

Abstract
A series of substituted 3,6-diphenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines were prepared and analyzed as inhibitors of phosphodiesterase 4 (PDE4). Synthesis, structure activity relationships and the selectivity of a highly potent analogue against related phosphodiesterase isoforms are presented.
Cyclic 3’, 5’ adenosine monophosphate (cAMP), a second messenger which mediates the actions of numerous cellular receptors, is a key element in the regulation of cell signaling1 and gene transcription.2 The control of intracellular cAMP levels is accomplished by a balance of cAMP synthesis by adenylate cyclase, and its degradation (hydrolysis) by a variety of phosphodiesterases (PDEs) (Figure 1). Cyclic guanosine monphosphate (cGMP) is controlled with similar mechanisms. The presence of these cyclic nucleotides have regulatory effects on protein kinase A (PKA) and protein kinase G (PKG), the guanine-nucleotide exchange factors (GEFs), and the cyclic-nucleotide gated (CNG) sodium and calcium channels. Manipulation of cAMP and cGMP levels in the cell represents a powerful mechanism for controlling cellular physiology, and small molecules modulators of adenylate cyclase, guanylate cyclase, and phosphodiesterases which are utilitzed as both research tools and as clinically used drugs.3
Figure 1.
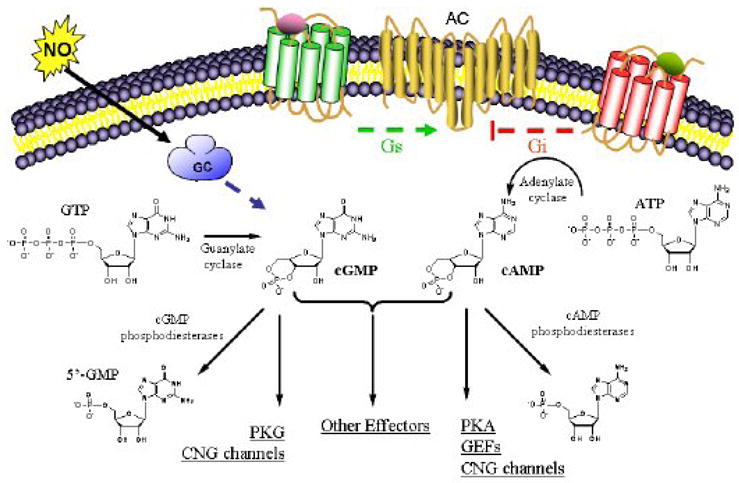
Cartoon description of Cyclic Nucleotide Regulation and Effect. Formation of cGMP via GC or NO stimulated guanylate cyclase activation and formation of cAMP via GPCR Gs stimulated adenylate cyclase activation. cGMP and cAMP regulate several effectors including PKA (protein kinase A), PKG (protein kinase G), GEF (guanine-nucleotide exchange factor) and CNG channels (cyclic-nucleotide gated ion channels). The action of numerous phosphodiesterases convert cAMP and cGMP to 5’-AMP and 5’-GMP respectively.
The PDE class of enzymes contains 11 principal isozymes with 21 characterized gene products.4 The majority of PDE family members and isoforms have been well characterized in terms of tissue distribution and substrate affinity.4 The pharmaceutical sector has focused particular attention on the PDE4 gene family,5,6 with inflammation as a primary therapeutic target.7,8 The role of PDE4 in the inflammatory responses associated with asthma and chronic obstructive pulmonary disease (COPD) has been widely studied.5,6,9,10 PDE4 modulation has also been pursued for memory and depressive disorders11, and more recently for inflammatory bowel disease.12
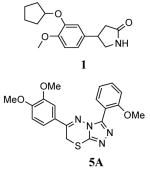
Due to the wide-ranging therapeutic interest in PDE4, numerous chemotypes capable of potent and selective PDE4 inhibition currently exist. Several PDE4 inhibitors have entered into clinical evaluation including rolipram (1)13, roflumilast (2)14, cilomilast (3)15, tofimilast (4)16 (Figure 2). Cilomilast has received an approval letter from the US FDA for use in maintenance of lung function in COPD, but is still under study due to prevalent gastrointestinal adverse effects (nausea/vomiting and abdominal pain).6 The potentially important clinical benefits of PDE4 inhibition, coupled with the limitations of current PDE4 inhibitors, highlight the need for novel PDE4 inhibitor chemotypes. Here we report a novel class of substituted 3,6-diphenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (represented by 5A (Figure 2)) as potent and selective inhibitors of PDE4.
Figure 2.

Structures of rolipram (1), roflumilast (2), cilomilast (3), tofimilast (4) and 6-(3,4-dimethoxyphenyl)-3-(2-methoxyphenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine 5A.
