res. 2005b;66:160–171. [Google Scholar]
- Elm JJ, Goetz CG, Ravina B, Shannon K, Wooten GF, Tanner CM, Palesch YY, Huang P, Guimaraes P, Kamp C, Tilley BC, Kieburtz K. A responsive outcome for Parkinson’s disease neuroprotection futility studies. Ann Neurol. 2005;57:197–203. doi: 10.1002/ana.20361. [DOI] [PubMed] [Google Scholar]
- Ernster L, Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta Mol Basis Dis. 1995;1271:195–204. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Doctor Jekyll and Mister Hyde: autophagy can promote both cell survival and cell death. Cell Death Differ. 2005;12(Suppl. 2):1468–1472. doi: 10.1038/sj.cdd.4401721. [DOI] [PubMed] [Google Scholar]
- Etminan M, Gill SS, Samii A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: a meta-analysis. Lancet Neurol. 2005;4:362–365. doi: 10.1016/S1474-4422(05)70097-1. [DOI] [PubMed] [Google Scholar]
- Faccio L, Fusco C, Chen A, Martinotti S, Bonventre JV, Zervos AS. Characterization of a novel human serine protease that has extensive homology to bacterial heat shock endoprotease HtrA and is regulated by kidney ischemia. J Biol Chem. 2000;275:2581–2588. doi: 10.1074/jbc.275.4.2581. [DOI] [PubMed] [Google Scholar]
- Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Chesselet MF. Alpha-synuclein and transgenic mouse models. Neurobiol Dis. 2004;17:123–130. doi: 10.1016/j.nbd.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Ferraro E, Cecconi F. Autophagic and apoptotic response to stress signals in mammalian cells. Arch Biochem Biophys. 2007;462:210–219. doi: 10.1016/j.abb.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Fernagut PO, Chesselet MF. Genetic mouse models of parkinsonism: strengths and limitations. NeuroRx. 2005;2:495–503. doi: 10.1602/neurorx.2.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, Shults C, Marder K, Conneally PM, Nichols WC. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology. 2003;60:796–801. doi: 10.1212/01.wnl.0000049470.00180.07. [DOI] [PubMed] [Google Scholar]
- Frasier M, Walzer M, McCarthy L, Magnuson D, Lee JM, Haas C, Kahle P, Wolozin B. Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp Neurol. 2005;192:274–287. doi: 10.1016/j.expneurol.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- Gal S, Zheng H, Fridkin M, Youdim MBH. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases. In vivo selective brain monoamine oxidase inhibition and prevention of MPTP-induced striatal dopamine depletion. J Neurochem. 2005;95:79–88. doi: 10.1111/j.1471-4159.2005.03341.x. [DOI] [PubMed] [Google Scholar]
- Galter D, Westerlund M, Carmine A, Lindqvist E, Sydow O, Olson L. LRRK2 expression linked to dopamine-innervated areas. Ann Neurol. 2006;59:714–719. doi: 10.1002/ana.20808. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Muqit MMK, Stanyer L, Healy DG, Abou-Sleiman PM, Hargreaves I, Heales S, Ganguly M, Parsons L, Lees AJ, Latchman DS, Holton JL, Wood NW, Revesz T. PINK1 protein in normal human brain and Parkinson’s disease. Brain. 2006;129:1720–1731. doi: 10.1093/brain/awl114. [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Cardona-Gomez P, Naftolin F, Chowen JA. Estradiol upregulates Bcl-2 expression in adult brain neurons. Neuroreport. 1998;9:593–597. doi: 10.1097/00001756-199803090-00006. [DOI] [PubMed] [Google Scholar]
- Gasser T, Muller-Myhsok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V, Bereznai B, Fabrizio E, Vieregge P, Horstmann RD. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat Genet. 1998;18:262–265. doi: 10.1038/ng0398-262. [DOI] [PubMed] [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the Zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Marelli C, Achilli A, et al. Mitochondrial DNA haplogroup K is associated with a lower risk of Parkinson’s disease in Italians. Eur J Hum Genet. 2005;13:748–752. doi: 10.1038/sj.ejhg.5201425. [DOI] [PubMed] [Google Scholar]
- Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T, Wood NW. Common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- Glinka Y, Tipton KF, Youdim MBH. Nature of inhibition of mitochondrial respiratory complex I by 6-hydroxydopamine. J Neurochem. 1996;66:2004–2010. doi: 10.1046/j.1471-4159.1996.66052004.x. [DOI] [PubMed] [Google Scholar]
- Glinka Y, Tipton KF, Youdim MBH. Mechanism of inhibition of mitochondrial respiratory complex I by 6-hydroxydopamine and its prevention by desferrioxamine. Eur J Pharmacol. 1998;351:121–129. doi: 10.1016/s0014-2999(98)00279-9. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O’Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Gong L, Daigneault EA, Acuff RV, Kostrzewa RM. Vitamin-E supplements fail to protect mice from acute MPTP neurotoxicity. Neuroreport. 1991;2:544–546. doi: 10.1097/00001756-199109000-00012. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alegre P. Therapeutic RNA interference for neurodegenerative diseases: from promise to progress. Pharmacol Ther. 2007;114:34–55. doi: 10.1016/j.pharmthera.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Irvani M, Cooper JM, King D, Jenner P, Schapira AHV. Pramipexole protects against apoptotic cell death by non-dopaminergic mechanisms. J Neurochem. 2004;91:1075–1081. doi: 10.1111/j.1471-4159.2004.02804.x. [DOI] [PubMed] [Google Scholar]
- Hague S, Peuralinna T, Eerola J, Hellstrom O, Tienari PJ, Singleton AB. Confirmation of the protective effect of iNOS in an independent cohort of Parkinson disease. Neurology. 2004;62:635–636. doi: 10.1212/01.wnl.0000110191.38152.29. [DOI] [PubMed] [Google Scholar]
