

Abstract
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors in human gastrointestinal tract. We first found that most GISTs expressed KIT, a receptor tyrosine kinase encoded by protooncogene c-kit and that approximately 90% of the sporadic GISTs had somatic gain-of-function mutations of the c-kit gene. Since both GISTs and interstitial cells of Cajal (ICCs) were double-positive for KIT and CD34, GISTs were considered to originate from ICCs or their precursor cells. We also found that germline gain-of-function mutations of the c-kit gene resulted in familial and multiple GISTs with diffuse hyperplasia of ICCs as the preexisting lesion. Moreover, we found that about half of the sporadic GISTs without c-kit gene mutations had gain-of-function mutations of platelet-derived growth factor receptor alpha (PDGFRA) gene that encodes another receptor tyrosine kinase. Imatinib which is known to inhibit constitutively activated BCR-ABL tyrosine kinase in chronic myelogenous leukemia also inhibits constitutive activation of mutated KIT and PDGFRA, and is now being used for metastatic or unresectable GISTs as a molecular target drug. Mutational analyses of c-kit and PDGFRA genes are considered to be significant for prediction of effectiveness of imatinib and newly developed/developing other agents on GISTs. Some mouse models of familial and multiple GISTs have been genetically created, and may be useful for further investigation of GIST biology.
Key Words: Gastrointestinal stromal tumors, gain-of-function mutation, c-kit gene, PDGFRA gene, molecular target therapy
INTRODUCTION
Most mesenchymal tumors arising from the gastrointestinal (GI) musculature in humans are gastrointestinal stromal tumors (GISTs) [1]. We first found that a large proportion of GISTs were positive for c-kit gene product, KIT receptor tyrosine kinase (TK) and had gain-of-function mutation of the c-kit gene [1]. These findings resulted in a general understanding of cellular origin and developmental mechanism of GIST, and then rapid progress concerning diagnosis and treatment of GISTs has been made. Immunohistochemistry of KIT is essential for pathological diagnosis of GISTs, and molecular target therapy using TK inhibitors such as imatinib is now being carried out for patients with metastatic or unrectable GISTs. Moreover, we found that the minority of GISTs had gain-of-function mutations of platelet-derived growth factor receptor alpha (PDGFRA) gene [2]. Here, we describe a compendium for developmental biology, diagnosis and therapy of GISTs.
HISTORY OF PATHOLOGICAL CLASSIFICATION OF GI MESENCHYMAL TUMORS
Previously, most of the GI mesenchymal tumors had been thought to originate from smooth muscle cells and they were pathologically diagnosed as leiomyomas, leiomyoblas-tomas or leiomyosarcomas. After electron microscopic and immunohistochemical analyses, however, many investigators have noticed that the tumor cells lacked differentiation features of smooth muscle cells or schwann cells [3–5]. They have also known that a small proportion of GI mesenchymal tumors were genuine smooth muscle tumors or neurogenic tumors [3–5]. Based on such recognition, the concept of GISTs was proposed for non-myogenic and non-neurogenic GI mesenchymal tumors [6].
In the first half of the 1990’s, it was reported that approximately 70% of non-myogenic and non-neurogenic GI mesenchymal tumors express CD34, which is one of the adhesion molecules [7–10]. But it was unclear whether approximately 30% of them which do not express CD34 but have similar histological findings should be included in the same tumor type, i.e. GISTs. Moreover, the CD34 expression in GISTs did not make a suggestion on cellular origin of GISTs. Therefore, a category of GISTs remained obscure. In 1998, we found that almost all non-myogenic and non-neurogenic GI mesenchymal tumors were positive for KIT by immunohistochemistry [1]. The discovery of common KIT expression in non-myogenic and non-neurogenic GI mesenchymal tumors contributed to understanding cellular origin of GISTs and to establishing the concept of GISTs [1]. At present, it is generally accepted that KIT-positive GI mesenchymal tumors are nearly equal to GISTs. Detailed description concerning cellular origin of GISTs appears in the later section.
c-KIT GENE, KIT AND RELATIONSHIP BETWEEN KIT AND PDGFRA
The c-kit gene was cloned in 1988 as a normal cellular homologue of v-kit oncogene in the genome of the Hardy Zuckermann 4 feline sarcoma virus [11–13]. It encodes KIT receptor TK, which molecular weight is approximately 145 Kd. KIT consists of extracellular (EC) domain, transmem-brane (TM) domain, juxtamembrane (JM) domain and TK domain Fig. (1). The EC domain is composed of five immu-noglobulin-like repeats and the TK domain is split into two domains (TK I and TK II domains) by a kinase insert Fig. (1). The JM domain is a portion between TM domain and TK domain, and is considered to play a role in regulation of KIT dimerization. KIT is classified into type III receptor TK subfamily as well as PDGFRs and colony-stimulating factor 1 receptor. Particularly, amino acid sequence of KIT shows high homology with that of PDGFRA, and c-kit gene and PDGFRA gene are located adjacently on human chromosome 4 and mouse chromosome 5 [14–16]. Gene duplication is considered to make these two similar genes. The ligand for KIT is stem cell factor (SCF) that was identified in 1990 [17–19]. Under unbound condition of KIT and SCF, the structure of JM domain is considered to inhibit dimerization of KIT. However, the conformational change of the JM domain by the binding of KIT and SCF appears to release the inhibitory effect. Thus, KIT forms dimer by binding of dimerized SCF. The KIT dimerization induces autophosphorylation of KIT on particular tyrosine residues, and the activated KIT receptor TK leads several signal transduction systems such as PI3K/Akt, Ras/MAPK and Stat pathways to activation Fig. (1) [20]. Two types of c-kit gene mutations are recognized. One is loss-of-function mutation in which the TK activity of KIT is lost. The other is gain-of-function mutation in which TK activity of KIT is constitutively increased without the binding of SCF. The c-kit gene resides in the mouse W locus and the rat Ws locus [21–24]. Since SCF-KIT signal pathways play a crucial role for the development of several cell types such as erythrocytes, mast cells, germ cells and mela-nocytes, these cells are deficient in mice and rats possessing two alleles of loss-of function mutant genes at W locus and Ws locus respectively [21–24]. On the other hand, the gain-of-function mutations of the c-kit gene were first reported in human mast cell leukemia cell line (HMC-1) by Furitsu et al. in 1993 [25]. They demonstrated that the cells had constitutive phosphorylation of KIT and two point mutations of c-kit gene resulting in amino acid substitution of Gly for Val at codon 560 (Val560Gly) in JM domain and Val for Asp at codon 816 (Asp816Val) in TK II domain [25]. The mutation corresponding to Asp816Val of the human HMC-1 cell line was found in the P-815 mouse mastocytoma cell line and the RBL-2H3 rat mast cell leukemia cell line [26,27]. In addition, the Asp816Val has been found in several clinical cases with mast cell neoplasms [28,29]. These results suggested that gain-of-function mutations of the c-kit gene were a cause of mast cell neoplasms.
