

Abstract
Two products of human cytomegalovirus (HCMV) UL111a gene have been previously identified to resemble human IL-10 (hIL-10). These viral IL-10s (vIL-10s) are able to induce signal transduction events and biological activities in a variety of cells. In this study, five novel vIL-10 transcripts were identified from HCMV AD169 infected MRC-5 cells. Some vIL-10 isoforms were post-translationally glycosylated, depending on the existence of a predicted N-linked glycosylation site. Similar to hIL-10, four of the vIL-10 isoforms apparently formed putative dimers. Among the different vIL-10 isoforms, vIL-10A significantly induced the phosphorylation of transcription factor STAT3 in THP-1 cells. All identified vIL-10 isoforms were able to form complexes with hIL-10, and enhanced hIL-10-induced STAT3 phosphorylation in different degrees. Identification of diverse forms of vIL-10 suggests that HCMV has developed a sophisticated mechanism to interfere with hIL-10 signaling pathway.
Keywords: cytomegalovirus, IL-10, vIL-10
1. INTRODUCTION
Human cytomegalovirus (HCMV), a member of β-herpesvirus family, causes severe diseases in neonates and in immunosuppressed individuals (Pass, 2001). In healthy individuals, HCMV is able to establish a persistent infection, usually without any symptoms, despite vigorous host antiviral immune responses. After the establishment of latency, HCMV periodically reactivates to produce infectious viral particles in infected hosts (Mocarski and Courcelle, 2001). Facing constant challenges from host immune responses, HCMV has developed various counteractive mechanisms that alter host antiviral immunity (Beisser et al., 2002; Michelson, 1999; van Cleef et al., 2006; Vink et al., 2001). The ability of HCMV to produce immune modulators, such as virus-encoded cytokines or cytokine receptors, apparently plays important roles in maintaining persistent viral infection in vivo. These viral proteins, by mimicking the sequences and structures of their human counterparts, are able to disrupt multiple signal transduction pathways that regulate cell growth, differentiation, and activation, contributing to viral evasion of constant host immune surveillance. For example, a protein encoded by HCMV UL146 is an α-chemokine that modulates host neutrophil activation (Penfold et al., 1999; Saederup and Mocarski, 2002). The product of US28 functions as a β-chemokine decoy receptor and disrupts β-chemokine signaling by binding and sequestering β-chemokines, such as RANTES (Beisser et al., 2002; Billstrom et al., 1998; Randolph-Habecker et al., 2002).
The interleukin-10 (IL-10) family belongs to the Class II cytokines and consists of several members, including IL-10, IL-19, IL-20, IL-22, IL-24, and IL-26. These cytokines function as important regulators to induce a wide range of biological effects in different cells (Conti et al., 2003; Fickenscher et al., 2002; Langer et al., 2004; Pestka et al., 2004; Wolk et al., 2002). IL-10 produced by both T and B cells exerts immune inhibitory as well as stimulatory effects. IL-10 inhibits the antigen-presenting capacity of macrophages and dendritic cells. IL-10 blocks cytokine production by many types of T cells. On the other hand, IL-10 promotes the development of certain types of B cells and stimulates the activities of NK cells (Conti et al., 2003; Fickenscher et al., 2002; Langer et al., 2004; Pestka et al., 2004; Wolk et al., 2002). An IL-10 isoform, IL-10δ3, was recently identified by Wu’s group (Wu et al., 2005). This IL-10 variant has an in-frame deletion of the entire exon 3. Although the exact function of IL-10δ3 is largely unknown, it is strongly associated with the good prognosis of acute lymphoblastic leukemia (ALL) in children.
The receptors of IL-10 family are called the Class II cytokine receptors (CRF2). The receptor subunits involved in the interaction with IL-10 family include IL-10R1, IL-10-R2, IL-20R1, IL-20R2, IL-22R1, and IL-22RB. Members of IL-10 family substantially share the receptor subunits for their signaling transductions (Conti et al., 2003; Fickenscher et al., 2002; Kotenko, 2002; Langer et al., 2004; Pestka et al., 2004; Wolk et al., 2002).
Several viruses, including HCMV, produce proteins that bear sequence homology to human IL-10 (hIL-10). However, the biological functions of HCMV vIL-10 are still poorly understood. The deletion of UL111a ORF appears to have no effect on viral replication in vitro (Chang et al., 2004). However, it is very likely that vIL-10 plays immune modulating roles in vivo when the virus is challenged by host immune responses. Because hIL-10 mainly functions as a potent suppressor of proinflammatory cytokine synthesis and antigen presentation, it is reasonable to speculate that vIL-10 plays a similar role in downregulating host immune responses against HCMV infection. More studies are needed to determine the exact role of vIL-10 in the maintenance of persistent viral infection under constant host immune surveillance.
In recent years, two vIL-10 gene products were identified from HCMV infected cells (Jenkins et al., 2004; Kotenko et al., 2000). To avoid confusion with the newly discovered vIL-10 isoforms described in this paper, the first (cmvIL-10) and the second (LA-cmv-IL-10) identified vIL-10 gene products are designated as vIL-10A and vIL-10B, respectively. The mRNA of vIL-10A and vIL-10B are different splicing products of the same precursor RNA transcribed from HCMV UL111a ORF (Jenkins et al., 2004; Kotenko et al., 2000).
vIL-10A was initially identified from HEL299 cells (a human diploid embryonic lung fibroblast-like cell line) infected by HCMV strain AD169. The vIL-10A is produced and secreted during the lytic phase of HCMV infection. The sequence of vIL-10A transcript reveals that the mRNA is derived from splicing of two introns from its precursor mRNA. The vIL-10A protein contains 175 amino acids and migrates around 21–30 kDa on SDS-PAGE. The vIL-10A has 27% amino acid sequence identity to hIL-10 (Kotenko et al., 2000). Several studies have shown that vIL-10A forms homo-dimers similar to hIL-10 and interacts with the IL-10 receptor complex to induce signal transduction (Jones et al., 2002; Kotenko et al., 2000; Yamamoto-Tabata et al., 2004). vIL-10A also inhibits proinflammatory cytokine production and the maturation of dendritic cells (Chang et al., 2004; Raftery et al., 2004).
Another viral IL-10 transcript, vIL-10B, was later identified from HCMV infected granulocyte-macrophage progenitors (GM-Ps) (Jenkins et al., 2004). Because GM-Ps have been used as models to study latent HCMV infection, the authors suggested that the expression of vIL-10B is associated with the latent phase of HCMV infection. The mRNA of vIL-10B results from single splicing of the first intron of vIL-10 gene, which produces a shorter product (Jenkins et al., 2004).
The identification of different vIL-10 isoforms from different HCMV infected cells indicates that the differential expression of vIL-10 may be influenced by the cellular factors. In this study, we investigated the expression profile of vIL-10 in HCMV AD169 infected MRC-5 and Bud8 cells and identified several new vIL-10 isoforms. We demonstrated that putative forms of vIL-10 proteins were detected from virus infected MRC-5 cells. It was shown that some vIL-10 isoforms were glycosylated as predicted and four of the isoforms were able to form putative dimers. Furthermore, we found that vIL-10 isoforms formed complexes with hIL-10 and modulated hIL-10 signaling, suggesting a previously unrecognized mechanism of interfering IL-10 signaling pathway by HCMV.