- Hague S, Rogaeva E, Hernandez D, Gulick C, Singleton A, Hanson M, Johnson J, Weiser R, Gallardo M, Ravina B, Gwinn-Hardy K, Crawley A, George-Hyslop PH, Lang AE, Heutink P, Bonifati V, Hardy J, Singleton A. Early-onset Parkinson’s disease caused by a compound heterozygous DJ-1 mutation. Ann Neurol. 2003;54:271–274. doi: 10.1002/ana.10663. [DOI] [PubMed] [Google Scholar]
- Halbig TD, Tse W, Olanow CW. Neuroprotective agents in Parkinson’s disease: clinical evidence and caveats. Neurol Clin. 2004;22:S1–S17. doi: 10.1016/j.ncl.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Ham J, Eilers A, Whitfield J, Neame SJ, Shah B. c-Jun and the transcriptional control of neuronal apoptosis. Biochem Pharmacol. 2000;60:1015–1021. doi: 10.1016/s0006-2952(00)00372-5. [DOI] [PubMed] [Google Scholar]
- Hancock DB, Martin ER, Fujiwara K, Stacy MA, Scott BL, Stajich JM, Jewett R, Li YJ, Hauser MA, Vance JM, Scott WK. NOS2A and the modulating effect of cigarette smoking in Parkinson’s disease. Ann Neurol. 2006;60:366–373. doi: 10.1002/ana.20915. [DOI] [PubMed] [Google Scholar]
- Hancock DB, Martin ER, Stajich JM, Jewett R, Stacy MA, Scott BL, Vance JM, Scott WK. Smoking, caffeine, and nonsteroidal anti-inflammatory drugs in families with Parkinson disease. Arch Neurol. 2007;64:576–580. doi: 10.1001/archneur.64.4.576. [DOI] [PubMed] [Google Scholar]
- Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–U40. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- Hara MR, Thomas B, Cascio MB, Bae BI, Hester LD, Dawson VL, Dawson TM, Sawa A, Snyder SH. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci U S A. 2006;103:3887–3889. doi: 10.1073/pnas.0511321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich K, Djarmati A, Schafer N, Hering R, Wellenbrock C, Weiss PH, Hilker R, Vieregge P, Ozelius LJ, Heutink P, Bonifati V, Schwinger E, Lang AE, Noth J, Bressman SB, Pramstaller PP, Riess O, Klein C. DJ-1 (PARK7) mutations are less frequent than Parkin (PARK2) mutations in early-onset Parkinson disease. Neurology. 2004a;62:389–394. doi: 10.1212/01.wnl.0000113022.51739.88. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Eskelson C, Wilmot B, Marder K, Harris J, Garrels J, Meija-Santana H, Vieregge P, Jacobs H, Bressman SB, Lang AE, Kann M, Abbruzzese G, Martinelli P, Schwinger E, Ozelius LJ, Pramstaller PP, Klein C, Kramer P. Distribution, type, and origin of Parkin mutations: review and case studies. Mov Disord. 2004b;19:1146–1157. doi: 10.1002/mds.20234. [DOI] [PubMed] [Google Scholar]
- Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, Alnemri ES. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem. 2002;277:432–438. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann Neurol. 2002;52:276–284. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- Hicks AA, Petursson H, Jonsson T, Stefansson H, Johannsdottir HS, Sainz J, Frigge ML, Kong A, Gulcher JR, Stefansson K, Sveinbjornsdottir S. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol. 2002;52:549–555. doi: 10.1002/ana.10324. [DOI] [PubMed] [Google Scholar]
- Holley CL, Olson MR, Colon-Ramos DA, Kornbluth S. Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nat Cell Biol. 2002;4:439–444. doi: 10.1038/ncb798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway R, Shoulson I, Kieburtz K, et al. Pramipexole vs levodopa as initial treatment for Parkinson disease – a randomized controlled trial. JAMA. 2000;284:1931–1938. doi: 10.1001/jama.284.15.1931. [DOI] [PubMed] [Google Scholar]
- Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, Rakic P, Flavell RA. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Iida M, Miyazaki I, Tanaka K, Kabuto H, Iwata-Ichikawa E, Ogawa N. Dopamine D2 receptor-mediated antioxidant and neuroprotective effects of ropinirole, a dopamine agonist. Brain Res. 1999;838:51–59. doi: 10.1016/s0006-8993(99)01688-1. [DOI] [PubMed] [Google Scholar]
- Iravani MM, Haddon CO, Cooper JM, Jenner P, Schapira AH. Pramipexole protects against MPTP toxicity in non-human primates. J Neurochem. 2006;96:1315–1321. doi: 10.1111/j.1471-4159.2005.03625.x. [DOI] [PubMed] [Google Scholar]
- Ishitani R, Kimura M, Sunaga K, Katsube N, Tanaka M, Chuang DM. An antisense oligodeoxynucleotide to glyceraldehyde-3-phosphate dehydrogenase blocks age-induced apoptosis of mature cerebrocortical neurons in culture. J Pharmacol Exp Ther. 1996;278:447–454. [PubMed] [Google Scholar]
- Itier JM, Ibanez P, Mena MA, et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12:2277–2291. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- Jiang H, Luan Z, Wang J, Xie J. Neuroprotective effects of iron chelator Desferal on dopaminergic neurons in the substantia nigra of rats with iron-overload. Neurochem Int. 2006;49:605–609. doi: 10.1016/j.neuint.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Jiang HB, Ren Y, Zhao JH, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745–1754. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnoczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature. 2003;425:721–727. doi: 10.1038/nature02052. [DOI] [PubMed] [Google Scholar]
- Joo WS, Jin BK, Park CW, Maeng SH, Kim YS. Melatonin increases striatal dopaminergic function in 6-OHDA-lesioned rats. Neuroreport. 1998;9:4123–4126. doi: 10.1097/00001756-199812210-00022. [DOI] [PubMed] [Google Scholar]