Fig. (1).
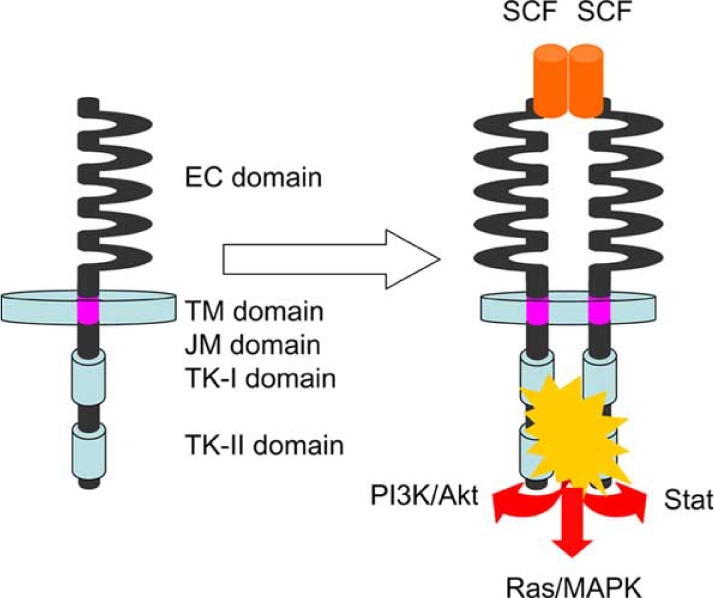
Schemas of KIT structure and of KIT activation by SCF binding. EC, extracellular; TM, transmembrane; JM, juxtamembrane; and TK, tyrosine kinase.
CELLULAR ORIGIN OF GISTs
Interstitial cells of Cajal (ICCs) were proved to play an important role as pacemakers of GI movement through the electrophysiological examination [30]. ICCs are distributed in musculature throughout the GI tract, and abundantly localize around myenteric plexus [31]. Since ICCs previously could not be easily identified by commonly used methods, ICC research had not fully progressed. In 1992, Maeda et al. [32] reported that there were KIT-positive cells in the GI musculature of mice and that the distribution of those cells was reminiscent of that of ICCs. They also showed that remarkable decrease of the KIT-positive cells was observed in the small intestine of W mutant mice [32]. We reported deficiency of c-kit mRNA-expressing cells in Ws mutant rats [33]. In 1995, Huizinga et al. [34] demonstrated that the KIT-positive cells were ICCs themselves. These results showed that ICCs could be easily and specifically identified by detection of KIT expression and that the SCF-KIT system was crucially required for the development of ICCs as well as erythrocytes, mast cells, germ cells and melanocytes [32–34]. During our ICC study, we hypothesized that ‘ICC tumors’ might be induced by gain-of-function mutation of c-kit gene, based on the facts that loss-of-function mutation of c-kit gene results in deficiency of ICCs and mast cells and that gain-of-function mutation of c-kit gene results in mast cell tumors as described above. Since the origin of GISTs had not been clarified at that time, we supposed that GISTs might originate from ICCs. KIT was used as a possible marker for identification of ‘ICC tumors’, and immunohisto-chemistry for KIT was performed on GI mesenchymal tumors. Although true smooth muscle tumors and true schwan- nomas were negative for KIT, almost all GISTs showed positive KIT staining Fig. (2) [1]. As described previously [10], approximately 70% of non-myogenic and non-neuroge- nic GI mesenchymal tumors, i.e, GISTs, expressed CD34 [1]. We also demonstrated that ICCs were positive for CD34 like GISTs [1]. We concluded that GISTs originate from ICCs because 1) both GISTs and ICCs are double-positive for KIT and CD34, 2) ICCs are the only cells that are double-positive for KIT and CD34 in the normal GI wall of humans [1]. The finding described by Mazur et al. in 1983 that the tumor cells of GISTs resembled the cells surrounding Auerbach’s ganglion cells in ultrastructure might indicate the ICC origin of GISTs [6]. Our conclusion was supported by some additional data that both GISTs and ICCs expressed the embryonic isoform of smooth muscle myosin heavy chains, intermediate filament nestin and protein kinase C theta [35–37]. After our discovery of KIT expression in GISTs, KIT became the most reliable marker for pathological diagnosis of GISTs.