High-throughput screening is commonly used to identify small molecule compounds that modulate biochemical or cellular processes.17 The NIH Molecular Libraries Initiative (MLI)18 has made available public sector screening, cheminformatics, and chemistry efforts on a large scale. Recently, we noted several substituted 3,6-diphenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines as potent inhibitors of PDE4. This core structure represents a novel chemotype capable of PDE4 inhibition. To explore this discovery further, novel analogues were prepared utilizing existing literature precedence to construct the heterocyclic framework (Scheme 1).19–22 Briefly, substituted benzoic acids were transformed to their analogous methyl esters and then to substituted benhydrazides (refluxing with hydrazine in ethanol). The hydrazides were treated with an ethanolic solution of potassium hydroxide to which carbon disulfide was added to produce the corresponding carbodithioates. The dithioates were heated (113 ºC) with hydrazine monohydrate and water then cooled and acidified to provide the substituted triazole in good yields. When necessary, α-bromoketones were produced in modest to good yields upon treatment of the corresponding acetophenones with bromine in chloroform. Condensation between the substituted triazole and substituted α–bromoketones was effected with heating in ethanol to provided the substituted 3,6-diphenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Mass directed preparative LC purification afforded pure samples in modest to good yields. All samples were characterized via proton NMR and HRMS.22 Analytical LC traces of standard 10 mM DMSO solutions confirmed that each product was >90% pure.
The primary leads were noted to have numerous methoxy substitutions on the adjunct 3-and 6-phenyl rings. Thus, our first exploration of structure activity relationships focused primarily on varied methoxy substitutions on these key moieties (compounds 5A-K, 6A-K, 7A-K, 8A-K, 9A-K, 10A-K and 11A-K shown in Figure 3). These derivatives were assayed against purified human PDE4A using IMAP technology (Molecular Devices, CA).24 The inhibitory effects (IC50 values) of these compounds are shown in Figure 3.
Figure 3.

Schematic representation of substituted 3,6-diphenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines 5A-K, 6A-K, 7A-K, 8A-K, 9A-K, 10A-K and 11A-K and each compounds apparent IC50 value versus PDE4A.
From this modest library it was obvious that the 3,4-dimethoxy phenyl substitution on the 5 position of the 3,6-dihydro-2H-1,3,4-thiadiazine ring is a critical functionality for potent PDE4 inhibition (compounds 5A-5K). All derivatives with this functionality had IC50 values in the low nanomolar range with the most potent being 3-(2,5-dimethoxyphenyl)-6-(3,4-dimethoxyphenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5F). The phenyl ring attached at the 3 position of the 1,2,4-triazole portion was seemingly less involved in defining the pharmacophore of this structure, as numerous methoxy substitutions had less obvious effects in terms of structure activity relationships.
Interestingly, the 3,4-alkoxy moiety (or more appropriately the 3,4-catechol diether) plays a key role in numerous PDE4 inhibitors [as illustrated by the structures of rolipram (1), roflumilast (2) and cilomilast (3) (Figure 2)]. Crystallographic analyses of several known inhibitors that share this feature show that a common hydrogen bond between this key functionality and a conserved glutamine residue provides the structural basis for inhibition.6,25 Importantly, this glutamine residue (Gln442 in PDEB and Gln369 in PDE4D) is within close proximity to the coordinated Zn2+ and Mg2+ ions that form the basis for the mechanism of cAMP hydrolysis by PDE4.25,26 Given the strong ability of 3,4-dimethoxy derivatives 5A-5K to inhibit PDE4 we surmise that this novel chemotype is inhibiting PDE4 via interaction at the same binding site. Interestingly, the similar 3,4-dimethoxy substitution pattern engineered upon the phenyl ring attached at the 3 position of the 1,2,4-triazole did not convey favorable affects on PDE4 inhibition as illustrated by compounds 6G, 7G, 8G, 9G, 10G and 11G. This suggests that the interaction between the 3,4 dimethoxy phenyl moiety attached at the 5 position of the 3,6-dihydro-2H-1,3,4-thiadiazine ring places the remainder of the molecule (i.e. the 1,2,4-triazole and the variously substituted phenyl ring attached at the 3 position) in an orientation that interrupts the binding of cAMP and subsequent hydrolysis.
Having defined this novel chemotype as capable of potent PDE4 inhibition, we next acsertained the selectivity of these compounds against other PDE enzymes. IC50’s of 5A and rolipram (1) were determined in biochemical assays for 12 PDE isoforms;27 results are shown in Table 1. Our lead compound 5A demonstrated nanomolar inhibitory potency against all three PDE4 isozymes (PDE4A, PDE4B and PDE4D) but 100-fold lower potency against the other the nine PDEs tested, including no activity against PDE7A and PDE7B, which are closely homologous to PDE4. Importantly, 5A showed potent inhibition of the therapeutically important PDE4B isoform, compared to the relatively low potency of rolipram at PDE4B.
Table 1.