- Junghanns S, Glockler T, Reichmann H. Switching and combining of dopamine agonists. J Neurol. 2004;251:19–23. doi: 10.1007/s00415-004-1605-7. [DOI] [PubMed] [Google Scholar]
- Kalivendi SV, Cunningham S, Kotamraju S, Joseph J, Hillard CJ, Kalyanaraman B. alpha-Synuclein up-regulation and aggregation during MPP+-induced apoptosis in neuroblastoma cells – intermediacy of transferrin receptor iron and hydrogen peroxide. J Biol Chem. 2004;279:15240–15247. doi: 10.1074/jbc.M312497200. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Scott WK, Li YJ, Hauser MA, van der Walt JM, Fujiwara K, Mayhew GM, West SG, Vance JM, Martin ER. Family-based case-control study of MAOA and MAOB polymorphisms in Parkinson disease. Mov Disord. 2006;21:2175–2180. doi: 10.1002/mds.21151. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NL, Jain S, Lynch JM, et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128:2786–2796. doi: 10.1093/brain/awh667. [DOI] [PubMed] [Google Scholar]
- Khan NL, Valente EM, Bentivoglio AR, Wood NW, Albanese A, Brooks DJ, Piccini P. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an F-18-dopa PET study. Ann Neurol. 2002;52:849–853. doi: 10.1002/ana.10417. [DOI] [PubMed] [Google Scholar]
- Khwaja M, McCormack A, McIntosh JM, Di Monte DA, Quik M. Nicotine partially protects against paraquat-induced nigrostriatal damage in mice; link to alpha6beta2* nAChRs. J Neurochem. 2007;100:180–190. doi: 10.1111/j.1471-4159.2006.04177.x. [DOI] [PubMed] [Google Scholar]
- Kienzl E, Puchinger L, Jellinger K, Linert W, Stachelberger H, Jameson RF. The role of transition metals in the pathogenesis of Parkinson’s disease. J Neurol Sci. 1995;134:69–78. doi: 10.1016/0022-510x(95)00210-s. [DOI] [PubMed] [Google Scholar]
- Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You T, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DF, Cooper JM, Schapira AHV. Pramipexole protects against MPP+ toxicity in SHSY5Y cells by maintaining mitochondrial membrane potential. Neurology. 2001;56:A377–A378. [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Kosaka T, Kakimura JI, Matsuoka Y, Kohno Y, Nomura Y, Taniguchi T. Protective effects of the antiparkinsonian drugs talipexole and pramipexole against 1-methyl-4-phenylpyridinium-induced apoptotic death in human neuroblastoma SH-SY5Y cells. Mol Pharmacol. 1998;54:1046–1054. [PubMed] [Google Scholar]
- Klein C. Implications of genetics on the diagnosis and care of patients with Parkinson disease. Arch Neurol. 2006;63:328–334. doi: 10.1001/archneur.63.3.328. [DOI] [PubMed] [Google Scholar]
- Kostrzewa RM. Review of apoptosis vs. necrosis of substantia nigra pars compacta in Parkinson’s disease. Neurotoxicol Res. 2000;2:239–250. doi: 10.1007/BF03033797. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Krueger MJ, Singer TP, Casida JE, Ramsay RR. Evidence that the blockade of mitochondrial respiration by the neurotoxin 1-methyl-4-phenylpyridinium (Mpp+) involves binding at the same site as the respiratory inhibitor, rotenone. Biochem Biophys Res Commun. 1990;169:123–128. doi: 10.1016/0006-291x(90)91442-u. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kupsch A, Sautter J, Gotz ME, Breithaupt W, Schwarz J, Youdim MBH, Riederer P, Gerlach M, Oertel WH. Monoamine oxidase-inhibition and MPTP-induced neurotoxicity in the non-human primate: comparison of rasagiline (TVP 1012) with selegiline. J Neural Transm. 2001;108:985–1009. doi: 10.1007/s007020170018. [DOI] [PubMed] [Google Scholar]
- Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. 2006;15:883–895. doi: 10.1093/hmg/ddl006. [DOI] [PubMed] [Google Scholar]
- Lan J, Jiang DH. Desferrioxamine and vitamin E protect against iron and MPTP-induced neurodegeneration in mice. J Neural Transm. 1997;104:469–481. doi: 10.1007/BF01277665. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Lesage S, Magali P, Lohmann E, Lacomblez L, Teive H, Janin S, Cousin PY, Durr A, Brice A. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum Mutat. 2007;28:27–32. doi: 10.1002/humu.20436. [DOI] [PubMed] [Google Scholar]
- Levecque C, Elbaz A, Clavel J, Richard F, Vidal JS, Amouyel P, Tzourio C, Alperovitch A, Chartier-Harlin MC. Association between Parkinson’s disease and polymorphisms in the nNOS and iNOS genes in a community-based case – control study. Hum Mol Genet. 2003;12:79–86. doi: 10.1093/hmg/ddg009. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo L, Wang XD. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang XD. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Li W, Srinivasula SM, Chai J, Li P, Wu JW, Zhang Z, Alnemri ES, Shi Y. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat Struct Biol. 2002;9:436–441. doi: 10.1038/nsb795. [DOI] [PubMed] [Google Scholar]
- Li Y, Tomiyama H, Sato K, Hatano Y, Yoshino H, Atsumi M, Kitaguchi M, Sasaki S, Kawaguchi S, Miyajima H, Toda T, Mizuno Y, Hattori N. Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology. 2005;64:1955–1957. doi: 10.1212/01.WNL.0000164009.36740.4E. [DOI] [PubMed] [Google Scholar]
- Li YJ, Scott WK, Zhang L, Lin PI, Oliveira SA, Skelly T, Doraiswamy MP, Welsh-Bohmer KA, Martin ER, Haines JL, Pericak-Vance MA, Vance JM. Revealing the role of glutathione S-transferase omega in age-at-onset of Alzheimer and Parkinson diseases. Neurobiol Aging. 2006;27:1087–1093. doi: 10.1016/j.neurobiolaging.2005.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linert W, Bridge MH, Huber M, Bjugstad KB, Grossman S, Arendash GW. In vitro and in vivo studies investigating possible antioxidant actions of nicotine: relevance to Parkinson’s and Alzheimer’s diseases. Biochim Biophys Acta Mol Basis Dis. 1999;1454:143–152. doi: 10.1016/s0925-4439(99)00029-0. [DOI] [PubMed] [Google Scholar]