Fig. (2).
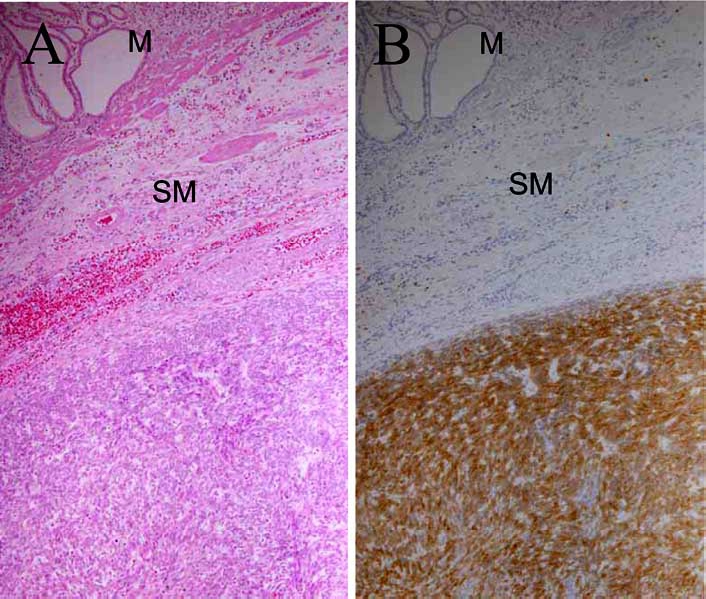
Histology (A) and KIT immunohistochemistry (B) of a GIST. M, mucosal layer; SM, submucosal layer.
SOMATIC GAIN-OF-FUNCTION MUTATIONS OF c-KIT GENE IN SPORADIC GISTs
We examined whether GISTs also had the gain-of-function mutations of c-kit gene like mast cell tumors. When sequencing of the whole coding region of the c-kit gene was carried out, mutations were detected in five of six GISTs [1]. Each mutation was different but all were located at JM domain encoded by exon 11 [1]. We examined whether the c-kit gene mutations found in GISTs were of gain-of-function. The mutant c-kit cDNAs were transiently introduced into 293T human embryonic kidney cell line, and the KIT auto-phosphorylation was analyzed. The mutant KIT found in GISTs showed constitutive tyrosine phosphorylation without SCF stimulation [1]. When the equivalent mutations were introduced into the mouse c-kit cDNA and they were stably transfected into the interleukin-3 (IL-3)-dependent Ba/F3 murine pro-B cell line, the Ba/F3 cells became to grow autonomously without IL-3 [1]. Transplanted Ba/F3 cells with mutated c-kit cDNA formed tumors in back of nude mice [1]. These results suggested that the c-kit gene mutations in GISTs are of gain-of-function and these mutations induce autonomous cell proliferation, i.e. development of GISTs.
Further analyses using many primary sporadic GIST cases revealed that 85 to 90% of all sporadic GISTs had c-kit gene mutations [38–41]. Seventy-five to eighty per cent of all sporadic GISTs have c-kit gene mutations at JM domain, 5 to 13% of them at EC domain encoded by exon 9, less than 4% at TK I domain encoded by exon13, and less than 4% at TK II domain encoded by exon17 Fig. (3) [39–45]. Various mutations are observed at JM domain, but the particular mutations are detected at EC, TK I, and TK II domains [38–45]. The EC domain mutation is usually duplication of codons 502 and 503, the JM I domain mutation Lys642Glu, and the TK II domain mutation Asp822Lys or Asp822His [42–45].
Fig. (3).

Locations and types of c-kit gene mutations and their frequency in sporadic GISTs. EC, extracellular; TM, transmembrane; JM, juxtamembrane; and TK, tyrosine kinase.
GERMLINE GAIN-OF-FUNCTION MUTATIONS OF c-KIT GENE IN FAMILIAL AND MULTIPLE GISTs
At least twelve families with multiple GISTs and germ-line c-kit gene mutations have been reported [46–58]. Patients of eight families have the mutation at JM domain [46–48,50,51,53–56], and those of one family at EC domain [57], those of one family at TK I domain [49], and those of two families at TK II domain [52,58]. The germline mutations are demonstrated to be of gain-of-function, and are considered to be the cause of multiple GIST development. Interestingly, remarkable diffuse proliferation of ICCs is observed as a preexisting lesion of multiple GISTs in small intestine of the patients [46,48,49,52]. Although mild symptom of acha-lasia-like dysphagia with manometrically abnormal simultaneous contraction of the esophagus has been reported in some patients [52], most of the patients do not appear to complain of any GI symptoms other than mild dysphagia. The c-kit gene mutation or ICC proliferation might not induce severely abnormal GI motility. Patients of some families show hyperpigmentation of perineal, perioral and digital area and/or mast cell neoplasms [46,50,51,53–55]. These findings might be attributable to the effect of germline gain-of-function mutation of c-kit gene on melanocytes and mast cells, but the reason why all of the patients with the mutation do not show the symptoms is not unclear.