PDE isoform selectivity data for 1 and 5A
| PDE isoform* | 1IC50(μM) | 5AIC50(μM) | |
|---|---|---|---|
|
PDE1A | >50 | >50 |
| PDE1B | inactive | >50 | |
| PDE3A | inactive | >50 | |
| PDE4A | 0.084 | 0.024 | |
| PDE4B | 16 | 0.54 | |
| PDE4D | 0.082 | 0.018 | |
| PDE5A | inactive | >50 | |
| PDE7A | inactive | inactive | |
| PDE7B | inactive | inactive | |
| PDE9A | inactive | inactive | |
| PDE10A | inactive | >50 | |
| PDE11A | inactive | >50 |
data represents the results from three seperate experiments.
Specific isoforms of PDE4 have been suggested as targets for a number of neuropsychiatric and immunologic disorders6 and small molecules capable of subtype selective inhibition, particularly of PDE4B over PDE4D, would therefore be extremely useful. For example, PDE4B knockout models demonstrate anxiogenic phenotypes28, and mutations in PDE4B-specific binding sites of DISC1 affect its binding to PDE4B and confer phenotypes related to schizophrenia and depression.29 Down-regulation of PDE4A and PDE4B are correlated with suppression of inflammatory cell function.30 By contrast, PDE4D is thought to play a role in emesis.6 The catalytic/binding domains of PDE4B and PDE4D are highly homologous, making structural insights of limited utility in designing more selective PDE4B inhibitors. 6
The novel structural class of PDE4 inhibitors reported here may provide a starting point for the design of subtype selective inhibitors. A unique feature of this chemotype is the extended phenyl ring attached at the 3 position of the 1,2,4-triazole. Given the likelihood that the catechol diether moiety interacts with the conserved glutamine residue, the use of molecular modeling and available structural information for both isoforms of PDE4 may allow for the design of novel analogues that favor individual PDE4 isoforms. Of particular importance is the presence of this seemingly modifiable phenyl ring. Additionally, it should be noted that the cLogP value for 5A (calculated value is 3.26) is reasonable for potential blood brain permeation rendering this novel PDE4 inhibitor an important chemotype for potential use in neuropsychiatric indications. In summary, we report a novel class of PDE4 inhibitors based upon a 3,6-diphenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine core structure. Initial SAR highly suggests that this chemotype is among the catechol diether class of PDE4 inhibitors. The most potent analogues maintained the presence of a 3,4-dimethoxy functionality located at the 5 position of the 3,6-dihydro-2H-1,3,4-thiadiazine ring but not the phenyl ring attached at the 3 position of the 1,2,4-triazole. A selected analogue (5A) was profiled for PDE isoform selectivity and showed selectivity for PDE4, with balanced inhibition of the PDE4A, 4B, and 4D isoforms. Synthesis of additional analogues aimed at exploiting the new chemotype as a potent and subtype-selective PDE4 inhibitor are currently underway.
Scheme 1.

a Reagents and conditions: (i) cat. H2SO4, MeOH, r.t. 12 h; (ii) hydrazine, EtOH, reflux 12h.; (iii) KOH, EtOH; then CS2 r.t. 12h; (iv) hydrazine monohydrate, H2O, reflux, 3h, then conc. HCl; (v) Br2, CHCl3, rt 5 min. then reflux 30 min to 4 hr; (vi) EtOH, 105 oC, 4 h.
Acknowledgments
The authors would like to thank Paul Shinn for assistance in compound management. The authors would like to thank Doug Auld for critical reading of this manuscript and technical assistance. This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research and the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Beavo JA, Brunton LL. Nat Rev Mol Cell Biol. 2002;3:710. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 2.Johannessen M, Delghandi MP, Moens U. Cellular Signaling. 2004;16:1211. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Menniti FS, Faraci WS, Schmidt CJ. Nat Rev Drug Discov. 2006;5:660. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 4.Bender AT, Beavo JA. Pharmacol Rev. 2006;58:488. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 5.McKenna JM, Muller GW. In: Cyclic Nucleotide Phosphodiesterases in Health and Disease. Beavo JA, Francis SH, Houslay MD, editors. CRC Press; 2006. p. 667. [Google Scholar]