2. MATERIALS AND METHODS
2.1 Virus strain and cells
AD169 strain of HCMV was obtained from NIH AIDS Research and Reference Reagent Program. Human fibroblast cells, MRC-5 and Bud8, were purchased from American Type Culture Collection (ATCC) and maintained in MEM supplemented with 10% FBS. 293FT cells were purchased from Invitrogen and maintained in DMEM supplemented with 10% FBS. THP-1 cells (ATCC) were maintained in RPMI supplemented with 10% FBS.
2.2 Infection of cultured cells
MRC-5 or Bud8 cells were cultured in a 10-cm dish until 70% confluence. HCMV AD169 was added to MRC-5 cells at MOI 0.1. The infected cells were incubated until 70% of the cells showed CPE (cytopathic effect). Cells were then collected for RNA preparation.
2.3 Clone HCMV vIL-10 gene
Total RNAs from the AD169-infected cells were purified with Trizol reagent (GIBCO) following the manufacturer’s instruction. 2 μg of RNA were reverse transcribed with a poly-T primer using SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). A 5′-end primer (5′-GATGGATCCATGCTGTCGGTGATGGTC-3′) and a 3′-end primer (5′-CTCGAATTCCTTTCTCGAGTGCAGAT-3′) were used to amplify vIL-10 cDNA. The synthesized cDNA was subjected to PCR amplification with pfu Turbo (Stratagene). The condition for the PCR reaction were: 94 °C for 1 min. followed by 30 cycles of 94 °C for 30 sec., 61 °C for 30 sec., and 72 °C for 1 min., and an additional incubation at 72 °C for 10 min. The PCR products were purified with a Qiagen PCR purification kit, and cloned into the BamHI and EcoRI sites of pBluescript KS II (+) vector. Clones of HCMV vIL-10 were sequenced. The sequences were then analyzed with Vector NTI software and compared with sequences in NCBI gene bank. The different forms of vIL-10 cDNA were also subcloned into pEF1A-myc-his vector for expressing tagged vIL-10 isoforms in mammalian cells.
2.4 Clone human IL-10 gene
Total RNA from TPA-treated BCBL-1 cells were extracted with Trizol reagent. The cDNA was synthesized as described above. The hIL-10 gene was PCR amplified from the cDNA with primers, 5′-GCGGGATCCATGCACAGCTCAGCACT-3′ and 5′-CGCGGATCCGTTTCGTATCTTCATTGT-3′. PCR reaction was performed with pfu (Stratagene). PCR condition were: 94 °C for 2 min., followed by 30 cycles of 92 °C for 30 sec., 54 °C for 30 sec., 72 °C for 1 min., and final extension of 72 °C for 10 min. The hIL-10 gene was first cloned into pSP72 vector and sequenced to ensure the overall sequence accuracy. The hIL-10 was then subcloned into pcDNA3.1-GST-Au1 plasmid for the expression of hIL-10-GST fusion protein.
2.5 Western blot analysis of vIL-10 expression in transfected 293FT and radioimmune precipitation of CMV AD169 infected MRC-5 cells
293FT cells were transfected with pEF-1/vIL-10 plasmids using Lipofectamin2000 (Invitrogen). Two days after transfection, cells were collected for Western blot analysis with an anti-myc antibody (Cell Signaling). In the dimerization study, cells were lysed with modified RIPA buffer (25mM Tris, pH6.8, 50mM NaCl, 1% NP-40, and 0.5% DOC) containing 20 mM of N-ethymaleimide (NEM). In the glycosylation study, two days after the transfection, cells were treated with 50 μg/ml tunicamycin for 24 hours. HCMV AD169 was used to infect MRC-5 cells at MOI 0.1. Eleven days post-infection, cells were rinsed with PBS and then labeled for 4h with 250 μCi/ml of 35S-Met/Cys in MEM medium containing 0.01% dialyzed serum. The cells were collected and lysed with modified RIPA buffer. The immunoprecipitated proteins were resolved on a 13.6 % SDS-PAGE gel and then autoradiographed. The goat anti-HCMV-vIL-10 antibody used in the experiment was purchased from R & D Systems.
2.6 GST pull-down assay
293FT cells were co-transfected with pcDNA3.1/hIL-10-GST and pEF-1/vIL-10 plasmids. Forty-eight hours after transfection, cells were lysed with lysis buffer (500mM NaCl, 50mM Tris, pH 8.0, 1% NP40). Cell lysates were mixed with GST beads to pull-down hIL-10-GST fusion protein and its associated complexes. The precipitated complexes were subjected to Western blot with an anti-myc antibody.
2.7 Phosphorylation of STAT3 in THP-1 cells
THP-1 cells were seeded in 6-well plates and maintained in the presence of 20 ng/ml TPA for 2 hours. The media were then replaced by conditioned media from 293FT cells transiently transfected with vIL-10 or hIL-10 plasmid. After being incubated for 1.5 hour, the THP-1 cells were collected and analyzed by Western blot with anti-STAT3 antibodies (Cell Signaling).
3. RESULTS
3.1 Diverse vIL-10 isoforms identified from HCMV infected MRC-5 and Bud8 cells
To investigate the expression profile of vIL-10, we examined the vIL-10 transcripts from HCMV infected MRC-5 fibroblast cells. Standard RT-PCR (reverse transcription-polymerase chain reaction) was performed to amplify vIL-10. The first strand cDNA synthesis was performed with an oligo-T primer to reverse-transcribe all polyadenylated cellular and viral mRNAs. The cDNAs of vIL-10 were then subjected to PCR amplification with vIL-10 specific primers, similar to what have been described by Pestka’s group (Kotenko et al., 2000). These primers were designed to amplify sequences between the 5′-end of the first exon and the 3′-end of the third exon of vIL-10 gene. To our surprise, the obtained vIL-10 PCR products appeared to contain DNA fragments of variable sizes when analyzed on an agarose gel (data not shown). We cloned the PCR products into the pBluescript vector. Twenty-eight clones containing vIL-10 inserts were collected and analyzed. We identified 8 different vIL-10 inserts, including an unspliced and five novel spliced UL111a transcripts as well as previously reported vIL-10A and vIL-10B. These novel vIL-10 transcripts are designated as vIL-10C, D, E, F, and G (The unspliced transcript was predicated to produce vIL-10D if being translated). In order to determine if multiple vIL-10 isoforms can also be detected from other HCMV infected cells, we performed RT-PCR with DNA from virus infected human Bud8 fibroblasts and THP-1 human monocytes. We identified 6 different vIL-10 transcripts in 12 clones from Bud8 cells, but none from THP-1 cells. The distribution of each vIL-10 isoform is summarized in Table 1.
Table 1.