- Liochev SI, Fridovich I. The role of O-2-center-dot- in the production of ho-center-dot – in-vitro and in-vivo. Free Radic Biol Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
- Litvan I, Chesselet MF, Gasser T, Di Monte DA, Parker D, Jr, Hagg T, Hardy J, Jenner P, Myers RH, Price D, Hallett M, Langston WJ, Lang AE, Halliday G, Rocca W, Duyckaerts C, Dickson DW, Ben Shlomo Y, Goetz CG, Melamed E. The etiopathogenesis of Parkinson disease and suggestions for future research. Part II. J Neuropathol Exp Neurol. 2007;66:329–336. doi: 10.1097/nen.0b013e318053716a. [DOI] [PubMed] [Google Scholar]
- Lockhart PJ, Lincoln S, Hulihan M, Kachergus J, Wilkes K, Bisceglio G, Mash DC, Farrer MJ. DJ-1 mutations are a rare cause of recessively inherited early onset parkinsonism mediated by loss of protein function. J Med Genet. 2004;41:222. doi: 10.1136/jmg.2003.011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 2002;9:1031–1042. doi: 10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- Lopez-Burillo S, Tan DX, Rodriguez-Gallego V, Manchester LC, Mayo JC, Sainz RM, Reiter RJ. Melatonin and its derivatives cyclic 3-hydroxymelatonin, N-1-acetyl-N (2)-formyl-5-methoxykynuramine and 6-methoxymelatonin reduce oxidative DNA damage induced by Fenton reagents. J Pineal Res. 2003;34:178–184. [Google Scholar]
- Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Maggi A, Ciana P, Belcredito S, Vegeto E. Estrogens in the nervous system: Mechanisms and nonreproductive functions. Annu Rev Physiol. 2004;66:291–313. doi: 10.1146/annurev.physiol.66.032802.154945. [DOI] [PubMed] [Google Scholar]
- Maker HS, Weiss C, Silides DJ, Cohen G. Coupling of dopamine oxidation (monoamine-oxidase activity) to glutathione oxidation via the generation of hydrogen-peroxide in rat-brain homogenates. J Neurochem. 1981;36:589–593. doi: 10.1111/j.1471-4159.1981.tb01631.x. [DOI] [PubMed] [Google Scholar]
- Manning-Bog AB, Langston JW. Model fusion, the next phase in developing animal models for Parkinson’s disease. Neurotoxicol Res. 2007;11:219–240. doi: 10.1007/BF03033569. [DOI] [PubMed] [Google Scholar]
- Mansson R, Hansson MJ, Morota S, Uchino H, Ekdahl CT, Elmer E. Re-evaluation of mitochondrial permeability transition as a primary neuroprotective target of minocycline. Neurobiol Dis. 2007;25:198–205. doi: 10.1016/j.nbd.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Elbaz A, et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, Xu ZH, Meyer SL, Savage MJ, Greene LA, Scott RW, Vaught JL. CEP–1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem. 2001;276:25302–25308. doi: 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP–1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marongiu R, Brancati F, Antonini A, Ialongo T, Ceccarini C, Scarciolla O, Capalbo A, Benti R, Pezzoli G, Dallapiccola B, Goldwurm S, Valente EM. Whole gene deletion and splicing mutations expand the PINK1 genotypic spectrum. Hum Mutat. 2007 doi: 10.1002/humu.9472. Mutation in Brief 943 online. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–444. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL, Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz JB, Mak T, Downward J. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Mata IF, Lockhart PJ, Farrer MJ. Parkin genetics: one model for Parkinson’s disease. Hum Mol Genet. 2004;13:R127–R133. doi: 10.1093/hmg/ddh089. [DOI] [PubMed] [Google Scholar]
- Mathiasen JR, Mckenna BAW, Saporito MS, Ghadge GD, Roos RP, Holskin BP, Wu ZL, Trusko SP, Connors TC, Maroney AC, Thomas BA, Thomas JC, Bozyczko-Coyne D. Inhibition of mixed lineage kinase 3 attenuates MPP+-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2004;1003:86–97. doi: 10.1016/j.brainres.2003.11.073. [DOI] [PubMed] [Google Scholar]
- Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol. 1999;157:142–149. doi: 10.1006/exnr.1999.7049. [DOI] [PubMed] [Google Scholar]
- Melrose HL, Lincoln SJ, Tyndall GM, Farrer MJ. Parkinson’s disease: a rethink of rodent models. Exp Brain Res. 2006;173:196–204. doi: 10.1007/s00221-006-0461-3. [DOI] [PubMed] [Google Scholar]
- Meulener M, Whitworth AJ, Armstrong-Gold CE, Rizzu P, Heutink P, Wes PD, Pallanck LJ, Bonini NM. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr Biol. 2005;15:1572–1577. doi: 10.1016/j.cub.2005.07.064. [DOI] [PubMed] [Google Scholar]
- Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Int Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- Mitsumoto A, Nakagawa Y. DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radic Res. 2001;35:885–893. doi: 10.1080/10715760100301381. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Yamada M, Mizuno Y. Parkin gene therapy for alpha-synucleinopathy: A rat model of Parkinson’s disease. Parkinsonism Relat Disord. 2006;12:S107–S109. doi: 10.1089/hum.2005.16.262. [DOI] [PubMed] [Google Scholar]
- Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci U S A. 1999;96:8867–8872. doi: 10.1073/pnas.96.16.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair VD, Olanow CW, Sealfon SC. Activation of phosphoinositide 3-kinase by D-2 receptor prevents apoptosis in dopaminergic cell lines. Biochem J. 2003;373:25–32. doi: 10.1042/BJ20030017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols WC, Pankratz N, Hernandez D, Paisan-Ruiz C, Jain S, Halter CA, Michaels VE, Reed T, Rudolph A, Shults CW, Singleton A, Foroud T. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet. 2005;365:410–412. doi: 10.1016/S0140-6736(05)17828-3. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Agid Y, Mizuno Y, et al. Levodopa in the treatment of Parkinson’s disease: current controversies. Mov Disord. 2004;19:997–1005. doi: 10.1002/mds.20243. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Hauser RA, Gauger L, Malapira T, Koller W, Hubble J, Bushenbark K, Lilienfeld D, Esterlitz J. The effect of deprenyl and levodopa on the progression of Parkinsons-disease. Ann Neurol. 1995;38:771–777. doi: 10.1002/ana.410380512. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Jankovic J. Neuroprotective therapy in Parkinson’s disease and motor complications: a search for a pathogenesis-targeted, disease-modifying strategy. Mov Disord. 2005;20:S3–S10. doi: 10.1002/mds.20457. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Schapira AHV, Lewitt PA, Kieburtz K, Sauer D, Olivieri G, Pohlmann H, Hubble J. TCH346 as a neuroprotective drug in Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2006;5:1013–1020. doi: 10.1016/S1474-4422(06)70602-0. [DOI] [PubMed] [Google Scholar]
- Oliveira SA, Scott WK, Martin ER, et al. Parkin mutations and susceptibility alleles in late-onset Parkinson’s disease. Ann Neurol. 2003;53:624–629. doi: 10.1002/ana.10524. [DOI] [PubMed] [Google Scholar]
- Ono K, Hirohata M, Yamada M. Anti-fibrillogenic and fibril-destabilizing activities of anti-Parkinsonian agents for alpha-synuclein fibrils in vitro. J Neurosci Res. 2007;85:1547–1557. doi: 10.1002/jnr.21271. [DOI] [PubMed] [Google Scholar]
- Orth M, Schapira AHV. Mitochondrial involvement in Parkinson’s disease. Neurochem Int. 2002;40:533–541. doi: 10.1016/s0197-0186(01)00124-3. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Pals P, Lincoln S, Manning J, Heckman M, Skipper L, Hulihan M, Van den BM, De Pooter T, Cras P, Crook J, Van Broeckhoven C, Farrer MJ. alpha-Synuclein promoter confers susceptibility to Parkinson’s disease. Ann Neurol. 2004;56:591–595. doi: 10.1002/ana.20268. [DOI] [PubMed] [Google Scholar]
- Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C, Conneally PM, Foroud T. Significant linkage of Parkinson disease to chromosome 2q36–37. Am J Hum Genet. 2003;72:1053–1057. doi: 10.1086/374383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadakis ES, Finegan KG, Wang X, Robinson AC, Guo C, Kayahara M, Tournier C. The regulation of Bax by c-Jun N-terminal protein kinase (JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway. FEBS Lett. 2006;580:1320–1326. doi: 10.1016/j.febslet.2006.01.053. [DOI] [PubMed] [Google Scholar]
- Papucci L, Schiavone N, Witort E, Donnini M, Lapucci A, Tempestini A, Formigli L, Zecchi-Orlandini S, Orlandini G, Carella G, Brancato R, Capaccioli S. Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J Biol Chem. 2003;278:28220–28228. doi: 10.1074/jbc.M302297200. [DOI] [PubMed] [Google Scholar]
- Park J, Kim SY, Cha GH, Lee SB, Kim S, Chung J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene. 2005;361:133–139. doi: 10.1016/j.gene.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch Neurol. 2004a;61:561–566. doi: 10.1001/archneur.61.4.561. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. The safety and tolerability of a mixed lineage kinase inhibitor (CEP-1347) in PD. Neurology. 2004b;62:330–332. doi: 10.1212/01.wnl.0000103882.56507.20. [DOI] [PubMed] [Google Scholar]
- Pearce RKB, Owen A, Daniel S, Jenner P, Marsden CD. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J Neural Transm. 1997;104:661–677. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- Petit A, Kawarai T, Paitel E, Sanjo N, Maj M, Scheid M, Chen F, Gu Y, Hasegawa H, Salehi-Rad S, Wang L, Rogaeva E, Fraser P, Robinson B, George-Hyslop P, Tandon A. Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J Biol Chem. 2005;280:34025–34032. doi: 10.1074/jbc.M505143200. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and Parkinson’s disease: a metaanalysis. Environ Res. 2001;86:122–127. doi: 10.1006/enrs.2001.4264. [DOI] [PubMed] [Google Scholar]
- Pyle A, Foltynie T, Tiangyou W, Lambert C, Keers SM, Allcock LM, Davison J, Lewis SJ, Perry RH, Barker R, Burn DJ, Chinnery PF. Mitochondrial DNA haplogroup cluster UKJT reduces the risk of PD. Ann Neurol. 2005;57:564–567. doi: 10.1002/ana.20417. [DOI] [PubMed] [Google Scholar]
- Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000;25:502–508. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Gruendemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- Ramirez AD, Liu XR, Menniti FS. Repeated estradiol treatment prevents MPTP-induced dopamine depletion in male mice. Neuroendocrinology. 2003;77:223–231. doi: 10.1159/000070277. [DOI] [PubMed] [Google Scholar]
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med. 2000;342:1484–1491. doi: 10.1056/NEJM200005183422004. [DOI] [PubMed] [Google Scholar]
- Ravina B, Kieburtz K, Tilley B, et al. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology. 2006;66:664–671. doi: 10.1212/01.wnl.0000201252.57661.e1. [DOI] [PubMed] [Google Scholar]
- Ravina BM, Fagan SC, Hart RG, Hovinga CA, Murphy DD, Dawson TM, Marler JR. Neuroprotective agents for clinical trials in Parkinson’s disease – a systematic assessment. Neurology. 2003;60:1234–1240. doi: 10.1212/01.wnl.0000058760.13152.1a. [DOI] [PubMed] [Google Scholar]