GAIN-OF-FUNCTION MUTATIONS OF PDGFRA GENE IN GISTs
As described above, c-kit gene mutations are not detected in 10 to 15% of sporadic GISTs. In 2003, Heinrich et al. and we reported that somatic mutations of PDGFRA gene were observed in sporadic GISTs lacking c-kit gene mutations [2,59]. The frequency reported by Heinrich et al. was 35% (14 of 40 GISTs without c-kit gene mutations), and that in our result was 62.5% (5 of 8 GISTs without c-kit gene mutations) [2,59]. The mutations were localized at JM domain encoded by exon 12 or TK II domain encoded by exon 18 of PDGFRA gene [2,59]. Recently, PDGFRA gene mutation of TK I domain encoded by exon 14 has been reported as a rare mutation-type [60,61]. KIT and PDGFRA have extremely similar amino acid sequence as described above, and exon 12 and exon 18 of PDGFRA gene are comparable to exon 11 and exon 17 of the c-kit gene respectively. Most frequent PDGFRA mutation appears to be Asp842Val at exon 18, and the mutation is structurally comparable to Asp816Val at exon 17 of c-kit gene. Interestingly, the Asp816Val is the mutation that commonly found in human mast cell tumors but never in primary GISTs [2,59]. The PDGFRA gene mutations were proved to result in constitutive tyrosine phos-phorylation of PDGFRA without PDGF-AA stimulation, and they were considered to be of gain-of-function [2,59]. These results suggested that the PDGFRA gene mutations are also another cause of GISTs. Sporadic GISTs with both c-kit and PDGFRA gene mutations have not been found, and these two types of mutations are considered to be mutually exclusive. Only one family with germline PDGFRA gene mutation of Asp846Tyr in the TK II domain has been reported [62].
MOLECULAR TARGET THERAPY FOR GISTs
Surgical resection is the basis of treatment for GISTs both in the past and now. However, the patients with unre-sectable or metastatic GISTs did not have practically remarkable effect of surgery and conventional chemotherapy. One of the TK inhibitors, imatinib, has completely revolutionized the status. Imatinib, a 2-phenylaminopyrimidine derivative, was initially developed as a specific inhibitor of PDGFRs and BCR-ABL [61]. BCR-ABL, constitutively activated non-receptor TK, is a product of gene fusion formed by translocation of chromosomes (Philadelphia chromosome) in patients with chronic myelogenous leukemia (CML) [63]. Its constitutive activation is the cause of CML, and imatinib is now successfully applied to patients with CML. In addition to the inhibitory effect of imatinib on PDGFR and BCR-ABL, Buchdunger et al. [64] found an inhibitory effect on wild-type KIT. Furthermore, it was confirmed by us [65] and others [66] that imatinib also inhibits various types of mutant KIT found in GISTs. These results suggested that imatinib might be effective for the treatment of patients with advanced GISTs, and in fact imatinib was first administered for a recurrent GIST patient with JM domain c-kit gene mutation in Finland [67]. Marvelous effect was observed in the patient, and multicenter trials of imatinib treatment for patients with unresectable and metastatic GISTs were performed thereafter. Successful clinical effect was confirmed in the majority of the patients, [68] and the agent has been approved as a therapeutic drug in many countries. During the trials, it became clear that the locations of c-kit and PDGFRA gene mutations were related to the effectiveness of imatinib on GIST patients [65,69-71]. The rate of partial response on GIST patients with JM domain mutations of the c-kit gene was 83.5%, whereas that with EC domain mutations was 47.8% [69]. GIST patients with PDGFRA gene mutation or with wild-type c-kit and wild-type PDGFRA gene appear to show lower rate of partial response. Mutational analyses in c-kit and PDGFRA genes are considered to be valuable for the prediction of imatinib effectiveness. As described above, imatinib is effective in the majority of GISTs, but there are some primarily resisitant GISTs for imatinib. Moreover, the secondarily resistant clones often develop after the initial successful treatment by imatinib. The main cause of the secondary resistance for imatinib is considered to be the secondary c-kit gene mutation on the identical gene [72–74]. The frequently observed secondary mutations are Val654Ala at exon 13, Thr670Ile at exon 14, Tyr823Asp and so on. The types of secondary mutations are basically not observed in primary GISTs. Interestingly, codon 820 and codon 822 that are the mutation sites observed in multiple and familial GISTs and sporadic GISTs respectively have been reported as the mutation sites in secondarily resistant GISTs. New agents against the imatinib-resistant tumors have been developed or are developing. Among these drugs, one of indolinone-derivative TK inhibitors, SUTENT/SU11248, has been clarified to show clinical effect for imatinib-resistant GISTs with exon 9 c-kit gene mutation, and in 2006 it has been approved by Food and Drug Administration in USA.
CONCLUSIONS
Although it is clear that gain-of-function mutations of c-kit and PDGFRA genes play an important role for the development of GISTs, unsolved subjects concerning the mechanism of GIST development still remain. Since multiple GISTs develop in later with ICC hyperplasia as a preexisting lesion [46,48,49,52], another event such as other gene mutations in addition to c-kit gene mutation appears to be required for neoplastic change from ICC hyperplasia. Such gene mutations have not been reported yet. In some NF1 patients, development of multiple GISTs has been reported [75,76], but we [77] and others [78,79] demonstrated that the rate of c-kit gene mutations was very low in NF1 GISTs. There may be an unknown mechanism for GIST development in NF1 patients. Further analyses for the development of GISTs without c-kit and PDGFRA gene mutations other than NF1 patients also have to be done.