Distribution of each vIL-10 isoforms in the clones sequenced.
| MRC-5 Cells | Bud 8 Cells | |||
|---|---|---|---|---|
| Isoforms | Number of Clones | Percentage | Number of Clones | Percentage |
| vIL-10A | 7 | 25% | 1 | 8.3% |
| vIL-10B | 4 | 14.3% | 3 | 25% |
| vIL-10C | 2 | 7.1% | 0 | 0 |
| vIL-10D | 1 | 3.6% | 1 | 8.3% |
| vIL-10E | 4 | 14.3% | 0 | 0 |
| vIL-10F | 3 | 10.7% | 1 | 8.3% |
| vIL-10G | 1 | 3.6% | 1 | 8.3% |
| Unspliced mRNA | 6 | 21.4% | 5 | 41.7 |
Our data indicated that these new vIL-10 isoforms are not rare products that may be produced during the viral infection. Although the majority of splicing events occurred at the sites previously identified in vIL-10A and B transcripts (Jenkins et al., 2004; Kotenko et al., 2000), new splicing donor and acceptor sites were found in mRNAs of vIL-10C, E, F and G. All different spliced products of vIL-10 are compared in Figure 1A. Interestingly, the first exon (nucleotide sequence 159678–159857) is conserved in all identified vIL-10 mRNAs. As a result, all vIL-10 isoforms contain an identical amino acid sequence from 1 to 60 at the N-terminus, as shown in Figure 1B. However, the C-termini of these vIL-10 isoforms vary in amino acid sequences and in lengths. We performed protein scan using web-based InterProScan program to compare the structure similarity of these vIL-10 isoforms. We found all vIL-10 isoforms maintain a domain located between the 20th and 60th amino acids. This structure represents a 4-helical bundle domain found predominantly in long- and short-chain cytokines, such as interleukins, interferons, growth hormones, and granulocyte colony stimulating factors. This core structure consists of a closed bundle of four helices in a left-handed twist, with two crossover connections, and is important for the cytokine/receptor interaction. The data suggested that all vIL-10 isoforms may have the ability to bind to the IL-10 receptor. In addition, we compared all vIL-10 isoforms with hIL-10 using Vector NTI software. Interestingly, vIL-10D (the shortest) had the highest homology to hIL-10, while vIL-10A (the longest) had the least homology (data not shown).
Figure 1.


(A) Schematic representation of HCMV vIL-10 transcripts. The nucleotide numbers correspond to DNA sequence of HCMV genome. (B) Amino acid sequences of all identified vIL-10 isoforms. Sequences of vIL-10 isoforms were aligned using MAFFT software (http://timpani.genome.ad.jp/~mafft/server/). The sequence identity (*) and similarity (: and ·) are indicated.
We examined vIL-10 proteins in HCMV AD169 infected MRC-5 cells. The viral infected cells were labeled with 35S methionine, and then subjected to radioimmune precipitation (RIP) with an anti-vIL-10 polyclonal antibody (R & D). The anti-vIL-10 antibody, elicited against the recombinant vIL-10A, recognizes a common sequence shared by all newly identified vIL-10 isoforms as well as the two previously described products. Each vIL-10 isoform was transiently expressed in 293FT cells, and analyzed side-by-side with the RIP samples by Western blot. Three major bands that apparently corresponded to vIL-10B, C and F were detected from HCMV infected MCR-5 cells, as shown in Figure 2. Because of the co-existence of glycosylated and non-glycosylated vIL-10 isoforms and similar molecular weights among some isoforms, it was impossible to determine the isoform identity of each individual band. Nevertheless, our data indicated multiple putative forms of vIL-10 expressed in HCMV infected MRC-5 cells. In addition, the RIP sample from viral infected cells showed three high molecular weight bands, ~90, 62, and 50 kDa. The 90 and 62 kDa bands presumably were IL-10R1 (90–110 kDa) and IL-10R2 (60 kDa). The nature of the 50 kDa band was unknown.
Figure 2.
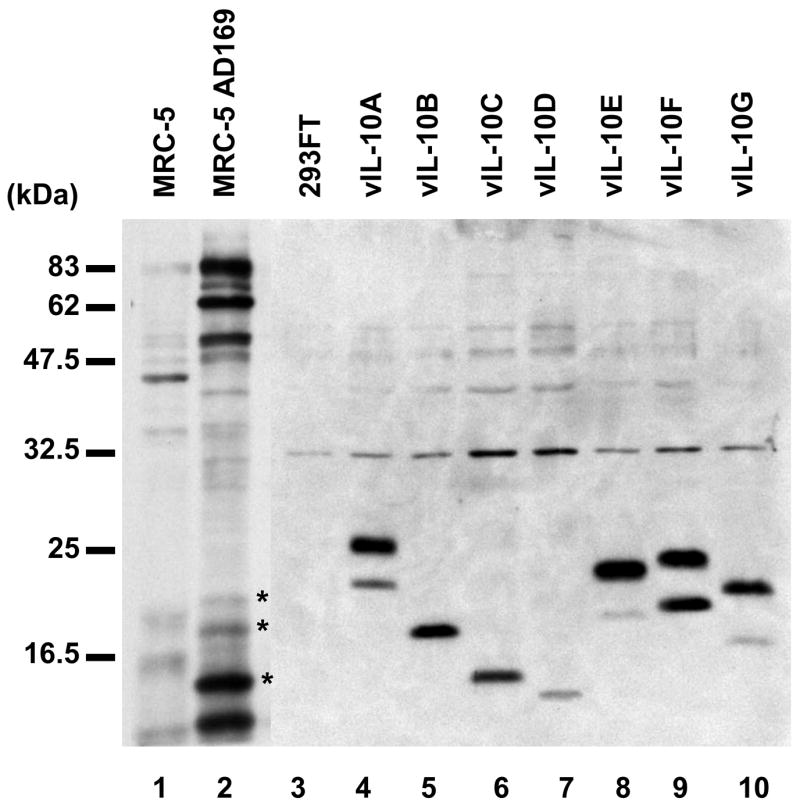
Detection of vIL-10 isoforms from HCMV AD169 infected MRC-5 cells. Samples of MRC-5 (Lane 1) and HCMV-infected MRC-5 cells (Lane 2) were analyzed by RIP. The specific bands precipitated from the HCMV infected sample by the anti-vIL-10 antibody were indicated as *. vIL-10 isoforms from transfected 293FT cells were separated side-by-side on the same gel and analyzed by Western blot with anti-myc antibody (Lane 3 to 10)
3.2 Expression and glycosylation of vIL-10 isoforms
We cloned all vIL-10 isoforms into pEF1A-myc-his vector. vIL-10 isoforms were then transiently expressed in 293FT cells. The expected products were analyzed by Western blot. As shown in Figure 3A, all vIL-10 isoforms were detected as expected. Each of the vIL-10 isoforms, A, E, F, and G showed two bands. The size difference between the doublelet was about 3 kDa, similar to the size of one N-linked oligosaccharide. We hypothesized that the higher bands represented glycosylated forms, while the lower bands were non-modified proteins. Previous study suggested that N-linked glycosylation likely occurred at the –Asn151-Gly152-Thr153- (Kotenko et al., 2000). Consistent with the presence of this predicted glycosylation site, vIL-10 isoforms A, E, F, and G (but not B, C, and D) showed double bands. To test this hypothesis, we treated the transfected cells with tunicamycin to inhibit N-linked glycosylation. In the tunicamycin treated cells, there was a significant reduction of the higher molecular weight band in isoform A, E, F, and G, as shown in Figure 3B. The sizes of isoform B, C, and D were not affected by tunicamycin, indicating that they were not glycosylated. Our results were consistent with the fact that vIL-10 isoforms underwent N-linked glycosylation depending on existence of the Asn-Gly-Thr site in their sequences.