- 6.Zhang KYJ, Ibrahim PN, Gillette S, Bollag G. Expert Opin Ther Targets. 2005;9:1283. doi: 10.1517/14728222.9.6.1283. [DOI] [PubMed] [Google Scholar]
- 7.Huang Z, Ducharme Y, Macdonald D, Robichaud A. Curr Opin Chem Biol. 2001;5:432. doi: 10.1016/s1367-5931(00)00224-6. [DOI] [PubMed] [Google Scholar]
- 8.Souness JE, Aldous D, Sargent C. Immunopharmacol. 2000;47:127. doi: 10.1016/s0162-3109(00)00185-5. [DOI] [PubMed] [Google Scholar]
- 9.Dyke HJ. Expert Opin Ther Patents. 2007;17:1183. doi: 10.1517/13543776.17.9.1183. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt DT, Watson N, Dent G, Rühlmann E, Branscheid D, Magnussen H, Rabe KF. Br J Pharmacol. 2000;131:1607. doi: 10.1038/sj.bjp.0703725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tully T, Bourtchouladze R, Scott R, Tallman J. Nat Rev Drug Discov. 2003;2:267. doi: 10.1038/nrd1061. [DOI] [PubMed] [Google Scholar]
- 12.Keshavarizian A, Mutla E, Guzman JP, Forsythe C, Banan A. Expert Opin Investig Drugs. 2007;16:1489. doi: 10.1517/13543784.16.9.1489. [DOI] [PubMed] [Google Scholar]
- 13.Kanes SJ, Tokarczyk J, Siegel SJ, Bilker W, Abel T, Kelly MP. Neuroscience. 2007;144:239. doi: 10.1016/j.neuroscience.2006.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boswell-Smith V, Page CP. Expert Opin Investig Drugs. 2006;15:1105. doi: 10.1517/13543784.15.9.1105. [DOI] [PubMed] [Google Scholar]
- 15.Kroegel C, Foerster M. Expert Opin Investig Drugs. 2007;16:109. doi: 10.1517/13543784.16.1.109. [DOI] [PubMed] [Google Scholar]
- 16.Duplantier AJ, Bachert EL, Cheng JB, Cohan VL, Jenkinson TH, Kraus KG, McKechney MW, Pillar JD, Watson JW. J Med Chem. 2007;50:344. doi: 10.1021/jm060904g. [DOI] [PubMed] [Google Scholar]
- 17.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS. Nature Chem Biol. 2007;3:466. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 18.Austin CP, Brady LS, Insel TR, Collins FS. Science. 2004;306:1138. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 19.Swamy SN, Basappa, Naveen S, Prabhuswamy B, Sridhar MA, Prasad JS, Rangappa KS. Struct Chem. 2006;17:91. [Google Scholar]
- 20.Reid JR, Heindel ND. J Heterocyclic Chem. 1976;13:925. [Google Scholar]
- 21.Jacob JN, Nichols DE. J Med Chem. 1981;24:1013. doi: 10.1021/jm00140a021. [DOI] [PubMed] [Google Scholar]
- 22.Moreno I, Tellitu I, Dominguez E, SanMartin R. Eur J Org Chem. 2002;13:2126. [Google Scholar]
- 23.General synthetic procedure and characterization data for selected compounds: Purchased reagents and anhydrous solvents were used without further purification. Reactions were conducted using oven-dried glassware under an atmosphere of nitrogen. All condensation reactions were performed in a Biotage 2.0-5.0 mL vessel with a stir bar and crimped top. Select products were purified via flash chromatography (where noted), using 40 – 63 μm silica gel. Ethyl acetate (EtOAc) and hexanes for chromatography were used as purchased. Purification of final products was performed on a Waters semi-preparative HPLC with mass detection, equipped with an Xterra C18 (5 micron, 19 x 100 mm) column and a 24 mL/min flow rate. A gradient was performed using an acetonitrile/water mobile phase (each containing 0.1% trifluoroacetic acid). Two different gradients (10% to 50% acetonitrile over 9 minutes or 30% to 70% acetonitrile