

Abstract
Plant growth and development are tightly linked to primary metabolism and are subject to natural variation. In order to obtain an insight into the genetic factors controlling biomass and primary metabolism and to determine their relationships, two Arabidopsis thaliana populations [429 recombinant inbred lines (RIL) and 97 introgression lines (IL), derived from accessions Col-0 and C24] were analyzed with respect to biomass and metabolic composition using a mass spectrometry-based metabolic profiling approach. Six and 157 quantitative trait loci (QTL) were identified for biomass and metabolic content, respectively. Two biomass QTL coincide with significantly more metabolic QTL (mQTL) than statistically expected, supporting the notion that the metabolic profile and biomass accumulation of a plant are linked. On the same basis, three out the six biomass QTL can be simulated purely on the basis of metabolic composition. QTL based on analysis of the introgression lines were in substantial agreement with the RIL-based results: five of six biomass QTL and 55% of the mQTL found in the RIL population were also found in the IL population at a significance level of P ≤ 0.05, with >80% agreement on the allele effects. Some of the differences could be attributed to epistatic interactions. Depending on the search conditions, metabolic pathway-derived candidate genes were found for 24–67% of all tested mQTL in the database AraCyc 3.5. This dataset thus provides a comprehensive basis for the detection of functionally relevant variation in known genes with metabolic function and for identification of genes with hitherto unknown roles in the control of metabolism.
Keywords: metabolic quantitative trait loci (mQTL), recombinant inbred line (RIL), introgression line (IL), Arabidopsis, GC-MS, metabolomics
Introduction
The phenotype displayed by an organism is the result of interaction between its genotype and the environment. Natural genetic variation is usually due to effects of multiple genes detectable as quantitative trait loci (QTL), and the expression of complex traits is the result of the contribution and interaction of numerous genes. One particular example of this is the growth of multicellular organisms, which has been shown to be governed by many genes that each contribute a small portion to the overall phenotype, for example in mouse (Rocha et al., 2004), chicken (Jacobsson et al., 2005), Arabidopsis (El-Lithy et al., 2004) or rice (Li et al., 2006).
In plants, numerous transgenic single-gene-driven attempts have been described with the goal of modifying growth and/or biomass. Many of these have targeted the production and/or distribution of primary metabolites within various parts of the plant such as the source and sink organs, i.e. the growing areas and storage organs, respectively (Sonnewald et al., 1994). However, it is fair to say that the success rate has been rather limited. On the other hand, numerous transgenic approaches have been utilized in an attempt to improve the metabolic composition of plants to meet requirements with respect to human food and animal feed. In such cases, the success rate has varied with the pathway targeted. Transgenic approaches have shown an impressively high success rate when applied to secondary metabolites such as carotenoids or flavonoids, or when applied to polymer quality (Lorberth et al., 1998; Mann et al., 2000; Muir et al., 2001). As a rule, the intended biochemical changes were achieved and were not accompanied by any major pleiotropic effects concerning growth and development. In contrast, when attempting to modify primary metabolism, such as sucrose biosynthesis or the tricarboxylic acid (TCA) cycle, major and mostly negative effects at the whole-plant level, specifically impaired growth and development, were observed in many cases (Trethewey et al., 1998).
Variation of growth and metabolic traits has been detected for a series of natural accessions and recombinant inbred lines (Cross et al., 2006; Meyer et al., 2007). Correlation analyses showed weak relationships between growth and the levels of individual metabolites, but a close and highly significant link between biomass and a specific combination of metabolites has been shown (Meyer et al., 2007). The observation of positive correlations of rosette weight with several enzyme activities indicated the importance of the catalytic activity of enzymes in central carbon and nitrogen metabolism and their effects on metabolic fluxes (Cross et al., 2006).
Taken together, these data indicate that primary metabolism, in contrast to secondary metabolism, is a network that is closely linked to plant growth and development, and that major perturbation of this network has strong detrimental effects on plant performance.
In order to obtain further insight into the genetic factors that control growth and metabolic traits and to elucidate their relationships, we performed a parallel QTL analysis for biomass and metabolic composition. To this end, metabolic profiling using GC-TOF mass spectrometry was applied to recombinant inbred line (RIL) and introgression line (IL) populations of Arabidopsis thaliana according to the concept of genetical genomics (Jansen and Nap, 2001). All plants were derived from a cross between the Arabidopsis thaliana accessions Col-0 and C24 (Törjék et al., 2006). Using the data obtained with regard to growth and metabolite composition, we addressed the following questions:
Is the heritable variation in these populations and its genetic basis sufficient to allow identification of biomass QTL and metabolic QTL?
Are metabolic QTL randomly distributed over the genome?
Can links between metabolism and growth be established on the basis of a statistically significant co-localization of shoot biomass and metabolic QTL?
What fraction of metabolic QTL regions contain candidate genes with related known or proposed metabolic function, and how many of these candidate genes show sequence variation leading to changes in the encoded protein?
As the Arabidopsis genome is fully sequenced (Arabidopsis Genome Initiative, 2000), well annotated (Haas et al., 2005) and was recently very thoroughly analyzed for its genetic diversity across 20 accessions (Clark et al., 2007), we were able to investigate 39 of the 85 metabolites of known chemical nature. An analysis was also performed to answer the question of whether analyses of RILs and ILs lead to similar or different results, and thus to what extent the two approaches can be considered complementary or redundant.
The results we present here demonstrate that, at least for a subset of the biomass QTL, there is substantial and significant overlap with metabolic QTL, suggesting a strong link between biomass and primary metabolism. In addition, QTL have been identified for multiple metabolites, and a candidate gene was identified for up to 67% of them. Five biomass QTL were identified in both the RIL and IL populations, and 55% of the mQTL identified in the RIL population were confirmed in the IL population.
Results
Analysis of the RIL population for biomass and metabolic QTL
Description of the RIL population and QTL mapping
The analyzed RIL population (Törjék et al., 2006) consisted of 429 lines from the reciprocal crosses Col-0 × C24 (n = 228) and C24 × Col-0 (n = 201) grown under controlled conditions in six consecutive experiments, in which each line was replicated at least three times. Plants harvested 15 days after sowing were used for shoot biomass determination, or were pooled and frozen, and subsequently subjected to metabolite profiling by GC–MS. We did not find significant differences in marker distribution between the two sub-populations (association between marker matrices estimated by Mantel test, P < 0.001). As we could not detect a significant difference in biomass between the two sub-populations either (Kolmogorov–Smirnov test, P = 0.180), we treated the RILs as one population in subsequent analyses.
The shoot biomass and metabolite data were used to map QTL based on a linkage map of 105 markers established for the Col-0/C24 RIL population (Törjék et al., 2006) by application of the software packages PLABQTL (Utz and Melchinger, 1996) and QTL Cartographer (Basten et al., 1994).
To identify the fraction of variation that is genetically determined and can potentially be mapped into mQTL, we estimated broad- and narrow-sense heritability for all metabolic traits as described in Experimental procedures. Broad-sense heritability was determined as H2 = 0.40, on average. Narrow-sense heritability was h2 = 0.08, with h2 = 0.16 for metabolites showing at least one QTL and h2 = 0.02 for the remaining metabolites. For biomass, h2 was determined to be 0.70.