Figure 3.

Expression and glycosylation of vIL-10 isoforms. A. The tagged vIL-10 isoforms transiently expressed in 293FT cells were detected by Western blot with an anti-myc antibody. B. The N-linked glycosylation of each vIL-10 isoform was analyzed after tunicamycin treatment. The vIL-10 isoforms from transfected cells treated without (Lane 1) or with tunicamycin (Lane 2) were analyzed by Western blot with an anti-myc antibody. The glycosylated and non-glycosylated vIL-10s are indicated as * and ** respectively.
3.3 Dimerization of vIL-10 isoforms
It has been shown that HCMV vIL-10A forms a homo-dimer similar to hIL-10 (Jones et al., 2002). The vIL-10A dimer is stabilized by an intramolecular disulfide bond that is formed between Cys78 on each of the vIL-10A molecule (Jones et al., 2002). Whether this is the only intramolecular disulfide bond in the vIL-10A dimer is unclear. Among the identified vIL-10 isoforms, vIL-10A, B, and F maintain the Cys78. In order to determine if all the vIL-10 isoforms are able to form dimers and if Cys78 is essential for their dimerization, we expressed myc-tagged vIL-10 isoforms in 293FT cells. The covalently linked protein dimers can be converted to monomer by the treatment of reducing agent β-mercaptoethanol to break intra- and inter-molecular disulfide bonds, resulting in changes of migration patterns on SDS-PAGE. The transfected cells were lysed in the presence of NEM to prevent artifactual disulfide-bond formation during the sample preparation. All vIL-10 samples analyzed on SDS-PAGE were pre-treated with or without β-mercaptoethanol (Figure 4). Under the reducing condition, the expected monomers of vIL-10 isoforms were detected. Under the non-reducing condition, additional high molecular weight masses were seen in vIL-10A, E, F, and G, apparently resulted from dimerization. Although, higher molecular weight masses were also detected in vIL-10B in the absence of β-mercaptoethanol, the expected dimmer bands appeared not to be the major bands. Thus, it was not clear these high molecular weight masses were dimers (oligomers) or aggregated products. Formation of dimer of vIL-10E suggested that Cys78 may be not required for the dimerization of some vIL-10s. Disulfide bond(s) between other Cys residues may also be involved in the formation of vIL-10 complexes.
Figure 4.

Dimerization of vIL-10 isoforms. vIL-10 isoforms were transiently expressed in 293FT cells. Cells were lysed in modified RIPA buffer containing 20 mM of NEM). Samples were then treated without (−) or with (+) β-mercaptoethanol before being analyzed by Western blot. The vIL-10 isofoms were indicated as *.
3.3 Inductions of STAT3 phosphorylation by vIL-10s and hIL-10
It has been reported that vIL-10A interacts with IL-10 receptor complexes. Such interaction induces signal transduction that leads to the phosphorylation of transcription factor STAT3. THP-1 cells has been used as a model to study IL-10 signaling pathway, and vIL-10 isoforms expressed in 293FT cells were readily secreted in the media (data not shown). In this study, we first compared the effects of different vIL-10 isoforms on the phosophorylation of STAT3. THP-1 cells were treated with media containing different vIL-10 isoforms for 1.5 hours. The THP-1 cell lysates were then analyzed by Western blot with antibodies against total STAT3 or phosphorylated STAT3. As shown in Figure 5, among seven vIL-10 isoforms, only vIL-10A treatment significantly induced the phosphorylation of STAT3 at Tyr705. Treatment of THP-1 cells with vIL-10 isoforms did not significantly alter the phosphorylation status of another site, Ser727, on STAT3 (Data not shown). The total amounts of STAT3 in each sample, and the vIL-10 inputs were also shown in Figure 5. Because vIL-10A is the longest vIL-10 identified, the result showed that “full-length” vIL-10 sequence was required for triggering IL-10 induced STAT3 phosphorylation in THP-1 cells. As previously described in Figure 1, all vIL-10 isoforms have identical first 60 amino acids at the N-terminus. This region contains a 4-helical bundle domain, which is a common structure for many cytokines to interact with their receptors, suggesting that all vIL-10s may still maintain the ability to bind to IL-10 receptor. However, sequence variations in other parts of the protein may affect the affinity of vIL-10 to its receptor, and consequently result in inducing different signal transductions.
Figure 5.
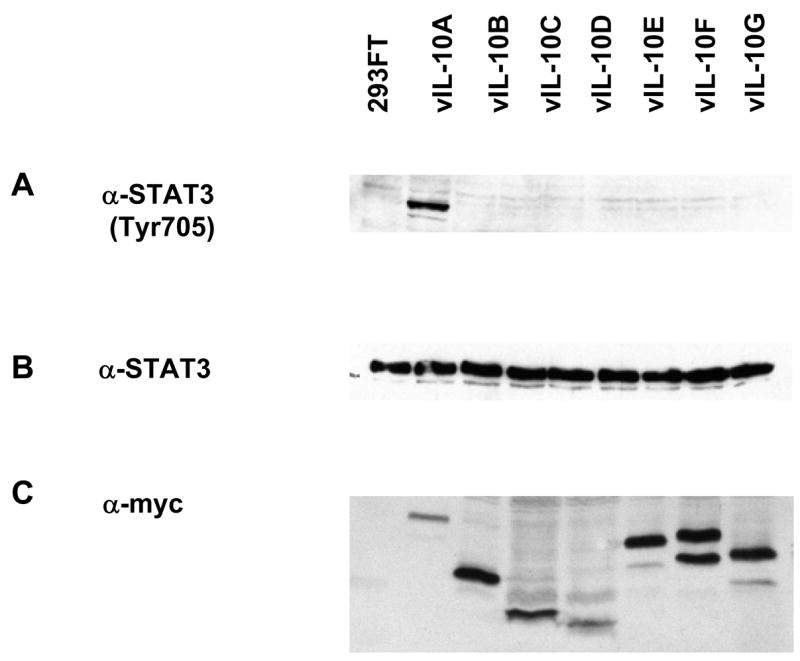
Phosphorylation of STAT3 induced by vIL-10 isoforms. Media from 293FT cells transfected with vIL-10 expressing plasmids were used to induce STAT3 phosphorylation in THP-1 cells. THP-1 cells were treated with the conditioned media for 1.5 hours, and then subjected to Western blot analysis with an anti-phosphorylated STAT3 (Tyr705) or an anti-STAT3 antibody. Expressions of vIL-10 isoforms were included to indicated the input of each protein.