ABBREVIATIONS
- CML
Chronic myelogenous leukemia
- EC
Extracellular
- GI
Gastrointestinal
- GIST
Gastrointestinal stromal tumor
- ICCs
Interstitial cells of Cajal
- IL-3
Interleukin-3
- JM
Juxtamembrane
- PDGFRA
Platelet-derived growth factor receptor alpha
- SCF
Stem cell factor
- TM
Transmembrane
- TK
Tyrosine kinase
REFERENCES
- 1.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Tunio GM, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 2.Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, Kitamura Y. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125:660–667. doi: 10.1016/s0016-5085(03)01046-1. [DOI] [PubMed] [Google Scholar]
- 3.Weiss RA, Mackay B. Malignant smooth muscle tumors of the gastrointestinal tract:an ultrastructural study of 20 cases. Ultrastruct Pathol. 1981;2:231–240. doi: 10.3109/01913128109048306. [DOI] [PubMed] [Google Scholar]
- 4.Saul SH, Mark L, Rast BS, Brooks JJ. The immuno-histochemistry of gastrointestinal stromal tumors. Am J Surg Pathol. 1987;11:464–473. doi: 10.1097/00000478-198706000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Fraquemont DW, Freierson HF. Muscle differentiation and clinicopathologic features of gastrointestinal stromal tumors. Am J Surg Pathol. 1992;16:947–954. doi: 10.1097/00000478-199210000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Mazur MT. Gastric stromal tumors. Am J Surg Pathol. 1983;7:507–519. doi: 10.1097/00000478-198309000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Traweek ST, Kandaraft PL, Mehta P, Battifora H. The human hematopoietic progenitor cell antigen (CD34) in vascular neoplasia. Am J Clin Pathol. 1991;96:25–31. doi: 10.1093/ajcp/96.1.25. [DOI] [PubMed] [Google Scholar]
- 8.van de Rijn M, Hendrickson MR, Rouse RV. CD34 expression by gastrointestinal tract stromal tumors. Hum Pathol. 1994;25:766–771. doi: 10.1016/0046-8177(94)90245-3. [DOI] [PubMed] [Google Scholar]
- 9.Monihan JM, Carr NJ, Sobin LH. CD34 immunoexpression in stromal tumours of the gastrointestinal tract and in mesenteric fibromatoses. Histopathology. 1994;25:469–473. doi: 10.1111/j.1365-2559.1994.tb00009.x. [DOI] [PubMed] [Google Scholar]
- 10.Miettinen M, Virolainen M, Maarit-Sarlomo-Rikala Gastrointestinal stromal tumors - value of CD34 antigen in their identification and separation from true leiomyomas and schwannomas. Am J Surg Pathol. 1995;19:207–216. doi: 10.1097/00000478-199502000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Besmer P, Murphy JE, George PC, Qiu F, Bergold PJ, Lederman L, Snyder HW, Jr, Broudeur D, Zuckerman EE, Hardy WD. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature. 1986;320:415–421. doi: 10.1038/320415a0. [DOI] [PubMed] [Google Scholar]
- 12.Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A. Human protooncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6:3341–3351. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu FH, Ray HP, Brown Y, Barker PE, Jhanwar S, Ruddle FH, Besmer P. Primary structure of c-kit: Relationship with the CSF-1/PDGF receptor kinase family-oncogenic activation of v-kit involves deletion of extracellular domain and C terminus. EMBO J. 1988;7:1003–1011. doi: 10.1002/j.1460-2075.1988.tb02907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reith AD, Bernstein A. In: Genes and phenotypes, genomic analysis. Davies KE, Tilgham SM, editors. Vol. 3. New York: Cold spring harbor laboratory Press; 1991. pp. 105–133. [Google Scholar]
- 15.Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:88–89. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- 16.Geissler EN, Ryan MA, Housman DE. The dominant-white spotting (W) locus of the mouse encodes the c-kit protooncogene. Cell. 1988;55:185–192. doi: 10.1016/0092-8674(88)90020-7. [DOI] [PubMed] [Google Scholar]
- 17.Williams DE, Eisenman J, Baird A, Rauch C, Ness KV, March CJ, Park LS, Martin U, Mochizuki DY, Boswell HS, Burgess GS, Cosman D, Lyman SD. Identification of a ligand for the c-kit proto-oncogene. Cell. 1990;63:167–174. doi: 10.1016/0092-8674(90)90297-r. [DOI] [PubMed] [Google Scholar]
- 18.Flanagan JG, Leder P. The kit ligand: A cell surface molecule altered in steel mutant fibroblasts. Cell. 1990;63:185–194. doi: 10.1016/0092-8674(90)90299-t. [DOI] [PubMed] [Google Scholar]
- 19.Zsebo KM, Williams DA, Geissler EN, Broudy YC, Martin FH, Atkins HL, Hsu RY, Birkitt NC, Okino KH, Murdock DC, Jacobson FW, Langley KE, Smith KA, Takeishi T, Cattanach BM, Galli SJ, Suggs SV. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63:213–224. doi: 10.1016/0092-8674(90)90302-u. [DOI] [PubMed] [Google Scholar]
- 20.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 21.Russell ES. Hereditary anemias of the mouse: a review for geneticists. Adv Genet. 1979;20:357–459. [PubMed] [Google Scholar]