Six biomass QTL explain 18% of the phenotypic variation
A complete list and description of QTL detected for shoot biomass is given in Table S1. The explained phenotypic (denoted by R2) and genotypic variation were determined from the final simultaneous fit of all putative QTL using PLABQTL. For biomass, six QTL explain 18.5 ± 3.4% of the phenotypic and 26.8 ± 4.9% of the genotypic variation. Individual QTL contributions range from 1.5 to 6.0% of the total variance. The mean R2 after cross-validation was 16.01% in the calibration and 8.92% in the validation, for a mean of six QTL.
Identification of mQTL for 84 metabolites
Samples taken from 369 of the 429 RILs were analyzed for their metabolic composition. A total of 181 compounds could be detected in more than 85% of all samples, and only those metabolites were taken into further consideration. The chemical nature is known for 85 of these compounds.
In total, we found 157 metabolic QTL for 84 metabolites, 50 of which are of known chemical structure. For 42 metabolites, only one QTL was identified, but a maximum of six QTL was found for tyrosine. The QTL are distributed unequally over marker positions, indicating ‘hot spots’ and empty regions (no metabolic QTL at 10 marker positions). The contribution of individual QTL to the phenotypic variation varied between 1.7 (unknown_092) and 52.1% (cellobiose).
A comparative overview of QTL for known metabolites and biomass is presented in Figure 1.
Figure 1.

Distribution of metabolic and biomass QTL. Significant metabolic QTL of metabolites known by structure are shown as black boxes at marker positions if covered by the support interval. For simplicity, the QTL of metabolites of unknown structure are omitted here. Information on all detected QTL is given in Supplementary Table S1. Metabolites are color-coded according to their chemical group as shown on the right. Vertical lines indicate marker positions, several of which are labeled with approximate distance in cM (top). Asterisks indicate QTL ‘hot spots’ (as determined using 1000 permutations at a 0.05 level).
Shared mQTL enriched for metabolites showing strong pairwise correlation
As a large fraction of the observed variation is due to genetic effects, concentrations of metabolites with shared QTL are expected to correlate, and with increasing numbers of co-located QTL the correlation may increase. Metabolite correlations may be caused by common genetic factors, e.g. regulatory or pathway genes. On the other hand, even if co-located QTL exist, the corresponding metabolites may be weakly correlated if their QTL show strong interaction with other loci that are different for the two metabolites in question. Alternatively, the metabolites may be subject to differential metabolic control, or may be differently affected by environmental influence. To test this, we plotted the number of QTL shared between two metabolites against the value for the Pearson correlation determined between the concentrations of the two metabolites measured in all RILs (Figure 2). The chance of sharing at least one QTL increases with stronger correlations, and overall the correlation increases with the number of shared QTL. However, examples of two deviant scenarios were found: (i) metabolite concentrations are highly correlated but are controlled by QTL at different positions [e.g. glucose and fructose (r = 0.849) show all together five individual QTL but none are shared], and (ii) metabolite concentrations are weakly correlated but they share common QTL (e.g. ethanolamine and fructose (r = 0.058) show three and four individual QTL, respectively, with two QTL in common).
Figure 2.

Dependency of shared QTL on data correlation. The number of overlapping QTL between two metabolites is plotted against the Pearson correlation value for the data vectors used for QTL calculation. Higher numbers of shared QTL are predominantly found for more strongly correlated traits. No normalization was applied with respect to the total number of determined QTL per trait.
Candidate genes involved in the biochemical pathways of the respective metabolite are identified for 24–67% of the metabolic QTL
Initial analyses of detected metabolic QTL with respect to underlying biochemical pathways show that it is possible to identify candidate genes even at the rather low mapping resolution that can be achieved using an RIL population. For example, inspection of the available information on pathways involving myo-inositol suggested candidate genes for three of four identified QTL (Table S1 and Figure 3). The AraCyc section of the TAIR database lists only 12 loci representing enzymes that catalyze reactions on myo-inositol. Three of these loci co-locate with determined mQTL: a myo-inositol oxygenase (AT1G14520, inositol oxidation pathway), a phosphatidyltransferase (AT4G38570, phospholipid biosynthesis pathway) and a stachyose synthase (AT4G01970, stachyose biosynthesis pathway). If all genes from pathways involving myo-inositol are considered, it is possible to find candidates for the remaining mQTL of this metabolite.
Figure 3.

myo-inositol QTL analysis reveals direct candidate genes for three of four determined QTL (1/18, 4/0 and 4/65). A LOD curve calculated using two independent programs (PLABQTL, red lines; QTL Cartographer, blue lines) is shown at the top. Horizontal lines indicate 0.05 (solid) and 0.25 (dotted) significance thresholds calculated based on 5000 permutations. Vertical lines indicate marker positions. At the bottom, the three relevant reaction steps according to the mQTL as connected by arrows are presented (pathways from left to right are inositol oxidation, stachyose biosynthesis and phospholipids biosynthesis). The pictograms in the center indicate the total number and location of genes known per pathway. Twelve genes (from six pathways) for enzymes catalyzing reactions in which myo-inositol is involved directly are known. The insert shows a comprehensive view of all AGI codes associated with myo-inositol (red, direct; black, pathway), indicating mQTL support intervals (blue), approximate LOD (number) and IL confirmation threshold reached (asterisk). A similar plot for all known metabolites is shown in Supplementary Figure S1.
We extended this analysis to all metabolites of known chemical structure, considering either (i) only the genes encoding enzymes that participate in a direct reaction with the respective metabolite, or (ii) all proteins assigned to pathways involving the metabolite. We were able to identify at least one candidate gene for 24% (direct reaction) and 67% (pathway enzymes) of all tested mQTL (Table S1).
In order to assess how much this coincidence of mQTL and enzyme genes relating to the respective metabolites deviates from the situation expected based on a random distribution of mQTL, we performed a permutation test in which we distributed for each of the 38 metabolites with assigned AGI codes all determined mQTL randomly over all chromosomes. We analyzed the overlap with potential candidate genes from AraCyc as described above, and compared the outcome with the results based on the measurement data. For 13 metabolites, we found more candidate genes than found on average in permutations, while three showed fewer genes. In most of the remaining cases, no candidate gene was assigned either in our experiment or in permutations. The number of identified candidate genes for myo-inositol, maltose and ethanolamine exceed the 95th perentile of the respective permutation results. However, although this analysis suggests that experimentally determined mQTL are enriched for corresponding pathway genes, the test statistic is not significant if multiple testing is considered.
For 10 of 20 mQTL, at least one of the direct candidate genes contains a polymorphism in the protein coding region leading to an amino acid exchange, according to recently published data on single nucleotide polymorphisms (SNPs) between C24 and Col-0 (Table s2; Clark et al., 2007).
Co-localization of biomass QTL with mQTL
One of the aims of this project was to determine the relationship between biomass QTL and mQTL, i.e. to what extent they co-localize. Inspection of the overlap between mQTL and biomass QTL in the RIL population shows that each biomass QTL coincides with several mQTL (with the number of mQTL per biomass QTL ranging from 5 to 12). However, due to the limited resolution of the QTL mapping, a considerable number of overlaps are expected to occur by chance. We therefore used a permutation test to identify statistically significant overlaps. This analysis showed that two of the six biomass QTL (1/88 and 4/0) co-locate with significantly more mQTL than expected by chance. Some metabolites (raffinose, tyrosine, serine, succinic acid) display up to two QTL co-localized with any of the biomass QTL. However, no enrichment for single compound classes or certain biochemical pathways was found amongst these metabolites.