over 9 minutes) were used during the purification. Fraction collection was triggered with mass spectrometry detection. Pure fractions were dried down using a Genevac rotary evaporator. Final yields after HPLC purification ranged from 20–75%.Samples were analyzed for purity on a an Agilent 1200 series LC/MS using a Zorbax Eclipse XBD-C8 reverse phase (5 micron, 4.6 x 150mm) column and a 1.1 mL/min flow rate. A gradient was performed using an acetonitrile/water mobile phase (each containing 0.1% trifluoroacetic acid). The gradient was 5% to 100% acetonitrile over 10 minutes. Purity of final compounds was determined to be >95%, using a five microliter injection with quantitation by AUC at 220 and 254 nanometers. HRMS were obtained by The Mass Spectrometry Core of the Research Technology Support Facility at Michigan State University on a Waters Q-Tof Ultima API mass spectrometer using the LockSpray method. 1HNMR spectroscopy was preformed on a 400 MHz Varian spectrometer. Chemical shifts were recorded on the ppm scale and were referenced to the appropriate residual solvent signal (as indicated). Formation of Substituted Benzoates: To a solution of benzoic acid (1 eq) in methanol (1.0 M) was added sulfuric acid (catalytic). The solution was stirred at room temperature for 12 h, at which time the solvent was removed by rotary evaporation. The crude reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was extracted twice with ethyl acetate and the organic extracts were combined, washed with water and brine, dried over Na2SO4, and concentrated by rotary evaporation. The crude product was purified by column chromatography to give the substituted benzoates in >80% yield. Formation of Substituted Aryldithiocarbazates: To a solution of methyl benzoate (1 eq) in ethanol (0.55 M) was added hydrazine (4 eq). The solution was heated to reflux with stirring until TLC showed full consumption of starting materials (12 h), then cooled. The solvent was removed by rotary evaporation and the crude reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was extracted twice with ethyl acetate and the organic extracts were combined, washed with water and brine, dried over Na2SO4, and concentrated by rotary evaporation. The crude hydrazide (1.0 eq) was taken up in ethanol (0.5 M). Potassium hydroxide (1.5 eq) was added, and stirred to dissolve. To this solution, carbon disulfide (1.5 eq) was added in a drop-wise fashion. Within a period of 1–10 min, the potassium salt precipitated from solution, and was allowed to stir as a suspension for 12 h. The suspension was filtered and dried to give the potassium aryldithiocarbazates as pale yellow powders in >85%. Formation of Substituted triazoles: To a mixture of aryldithiocarbazate (1 eq) in water (10.0 M) was added hydrazine monohydrate (2 eq). The mixture was heated to 113 °C to induce cyclization to the triazole with formation of hydrogen sulfide gas (reaction mixture turned greenish brown). After 0.75 h, the reaction mixture was cooled and ice chips were added. Acidification with conc. hydrochloric acid precipitated a white solid. The product was filtered and washed with 2 x 20 mL portions of cold water to give the triazoles. If necessary, recrystallization from 95% ethanol garnered analytically pure products. Final yields ranged from 75–90%.Formation of Substituted 2-bromoacetophenones: To a solution of substituted acetophenone in chloroform (0.35 M) was added bromine (1.2 eq). The solution was stirred at room temperature for 0.5 h, then heated to reflux for another 0.5-2 h until TLC showed full consumption of starting materials. The reaction mixture was concentrated by rotary evaporation and the crude product was purified by column chromatography. Final