- 22.Nocka K, Tan JC, Chiu E, Chu TY, Ray P, Traktman P, Besmer P. Molecular bases of dominant negative and loss of function mutation at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990;9:1805–1813. doi: 10.1002/j.1460-2075.1990.tb08305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsujimura T, Hirota S, Nomura S, Niwa Y, Yamazaki M, Tono T, Morii E, Kim HM, Kondo K, Nishimune Y, Kitamura Y. Characterization of Ws mutant allele of rats :a 12-base deletion in tyrosine kinase domain of c-kit gene. Blood. 1991;78:1942–1946. [PubMed] [Google Scholar]
- 24.Tei H, Kasugai T, Tsujimura T, Adachi S, Furitsu T, Tohya K, Kimura M, Zsebo KM, Newlands GF, Miller HRP, Kanakura Y, Kitamura Y. Characterization of cultured mast cells derived from Ws/Ws mast cell-deficient rats with a small deletion at tyrosine kinase domain of c-kit. Blood. 1994;83:916–925. [PubMed] [Google Scholar]
- 25.Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, Sugahara H, Butterfield JH, Ashman LK, Kanayama Y, Matsuzawa Y, Kitamura Y, Kanakura Y. Identification of mutations in the coding sequence of the protooncogene c-kit in a human mast cell leukemia cell line causing ligand- independent activation of c-kit product. J Clin Invest. 1993;92:1736–1744. doi: 10.1172/JCI116761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsujimura T, Furitsu T, Morimoto M, Isozaki K, Nomura S, Matsuzawa Y, Kitamura Y, Kanakura Y. Ligand-independent activation of c-kit receptor tyrosine kinase in a murine mastocytoma cell line P-815 generated by a point mutation. Blood. 1994;83:2619–2626. [PubMed] [Google Scholar]
- 27.Tsujimura T, Furitsu T, Morimoto M, Kanayama Y, Nomura S, Matsuzawa Y, Kitamura Y, Kanakura Y. Substitution of an aspartic acid results in constitutive activation of c-kit receptor tyrosine kinase in a rat tumor mast cell line RBL-2H3. Int Arch Allergy Immunol. 1995;106:377–385. doi: 10.1159/000236870. [DOI] [PubMed] [Google Scholar]
- 28.Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, Metcalfe DD. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA. 1995;92:10560–10564. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Longley BJ, Tyrrell L, Lu SZ, Ma YS, Langley K, Ding TG, Duffy T, Jacobs P, Tang LH, Modlin I. Somatic c-kit activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312–314. doi: 10.1038/ng0396-312. [DOI] [PubMed] [Google Scholar]
- 30.Barajas-Lopetz C, Berezin I, Daniel EE, Huizinga JD. Pacemaker activity recorded in interstitial cells of Cajal of the gastrointestinal tract. Am J Physiol. 1989;257:C830–835. doi: 10.1152/ajpcell.1989.257.4.C830. [DOI] [PubMed] [Google Scholar]
- 31.Christensen J. The enteric nervous system. In: Kumar D, Wingate D, editors. An illustrated guide to gastrointestinal motility. 2nd. Edinburgh, England: Churchill Livingstone; 1993. pp. 10–31. [Google Scholar]
- 32.Maeda H, Yamagata A, Nishikawa S, Yoshinaga K, Kobayashi S, Nishi K, Nishikawa SI. Requirement of c-kit for development of intestinal pacemaker system. Development. 1992;116:369–375. doi: 10.1242/dev.116.2.369. [DOI] [PubMed] [Google Scholar]
- 33.Isozaki K, Hirota S, Nakama A, Miyagawa JI, Shinomura Y, Xu Z, Nomura S, Kitamura Y. Disturbed intestinal movement, bile reflux to the stomach, and deficiency of c-kit expressing cells in Ws/Ws mutant rats. Gastroenterology. 1995;109:456–464. doi: 10.1016/0016-5085(95)90333-x. [DOI] [PubMed] [Google Scholar]
- 34.Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373:347–349. doi: 10.1038/373347a0. [DOI] [PubMed] [Google Scholar]
- 35.Sakurai S, Fukasawa T, Chong JM, Tanaka A, Fukayama M. Embryonic form of smooth musle myosin heavy chain (SMemb/MHC-B) in gastrointestinal stromal tumor and interstitial cells of Cajal. Am J Pathol. 1999;154:23–28. doi: 10.1016/S0002-9440(10)65246-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsujimura T, Makiishi-Shimobayashi C, Lundkvist J, Lendahl U, Nakasho K, Sugihara A, Iwasaki T, Mano M, Yamada N, Yamashita K, Toyosaka A, Terada N. Expression of the intermediate filament nestin in gastrointestinal stromal tumors and interstitial cells of Cajal. Am J Pathol. 2001;158:817–823. doi: 10.1016/S0002-9440(10)64029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blay P, Astudillo A, Buesa JM, Campo E, Abad M, Garcia-Garcia J, Miquel R, Marco V, Sierra M, Losa R, Lacave A, Brana A, Balbin M, Freije JM. Protein kinase C theta is highly expressed in gastrointestinal stromal tumors but not in other mesenchymal neoplasias. Clin Cancer Res. 2004;10:4089–4095. doi: 10.1158/1078-0432.CCR-04-0630. [DOI] [PubMed] [Google Scholar]
- 38.Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M. Mutations in exon 11 of c-kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol. 1999;154:53–60. doi: 10.1016/S0002-9440(10)65250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moskaluk CA, Tian Q, Marshall CR, Rumpel CA, Franquemont DW, Frierson HF., Jr Mutations of c-kit JM domain are found in a minority of human gastrointestinal stromal tumors. Oncogene. 1999;18:1897–1902. doi: 10.1038/sj.onc.1202496. [DOI] [PubMed] [Google Scholar]