Epistasis
Due to the nature of metabolic networks consisting of multiple interconnected metabolic pathways, prevalent epistatic interactions are expected to occur among mQTL, influencing various steps within single or among multiple pathways. Therefore, we conducted a full scan for all possible digenic maker interactions using the PLABBIC version of PLABQTL, but no significant digenic marker epistasis for the biomass and metabolite traits was detected. To reduce the multiple-testing problem inherent in this approach, we adjusted the procedure to test only previously determined mQTL of known metabolites against markers located elsewhere in the genome. From the resulting likelihood profile, we kept the maxima and evaluated per se mQTL and epistatic effect maxima in a final model. In this last step, non-significant effects were dropped according to a Bayesian information criterion. Following this procedure, one significant epistatic effect was detected for biomass (1/88 × 3/82, R2 = 2.17%) and a further 38 such interactions were identified for 27 of the 50 known metabolites taken into consideration. However, these effects are rather small if compared to per se mQTL, explaining only 2.72% of the phenotypic variation on average. The strongest interaction (R2 = 4.92%) was determined between two tyrosine QTL (4/42 and 5/74). Other substantial effects were identified for glycerate (2/61, R2 = 4.43%) and maltose (4/38, R2 = 3.69%), which exhibited additive interactions with genomic positions to which no mQTL had been previously assigned (Table S3).
Analysis of the IL populations and comparison with the RIL-based data
Five common biomass QTL detected in IL and RIL populations
A QTL analysis was also carried out using 97 lines of two corresponding reciprocal IL populations of the crosses Col-0 × C24 and C24 × Col-0 (Törjék et al., unpublished data) in order to verify the QTL detected in the RIL population. Biomass data were analyzed using the appropriate contrasts in anova. Twenty-six IL were significantly different (P < 0.05) from the recurrent parental line and thus identified biomass QTL. The six biomass QTL regions previously identified by the RIL analysis were covered by another set of 26 ILs. The intersection of the two sets consists of 13 ILs. To compare this result to a random intersection, we calculated the probability of identifying 13 or more of 26 fixed ILs when 26 are drawn randomly from 97. This probability is given by the hypergeometric distribution. Its value of < 0.003 demonstrates the significance of the finding. By means of the IL analysis, five (of six) biomass QTL detected by the RIL analysis were verified at positions 1/88, 3/13, 3/59, 4/47 and 5/86 (Table S1). In addition, the IL analysis revealed a further four regions with an effect on biomass at positions 1/10, 2/72, 3/46 and 5/62-67. Detailed analyses of the individual ILs indicated complex situations, e.g. on chromosome 1 with potentially two QTL of opposing effects located very closely to each other.
RIL-based metabolic QTL are also detected in the introgression lines
Detection of changes in metabolite concentrations due to introgression of a donor genotype in the background was used to confirm mQTL determined in the RIL population. For 94 of the ILs, six replicate GC–MS measurements were carried out, and compared against the metabolite values determined in up to 30 measurements for the respective parental lines. Due to the different population size, P-values were estimated as described in Experimental procedures.
At a level of P≤ 0.05 (not multiple-testing-corrected), 55% of the RIL mQTL are confirmed within the IL population. In 82% of the cases, the positive-effect allele was also confirmed, i.e. if the C24 genotype in the RIL population showed an increased metabolite level compared to Col-0, the same was true for the respective IL. This high level of allele confirmation is independent of the P-value applied (see Table 1).
Table 1.
Estimated P-values for IL–parent comparisons
| Significance level | Number of significant changes | FDR (%) | Number of confirmed RIL QTL | Confirmed RIL QTL (%) | Average R2 of confirmed RIL QTL (%) | Average R2 of non confirmed RIL QTL (%) | Confirmed allelic effect | Confirmed allelic effect (%) |
|---|---|---|---|---|---|---|---|---|
| 0.001 | 177 | 9.61 | 17 | 11.33 | 11.62 | 6.67 | 16 | 94 |
| 0.01 | 773 | 22.01 | 41 | 27.33 | 10.17 | 6.12 | 38 | 93 |
| 0.05 | 2511 | 33.88 | 83 | 55.33 | 7.79 | 6.54 | 68 | 82 |
| 0.1 | 3941 | 43.17 | 99 | 66.00 | 7.45 | 6.79 | 80 | 81 |
Significant results and RIL QTL confirmation at various threshold levels. The false dicovery rate (FDR) is defined as the expectation of the ratio of false positives to the sum of false and true positives. We estimated the FDR by (significance level × number of observations)/(number of significant changes). R2, phenotypic explained variance.
RIL-based mQTL explaining a large part of the phenotypic variance were confirmed preferentially in the IL population. This becomes even more evident when the significance threshold is lowered. At P-values ≤ 0.001, differences between ILs and parents were detected for 177 metabolite/IL combinations (equivalent to 177 mQTL). Of these, 17 had been observed previously in the RILs (11% confirmation). The mean contribution to phenotypic variance of confirmed QTL is 11.6%, compared with 6.4% for non-confirmed QTL.
Prediction of biomass QTL via a combination of mQTL
We are interested in determining the link between biomass QTL and mQTL/metabolic composition. We have previously shown that, for this RIL population, a canonical combination of metabolites can be used to predict the biomass (Meyer et al., 2007). We therefore performed a meta-QTL search using the predicted dry biomass vector (canonical variate) as a new trait to determine whether or not any of the biomass QTL can be predicted based on the metabolic composition (Figure 4). Of the six biomass QTL, three (1/88, 3/13 and 5/86) could be predicted by the metabolic composition, three were not predicted, and two new QTL appeared in the predicted pattern (1/10 and 3/82). One of these new peaks (1/10) corresponds to a QTL that was also identified in ILs.
Figure 4.

Meta-QTL analysis. Meta-QTL analysis using the measured biomass (blue line) and the canonical variate (predicted biomass, red line) calculated from the metabolic profiles as described by Meyer et al. (2007). Horizontal lines indicate 0.05 (solid) and 0.25 (dotted) significance thresholds calculated based on 5000 permutations. Chromosomal length is given in cM.
Discussion
Several successful studies have been conducted to date to identify novel genes based on QTL analysis (Kliebenstein et al., 2001; Kroymann et al., 2003; Werner et al., 2005; Zhang et al., 2006). However, with a few exceptions (Keurentjes et al., 2006; Schauer et al., 2006; West et al., 2007), only a limited number of traits (usually less than 20) have been assessed.
The parallel analysis of IL and RIL populations of Arabidopsis thaliana for biomass and 181 individual metabolites (for which the chemical structure is known for 85) described here is a unique data set. Together with the available information from the fully sequenced and thoroughly annotated Arabidopsis genome, it provides a direct method for detection of functionally relevant variation in known genes with metabolic function and the identification of genes hitherto not assigned to metabolic functions, and emphasizes the link between metabolism and growth/biomass accumulation.
Comparison of ILs versus RILs
Introgression lines are widely used to test for changes in various traits when compared to a parental line. Lines of an introgression population have a common genetic background and various short donor segments from another line, thus allowing focus on a specific region of the genome (Eshed and Zamir, 1995). Several studies have demonstrated the possibility of fine-mapping single RIL QTL in Arabidopsis using ILs (Alonso-Blanco et al., 1998; Bentsink et al., 2003; Swarup et al., 1999). Recently Keurentjes et al. (2007) described an exhaustive analysis on the overlap between QTL based on RIL and IL populations derived from the Arabidopsis thaliana accessions Cvi and Ler. Comparing QTL detected for six developmental traits analyzed in a RIL population of 167 lines and an IL population of 92 lines, 58% of 33 RIL QTL were confirmed. Although Keurentjes et al. (2007) tested up to 116 replicas per IL in a BIN approach, allowing a stricter significance level, this figure is largely in agreement with our findings. Applying a P-value threshold of 0.05, 83 metabolic QTL detected in the RIL population (equivalent to 55%) were confirmed in the IL population. In 82% of the cases, the direction of the effect was the same in RILs and ILs, independent of the applied threshold.