3.5 Interactions of vIL-10s with hIL-10 and alterations of hIL-10 induced signal transduction
Both vIL-10A and hIL-10 form homo-dimers. Because of the sequence and structure similarities between vIL-10A and hIL-10 (Jones et al., 2002), we hypothesized that hetero-dimers may form between hIL-10 and vIL-10s. To test our hypothesis, we made a plasmid construct that expressed hIL-10-GST fusion protein. The GST domain was fused to the C-terminus of hIL-10. Because the N-terminal signal peptide of hIL-10 remains intact, the hIL-10-GST fusion protein was presumably synthesized and processed through ER and Golgi complex in a similar way as hIL-10. The secretion of hIL-10-GST into the media of the transfected 293FT cells was confirmed by Western blot (data not shown). We co-expressed hIL-10-GST with each vIL-10 isoform in 293FT cells. A vector expressing GST protein served as a negative control in the experiments. Complexes formed with hIL-10-GST were pulled-down with GST beads, and analyzed with Western blot using an anti-myc antibody. As seen in Figure 6, all isoforms of vIL-10 were detected from the hIL-10-GST complexes, while GST alone was unable to pull-down these vIL-10 isoforms. The lower panels show the presence of each vIL-10 isoforms in the lysates used for the pull-down assays. The amount of vIL-10D pulled-down by hIL-10-GST was less compared to the other isoforms, indicating the vIL-10D/hIL-10-GST complexes were less stable or less efficiently formed. The data showed that both glycosylated and non-glycosylated forms of vIL-10s can be pulled-down by hIL-10-GST fusion protein, suggesting that glycosylation is not essential for the complex formations. We also performed co-immumoprecipitation with hIL-10 that lacks of its signal peptide, and we were unable to detect the interaction of hIL-10ΔSP and vIL-10 (data not shown). It suggested that the interaction may require both proteins to be synthesized and processed through endoplasmic reticulum (ER) and Golgi systems.
Figure 6.

Interaction of vIL-10 isoforms and hIL-10. Each vIL-10 isoforms was co-expressed with GST or hIL-10-GST in 293FT cells. Cell lysates were subjected to a GST pull-down assay. Samples were then analyzed by Western blot with an anti-myc antibody (upper panels). The whole cell lysates (WCL) were also analyzed to show equal amounts of vIL-10 isoform in each sample (lower panels).
Based on the result that the vIL-10 isoforms interacted with hIL-10, we investigated if vIL-10 altered hIL-10 signaling. hIL-10-GST and different vIL-10 isoforms were co-expressed in 293FT cells. Media containing hIL-10-GST and vIL-10 complexes were first confirmed by Western blot and then used to treat THP-1 cells. As shown in Figure 7, hIL-10-GST alone was able to induce STAT3 phosphorylation (panel A). However, the phosphorylation of STAT3 was greatly enhanced in the presence of vIL-10A. Other vIL-10 isoforms (B to G) slightly increased the hIL-10-GST induced STAT3 phosphorylation. The panel B and C show levels of total STAT3 and hIL-10-GST in each sample respectively. In this experiment, we used media from mock transfected 293FT cells as a negative control for the effect of hIL-10-GST and vIL-10s. Based on previously published results, the effects on STAT3 phosphorylation were very likely caused by these proteins (Ji et al., 2005; Tanaka et al., 2005; Wise et al., 2007). However, we cannot rule out the possibility that some cellular factors in the hIL-10-GST or vIL-10 containing media might contribute to the STAT3 phosphorylation. Further confirmation with purified hIL-10 and vIL-10s is needed in the future.
Figure 7.
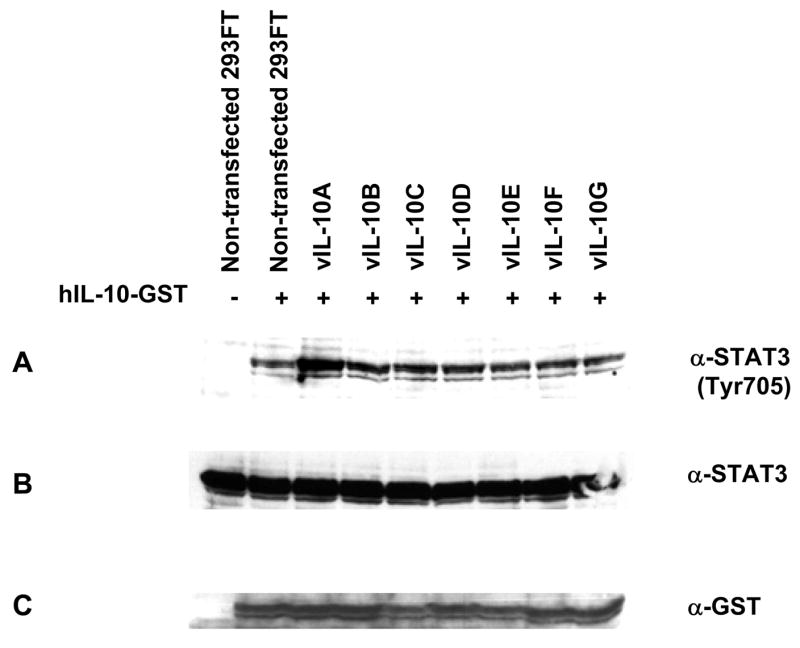
Effects of vIL-10 isoforms on hIL-10-GST induced STAT3 phosphorylation. hIL-10-GST was co-expressed with each vIL-10 isoform in 293FT cells. The conditioned media were collected and used to induce STAT3 phosphorylation in THP-1 cells. Levels of phosphorylated STAT3 were compared by Western blot. Total amounts of STAT3 and hIL-10-GST inputs were shown as controls.
4. DISCUSSION
Many cytokines, include IL-4, IL-6, IL-10 and IL-15, are found to have multiple isoforms derived from different spliced mRNA transcripts (Bihl et al., 2002; Ledru et al., 2003; Meazza et al., 1996; Nishimura et al., 2005; Nishimura et al., 1998; Tan and Lefrancois, 2006; Vasiliev et al., 2003; Wu et al., 2005; Yatsenko et al., 2004). These cytokine isoforms generally have different affinities to their receptors, contributing to generating different biological signals. In the case of IL-10 and its isoform IL-10δ3, both cytokines can be detected from ALL samples. It is found that IL-10 is associated with the progression of ALL, while IL-10δ3 is linked to good response to therapy and better clinical outcomes (Wu et al., 2005). These data demonstrate different roles of cytokine isoforms in modulating signaling transduction.
Two products of HCMV UL111a gene that bear sequence homology to hIL-10 were recently been identified by two groups. Pestka’s group identified vIL-10A from HCMV infected HEL 299 cells, and Slobedman’s group found vIL-10B from GM-Ps (Jenkins et al., 2004; Kotenko et al., 2000). It should be mentioned that HCMV behaves very differently in these two culturing systems. HEL 299 cells support HCMV for lytic replication while GM-Ps have been used as models for studying the viral latent infection. Results from Slobedman’s group suggested vIL-10B is a latency-associated product. Interestingly, in their paper, they also mentioned the detection of vIL-10B from HCMV infected primary HFF cells. Thus, the association of vIL-10 expressions and viral replication cycle is still worth further investigation.