- 40.Sakurai S, Fukasawa T, Chon JM, Tanaka A, Fukayama M. c-kit gene abnormality in gastrointestinal stromal tumors (tumors of interstitial cells of Cajal) Jpn J Cancer Res. 1999;90:1321–1328. doi: 10.1111/j.1349-7006.1999.tb00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakahara M, Isozaki K, Hirota S, Miyagawa JI, HaseSawada N, Taniguchi M, Nishida T, Kanayama S, Kitamura Y, Shinomura Y, Matsuzawa Y. A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology. 1998;115:1090–1095. doi: 10.1016/s0016-5085(98)70079-4. [DOI] [PubMed] [Google Scholar]
- 42.Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, Xiao S, Singer S, Fletcher CDM, Fletcher JA. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol. 2000;156:791–795. doi: 10.1016/S0002-9440(10)64946-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lasota J, Wozniak A, Sarlomo-Rikala M, Rys J, Kordek R, Nassar A, Sobin LH, Miettinen M. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. Am J Pathol. 2000;157:1091–1095. doi: 10.1016/S0002-9440(10)64623-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirota S, Nishida T, Isozaki K, Taniguchi M, Nakamura J, Okazaki T, Kitamura Y. Gain-of-function mutation at the extracellular domain of KIT in gastrointestinal stromal tumours. J Pathol. 2001;193:505–510. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH818>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 45.Kinoshita K, Isozaki K, Hirota S, Nishida T, Chen H, Nakahara M, Nagasawa Y, Ohashi A, Shinomura Y, Kitamura Y, Matsuzawa Y. c-kit gene mutation at exon 17 or 13 is very rare in sporadic gastrointestinal stromal tumors. J Gastroenterol Hepatol. 2003;18:147–151. doi: 10.1046/j.1440-1746.2003.02911.x. [DOI] [PubMed] [Google Scholar]
- 46.Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T, Takabayashi A, Matsuda H, Kitamura Y. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nature Genet. 1998;19:323–324. doi: 10.1038/1209. [DOI] [PubMed] [Google Scholar]
- 47.O’Brien P, Kapusta L, Dardick I, Axler J, Gnidec A. Multiple familial gastrointestinal autonomic nerve tumors and small intestinal neuronal dysplasia. Am J Surg Pathol. 1999;23:198–204. doi: 10.1097/00000478-199902000-00009. [DOI] [PubMed] [Google Scholar]
- 48.Hirota S, Okazaki T, Kitamura Y, O'Brien P, Kapusta L, Dardick I. Cause of familial and multiple gastrointestinal autonomic nerve tumors with hyperplasia of interstitial cells of Cajal is germline mutation of the c-kit gene. Am J Surg Pathol. 2000;24:326–327. doi: 10.1097/00000478-200002000-00045. [DOI] [PubMed] [Google Scholar]
- 49.Isozaki K, Terris B, Belghiti J, Schiffmann S, Hirota S, Vanderwinden JM. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am J Pathol. 2000;157:1581–1585. doi: 10.1016/S0002-9440(10)64795-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maeyama H, Hidaka E, Ota H, Minami S, Kajiyama M, Kuraishi A, Mori H, Matsuda Y, Wada S, Sodeyama H, Nakata S, Kawamura N, Hata S, Watanabe M, Iijima Y, Katsuyama T. Familial gastrointestinal stromal tumor with hyperpigmentation : association with a germline mutation of the c-kit gene. Gastroenterology. 2001;120:210–215. doi: 10.1053/gast.2001.20880. [DOI] [PubMed] [Google Scholar]
- 51.Beghini A, Tibiletti MG, Roversi G, Chiaravalli AM, Serio G, Capella C, Larizza L. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer. 2001;92:657–662. doi: 10.1002/1097-0142(20010801)92:3<657::aid-cncr1367>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 52.Hirota S, Nishida T, Isozaki K, Taniguchi M, Nishikawa K, Ohashi A, Takabayashi A, Obayashi T, Okuno T, Kinoshita K, Chen H, Shinomura Y, Kitamura Y. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology. 2002;122:1493–1499. doi: 10.1053/gast.2002.33024. [DOI] [PubMed] [Google Scholar]
- 53.Robson ME, Glogowski E, Sommer G, Antonescu CR, Nafa K, Maki RG, Ellis N, Besmer P, Brennan M, Offit K. Pleomorphic characteristics of a germ-line KIT mutation in a large kindred with gastrointestinal stromal tumors, hyperpigmentation, and dysphagia. Clin Cancer Pathol. 2004;10:1250–1254. doi: 10.1158/1078-0432.ccr-03-0110. [DOI] [PubMed] [Google Scholar]
- 54.Carballo M, Aguilar F, Pol MA, Gamundi MJ, Hernan I, Martinez-Gimeno M. Novel c-KIT germline mutation in a family with gastrointestinal stromal tumors and cutaneous hyperpigmentation. Am J Med Genet A. 2005;132:361–364. doi: 10.1002/ajmg.a.30388. [DOI] [PubMed] [Google Scholar]
- 55.Li FP, Fletcher JA, Heinrich MC, Garber JE, Sallan SE, Curiel-Lewandrowski C, Duensing A, van de Rijn, Schnipper LE, Demetri GD. Familial gastrointestinal stromal tumor syndrome: phenotype and molecular features in a kindred. J Clin Oncol. 2005;23:2735–2743. doi: 10.1200/JCO.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 56.Kim HJ, Lim SJ, Park K, Yuh YJ, Jang SJ, Choi J. Multipe gastrointestinal stromal tumors with a germline c-kit mutation. Pathol Int. 2005;55:655–659. doi: 10.1111/j.1440-1827.2005.01885.x. [DOI] [PubMed] [Google Scholar]