However, not all QTL identified in the RIL population were confirmed using the ILs, and additional QTL were detected using the latter. The differences between RIL- and IL-based QTL (QTLRIL and QTLIL, respectively) can be explained to some extent by epistatic effects. Although no significant digenic marker epistasis was found when all possible marker interactions were considered, probably due to the high number of hypotheses to test, we did identify 38 epistatic interactions using the more targeted approach of testing only the previously detected QTL against the genetic background. With respect to epistasis and effect confirmation, four possible scenarios can be distinguished.
An mQTLRIL is not confirmed by an mQTLIL and shows significant epistatic interactions. This is the case for many of the stronger epistatic effects (e.g. tyrosine 4/42 × 5/74, R2 = 4.92%; glycerate 2/61 × 3/67, R2 = 4.43%), and is consistent with the assumption that loss of the epistatic interaction prevented its identification in ILs.
An mQTLRIL is confirmed by an mQTLIL and lacks epistasis (as observed for threonic acid 4/63 and urea 1/76). This suggests that such mQTL act as single-effect loci independently of other genetic factors. The existence of examples for these two cases supports the hypothesis that epistasis is a major cause of differences between sets of QTLRIL and QTLIL.
mQTLRIL that are confirmed by mQTLIL but show significant epistasis. This was true for three of four raffinose QTL. However, here the variance explained by the epistatic effects is low (R2 = 1.5–2.6%) especially with respect to the variance explained by the QTL per se (R2 = 4.7–6.4%). Furthermore, epistatic interactions between a C24 and Col-0 allele will be retained in ILs and hence contribute to effects detectable in both analyses.
An mQTLRIL is not confirmed by an mQTLIL but no epistasis can be detected, as exemplified by the five serine mQTL. Here, a more complex situation such as multi-way interactions between several loci, which escape detection in the epistasis analysis, can be assumed.
Very complex epistatic interactions may strongly interfere with QTL detection in RIL populations. Depending on the specific allele combination of the interacting loci necessary to elicit a strong effect, the uniformity of the genetic background in ILs may be an advantage, allowing identification of QTL that are not detectable in a RIL population. In addition, opposing-effect QTL present in close vicinity to each other may also interfere with QTL detection in RILs. Such arrangements have been shown to exist by Kroymann and Mitchell-Olds (2005), and may by chance be broken up upon creation of a particular IL through recombination between the QTL linked in repulsion. Events such as this will result in QTL detection in ILs but not in RILs, with the IL approach being favored by the fact that more replication can be afforded in IL analyses (due to a more limited number of individual lines), with a concomitant increase in the precision of the trait expression measurement. Thus, while QTL detected in both analyses may be preferred for follow-up studies, QTL detected in only one of the two populations should not be generally dismissed, and the two approaches may be considered complementary. Taking into account the amount of work that we invested in the generation and evaluation of both populations, they yielded a comparable level of information. However, if it is not the genetic architecture of a trait that is of major interest but its modification in a more applied approach, ILs would be favored for use because advantageous genome segments may be identified in a genetic background close to an elite variety, und thus may be integrated into a breeding program more quickly.
Number and contribution of biomass and mQTL compared with other studies
As described in Results, the variance of both the RIL and IL populations allows identification of QTL for shoot biomass. Six biomass QTL explaining 18% of the total variation, with individual contributions varying from 1.5 to 6%, were identified in the RIL population, and nine biomass QTL were observed in the IL population. Five of the biomass QTL were detected in both populations. These results for biomass are similar to those for other Arabidopsis RIL populations used to detect QTL for aerial/shoot mass, with up to eight QTL detected (El-Lithy et al., 2004; Loudet et al., 2003; Rauh et al., 2002; Ungerer and Rieseberg, 2003). Both RIL QTL on chromosome 3 and two effects in ILs on chromosomes 2 and 4 overlap with results obtained by Loudet et al.(2003) (3/49, 2/72 and 5/62) and El-Lithy et al. (2004) (3/13).
In a comparable study of biomass at an early developmental stage in Aegilops tauschii (ter Steege et al., 2005), only two putative QTL were detected. In seedling-stage maize, three QTL for shoot dry weight, each explaining 11–15% of phenotypic variance, were detected in a F2:F3 population of 226 families (Jompuk et al., 2005). Further biomass QTL analyses, e.g. of poplar (Wullschleger et al., 2005), rice (Hittalmani et al., 2002; Li et al., 2006) and Miscanthus sinensis (Atienza et al., 2003), each revealed a limited number of QTL, usually with a restricted fraction of the phenotypic variance explained. Even in a very large QTL mapping experiment in maize (Schön et al., 2004), in which more than 30 growth-related QTL were identified, only about 50% of the genetic variance was explained. The effects of individual QTL on the phenotypic variance were generally small. Thus, the individual contribution of shoot biomass QTL in the Arabidopsis RIL/IL populations analyzed here is very similar to the situation described for other species, including crops such as maize. The observed heritability (0.71) of the biomass trait in the analyzed RIL population and the rather limited fraction of the genetic variation explained jointly or individually by the detected QTL indicate that biomass accumulation is probably affected by a very large number of small-effect QTL. This is consistent with the conclusions of Kroymann and Mitchell-Olds (2005).
In the RIL population, a total of 157 metabolic QTL were identified, with at least one metabolic QTL for approximately half of the compounds analyzed (181 compounds were analyzed, and at least one QTL was identified for 84 compounds). The contribution of individual QTL to the total phenotypic variance ranged from 1.7% to more than 50%. Analysis of the IL population resulted in numerous QTL, the specific numbers ranging from 177 for P≤ 0.001 to 2511 for P≤ 0.05. These numbers are in the same range as described in two previous reports on identification of metabolic QTL using RIL and IL populations. Schauer et al. (2006) identified 889 mQTL in a tomato population of 76 ILs, monitoring 74 metabolites at a significance level of 0.05. Using 2129 mass signals (with an unknown number of underlying chemical compounds), Keurentjes et al. (2006) identified 4213 metabolic QTL at a P-value threshold of 0.0001 in an Arabidopsis Cvi × Ler RIL population. In a recent QTL study using a Bay-0 × Sha Arabidopsis RIL population, Calenge et al. (2006) detected a total of 39 QTL for starch, glucose, fructose and sucrose contents at 14 distinct loci, which co-localize with QTL for other physiological traits.
The findings that two biomass QTL co-locate with significantly more mQTL than expected from a random distribution, and, furthermore, that some of the biomass QTL can be simulated by QTL mapping of a certain linear combination of metabolite levels, fit into the emerging picture that metabolic composition is related to growth/biomass accumulation, as also shown previously (Meyer et al., 2007). While some metabolites such as ethanolamine, raffinose and tyrosine, which contributed strongly to the metabolic signature identified in the previous work, also show co-localized QTL with biomass, others do not. However, this finding is not unexpected considering the small amount of variation that is explained by any individual metabolite or metabolic QTL.