In this study, we investigated the expression profile of vIL-10 isoforms in HCMV AD169 infected MRC-5 and Bud8 fibroblast cells. Despite that HEL 299, MRC-5 and Bud8 cells are all HCMV-permissive human fibroblasts, the expression profile of vIL-10 isoforms was apparently different. The RT-PCR result of HCMV infected HEL 299 cells shown by Pestka’s group demonstrated a single product of vIL-10A (Kotenko et al., 2000). However, our result obtained from HCMV infected MRC-5 and Bud8 cells indicated that there are multiple RT-PCR products (data not shown). This led to the identification of five novel vIL-10 isoforms in the study. The newly identified vIL-10C, D, E, F, and G, resulted from differently spliced mRNA transcripts. The previously described splicing donor sites (159857 and 160134) and splicing acceptor sites (159934 and 160218) in vIL-10A appeared to be the most common sites for splicing (Figure 1). However, alternative splicing donor and acceptor sites were seen in vIL-10C, E, F, and G isoforms. As a result, vIL-10 isoforms with different lengths, deletions and C-terminal sequences were generated. Our result indicated that these newly identified vIL-10s were not derived from some infrequent splicing events during the viral infection (Table 1). In HCMV infected MRC-5 cells, we immunoprecipitated multiple bands with anti-vIL-10 specific antibody, which were presumably different vIL-10 isoforms. It should be noticed that we could not exclude the possibility that some bands were degraded products of full-length vIL-10A. However, our observation in the transfected 293FT cells and from the conditioned media did not indicate unusually high instability and degradation of vIL-10 isoforms. Together with our RT-PCR data, it is likely that those detected bands were different vIL-10 isoforms.
We compared the expression profiles of vIL-10 isoforms in MRC-5, Bud8, and THP-1 cells. THP-1 cells has been used as a model for HCMV latent infection, we were unable to detect any expression of all vIL-10 isoforms. The expression patterns of vIL-10 isoforms in HCMV infected MRC-5 and Bud8 cells were noticeably different (Table 1), suggesting that the differential expression of vIL-10 isoforms may be influenced by cellular factors. However, we cannot exclude possible roles of viral factors because HCMV may replicate differently in different permissive cell lines. The ability of HCMV to produce multiple vIL-10 isoforms indicates that the virus has evolved a sophisticated mechanism to interfere IL-10 signaling pathway. Modulating host immune responses is a key for a virus to evade host immune surveillance, and to establish persistent infection. Based on our and other groups’ findings, it is very likely that additional vIL-10 isoforms of HCMV may exist. It will be interesting to know whether IL-10 homologs of other viruses have multiple isoforms.
Interestingly, all vIL-10 isoforms have an identical 60-amino acid sequence at the N-terminus. Thus, the intact signal peptide (the first 19 amino acids) is likely to be functional to direct the glycosylation and the transportation of the isoforms through ER and Golgi for secretion (Kotenko et al., 2000). vIL-10A, E, F, and G (but not B, C, and D) contain the predicted N-linked glycosylation site, –Asn151-Gly152-Thr153- site. As expected, vIL-10A, E, and F were found glycosylated, but not B, C, and D. Glycosylation of vIL-10s may influence their biological activities, as seen in the case of IL-22, a member of the IL-10 family (Logsdon et al., 2004).
The crystallography data of vIL-10A showed that vIL-10A forms homo-dimers which structurally resemble to hIL-10 homo-dimers. It has been shown that Cys78 of vIL-10A is involved in stabilizing the vIL-10A dimers through an intra-molecular disulfide bond between individual vIL-10A polypeptides (Jones et al., 2002). Among all isoforms identified, only vIL-10A, B, and F maintain the Cys78 residue. However, our data showed that vIL-10A, E, F, and G formed dimers under the non-reducing condition, suggesting that Cys78 may not be essential for dimerization. vIL-10B formed high molecular masses under the non-reducing condition. The nature of these masses was not clear. The result indicates that disulfide bond(s) between Cys residues may be involved in forming high molecular weight vIL-10 complexes. In this study, we did not investigate whether different vIL-10 isoforms were able to bind to each other. We speculated that complexes between different vIL-10 isoforms may also be formed in the AD169 infected MRC-5 cells.
Several groups have shown that vIL-10A forms homo-dimer and interacts with IL-10R. The ability of forming homo-dimers or possible hetero-dimers by different vIL-10 isoforms may enable HCMV to post-translationally generate diverse IL-10R ligands that interfere with the hIL-10 signaling pathway. In this study, we use THP-1 cells as a model to investigate the effect of vIL-10 isoforms on the IL-10 signal pathway. It has been shown that hIL-10 interacts with IL-10R on THP-1 cells, resulting in the phosphorylation of transcription factor STAT3 (Ji et al., 2005). We found vIL-10A was capable of inducing STAT3 phosphorylation in THP-1 cells, while other isoforms, B to G did not show significant induction. It is not surprised that different vIL-10s induce different biological consequences. A recent report show that in contrast to vIL-10A, vIL-10B does not alter CD40, CD80, CD86 and CD83 on dendritic cells, nor inhibit pro-inflammatory cytokine production after LPS stimulation. However, vIL-10B downregulates the expression of MHC class II on GM-Ps. Whether the biological effects of vIL-10A and vIL-10B were mediated through the STAT3 phosphorylation remains unclear. These findings indicate that vIL-10 isoforms may induce different biological effects through different mechanisms (Godwin et al., 2006).
Because of the sequence and structure similarities between vIL-10A and hIL-10, we investigated whether vIL-10 isoforms were able to form complexes with hIL-10. We performed GST pull-down assay. Our results showed that all identified vIL-10 isoforms interacted with hIL-10-GST fusion protein. This is the first report suggesting that vIL-10s and hIL-10 could interact. We further attempted to investigate the biological consequences of these interactions. In the presence of vIL-10A, the hIL-10 induced STAT3 phosphorylation was significantly increased. Because both hIL-10 and vIL-10A can induce STAT3 phosphorylation, whether such elevation resulted from additive or agonistic effect needs to be further investigated. Interestingly, despite all other vIL-10 isoforms (B through G) did not significantly induce STAT3 phosphorylation by themselves, they slightly increased the effect of hIL-10 when co-expressed. The data suggest the complicated nature of vIL-10s in modulating host IL-10 signaling in the context of HCMV infection.
In addition to the potential for interfering with cellular IL-10 signaling pathways, it should be mentioned that vIL-10s may be directly involved in the deregulation of cell growth control. A viral transforming gene, called MtrII, is located within the region of map units 0.685–0.770 in HCMV genome. The expression of MtrII results in transformation of NIH 3T3 cells (Choudhury et al., 1992; Doniger et al., 1999; Jahan et al., 1989; Muralidhar et al., 1996; Razzaque et al., 1988; Razzaque et al., 1991; Thompson et al., 1994; Wang and Razzaque, 1993). Sequence analysis revealed that the MtrII gene is in fact the first exon of vIL-10A. The predicted product of MtrII is identical to vIL-10D. The transforming activity of MtrII is apparently associated with its ability to bind and inhibit tumor suppressor p53 (Muralidhar et al., 1996). However, in the published papers, MtrII was studied by exogenous expressions of this protein in various cells. We were unable to find any study examining the presence of endogenous mRNA or protein of MtrII in HCMV-infected cells. Thus, it has not been clear whether MtrII is indeed expressed in HCMV infected cells. In this paper, we provide evidence showing that MtrII or vIL-10D is expressed in virally infected cells based on our RT-PCR results. The vIL-10D may not be the only isoform that has cell transforming activity. The minimum domain for the cell transforming activity on MtrII is mapped within the first 49 amino acids at the N-terminus (Thompson et al., 1994). All vIL-10 isoforms contain this domain, suggesting their potential roles in disrupting cell cycle control. Thus, it is certainly interesting to know if vIL-10s are involved in regulating viral replication by modulating cell growth.