- Reiter RJ, Poeggeler B, Tan D, Chen LD, Manchester LC, Guerrero JM. Antioxidant capacity of melatonin – a novel action not requiring a receptor. Neuroendocrinol Lett. 1993;15:103–116. [Google Scholar]
- Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K, Youdim MBH. Transition-metals, ferritin, glutathione, and ascorbic-acid in parkinsonian brains. J Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- Rogaeva E, Johnson J, Lang AE, et al. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol. 2004;61:1898–1904. doi: 10.1001/archneur.61.12.1898. [DOI] [PubMed] [Google Scholar]
- Roghani M, Behzadi G. Neuroprotective effect of vitamin E on the early model of Parkinson’s disease in rat: behavioral and histochemical evidence. Brain Res. 2001;892:211–217. doi: 10.1016/s0006-8993(00)03296-0. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Sangchot P, Sharma S, Chetsawang B, Porter J, Govitrapong P, Ebadi M. Deferoxamine attenuates iron-induced oxidative stress and prevents mitochondrial aggregation and alpha-synuclein translocation in SK-N-SH cells in culture. Dev Neurosci. 2002;24:143–153. doi: 10.1159/000065700. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Brown EM, Miller MS, Carswell S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo. J Pharmacol Exp Ther. 1999;288:421–427. [PubMed] [Google Scholar]
- Sapru MK, Yates JW, Rogan S, Jiang LX, Halter J, Bohn MC. Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp Neurol. 2006;198:382–390. doi: 10.1016/j.expneurol.2005.12.024. [DOI] [PubMed] [Google Scholar]
- Saunders-Pullman R, Gordon-Elliott J, Parides M, Fahn S, Saunders HR, Bressman S. The effect of estrogen replacement on early Parkinson’s disease. Neurology. 1999;52:1417–1421. doi: 10.1212/wnl.52.7.1417. [DOI] [PubMed] [Google Scholar]
- Sawa A, Khan AA, Hester LD, Snyder SH. Glyceraldehyde-3-phosphate dehydrogenase: Nuclear translocation participates in neuronal and nonneuronal cell death. Proc Natl Acad Sci U S A. 1997;94:11669–11674. doi: 10.1073/pnas.94.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada H, Ibi M, Kihara T, Honda K, Nakamizo T, Kanki R, Nakanishi M, Sakka N, Akaike A, Shimohama S. Estradiol protects dopaminergic neurons in a MPP(+)Parkinson’s disease model. Neuropharmacology. 2002;42:1056–1064. doi: 10.1016/s0028-3908(02)00049-7. [DOI] [PubMed] [Google Scholar]
- Schapira AHV. Neuroprotection in PD – a role for dopamine agonists? Neurology. 2003;61:S34–S42. doi: 10.1212/wnl.61.6_suppl_3.s34. [DOI] [PubMed] [Google Scholar]
- Schapira AHV. Present and future drug treatment for Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2005;76:1472–1478. doi: 10.1136/jnnp.2004.035980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AHV, Gu M, King DF, Cooper JM, Jenner P. Pramipexole protects against rotenone toxicity: mechanisms and implications. Neurology. 2002;58:A495–A495. [Google Scholar]
- Schroeter H, Boyd CS, Ahmed R, Spencer JPE, Duncan RF, Rice-Evans C, Cadenas E. c-Jun N-terminal kinase (JNK)-mediated modulation of brain mitochondria function: new target proteins for JNK signalling in mitochondrion-dependent apoptosis. Biochem J. 2003;372:359–369. doi: 10.1042/BJ20030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shoulson I, Fahn S, Oakes D, Kieburtz K, Lang A, Langston JW, LeWitt P, Olanow CW, Penney JB, Tanner C, Rudolph A, Pelusio RM. Effects of tocopherol and deprenyl on the progression of disability in early Parkinsons-disease. N Engl J Med. 1993;328:176–183. doi: 10.1056/NEJM199301213280305. [DOI] [PubMed] [Google Scholar]
- Shoulson I, Oakes D, Fahn S, Lang A, Langston JW, LeWitt P, Olanow CW, Penney JB, Tanner C, Kieburtz K, Rudolph A. Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson’s disease: a randomized placebo-controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Ann Neurol. 2002;51:604–612. doi: 10.1002/ana.10191. [DOI] [PubMed] [Google Scholar]
- Shoulson I. Parkinson Study Group (PSG) & PRECEPT Investigators. CEP–1347 treatment fails to favorably modify the progression of Parkinson’s disease (PRECEPT) study. Neurology. 2006;67(1):185–185. [Google Scholar]
- Shults CW, Beal MF, Fontaine D, Nakano K, Haas RH. Absorption, tolerability, and effects on mitochondrial activity of oral coenzyme Q(10) in parkinsonian patients. Neurology. 1998;50:793–795. doi: 10.1212/wnl.50.3.793. [DOI] [PubMed] [Google Scholar]
- Shults CW, Haas RH, Passov D, Beal MF. Coenzyme Q(10) levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann Neurol. 1997;42:261–264. doi: 10.1002/ana.410420221. [DOI] [PubMed] [Google Scholar]
- Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, Kompoliti K, Perlmutter JS, Reich S, Stern M, Watts RL, Kurlan R, Molho E, Harrison M, Lew M. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59:1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- Siderowf A, Stern M. Clinical trials with rasagiline – evidence for short-term and long-term effects. Neurology. 2006;66:S80–S88. doi: 10.1212/wnl.66.10_suppl_4.s80. [DOI] [PubMed] [Google Scholar]
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14:3477–3492. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- Simon DK, Pulst SM, Sutton JP, Browne SE, Beal MF, Johns DR. Familial multisystem degeneration with parkinsonism associated with the 11778 mitochondrial DNA mutation. Neurology. 1999;53:1787–1793. doi: 10.1212/wnl.53.8.1787. [DOI] [PubMed] [Google Scholar]