Derivation of metabolites sharing mQTL from either the same or widely divergent pathways
A number of mQTL are shared between metabolites. Two principal classes can be distinguished:
Metabolites sharing a QTL are derived from the same biochemical pathway or from related pathways, as observed for ornithine/proline (position 4/66; common pathway: proline biosynthesis). This identifies the shared QTL as a candidate for a pathway QTL, which could be either a gene controlling the formation of a rate-limiting precursor or a higher-hierarchy controller of the entire pathway such as a transcription factor.
In other cases (e.g. position 3/14), metabolites with common QTL are derived from widely divergent pathways, which could be due to a major controller of several pathways or a small molecule produced in one pathway and controlling the other pathway. However, it should be kept in mind that, at present, the limited genetic resolution does not allow exclusion of the much more trivial possibility of the shared genomic regions actually being composed of several linked genes with enzymatic functions in different pathways.
mQTL cover both biosynthetic and regulatory genes
A comprehensive overview of all mQTL observed in the RIL population for known metabolites, including their effect, confirmation in the IL population and the chromosomal localization of all associated genes is shown in Figure S1.
The observation that a pathway-associated gene could be localized in the mQTL region for 24–67% of all metabolic QTL can be exploited in a number of ways. One exciting possibility is the use of this dataset as a source for identifying novel functionally relevant polymorphisms in the genome by comparative sequencing of both alleles of candidate genes. In agreement with this, comparison with recently published data on SNPs between C24 and Col-0 (Clark et al., 2007) showed that, for 10 out of 20 mQTL, at least one of the direct candidate genes contains a polymorphism in the protein coding region leading to an amino acid exchange. Obviously this is only a first indication, and does not prove that this amino acid exchange is responsible for the mQTL. Furthermore, it should be kept in mind that we did not observe significant enrichment of pathway genes within experimentally determined mQTL (see Results). This is mainly due to the fact that, for 18 of 39 metabolites, 15 or more (up to 130) direct candidate genes are known, which appear to be uniformly distributed by visual inspection (Figure S1). Due to the relatively large confidence intervals, random distribution of mQTL over chromosomes will, in such cases, always lead to successful candidate gene identification hampering a permutation test. If we exclude these 18 metabolites, a P-value of 0.08 is obtained in the permutation test, indicating that mQTL are possibly enriched for pathway-related genes, and that comparative analyses of the alleles would be worthwhile.
A level of up to 67% coverage of mQTL by biosynthetic candidate genes also implies that at least 33% of the mQTL probably harbor genes of hitherto unknown metabolic functions (e.g. as regulators), a rather large and at first unexpectedly high fraction. Although the presence of biosynthetic genes in C24 only (and thus not in AraCyc) could explain some of these mQTL, the above conclusion is supported by a seemingly unrelated observation, i.e. the unequal distribution of mQTL over the genome. The chromosomal distribution of the total 157 mQTL differs in a statistically significant manner from a random distribution, with two significant hot spots (up to 16 QTL at the top of chromosome 4, and 12 QTL at 5/75) and other areas lacking mQTL (no mQTL detected for 38 marker positions). There are two possible explanations for this uneven distribution: either it is a reflection of the uneven distribution of biosynthetic genes in the Arabidopsis genome, or a larger proportion of mQTL detected do not correspond to genes with known metabolic function (mostly enzyme genes) but represent regulatory genes of a higher hierarchical order that thus control more than one metabolite. To distinguish these two possibilities, we compared the distribution of metabolic genes in the genome with the mQTL distribution. The results of this analysis showed that the clustering of mQTL does not correlate significantly with the distribution of metabolic genes over the Arabidopsis genome, irrespective whether all metabolic genes or only metabolic genes from biosynthetic pathways covered in our analysis are taken into account (data not shown). This suggests that the uneven distribution of mQTL is due to the second explanation, i.e. a large proportion of mQTL detected identify hitherto unknown metabolic functions, most likely regulatory genes controlling primary metabolism and thus probably having a strong influence on biomass formation. The available ILs that confirmed such mQTL enable positional cloning of the corresponding novel metabolic function genes.
Experimental procedures
Creation of recombinant inbred (RIL) and introgression line (IL) populations
Two reciprocal sets of RILs were developed from a cross between the two A. thaliana accessions C24 and Col-0. F2 plants were propagated by controlled self-pollination using the single-seed descent method to the F8 generation, at which stage genotyping and bulk amplification was performed. The mapping population consisted of 228 Col-0 × C24 F8 and 201 C24 × Col-0 F8 individual lines. The RIL population was genotyped using a set of 110 framework SNP markers (Törjék et al., 2003) as described previously (Törjék et al., 2006). Marker distributions per chromosome in the two sub-populations were compared using Mantel tests (1000 permutations) of the corresponding similarity matrices obtained by simple matching, using the statistical software package Genstat for Windows version 6.1 (Payne et al., 2002).
As a base population for IL development, two sets of reciprocal BC3 F1 lines were created from the F2 of a reciprocal cross between the two A. thaliana accessions C24 and Col-0, through three cycles of backcrossing followed by one cycle of selfing using the single-seed descent method (Törjék et al., in press). The BC3 F1 lines were genotyped using the same set of 110 framework markers (Törjék et al., 2003). Lines with positive-effect segments were subjected to further cycles of backcrossing and selfing to produce substitutions in both the Col-0 and C24 genomic backgrounds using marker-assisted selection. The average introgression lengths are 17.3 and 19.3 cM in ILs with Col-0 and C24 backgrounds, respectively.
Plant cultivation
The RIL population was cultivated in at least three experiments using a split-plot design. The growth room was declared as the whole plot with two factors (chamber 1 and chamber 2). Each sub-plot contained the entire RIL population and the controls (C24, Col-0, C24 × Col F1, Col × C24 F1). Plants were grown in a 1:1 mixture of GS 90 soil (Gebrüder Patzer, Sinntal-Jossa, Germany) and vermiculite (Deutsche Vermiculite Dämmstoff-GmbH; http://www.vermiculite.de) in 96-well trays. Six plants of the same line were grown per well. Seeds were germinated in a growth chamber at 6°C for 2 days before transfer to a long-day regime [16 h fluorescent light (120 μmol m−2 sec−1) at 20°C and 60% relative humidity/8 h dark at 18°C and 75% relative humidity]. To avoid position effects, trays were rotated around the growth chamber every 2 days.
ILs were selected to cover the QTL regions determined in the RIL experiment (26 ILs for the six biomass QTL, 16 for other traits), and plants were grown in two blocks with 12 sub-plots each. Each subplot contained 42 ILs, 42 test crosses (IL TCs) to the recurrent parent, and the controls twice (C24, Col-0, C24 × Col, Col × C24). The position within the sub-plot was random. In addition, ‘unselected’ ILs without IL TCs were grown in the same experiment. In this case, each sub-plot consisted of 56 ILs and 36 controls. Growing conditions were identical to those used for the RILs.
Shoot dry biomass
Shoot dry biomass was determined 15 days after sowing. Plants from the same well were harvested together and placed in a vacuum oven at 80°C for 48 h. Dry biomass was measured using an analysis balance. Mean shoot dry biomass (mg/plant) was estimated using a linear mixed model (Piepho et al., 2003) as described by Meyer et al. (2007). Biomass in the two RIL sub-populations was compared by a Kolmogorov–Smirnov test using Genstat for Windows version 6.1 (Payne et al., 2002).