yields ranged from 50–95%. Formation of Substituted 3,6-diphenyl-7H-[1, 2, 4]-triazolo[3, 4-b] [1, 3, 4] thiadiazines: To a mixture of triazole (1.0 eq) and substituted 2-bromoacetophenone (1.0 eq) was added ethanol (0.1 M). The reaction mixture was sealed in a crimp-top high pressure vessel and stirred at 105 °C for 4 h. The crude reaction mixture was partitioned between methylene chloride and water. The aqueous layer was removed and the organic layer was washed with a mixture of water and brine, then concentrated by rotary evaporation. The crude product was purified by preparative HPLC (see General Experimental for details). 3-(2-methoxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5A):1H NMR (d6-DMSO, 400 MHz) 7.61-7.55 (m, 2H), 7.50 (dd, 2.0, 8.4 Hz, 1H), 7.41 (d, 2.0 Hz, 1H), 7.24 (d, 8.0 Hz, 1H), 7.15-7.07 (m, 2H), 4.22 (s, 2H), 3.80 (s, 3H), 3.77 (s, 3H), 3.73 (s, 3H); 13C NMR (d6-DMSO, 100 MHz) 158.3, 156.3, 152.9, 150.5, 149.5, 142.9, 6, 132.2, 125.9, 122.4, 121.0, 114.3, 112.6, 112.2, 110.6, 56.5, 56.4, 56.2, 23.5; LC-MS: RT (min) = 6.19; [M + H]+ 383.1; HRMS calcd for C19H19N4O3S (M + H) 383.1100 , found 383.1181. 3-(3-methoxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5B):1H NMR (CDCl3, 400 MHz) 7.74-7.70 (m, 2H), 7.59 (d, 2.0 Hz, 1H), 7.43-7.35 (m, 2H), 7.02 (dd, 2.8, 8.4 Hz, 1H), 6.94 (d, 8.4 Hz, 1H), 4.00 (s, 2H), 3.95 (s, 3H), 3.93 (s, 3H), 3.84 (s, 3H); LC-MS: RT (min) = 6.77; [M + H]+ 383.0; HRMS calcd for C19H19N4O3S (M + H) 383.1100 , found 383.1184. 3-(4-methoxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5C):1H NMR (CDCl3, 400 MHz) 8.09 (dd, 2.4, 6.8 Hz, 2H), 7.57 (d, 2.4 Hz, 1H), 7.42 (dd, 2.0, 8.4 Hz, 1 H), 6.99 (dd, 2.0, 6.8 Hz, 2H), 6.96 (d, 8.4 Hz), 3.97 (s, 2H), 3.96 (s, 3H), 3.93 (s, 3H), 3.87 (s, 3H); LC-MS: RT (min) = 6.60; [M + H]+ 383.1;HRMS calcd for C19H19N4O3S (M + H) 383.1100 , found 383.1176. 3-(2, 3-dimethoxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5D):1H NMR (CDCl3, 400 MHz) 7.42 (d, 2.0 Hz, 1H), 7.33 (dd, 2.0, 8.4 Hz, 1H), 7.21 (dd, 1.6, 7.6 Hz, 1H), 7.16 (t, 7.6 Hz), 7.08 (dd, 1.6, 7.6 Hz, 1H), 6.89 (d, 8.4 Hz, 1H), 3.97 (s, 2H), 3.92 (s, 3H), 3.89 (s, 3H), 3.84 (s, 3H), 3.75 (s, 3H); LC-MS: RT (min) = 6.35; [M + H]+ 413.1; HRMS calcd for C20H21N4O4S (M + H) 413.1205, found 413.1281. 3-(2, 4-dimethoxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5E):1H NMR (CDCl3, 400 MHz) 7.59 (d, 8.8 Hz, 1H), 7.43-7.41 (m, 2H), 6.93 (d, 8.8 Hz, 1H), 6.60 (dd, 2.4, 8.8 Hz, 1H), 6.54 (d, 2.4 Hz), 4.06 (s, 2H), 3.93 (s, 3H), 3.87 (s, 3H), 3.86 (s, 3H), 3.75 (s, 3H); LC-MS: RT (min) = 6.18; [M + H]+ 413.1; HRMS calcd for C20H21N4O4S (M + H) 413.1205, found 413.1288. 3-(2, 5-dimethoxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5F):1H NMR (CDCl3, 400 MHz) 7.43 (d, 2.0, 1H), 7.40 (dd, 2.0, 8.4 Hz, 1H), 7.23 (d, 3.2 Hz, 1H), 7.06 (dd, 3.2, 9.2 Hz, 1H), 6.93 (dd, 3.2, 12 Hz, 2H), 4.04 (s, 3H), 3.93 (s, 3H), 3.84 (s, 3H), 3.79 (s, 3H), 3.69 (s, 3H); LC-MS: RT (min) = 6.29; [M + H]+ 413.1; HRMS calcd for C20H21N4O4S (M + H) 413.1205, found 413.1278. 3-(3, 4-dimethoxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5G): 1H NMR (CDCl3, 400 MHz) 7.76-7.73 (m, 2H), 7.56 (d, 2.4 Hz, 1H), 7.44 (dd, 2.0, 8.4 Hz, 1H), 6.98-6.94 (m, 2H), 3.98 (s, 2H), 3.97 (s, 3H), 3.95 (s, 3H), 3.94 (s, 3H), 3.93 (s, 3H); LC-MS: RT (min) = 6.22; [M + H]+ 413.1; HRMS calcd for C20H21N4O4S (M + H) 413.1205, found 413.1279. 3-(3, 5-dimethoxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5H):1H NMR (CDCl3, 400 MHz) 7.65 (d, 1.6 Hz, 1H), 7.41 (d, 2.0 Hz, 1H), 7.39 (d, 2.4 Hz, 2H), 6.95 (d, 8.8 Hz, 1H), 6.58 (t, 2.4 Hz, 1H), 3.98 (s, 2H), 3.97 (s, 3H), 3.95 (s, 3H), 3.83 (s, 6H); LC-MS: RT (min) = 6.95; [M + H]+ 413.1; HRMS calcd for C20H21N4O4S (M + H) 413.1205, found 413.1278. 