- Singh N, Pillay V, Choonara YE. Advances in the treatment of Parkinson’s disease. Prog Neurobiol. 2007;81:29–44. doi: 10.1016/j.pneurobio.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang HB, Moore DJ, Liang YD, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofic E, Paulus W, Jellinger K, Riederer P, Youdim MBH. Selective increase of iron in substantia-nigra zona compacta of parkinsonian brains. J Neurochem. 1991;56:978–982. doi: 10.1111/j.1471-4159.1991.tb02017.x. [DOI] [PubMed] [Google Scholar]
- Sohmiya M, Tanaka M, Tak NW, Yanagisawa M, Tanino Y, Suzuki Y, Okamoto K, Yamamoto Y. Redox status of plasma coenzyme Q10 indicates elevated systemic oxidative stress in Parkinson’s disease. J Neurol Sci. 2004;223:161–166. doi: 10.1016/j.jns.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Solano SM, Miller DW, Augood SJ, Young AB, Penney JB. Expression of alpha-synuclein, parkin, and ubiquitin carboxy-terminal hydrolase L1 mRNA in human brain: Genes associated with familial Parkinson’s disease. Ann Neurol. 2000;47:201–210. [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Stokes AH, Hastings TG, Vrana KE. Cytotoxic and genotoxic potential of dopamine. J Neurosci Res. 1999;55:659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Muller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Kruger R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- Sun M, Latourelle JC, Wooten F, et al. Influence of heterozygosity for Parkin mutation on onset age in familial Parkinson disease The GenePD study. Arch Neurol. 2006;63:826–832. doi: 10.1001/archneur.63.6.826. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP, Davis RE, Parker WD. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann Neurol. 1996;40:663–671. doi: 10.1002/ana.410400417. [DOI] [PubMed] [Google Scholar]
- Tan DX, Manchester LC, Reiter RJ, Qi WB, Karbownik M, Calvo JR. Significance of melatonin in antioxidative defense system: reactions and products. Biol Signals Recept. 2000a;9:137–159. doi: 10.1159/000014635. [DOI] [PubMed] [Google Scholar]
- Tan EK, Khajavi M, Thornby JI, Nagamitsu S, Jankovic J, Ashizawa T. Variability and validity of polymorphism association studies in Parkinson’s disease. Neurology. 2000b;55:533–538. doi: 10.1212/wnl.55.4.533. [DOI] [PubMed] [Google Scholar]
- Tan EK, Yew K, Chua E, Puvan K, Shen H, Lee E, Puong KY, Zhao Y, Pavanni R, Wong MC, Jamora D, de Silva D, Moe KT, Woon FP, Yuen Y, Tan L. PINK1 mutations in sporadic early-onset Parkinson’s disease. Mov Disord. 2006;21:789–793. doi: 10.1002/mds.20810. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson VL, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- Tatton NA. Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson’s disease. Exp Neurol. 2000;166:29–43. doi: 10.1006/exnr.2000.7489. [DOI] [PubMed] [Google Scholar]
- Tatton W, Chalmers-Redman R, Tatton N. Neuroprotection by deprenyl and other propargylamines: glyceraldehyde-3-phosphate dehydrogenase rather than monoamine oxidase B. J Neural Transm. 2003a;110:509–515. doi: 10.1007/s00702-002-0827-z. [DOI] [PubMed] [Google Scholar]
- Tatton WG, Chalmers-Redman R, Brown D, Tatton N. Apoptosis in Parkinson’s disease: signals for neuronal degradation. Ann Neurol. 2003b;53:S61–S70. doi: 10.1002/ana.10489. [DOI] [PubMed] [Google Scholar]
- The NINDS NET-PD Investigators. A randomized clinical trial of coenzyme Q10 and GPI–1485 in early Parkinson disease. Neurology. 2007;68:20–28. doi: 10.1212/01.wnl.0000250355.28474.8e. [DOI] [PubMed] [Google Scholar]
- Thomas B, Mohanakumar KP. Melatonin protects against oxidative stress caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the mouse nigrostriatum. J Pineal Res. 2004;36:25–32. doi: 10.1046/j.1600-079x.2003.00096.x. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol Lond. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Vatassery GT, Smith WE, Quach HT, Lai JCK. In-vitro oxidation of vitamin-e, vitamin-c, thiols and cholesterol in rat-brain mitochondria incubated with free-radicals. Neurochem Int. 1995;26:527–535. doi: 10.1016/0197-0186(94)00147-m. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C, Moritz RL, Simpson RJ, Vaux DL. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem. 2002;277:445–454. doi: 10.1074/jbc.M109891200. [DOI] [PubMed] [Google Scholar]
- Wakamatsu K, Fujikawa K, Zucca FA, Zecca L, Ito S. The structure of neuromelanin as studied by chemical degradative methods. J Neurochem. 2003;86:1015–1023. doi: 10.1046/j.1471-4159.2003.01917.x. [DOI] [PubMed] [Google Scholar]
- Waldmeier P, Bozyczko-Coyne D, Williams M, Vaught JL. Recent clinical failures in Parkinson’s disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neurodegenerative diseases. Biochem Pharmacol. 2006;72:1197–1206. doi: 10.1016/j.bcp.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine-kinase isoenzymes in tissues with high and fluctuating energy demands – the phosphocreatine circuit for cellular-energy homeostasis. Biochem J. 1992;281:21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt JM, Nicodemus KK, Martin ER, et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet. 2003;72:804–811. doi: 10.1086/373937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Ko HS, Thomas B, Tsang F, Chew KCM, Tay SP, Ho MWL, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum Mol Genet. 2005;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- Wang DL, Qian L, Xiong H, Liu JD, Neckameyer WS, Oldham S, Xia K, Wang JZ, Bodmer R, Zhang ZH. Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci U S A. 2006;103:13520–13525. doi: 10.1073/pnas.0604661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Wei QQ, Wang CY, Hill WD, Hess DC, Dong Z. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004;279:19948–19954. doi: 10.1074/jbc.M313629200. [DOI] [PubMed] [Google Scholar]