Metabolite data
Sample preparation, measurement and data processing
Samples for the analysis of metabolic composition were collected together with the material for dry biomass analysis at 15 days after sowing. Harvested material (shoot and leaf) was frozen at−80°C immediately, and kept at this temperature until further processing. Between two and six plants were pooled per sample. One replicate for each RIL and six replicates for each IL were measured. Extraction, derivatization, GC–MS analysis and data processing were performed as described previously (Lisec et al., 2006). A targeted metabolomics approach was used, based on a reference library containing 181 compounds.
The resulting data consist of intensity values for each referenced compound and measurement, respectively. These raw data were normalized (see below) before QTL analysis.
Normalization
All samples were measured in groups of 30–50, equivalent to one measurement day. The huge number of samples led to measurement periods of several weeks per experiment. It is therefore necessary to correct for variation in detector sensitivity over this time, which otherwise causes artificial differences in absolute intensity depending on the measurement day. The samples for the two experiments (RILs and ILs) were measured using different set-ups (see below), and were therefore normalized using different strategies.
For the RIL experiment, all samples were measured over a measurement period of 26 days. Samples from different genetic backgrounds were distributed in equal proportions per day and otherwise completely randomized. Hence we assumed that the genetic and phenotypic variance covered by all samples of a set (approximately 40) is comparable between days. Therefore, metabolite data were normalized by dividing the intensity of the metabolite i by the median of all measurements of i per measurement day.
IL samples were measured in groups consisting of genotypes related to either of the parents [C24 and M lines (C24 with Col-0 introgressions) or Col-0 and N lines (Col-0 with C24 introgressions)] in an attempt to reduce the technical error for our comparisons of interest (parent versus corresponding ILs). Six replicates per genotype were measured in total on 6 days, always together with the same set of genotypes including four to six replicates of the respective parent. Samples were randomized within days.
Two normalization steps were applied to IL samples. To account for intensity differences, we normalized each metabolite profile (sample) by its mean trimmed 20% (the vector sum between the 10th and 90th percentiles, k = 0.1 n [n=metabolite number]).
This was determined to be more robust than using an internal standard (data not shown). However, it assumes that the same amount of material is applied to the column for each sample, and that the variation in total peak area of the analyzed metabolic subset is low. As only a low correlation (r < 0.05) between trimmed mean and biomass was observed for the IL data, this seems to be a fair assumption. In a second step, we improved normality by dividing each metabolite intensity value by the median of all values for this metabolite i from the same measurement set j and applying the logarithm:
Candidate gene identification
To identify possible candidate genes for mQTL, the AraCyc 3.5 database was downloaded from TAIR (Arabidopsis Information Resource, http://www.arabidopsis.org). For each mQTL, a search window was determined according to the presence of markers within its 1-LOD support interval (Table S1). The resulting AGI codes were tested for either direct association with the metabolite or association with one of the pathways in which the metabolite is involved.
To compare the distribution of metabolic genes over the Arabidopsis genome against the mQTL distribution, we counted all genes around each marker, i.e. the interval
where Mk is the position of marker k (in bp). This approach was followed for the complete AraCyc data set and for a selection containing only information on pathways in which metabolites measured in this study are present. A Pearson correlation value was calculated between the mQTL distribution and both gene distributions separately.
For permutation tests in the candidate gene approach, all QTL of a single metabolite were combined and randomly distributed over the five chromosomes 10 000 times. The total number of overlapping candidate genes was recorded in each permutation, and the final distribution of these values was compared against the outcome for the actual data.
Estimation of heritability
Broad-sense heritability (H2) is defined as the part of phenotypic variation that is explained by the genotype. We used a similar approach to that of Keurentjes et al. (2007), and estimated within-line variance (VP) based on replicate measurements of both parents and the reciprocal F1 hybrids (10 replicates each). To account for various measurement levels, we normalized the calculated variance (s2) of each genotype (Gk) using the squared mean before averaging:
 |
After an equivalent transformation of the RIL values, we calculated broad-sense heritability as:
To prevent over-estimation, we removed outliers more than three standard deviations away from the mean. In the case of negative values, we assumed the heritability to be zero.
Narrow-sense heritability (h2) was estimated by parent–offspring regression according the method described by Falconer and Mackay (1996). Here we made use of available RIL parent test crosses, which were measured together with the RIL samples.
QTL analyses
Recombinant inbred lines
For QTL analyses, a map containing 105 markers was used, on which only one representative (with fewest missing values) of very tightly linked markers was integrated. Two software packages implementing different detection algorithms [PLABQTL, multiple regression (Utz and Melchinger, 1996); QTL Cartographer, maximum-likelihood methods (Basten et al., 1994)] were combined to obtain robust QTL estimates. Composite interval mapping (CIM) was performed on an RIL population of 429 lines (dry biomass) or 369 lines (metabolites) with 1 cM increments. Co-factors were automatically selected by forward stepwise regression. Significant LOD thresholds were determined using 5000 permutations. QTL were regarded as significant if they were detected using LOD0.05 in one package, and reached at least LOD0.25 in the other. QTL location and partial R2 were further validated using 1000 runs of the fivefold cross-validation procedure implemented in PLABQTL. Given a population size of 429 and a significance level of 0.05, it can be shown (Hackett, 2002) that 99% of all QTL that contribute more than 5% to the total variance and more than 50% of those that contribute more than 1% will be detected. Most of the undetected QTL will be below the 1% line. To identify 50% of QTL that have a contribution of 0.5%, we would have to double our population size.
Co-localizations of QTL from different traits are expected given the high number of traits and the limited number of markers. The deviation from the random number of co-localizations was calculated as follows. The QTL of each metabolite were randomly distributed over the 105 marker positions. We then counted the number of co-localizations with each of the dry biomass QTL or with other metabolite QTL. This procedure was repeated 1000 times, yielding a distribution of the maximum numbers of co-localizations. The 95% quantile of the distribution for metabolite–biomass QTL co-localization was eight, hence eight or more QTL at one genome position are regarded as significantly co-localized. The corresponding 95% quantile for the metabolite–metabolite QTL co-localization was ten.
Introgression lines
To identify metabolites with a significantly altered intensity in a certain genotype, we compared metabolite values of ILs (six replicates, IL) against all parental line samples measured within the same set (approximately 30 replicates, P). To estimate a P-value empirically, we compared the true mean difference  in k permutations (k = 10 000) with the calculated difference
in k permutations (k = 10 000) with the calculated difference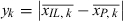 , where
, where  is the mean of a sample (of size six) drawn from the set union of IL and P, and
is the mean of a sample (of size six) drawn from the set union of IL and P, and  is the mean of the remaining values of this set union:
is the mean of the remaining values of this set union:
Hence, if the measured mean difference was higher than the mean differences calculated for 9500 of the 10 000 permutations, we obtain a P-value estimate of 0.05.
Epistasis
The software package PLABQTL (version 1.2BIC was used to estimate epistatic interactions. In an initial screening for digenic epistatic effects by two-way anova between all pairs of marker loci, no significant effects were determined using the integrated scanning function.
In the following analysis, every mQTL of a known metabolite was tested for additive × additive effects against the genetic background at intervals of 2 cM. A range of 10 cM around the actual QTL position was blocked during this analysis. The resulting likelihood profiles for all mQTL of a metabolite were overlaid and inspected visually for maximum LOD estimates. A full model for each metabolite containing all per se QTL and their putative epistatic interactions was set up (one epistatic interaction per mQTL was usually included, none if the maximum effect coincided with another per se QTL, and two if equivalent interactions were present). From this full model, non-significant effects were omitted in a backward elimination step using a Bayesian information criterion (Kusterer et al., 2007) before re-estimating all remaining parameters simultaneously.