3-(2-methylpheny)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5I):1H NMR (CDCl3, 400 MHz) 7.99 (s, 1H), 7.92 (d, 7.6 Hz, 1H), 7.60 (d, 2.0 Hz, 1H), 7.42-7.35 (m, 2H), 7.30 (d, 7.6 Hz, 1H), 6.96 (d, 8.8 Hz, 1H), 3.98 (s, 2H), 3.97 (s, 3H), 3.93 (s, 3H), 2.42 (s, 3H); LC-MS: RT (min) = 7.04; [M + H]+ 367.1; HRMS calcd for C19H19N4O2S (M + H) 367.1150, found 367.1224. 3-(2-ethoxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5J):1H NMR (CDCl3, 400 MHz) 7.65 (d, 6.8 Hz, 1H), 7.52 (t, 8.4 Hz, 1H), 7.41 (d, 8.8 Hz, 1H), 7.07 (t, 7.2 Hz, 1H), 6.99 (d, 8.4 Hz, 1H), 6.92 (d, 8.0 Hz, 1H), 4.04 (s, 2H), 3.97 (q, 6.8 Hz, 2H), 3.92 (s, 3H), 3.81 (s, 3H), 1.18 (t, 6.8 Hz, 3H); LC-MS: RT (min) = 6.55; [M + H]+ 397.1; HRMS calcd for C20H21N4O3S (M + H) 397.1256, found 397.1337. 3-(2-hydroxyphenyl)-6-(3, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (5K):1H NMR (CDCl3, 400 MHz) 8.34 (dd, 1.6 Hz, 1H), 7.63 (d, 1.6 Hz, H), 7.44 (dd, 2.4, 8.4 Hz, 1H), 7.36 (ddd, 1.6, 7.2, 12 Hz, 1H), 7.13 (dd, 1.2, 8.4 Hz, 1H), 6.92 (ddd, 1.2, 8.4, 15.2 Hz, 1H), 4.00 (s, 2H), 3.98 (s, 3H), 3.97 (s, 3H); LC-MS: RT (min) = 7.63; [M + H]+ 369.0; HRMS calcd for C18H17N4O3S (M + H) 369.0943, found 369.1018. 3-(2-methoxyphenyl)-6-(2, 5-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (10A):1H NMR (CDCl3, 400 MHz) 7.60 (d, 7.6 Hz, 1H), 7.49 (app. t, 8.8 Hz, 1H), 7.07-6.98 (m, 4H), 6.91 (app. d, 10.4 Hz, 1H), 4.00 (s, 2H), 3.85 (s, 3H), 3.77 (s, 3H), 3.72 (s, 3H); LC-MS: RT (min) = 6.79; [M + H]+383.1; HRMS calcd for C19H19N4O3S (M + H) 383.1100 , found 383.1176. 3-(3-methoxyphenyl)-6-(3-methoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (7B):1H NMR (CDCl3, 400 MHz) 7.85 (d, 8.8 Hz, 2H), 7.67 (d, 7.2 Hz, 2H), 7.36 (t, 8.0 Hz, 1H), 7.01-6.96 (m, 3H), 3.94 (s, 2H), 3.85 (s, 3H), 3.83 (s, 3H); LC-MS: RT (min) = 7.21; [M + H]+ 353.1; HRMS calcd for C18H17N4O2S (M + H) 353.0994, found 353.1068. 3-(4-methoxyphenyl)-6-(4-methoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (8C):1H NMR (CDCl3, 400 MHz) 8.01 (d, 8.8 Hz, 2H), 7.84 (d, 8.4 Hz, 2H), 6.98 (d, 8.4 Hz, 4H), 3.97 (s, 2H), 3.86 (s, 3H), 3.84 (s, 3H); LC-MS: RT (min) = 7.02; [M + H]+ 353.1; HRMS calcd for C18H17N4O2S (M + H) 353.0994, found 353.1075. 3-(2, 3-dimethoxyphenyl)-6-(2, 5-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (10D): 1H NMR (CDCl3, 400 MHz) 7.20 (dd, 1.6, 7.6 Hz, 1H), 7.14 (t, 8.0 Hz, 1H), 7.10 (d, 3.2 Hz, 1H), 7.06 (dd, 1,6, 8.0 Hz, 1H), 6.99 (dd, 3.2, 9.2 Hz, 1H), 6.88 (d, 9.2 Hz, 1H), 3.98 (s, 2.0), 3.87 (s, 3H), 3.85 (s, 3H), 3.74 (s, 3H), 3.79 (s, 3H); LC-MS: RT (min) = 6.93; [M + H]+ 413.1; HRMS calcd for C20H21N4O4S (M + H) 413.1205, found 413.1288. 3-(2, 4-dimethoxyphenyl)-6-(2, 5-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (10E): 1H NMR (CDCl3, 400 MHz) 7.52 (d, 8.4 Hz, 1H), 7.04 (d, 3.2 Hz, 1H), 6.98 (dd, 3.2, 9.2 Hz, 1H), 6.90 (d, 8.8 Hz, 1H), 6.57 (2.4, 8.4 Hz, 1H), 6.51 (d, 2.4 Hz, 1H), 3.93 (s, 2H), 3.85 (s, 3H), 3.83 (s, 3H), 3.75 (s, 6H); LC-MS: RT (min) = 6.70; [M + H]+ 413.0; HRMS calcd for C20H21N4O4S (M + H) 413.1205, found 413.1285. 3-(2, 5-dimethoxyphenyl)-6-(4-methoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (8F):1H NMR (CDCl3, 400 MHz) 7.74 (d, 8.8 Hz, 2H), 7.16 (d, 2.8 Hz, 1H), 7.01 (dd, 3.2, 9.2 Hz, 1H), 6.90 (dd, 3.6, 9.2 Hz, 3H), 3.94 (s, 2H), 3.81 (s, 3H), 3.76 (s, 3H), 3.63 (s, 3H); LC-MS: RT (min) = 6.75; [M + H]+ 383.1; HRMS calcd for C19H19N4O3S (M + H) 383.1100, found 383.1174. 3-(3, 4-dimethoxyphenyl)-6-(2, 4-dimethoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (9G):1H NMR (CDCl3, 400 MHz) 7.75 (d, 1.6 Hz, 1H), 7.69 (dd, 1.6, 8.4 Hz, 1H), 7.61 (d, 8.4 Hz, 1H), 6.88 (d, 8.8 Hz, 1H), 6.54 (ddd, 2.0, 8.4, 18 Hz), 3.94 (s, 2H), 3.88 (s, 9H), 3.84 (s, 3H)=; LC-MS: RT (min) =6.78; [M + H]+ 413.1; HRMS calcd for C20H21N4O4S (M + H) 413.1205, found 413.1208. 