In summary, the vIL-10 expression profiles were apparently different among different human fibroblasts. We have identified five new vIL-10 isoforms from HCMV AD169 infected MRC-5 cells. The newly identified vIL-10 isoforms underwent expected post-translational modification similar to their cellular counterpart hIL-10. Unlike previously discovered vIL-10A, these vIL-10 isoforms did not significantly induce STAT3 phosphorylation in THP-1 cells. vIL-10 isoforms can form complexes with hIL-10, suggesting their roles in interfering with hIL-10 signaling.
Acknowledgments
We thank Drs. Craig S. Miller and Robert J. Jacob for critically reading the manuscript and for helpful discussions. We also thank Jingchun Chen and Dr. Bo Yuan at the Ohio State University for the discussion of sequence comparison of vIL-10 isoforms. This project described was supported by Grant RFA-RR-03-014 from the National Center for Research Resources (NCRR), a component of National Institutes of Health (NIH), and its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.
Footnotes
The GenBank/EMBL/DDBJ Accession Numbers: cmvIL-10C (GenBank accession no. DQ924564); cmvIL-10D (GenBank accession no. DQ924565); cmvIL-10E (GenBank accession no. DQ924566); cmvIL-10F (GenBank accession no. DQ924567); and cmvIL-10G (GenBank accession no. DQ924568).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beisser PS, Goh CS, Cohen FE, Michelson S. Viral chemokine receptors and chemokines in human cytomegalovirus trafficking and interaction with the immune system. CMV chemokine receptors. Curr Top Microbiol Immunol. 2002;269:203–34. doi: 10.1007/978-3-642-59421-2_13. [DOI] [PubMed] [Google Scholar]
- Bihl MP, Heinimann K, Rudiger JJ, Eickelberg O, Perruchoud AP, Tamm M, Roth M. Identification of a novel IL-6 isoform binding to the endogenous IL-6 receptor. Am J Respir Cell Mol Biol. 2002;27(1):48–56. doi: 10.1165/ajrcmb.27.1.4637. [DOI] [PubMed] [Google Scholar]
- Billstrom MA, Johnson GL, Avdi NJ, Worthen GS. Intracellular signaling by the chemokine receptor US28 during human cytomegalovirus infection. J Virol. 1998;72(7):5535–44. doi: 10.1128/jvi.72.7.5535-5544.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WL, Baumgarth N, Yu D, Barry PA. Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J Virol. 2004;78(16):8720–31. doi: 10.1128/JVI.78.16.8720-8731.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury S, Woodworth CD, Inamdar A, el-Beik T, DiPaolo JA, Rosenthal LJ. Differences in retention and expression of transfected human cytomegalovirus Towne XbaI-E transforming fragment in human cervical and NIH 3T3 lines. Intervirology. 1992;33(4):187–96. doi: 10.1159/000150250. [DOI] [PubMed] [Google Scholar]
- Conti P, Kempuraj D, Frydas S, Kandere K, Boucher W, Letourneau R, Madhappan B, Sagimoto K, Christodoulou S, Theoharides TC. IL-10 subfamily members: IL-19, IL-20, IL-22, IL-24 and IL-26. Immunol Lett. 2003;88(3):171–4. doi: 10.1016/s0165-2478(03)00087-7. [DOI] [PubMed] [Google Scholar]
- Doniger J, Muralidhar S, Rosenthal LJ. Human cytomegalovirus and human herpesvirus 6 genes that transform and transactivate. Clin Microbiol Rev. 1999;12(3):367–82. doi: 10.1128/cmr.12.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickenscher H, Hor S, Kupers H, Knappe A, Wittmann S, Sticht H. The interleukin-10 family of cytokines. Trends Immunol. 2002;23(2):89–96. doi: 10.1016/s1471-4906(01)02149-4. [DOI] [PubMed] [Google Scholar]
- Godwin M, Cheung A, Jenkins C, Lewis Stern J, Plachter B, Pepperl-Klindworth S, Abendroth A, Slobedman B. Immunomodulation by a Viral Homologue Of IL10 Expressed by Human Cytomegalovirus during the Latent Phase of Infection. 31st International Herpesvirus Workshop. 2006:111. [Google Scholar]
- Jahan N, Razzaque A, Brady J, Rosenthal LJ. The human cytomegalovirus mtrII colinear region in strain Tanaka is transformation defective. J Virol. 1989;63(6):2866–9. doi: 10.1128/jvi.63.6.2866-2869.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins C, Abendroth A, Slobedman B. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J Virol. 2004;78(3):1440–7. doi: 10.1128/JVI.78.3.1440-1447.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji JD, Kim HJ, Rho YH, Choi SJ, Lee YH, Cheon HJ, Sohn J, Song GG. Inhibition of IL-10-induced STAT3 activation by 15-deoxy-Delta12,14-prostaglandin J2. Rheumatology (Oxford) 2005;44(8):983–8. doi: 10.1093/rheumatology/keh657. [DOI] [PubMed] [Google Scholar]
- Jones BC, Logsdon NJ, Josephson K, Cook J, Barry PA, Walter MR. Crystal structure of human cytomegalovirus IL-10 bound to soluble human IL-10R1. Proc Natl Acad Sci U S A. 2002;99(14):9404–9. doi: 10.1073/pnas.152147499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotenko SV. The family of IL-10-related cytokines and their receptors: related, but to what extent? Cytokine Growth Factor Rev. 2002;13(3):223–40. doi: 10.1016/s1359-6101(02)00012-6. [DOI] [PubMed] [Google Scholar]
- Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10) Proc Natl Acad Sci U S A. 2000;97(4):1695–700. doi: 10.1073/pnas.97.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer JA, Cutrone EC, Kotenko S. The Class II cytokine receptor (CRF2) family: overview and patterns of receptor-ligand interactions. Cytokine Growth Factor Rev. 2004;15(1):33–48. doi: 10.1016/j.cytogfr.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Ledru E, Fevrier M, Lecoeur H, Garcia S, Boullier S, Gougeon ML. A nonsecreted variant of interleukin-4 is associated with apoptosis: implication for the T helper-2 polarization in HIV infection. Blood. 2003;101(8):3102–5. doi: 10.1182/blood-2002-08-2499. [DOI] [PubMed] [Google Scholar]
- Logsdon NJ, Jones BC, Allman JC, Izotova L, Schwartz B, Pestka S, Walter MR. The IL-10R2 binding hot spot on IL-22 is located on the N-terminal helix and is dependent on N-linked glycosylation. J Mol Biol. 2004;342(2):503–14. doi: 10.1016/j.jmb.2004.07.069. [DOI] [PubMed] [Google Scholar]
- Meazza R, Verdiani S, Biassoni R, Coppolecchia M, Gaggero A, Orengo AM, Colombo MP, Azzarone B, Ferrini S. Identification of a novel interleukin-15 (IL-15) transcript isoform generated by alternative splicing in human small cell lung cancer cell lines. Oncogene. 1996;12(10):2187–92. [PubMed] [Google Scholar]