Acknowledgments
We are grateful to B. Kusterer, A.E. Melchinger and H.F. Utz (Institute of Plant Breeding, Seed Science and Population Genetics, University of Hohenheim, Stuttgart, Germany) for their support in the use of the PLAPQTL package and making the most recent version available prior to publication. We thank H.-P. Piepho (Institute of Crop Production and Grassland Research and State Plant Breeding Institute, University of Hohenheim, Stuttgart, Germany) for developing a mixed model for mean dry mass estimation, and C.A. Hackett (Scottish Crop Research Institute [SCRI], Invergowie Dundee, UK) for useful discussions on QTL analyses. We thank Änne Eckardt, Anke Kalkbrenner, Cindy Marona, Melanie Teltow, Gudrun Wolter and Monique Zeh for excellent technical assistance, and Katrin Seehaus and Dirk Zerning for plant cultivation. This work was supported by a grant from the Deutsche Forschungsgemeinschaft to T.A. and R.C.M. (AL 387/6-1, 6-2), a grant from the EC to T.A. (QLG2-CT-2001-01097), grants from the Deutsche Forschungsgemeinschaft to O.F. and L.W. (FI 842/2-1,WI 550/3-2), AGRON-OMICS (http://www.agron-omics.eu) and by the Max Planck Society.
Supplementary material
The following supplementary material is available for this article online:
Comprehensive mQTL overview for known metabolites.
QTL analysis results for biomass and metabolic traits.
SNP positions and resulting amino acid changes for mQTL candidate genes according to data published by Clark et al. 2007).
Epistatic interactions (additive × additive) for mQTL of 50 known metabolites.
This material is available as part of the online article from http://www.blackwell-synergy.com
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alonso-Blanco C, El-Assal SE, Coupland G, Koornneef M. Analysis of natural allelic variation at flowering time loci in the Landsberg erecta and Cape Verde Islands ecotypes of Arabidopsis thaliana. Genetics. 1998;149:749–764. doi: 10.1093/genetics/149.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Atienza SG, Satovic Z, Petersen KK, Dolstra O, Martín A. Identification of QTLs influencing agronomic traits in Miscanthus sinensis Anderss. I. Total height, flag-leaf height and stem diameter. Theor. Appl. Genet. 2003;107:123–129. doi: 10.1007/s00122-003-1220-5. [DOI] [PubMed] [Google Scholar]
- Basten CJ, Weir BS, Zeng ZB. Zmap – A QTL Cartographer. Vol. 22. Guelph, Ontario, Canada: 1994. pp. 65–66. [Google Scholar]
- Bentsink L, Yuan K, Koornneef M, Vreugdenhil D. The genetics of phytate and phosphate accumulation in seeds and leaves of Arabidopsis thaliana, using natural variation. Theor. Appl. Genet. 2003;106:1234–1243. doi: 10.1007/s00122-002-1177-9. [DOI] [PubMed] [Google Scholar]
- Calenge F, Saliba-Colombani V, Mahieu S, Loudet O, Daniel-Vedele F, Krapp A. Natural variation for carbohydrate content in Arabidopsis. Interaction with complex traits dissected by quantitative genetics. Plant Physiol. 2006;141:1630–1643. doi: 10.1104/pp.106.082396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RM, Schweikert G, Toomajian C, et al. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science. 2007;317:338–342. doi: 10.1126/science.1138632. [DOI] [PubMed] [Google Scholar]
- Cross JM, von Korff M, Altmann T, Bartzetko L, Sulpice R, Gibon Y, Palacios N, Stitt M. Variation of enzyme activities and metabolite levels in 24 Arabidopsis accessions growing in carbon-limited conditions. Plant Physiol. 2006;142:1574–1588. doi: 10.1104/pp.106.086629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Lithy ME, Clerkx EJM, Ruys GJ, Koornneef M, Vreugdenhil D. Quantitative trait locus analysis of growth-related traits in a new Arabidopsis recombinant inbred population. Plant Physiol. 2004;135:444–458. doi: 10.1104/pp.103.036822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshed Y, Zamir D. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics. 1995;141:1147–1162. doi: 10.1093/genetics/141.3.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Quantitative Genetics. 4. Harlow, UK: Pearson Education Limited; 1996. [Google Scholar]
- Haas BJ, Wortman JR, Ronning CM, et al. Complete reannotation of the Arabidopsis genome: methods, tools, protocols and the final release. BMC Biol. 2005;3:7. doi: 10.1186/1741-7007-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett CA. Statistical methods for QTL mapping in cereals. Plant Mol. Biol. 2002;48:585–599. doi: 10.1023/a:1014896712447. [DOI] [PubMed] [Google Scholar]
- Hittalmani S, Shashidhar HE, Bagali PG, Huang N, Sidhu JS, Singh VP, Khush GS. Molecular mapping of quantitative trait loci for plant growth, yield and yield related traits across three diverse locations in a doubled haploid rice population. Euphytica. 2002;125:207–214. [Google Scholar]
- Jansen RC, Nap JP. Trends. Genet. Vol. 17. 2001. Genetical genomics: the added value from segregration; pp. 388–391. [DOI] [PubMed] [Google Scholar]
- Jacobsson L, Park H-B, Wahlberg P, Fredriksson R, Perez-Enciso M, Siegel PB, Andersson L. Many QTLs with minor additive effects are associated with a large difference in growth between two selection lines in chickens. Genet. Res. 2005;86:115–125. doi: 10.1017/S0016672305007767. [DOI] [PubMed] [Google Scholar]
- Jompuk C, Fracheboud Y, Stamp P, Leipner J. Mapping of quantitative trait loci associated with chilling tolerance in maize (Zea mays L.) seedlings grown under field conditions. J. Exp. Bot. 2005;56:1153–1163. doi: 10.1093/jxb/eri108. [DOI] [PubMed] [Google Scholar]
- Keurentjes JJB, Fu J, de Vos CHR, Lommen A, Hall RD, Bino RJ, van der Plas LHW, Jansen RC, Vreugdenhil D, Koornneef M. The genetics of plant metabolism. Nat. Genet. 2006;38:842–849. doi: 10.1038/ng1815. [DOI] [PubMed] [Google Scholar]
- Keurentjes JJB, Bentsink L, Alonso-Blanco C, Hanhart CJ, Vries HB-D, Effgen S, Vreugdenhil D, Koornneef M. Development of a near-isogenic line population of Arabidopsis thaliana and comparison of mapping power with a recombinant inbred line population. Genetics. 2007;175:891–905. doi: 10.1534/genetics.106.066423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein DJ, Lambrix VM, Reichelt M, Gershenzon J, Mitchell-Olds T. Gene duplication in the diversification of secondary metabolism: tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell. 2001;13:681–693. doi: 10.1105/tpc.13.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroymann J, Mitchell-Olds T. Epistasis and balanced polymorphism influencing complex trait variation. Nature. 2005;435:95–98. doi: 10.1038/nature03480. [DOI] [PubMed] [Google Scholar]