3-(3, 5-dimethoxyphenyl)-6-(4-methoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (8H): 1H NMR (CDCl3, 400 MHz) 7.87 (d, 8.8 Hz, 2H), 7.32 (d, 2.4 Hz, 2H), 6.98 (d, 8.8 Hz, 2H), 6.56 (app. t, 2.4 Hz, 1H), 3.94 (s, 2H), 3.86 (s, 3H), 3.81 (s, 6H); LC-MS: RT (min) = 7.36; [M + H]+ 383.1; HRMS calcd for C19H19N4O3S (M + H) 383.1100 , found 383.1181. 3-(2-methylphenyl)-6-(4-methoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (8I): 1H NMR (CDCl3, 400 MHz) 7.95 (s, 1H), 7.88-7.84 (m, 3H), 7.37 (t, 7.6 Hz, 1H), 7.27 (app. t, 7.6 Hz, 1H), 7.01-6.97 (m, 2H), 3.95 (s, 2H), 3.87 (s, 3H), 2.41 (s, 3H); LC-MS: RT (min) = 7.56; [M + H]+ 337.1; HRMS calcd for C18H17N4OS (M + H) 337.1045 , found 337.1119. 3-(2-ethoxyphenyl)-6-(4-methoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (8J): 1H NMR(CDCl3, 400 MHz) 7.77 (d, 8.8 Hz, 2H), 7.65 (dd, 1.2, 7.6 Hz, 1H), 7.50 (app. t, 7.2 Hz, 1H), 7.09 (t, 7.6 Hz, 1H), 6.96 (dd, 3.6, 8.8 Hz, 3H), 3.96 (q, 6.8 Hz, 2H), 3.95, (s, 2H), 3.85 (s, 3H), 1.12 (t, 6.8, 3H); LC-MS: RT (min) = 7.06; [M + H]+ 367.1; HRMS calcd for C19H19N4O2S (M + H) 367.4368, found 367.1226. 3-(2-hydroxyphenyl)-6-(4-methoxyphenyl)-7H-[1, 2, 4]-triazolo-[3, 4b]-[1, 3, 4]-thiadiazine (8K): 1H NMR (CDCl3, 400 MHz) 8.33 (d, 8.0 Hz, 1H), 7.91 (d, 8.8 Hz, 2H), 7.35 (t, 7.2 Hz, 1H), 7.12 (d, 8.0 Hz, 1H), 7.05 (d, 8.8 Hz, 2H), 6.95 (t, 7.6 Hz, 1H), 3.97 (s, 2H), 3.90 (s, 3H); LC-MS: RT (min) = 8.12; [M + H]+ 339.0; HRMS calcd for C17H15N4O2S (M + H) 339.0837, found 339.0918.
- 24.PDE assay: Inhibition of PDE4A was performed using IMAP technology (Molecular Devices, CA). Briefly, two μl/well of PDE4A1A (BPS Bioscience, CA) mixture (0.05ng/μl PDE4A1A, 10 mM Tris pH 7.2, 0.1% BSA, 10 mM MgCl2, 1 mM DTT, and 0.05% NaN3, final concentration) was dispensed into 1536-well black/solid bottom assay plates (Greiner Bio-One North America, NC) using a Flying Reagent Dispenser (Aurora Discovery, CA). The plates were centrifuged at 1000 rpm for 30s and then 23 nL compound was transferred to the assay plate using a Kalypsys pin tool. After incubation at room temperature for 5 min, 2 μL/well of cAMP (100 nM, final concentration) was dispensed for a final assay volume of 4 μL /well. The plates were centrifuged at 1000 rpm for 30s, incubated for 40 min at room temperature, and then 4 μL of IMAP binding reagent were added to the assay plate. After 1 to 4 hr incubation at room temperature, the fluorescence polarization (FP) signal (Ex= 485 nm, Em= 530 nm) was measured on Viewlux plate reader (Perkin Elmer, MA).
- 25.Lee ME, Markowitz J, Lee JO, Lee H. FEBS Lett. 2002;530:53. doi: 10.1016/s0014-5793(02)03396-3. [DOI] [PubMed] [Google Scholar]
- 26.Xu RX, Hassell AM, Vanderwall D, Lambert MH, Holmes WD, Luther MA, Rocque WJ, Milburn MV, Zhao Y, Ke H, Nolte RT. Science. 2000;288:1822. doi: 10.1126/science.288.5472.1822. [DOI] [PubMed] [Google Scholar]
- 27.BPS Bioscience Inc. 11526 Sorrento Valley Rd. Ste. A2; San Diego, CA 92121. www.bpsbioscience.com.
- 28.Zhang HT, Huang Y, Masood A, Stolinski LR, Li Y, Zhang L, Dlaboga D, Jin SL, Conti M, O'Donnell JM. Neuropsychopharmacology. 2007 doi: 10.1038/sj.npp.1301537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murdoch H, Mackie S, Collins DM, Hill EV, Bolger GB, Klussmann E, Porteous DJ, Millar JK, Houslay MD. J Neurosci. 2007;27:9513. doi: 10.1523/JNEUROSCI.1493-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manning CD, Burman M, Christensen SB, Cieslinski LB, Essayan DM, Grous M, Torphy TJ, Barnette MS. Br J Pharmacol. 1999;128:1393. doi: 10.1038/sj.bjp.0702911. [DOI] [PMC free article] [PubMed] [Google Scholar]