- Michelson S. Human cytomegalovirus escape from immune detection. Intervirology. 1999;42(5–6):301–7. doi: 10.1159/000053964. [DOI] [PubMed] [Google Scholar]
- Mocarski E, Courcelle CT. Cytomegaloviruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4. The Williams & Wilkins Co; Philadelphia, PA: 2001. pp. 2629–2673. [Google Scholar]
- Muralidhar S, Doniger J, Mendelson E, Araujo JC, Kashanchi F, Azumi N, Brady JN, Rosenthal LJ. Human cytomegalovirus mtrII oncoprotein binds to p53 and down-regulates p53-activated transcription. J Virol. 1996;70(12):8691–700. doi: 10.1128/jvi.70.12.8691-8700.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura H, Fujimoto A, Tamura N, Yajima T, Wajjwalku W, Yoshikai Y. A novel autoregulatory mechanism for transcriptional activation of the IL-15 gene by a nonsecretable isoform of IL-15 generated by alternative splicing. Faseb J. 2005;19(1):19–28. doi: 10.1096/fj.04-2633com. [DOI] [PubMed] [Google Scholar]
- Nishimura H, Washizu J, Nakamura N, Enomoto A, Yoshikai Y. Translational efficiency is up-regulated by alternative exon in murine IL-15 mRNA. J Immunol. 1998;160(2):936–42. [PubMed] [Google Scholar]
- Pass R. Cytomegalovirus. In: Knipe DM, Howley PM, editors. Fields Virology. The Williams & Wilkins Co; Philadelphia, PA: 2001. pp. 2675–2705. [Google Scholar]
- Penfold ME, Dairaghi DJ, Duke GM, Saederup N, Mocarski ES, Kemble GW, Schall TJ. Cytomegalovirus encodes a potent alpha chemokine. Proc Natl Acad Sci U S A. 1999;96(17):9839–44. doi: 10.1073/pnas.96.17.9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–79. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- Raftery MJ, Wieland D, Gronewald S, Kraus AA, Giese T, Schonrich G. Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus-encoded IL-10. J Immunol. 2004;173(5):3383–91. doi: 10.4049/jimmunol.173.5.3383. [DOI] [PubMed] [Google Scholar]
- Randolph-Habecker JR, Rahill B, Torok-Storb B, Vieira J, Kolattukudy PE, Rovin BH, Sedmak DD. The expression of the cytomegalovirus chemokine receptor homolog US28 sequesters biologically active CC chemokines and alters IL-8 production. Cytokine. 2002;19(1):37–46. doi: 10.1006/cyto.2002.0874. [DOI] [PubMed] [Google Scholar]
- Razzaque A, Jahan N, McWeeney D, Jariwalla RJ, Jones C, Brady J, Rosenthal LJ. Localization and DNA sequence analysis of the transforming domain (mtrII) of human cytomegalovirus. Proc Natl Acad Sci U S A. 1988;85(15):5709–13. doi: 10.1073/pnas.85.15.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque A, Zhu F, Jones C. Functional analysis of human cytomegalovirus morphological transforming region II (mtrII) Virology. 1991;181(1):399–402. doi: 10.1016/0042-6822(91)90513-b. [DOI] [PubMed] [Google Scholar]
- Saederup N, Mocarski ES., Jr Fatal attraction: cytomegalovirus-encoded chemokine homologs. Curr Top Microbiol Immunol. 2002;269:235–56. doi: 10.1007/978-3-642-59421-2_14. [DOI] [PubMed] [Google Scholar]
- Tan X, Lefrancois L. Novel IL-15 isoforms generated by alternative splicing are expressed in the intestinal epithelium. Genes Immun. 2006;7(5):407–16. doi: 10.1038/sj.gene.6364314. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Hoshino Y, Gold J, Hoshino S, Martiniuk F, Kurata T, Pine R, Levy D, Rom WN, Weiden M. Interleukin-10 induces inhibitory C/EBPbeta through STAT-3 and represses HIV-1 transcription in macrophages. Am J Respir Cell Mol Biol. 2005;33(4):406–11. doi: 10.1165/rcmb.2005-0140OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, Doniger J, Rosenthal LJ. A 79 amino acid oncogene is responsible for human cytomegalovirus mtrII induced malignant transformation. Arch Virol. 1994;136(1–2):161–72. doi: 10.1007/BF01538825. [DOI] [PubMed] [Google Scholar]
- van Cleef KW, Smit MJ, Bruggeman CA, Vink C. Cytomegalovirus-encoded homologs of G protein-coupled receptors and chemokines. J Clin Virol. 2006;35(3):343–8. doi: 10.1016/j.jcv.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Vasiliev AM, Vasilenko RN, Kulikova NL, Andreev SM, Chikileva IO, Puchkova GY, Kosarev IV, Khodyakova AV, Khlebnikov VS, Ptitsyn LR, Shcherbakov GY, Uversky VN, DuBuske LM, Abramov VM. Structural and functional properties of IL-4delta2, an alternative splice variant of human IL-4. J Proteome Res. 2003;2(3):273–81. doi: 10.1021/pr025586y. [DOI] [PubMed] [Google Scholar]
- Vink C, Smit MJ, Leurs R, Bruggeman CA. The role of cytomegalovirus-encoded homologs of G protein-coupled receptors and chemokines in manipulation of and evasion from the immune system. J Clin Virol. 2001;23(1–2):43–55. doi: 10.1016/s1386-6532(01)00184-6. [DOI] [PubMed] [Google Scholar]
- Wang J, Razzaque A. Mapping and DNA sequence analysis of the cytomegalovirus transforming domain III (mtrIII) Virus Res. 1993;30(3):221–38. doi: 10.1016/0168-1702(93)90092-2. [DOI] [PubMed] [Google Scholar]
- Wise L, McCaughan C, Tan CK, Mercer AA, Fleming SB. Orf virus interleukin-10 inhibits cytokine synthesis in activated human THP-1 monocytes, but only partially impairs their proliferation. J Gen Virol. 2007;88(Pt 6):1677–82. doi: 10.1099/vir.0.82765-0. [DOI] [PubMed] [Google Scholar]
- Wolk K, Kunz S, Asadullah K, Sabat R. Cutting edge: immune cells as sources and targets of the IL-10 family members? J Immunol. 2002;168(11):5397–402. doi: 10.4049/jimmunol.168.11.5397. [DOI] [PubMed] [Google Scholar]
- Wu S, Gessner R, Taube T, von Stackelberg A, Henze G, Seeger K. Expression of interleukin-10 splicing variants is a positive prognostic feature in relapsed childhood acute lymphoblastic leukemia. J Clin Oncol. 2005;23(13):3038–42. doi: 10.1200/JCO.2005.00.885. [DOI] [PubMed] [Google Scholar]
- Yamamoto-Tabata T, McDonagh S, Chang HT, Fisher S, Pereira L. Human cytomegalovirus interleukin-10 downregulates metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J Virol. 2004;78(6):2831–40. doi: 10.1128/JVI.78.6.2831-2840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsenko OP, Filipenko ML, Khrapov EA, Voronina EN, Kozlov VA, Sennikov SV. Alternative splicing of mRNA of mouse interleukin-4 and interleukin-6. Cytokine. 2004;28(4–5):190–6. doi: 10.1016/j.cyto.2004.08.009. [DOI] [PubMed] [Google Scholar]