- 57.Hartmann K, Wardelmann E, Ma Y, Merkelbach-Bruse S, Preussner LM, Woolery C, Baldus SE, Heinicke T, Thiele J, Buettner R, Longley BJ. Novel germline mutation of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology. 2005;129:1042–1046. doi: 10.1053/j.gastro.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 58.O’Riain C, Corless CL, Heinrich MC, Keegan D, Vioreanu M, Maguire D, Sheahan K. Gastrointestinal stromal tumors: insights from a new familial GIST kindred with unusual genetic and pathologic features. Am J Surg Pathol. 2005;29:1680–1683. doi: 10.1097/01.pas.0000173024.79852.08. [DOI] [PubMed] [Google Scholar]
- 59.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 60.Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J, Heinrich MC. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23:5357–5364. doi: 10.1200/JCO.2005.14.068. [DOI] [PubMed] [Google Scholar]
- 61.Lasota J, Stachura J, Miettinen M. GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumors with epithelioid morphology. Lab Invest. 2006;86:94–100. doi: 10.1038/labinvest.3700360. [DOI] [PubMed] [Google Scholar]
- 62.Chompret A, Kannengiesser C, Barrois M, Terrier P, Dahan P, Tursz T, Lenoir GM, Bressac-De Paillerets B. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology. 2004;126:318–321. doi: 10.1053/j.gastro.2003.10.079. [DOI] [PubMed] [Google Scholar]
- 63.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 64.Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, Lydon NB. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- 65.Chen H, Isozaki K, Kinoshita K, Ohashi A, Shinomura Y, Matsuzawa Y, Kitamura Y, Hirota S. Imatinib inhibits various types of activating mutant KIT found in gastrointestinal stromal tumors. Int J Cancer. 2003;105:130–135. doi: 10.1002/ijc.11025. [DOI] [PubMed] [Google Scholar]
- 66.Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD, Fletcher JA, Demetri GD. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20:5054–5058. doi: 10.1038/sj.onc.1204704. [DOI] [PubMed] [Google Scholar]
- 67.Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, Silberman S, Capdeville R, Dimitrijevic S, Druker B, Demetri GD. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344:1052–1056. doi: 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- 68.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 69.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CDM, Silberman S, Dimitrijevic S, Flecther JA. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 70.Ma Y, Zeng S, Metcalfe DD, Akin C, Dimitrijevic S, Butterfield JH, McMahon G, Longley BJ. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. 2002;99:1741–1744. doi: 10.1182/blood.v99.5.1741. [DOI] [PubMed] [Google Scholar]
- 71.Ohashi A, Kinoshita K, Isozaki K, Nishida T, Shinomura Y, Kitamura Y, Hirota S. Different inhibitory effect of Imatinib on phosphorylation of MAPK and Akt, and cell proliferation in cells expressing different types of mutant PDGF receptor alpha. Int J Cancer. 2004;111:317–321. doi: 10.1002/ijc.20305. [DOI] [PubMed] [Google Scholar]
- 72.Wakai T, Kanda T, Hirota S, Ohashi A, Shirai Y, Hatakeyama K. Late resistance to imatinib therapy associated with a second KIT mutation in metastatic gastrointestinal stromal tumour. Br J Cancer. 2004;90:2059–2061. doi: 10.1038/sj.bjc.6601819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, Vranckx H, Wasag B, Prenen H, Roesel J, Hagemeijer A, Van Oosterom A, Marynen P. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinibresistant mutants. Gastroenterology. 2005;128:270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 74.Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–4190. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 75.Min KW, Balaton AJ. Small intestinal stromal tumors with skeinoid fibers in neurofibromatosis: report of four cases with ultrastructural study of skeinoid fibers from paraffin blocks. Ultrastruct Pathol. 1993;17:307–314. doi: 10.3109/01913129309027777. [DOI] [PubMed] [Google Scholar]
- 76.Ishida T, Wada I, Horiuchi H, Oka T, Machinami R. multiple small intestinal stromal tumors with skeinoid fibers in association with neurofibromatosis 1 (von Recklinghausen’s disease) Pathol Int. 1996;46:689–695. doi: 10.1111/j.1440-1827.1996.tb03673.x. [DOI] [PubMed] [Google Scholar]
- 77.Kinoshita K, Hirota S, Isozaki K, Ohashi A, Nishida T, Kitamura Y, Shinomura Y, Matsuzawa Y. Absence of c-kit gene mutations in gastrointestinal stromal tumours of neurofibromatosis type I patients. J Pathol. 2004;202:80–85. doi: 10.1002/path.1487. [DOI] [PubMed] [Google Scholar]
- 78.Yantiss RK, Rosenberg AE, Sarran L, Besmer P, Antonescu CR. Multiple gastrointestinal stromal tumors in type I neurofibromatosis: a pathologic and molecular study. Mod Pathol. 2005;18:475–484. doi: 10.1038/modpathol.3800334. [DOI] [PubMed] [Google Scholar]
- 79.Takazawa Y, Sakurai S, Sakuma Y, Ikeda T, Yamaguchi J, Hashizume Y, Yokoyama S, Motegi A, Fukayama M. Gastrointestinal stromal tumors of neurofibromatosis type I (von Recklinghausen’s disease) Am J Surg Pathol. 2005;29:755–763. doi: 10.1097/01.pas.0000163359.32734.f9. [DOI] [PubMed] [Google Scholar]