- West A, Periquet M, Lincoln S, Lucking CB, Nicholl D, Bonifati V, Rawal N, Gasser T, Lohmann E, Deleuze JF, Maraganore D, Levey A, Wood N, Durr A, Hardy J, Brice A, Farrer M. Complex relationship between parkin mutations and Parkinson disease. Am J Med Genet. 2002;114:584–591. doi: 10.1002/ajmg.10525. [DOI] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth AJ, Wes PD, Pallanck LJ. Drosophila models pioneer a new approach to drug discovery for Parkinson’s disease. Drug Discov Today. 2006;11:119–126. doi: 10.1016/S1359-6446(05)03693-7. [DOI] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RM, Chen RC, Chiueh CC. Effect of MAO-B inhibitors on MPP+ toxicity in vivo. Reactive Oxygen Species Radiat Mol Biol. 2000;899:255–261. doi: 10.1111/j.1749-6632.2000.tb06191.x. [DOI] [PubMed] [Google Scholar]
- Xie YX, Bezard E, Zhao BL. Investigating the receptor-independent neuroprotective mechanisms of nicotine in mitochondria. J Biol Chem. 2005;280:32405–32412. doi: 10.1074/jbc.M504664200. [DOI] [PubMed] [Google Scholar]
- Yang LC, Sugama S, Chirichigno JW, Gregorio J, Lorenzl S, Shin DH, Browne SE, Shimizu Y, Joh TH, Beal MF, Albers DS. Minocycline enhances MPTP toxicity to dopaminergic neurons. J Neurosci Res. 2003;74:278–285. doi: 10.1002/jnr.10709. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Haque ME, Imai Y, Kosek J, Yang L, Beal MF, Nishimura I, Wakamatsu K, Ito S, Takahashi R, Lu B. Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci U S A. 2005;102:13670–13675. doi: 10.1073/pnas.0504610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YF, Gehrke S, Imai Y, Huang ZN, Ouyang Y, Wang JW, Yang LC, Beal MF, Vogel H, Lu BW. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused inactivation of Drosophila Pink1 is rescued by by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota T, Sugawara K, Ito K, Takahashi R, Ariga H, Mizusawa H. Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem Biophys Res Commun. 2003;312:1342–1348. doi: 10.1016/j.bbrc.2003.11.056. [DOI] [PubMed] [Google Scholar]
- Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3:744–751. doi: 10.1016/S1474-4422(04)00937-8. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Ben Shachar D, Riederer P. The possible role of iron in the etiopathology of Parkinson’s disease. Mov Disord. 1993;8:1–12. doi: 10.1002/mds.870080102. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Stephenson G, Ben Shachar D. Ironing iron out in Parkinson’s disease and other neurodegenerative diseases with iron chelators: a lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28. Ann N Y Acad Sci. 2004;1012:306–325. doi: 10.1196/annals.1306.025. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Weinstock M. Therapeutic applications of selective and non-selective inhibitors of monoamine oxidase A and B that do not cause significant tyramine potentiation. Neurotoxicology. 2004;25:243–250. doi: 10.1016/S0161-813X(03)00103-7. [DOI] [PubMed] [Google Scholar]
- Youdim MBH, Buccafusco JJ. Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol Sci. 2005;26:27–35. doi: 10.1016/j.tips.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Youdim MBH, Fridkin M, Zheng HL. Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech Ageing Dev. 2005;126:317–326. doi: 10.1016/j.mad.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Youdim MBH, Weinstock M. Molecular basis of neuroprotective activities of rasagiline and the anti-Alzheimer drug TV3326 [(N-propargyl-3R)aminoindan-5-YL)-ethyl methyl carbamate. Cell Mol Neurobiol. 2001;21:555–573. doi: 10.1023/A:1015131516649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez TE, Del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Manzino L, Sonsalla PK, Bernard LP. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson’s disease. Exp Neurol. 2007;203:512–520. doi: 10.1016/j.expneurol.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Shimoji M, Thomas B, Moore DJ, Yu SW, Marupudi NI, Torp R, Torgner IA, Ottersen OP, Dawson TM, Dawson VL. Mitochondrial localization of the Parkinson’s disease related protein DJ-1: implications for pathogenesis. Hum Mol Genet. 2005a;14:2063–2073. doi: 10.1093/hmg/ddi211. [DOI] [PubMed] [Google Scholar]
- Zhang X, Xie W, Qu S, Pan T, Wang X, Le W. Neuroprotection by iron chelator against proteasome inhibitor-induced nigral degeneration. Biochem Biophys Res Commun. 2005b;333:544–549. doi: 10.1016/j.bbrc.2005.05.150. [DOI] [PubMed] [Google Scholar]
- Zheng H, Gal S, Weiner LM, Bar-Am O, Warshawsky A, Fridkin M, Youdim MBH. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: in vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J Neurochem. 2005;95:68–78. doi: 10.1111/j.1471-4159.2005.03340.x. [DOI] [PubMed] [Google Scholar]
- Zhou H, Falkenburger BH, Schulz JB, Tieu K, Xu Z, Xia XG. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci. 2007;3:242–250. doi: 10.7150/ijbs.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BYS, Ona V, Li MW, Sarang S, Liu AS, Hartley DM, Du CW, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]