- Kroymann J, Donnerhacke S, Schnabelrauch D, Mitchell-Olds T. Evolutionary dynamics of an Arabidopsis insect resistance quantitative trait locus. Proc. Natl Acad. Sci. USA. 2003;100:14587–14592. doi: 10.1073/pnas.1734046100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusterer B, Piepho H-P, Utz HF, Schön CC, Muminovic J, Meyer RC, Altmann T, Melchinger AE. Heterosis for biomass-related traits in Arabidopsis investigated by QTL analysis of the triple test cross design with recombinant inbred lines. Genetics. 2007;177:1839–1850. doi: 10.1534/genetics.107.077628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S-B, Zhang Z-H, Ying H, Li C-Y, Xuan J, Ting M, Li Y-S, Zhu Y-G. Genetic dissection of developmental behavior of crop growth rate and its relationships with yield and yield related traits in rice. Plant Sci. 2006;170:911–917. [Google Scholar]
- Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR. Gas chromatography mass spectrometry based metabolite profiling in plants. Nat. Protocols. 2006;1:387–396. doi: 10.1038/nprot.2006.59. [DOI] [PubMed] [Google Scholar]
- Lorberth R, Ritte G, Willmitzer L, Kossmann J. Inhibition of a starch-granule-bound protein leads to modified starch and repression of cold sweetening. Nat. Biotechnol. 1998;16:473–477. doi: 10.1038/nbt0598-473. [DOI] [PubMed] [Google Scholar]
- Loudet O, Chaillou S, Merigout P, Talbotec Jl, Daniel-Vedele FO. Quantitative trait loci analysis of nitrogen use efficiency in Arabidopsis. Plant Physiol. 2003;131:345–358. doi: 10.1104/pp.102.010785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann V, Harker M, Pecker I, Hirschberg J. Metabolic engineering of astaxanthin production in tobacco flowers. Nat. Biotechnol. 2000;18:888–892. doi: 10.1038/78515. [DOI] [PubMed] [Google Scholar]
- Meyer RC, Steinfath M, Lisec J, et al. The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA. 2007;104:4759–4764. doi: 10.1073/pnas.0609709104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir SR, Collins GJ, Robinson S, Hughes S, Bovy A, Vos CHRD, van Tunen AJ, Verhoeyen ME. Overexpression of petunia chalcone isomerase in tomato results in fruit containing increased levels of flavonols. Nat. Biotechnol. 2001;19:470–474. doi: 10.1038/88150. [DOI] [PubMed] [Google Scholar]
- Payne RW, Baird DB, Cherry M, et al. Genstat Release 6.1 Reference Manual. Oxford, UK: VSN International; 2002. [Google Scholar]
- Piepho HP, Büchse A, Emrich K. A hitchhiker’s guide to mixed models for randomized experiments. J. Agron. Crop Sci. 2003;189:310–322. [Google Scholar]
- Rauh L, Basten C, Buckler S. Quantitative trait loci analysis of growth response to varying nitrogen sources in Arabidopsis thaliana. Theor. Appl. Genet. 2002;104:743–750. doi: 10.1007/s00122-001-0815-y. [DOI] [PubMed] [Google Scholar]
- Rocha JL, Eisen EJ, Vleck LDV, Pomp D. A large-sample QTL study in mice: I. Growth. Mamm. Genome. 2004;15:83–99. doi: 10.1007/s00335-003-2312-x. [DOI] [PubMed] [Google Scholar]
- Schauer N, Semel Y, Roessner U, et al. Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 2006;24:447–454. doi: 10.1038/nbt1192. [DOI] [PubMed] [Google Scholar]
- Schön CC, Utz HF, Groh S, Truberg B, Openshaw S, Melchinger AE. Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics for complex traits. Genetics. 2004;167:485–498. doi: 10.1534/genetics.167.1.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnewald U, Lerchl J, Zrenner R, Frommer W. Manipulation of sink–source relations in transgenic plants. Plant Cell Environ. 1994;17:649–658. [Google Scholar]
- ter Steege MW, den Ouden FM, Lambers H, Stam P, Peeters AJM. Genetic and physiological architecture of early vigor in Aegilops tauschii, the D-genome donor of hexaploid wheat. A quantitative trait loci analysis. Plant Physiol. 2005;139:1078–1094. doi: 10.1104/pp.105.063263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup K, Alonso-Blanco C, Lynn JR, Michaels SD, Amasino RM, Koornneef M, Millar AJ. Natural allelic variation identifies new genes in the Arabidopsis circadian system. Plant J. 1999;20:67–77. doi: 10.1046/j.1365-313x.1999.00577.x. [DOI] [PubMed] [Google Scholar]
- Törjék O, Berger D, Meyer RC, Müssig C, Schmid KJ, Sörensen TR, Weisshaar B, Mitchell-Olds T, Altmann T. Establishment of a high-efficiency SNP-based framework marker set for Arabidopsis. Plant J. 2003;36:122–140. doi: 10.1046/j.1365-313x.2003.01861.x. [DOI] [PubMed] [Google Scholar]
- Törjék O, Witucka-Wall H, Meyer RC, von Korff M, Kusterer B, Rautengarten C, Altmann T. Segregation distortion in Arabidopsis C24/Col-0 and Col-0/C24 recombinant inbred line populations is due to reduced fertility caused by epistatic interaction of two loci. Theor. Appl. Genet. 2006;113:1551–1561. doi: 10.1007/s00122-006-0402-3. [DOI] [PubMed] [Google Scholar]
- Trethewey RN, Geigenberger P, Riedel K, Hajirezaei M-R, Sonnewald U, Stitt M, Riesmeier JW, Willmitzer L. Combined expression of glucokinase and invertase in potato tubers leads to a dramatic reduction in starch accumulation and a stimulation of glycolysis. Plant J. 1998;15:109–118. doi: 10.1046/j.1365-313x.1998.00190.x. [DOI] [PubMed] [Google Scholar]
- Ungerer MC, Rieseberg LH. Genetic architecture of a selection response in Arabidopsis thaliana. Evolution Int. J. Org. Evolution. 2003;57:2531–2539. doi: 10.1111/j.0014-3820.2003.tb01497.x. [DOI] [PubMed] [Google Scholar]
- Utz HF, Melchinger AE. PLABQTL: a program for composite interval mapping of QTL. J. Quant. Trait Loci. 1996;2 [Google Scholar]
- Werner JD, Borevitz JO, Warthmann N, Trainer GT, Ecker JR, Chory J, Weigel D. Quantitative trait locus mapping and DNA array hybridization identify an FLM deletion as a cause for natural flowering-time variation. Proc. Natl Acad. Sci. USA. 2005;102:2460–2465. doi: 10.1073/pnas.0409474102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MAL, Kim K, Kliebenstein DJ, van Leeuwen H, Michelmore RW, Doerge RW, Clair DAS. Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics. 2007;175:1441–1450. doi: 10.1534/genetics.106.064972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger SD, Yin TM, Difazio SP, Tschaplinski TJ, Gunter LE, Davis MF. Phenotypic variation in growth and biomass distribution for two advanced-generation pedigrees of hybrid poplar. Can. J. For. Res. 2005;35:1779–1789. [Google Scholar]
- Zhang Z, Ober JA, Kliebenstein DJ. The gene controlling the quantitative trait locus EPITHIOSPECIFIER MODIFIER1 alters glucosinolate hydrolysis and insect resistance in Arabidopsis. Plant Cell. 2006;18:1524–1536. doi: 10.1105/tpc.105.039602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comprehensive mQTL overview for known metabolites.
QTL analysis results for biomass and metabolic traits.
SNP positions and resulting amino acid changes for mQTL candidate genes according to data published by Clark et al. 2007).
Epistatic interactions (additive × additive) for mQTL of 50 known metabolites.