

Abstract
Mechanisms of cellular adaptation may have some commonalities across different organisms. Revealing these common mechanisms may provide insight in the organismal level of adaptation and suggest solutions to important problems related to the adaptation. An increased rate of mutations, referred as the mutator phenotype, and beneficial nature of these mutations are common features of the bacterial stationary-state mutagenesis and of the tumorigenic transformations in mammalian cells. We argue that these commonalities of mammalian and bacterial cells result from their stress-induced adaptation that may be described in terms of a common model. Specifically, in both organisms the mutator phenotype is activated in a subpopulation of proliferating stressed cells as a strategy to survival. This strategy is an alternative to other survival strategies, such as senescence and programmed cell death, which are also activated in the stressed cells by different subpopulations. Sustained stress-related proliferative signalling and epigenetic mechanisms play a decisive role in the choice of the mutator phenotype survival strategy in the cells. They reprogram cellular functions by epigenetic silencing of cell-cycle inhibitors, DNA repair, programmed cell death, and by activation of repetitive DNA elements. This reprogramming leads to the mutator phenotype that is implemented by error-prone cell divisions with the involvement of Y family polymerases. Studies supporting the proposed model of stress-induced cellular adaptation are discussed. Cellular mechanisms involved in the bacterial stress-induced adaptation are considered in more detail.
Key Words: Adaptive mutagenesis, bacteria, cancer, stress, histone-like proteins, epigenetic alterations
INTRODUCTION
Adaptation is a fundamental process that occurs not only at the level of organisms, but also at the level of individual cells. Although adaptation is usually referred to a trait of a living organism, cellular level of adaptation may share more commonalities across different organisms. Understanding such common mechanisms may not only explain some cellular phenomena, but also suggest solutions to important problems related to organismal adaptation, including mammalian life-style diseases, bacterial resistance to antibiotics, and others. It is the focus of this paper to review experimental studies that support the common mechanisms of adaptation in mammalian and bacterial cells.
There is a striking similarity in the high adaptation potential of the bacterial cell and that of the mammalian tumor cell [1–3]. This similarity suggests that stationary-state, or adaptive, mutagenesis1 in bacteria and tumorigenic transformations in mammals may represent an adaptation strategy that is common for cells of the organisms. In both cases, the adaptive potential is acquired in response to a sustained stress environment1, and is promoted by the increased rate of genetic mutations that permit the cells to escape the restrictions that limit growth of normal cells. These mutations are beneficial at the level of an individual mammalian and bacterial cell.
In bacteria, the increased rate of mutations is observed in different stressful environments during the stationary state1 or stationary phase1 growth [4–6]. During this phase there is little or no increase in bacterial biomass, because the continuing cell division is balanced by the continuing cell death. During the stationary phase, the long-term survival of bacteria results in the appearance of cells with a high mutation rate that express a growth advantage phenotype.
In mammals, most malignant transformations also take place in response to a sustained stress environment1. Similar to the bacterial stationary state, this environment is characterized by an increase in continuing cell death and cell renewal. This change may be induced by a variety of chemical and physical agents, inflammation, and even cytokines. The exposure to such factors may lead to a loss of genetic stability and to mutations in tumour-suppressor genes and proto-oncogenes. The mutations may release mammalian cells from growth constraints and allow the cells to adapt quickly to a new stress in the tumour environment.
Thus, in both bacterial cells and mammalian tumour cells, a mutational change has an adaptive nature and is beneficial at the cellular level. Mutations are specific to the environmental challenge that causes the stress and occur in genes that are under the selection pressure in the environment.
Epigenetic mechanisms play a key role in the adaptation of mammalian cells to a sustained stress that ultimately leads to tumorigenic transformations of the cells [3]. Epigenetic alterations1 are inherited modifications of the genome that occur without changes in the base sequence of DNA. These alterations create an inherited pattern of gene expressions in the genome realized as a set of repeatedly executing cellular functions. In the sustained stress environment, a continuing proliferation of a subpopulation of mammalian cells induces their stress-related epigenetic alterations1 [3]. These epige-netic alterations cause silencing of genes that suppress proliferation through activation of cell-cycle arrest, apoptosis, DNA repair, cytokine signalling, and cell differentiation. On the other hand there is epigenetic activation of genes involved in promotion of proliferation and DNA repeats in the proliferating subpopulation. Together these epigenetic alterations prime the cells to activate the mutator phenotype1, which, in turn, transforms the epigenetically reprogrammed proliferating cells into tumour cells. Thus, mammalian tu-morigenesis may be considered as a process of cellular adaptation to a sustained stress environment by means of epige-netic alterations in the mammalian genome, subsequent mutations of the base sequence of DNA, and natural selection of emerging mutant cells via apoptosis.
The role of epigenetic mechanisms in bacterial adaptive mutagenesis has not been previously reported, to the best of our knowledge. The similarities in phenotypic traits (e.g. stress-induced genetic instability, growth advantage) expressed in tumorigenically transformed mammalian cells and stationary-phase bacterial cells may provide the basis for defining such a role.
In this study, we propose that epigenetic alterations similar to those described in mammalian stressed cells may be induced in stressed bacterial cells and prime them for activation of the mutator phenotype. Specifically, we introduce the model for this activation Fig. (1) through the stress-induced adaptation of bacterial cells that requires their epigenetic reprogramming1. According to this model, in the stationary phase environment, bacterial cells sense continuing stress-related survival and proliferative signals. These sustained signals cause epigenetic reprogramming of the cells. The reprogramming involves silencing or activation of the proper genes and thus the induction of the mutator phenotype as an alternative to other survival strategies such as programmed cell death, senescence, and differentiation.
Fig. (1).
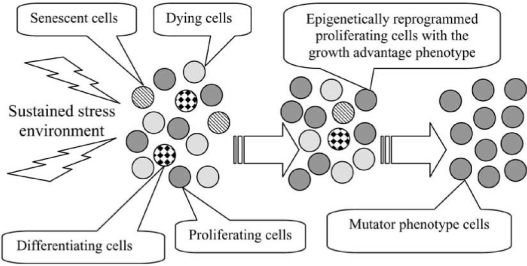
Activation of the mutator phenotype strategy in a subpopulation of proliferating cells by epigenetic reprogramming. Sustained stress environment may lead to the separation of the cell population into subpopulations activating different survival strategies. The epigenetically reprogrammed proliferating subpopulation acquires the growth advantage phenotype, outgrows the other subpopulations, and activates the mutator phenotype.
The main objective of the paper is to review experimental evidence supporting the proposed model of activation of bacterial mutator phenotype though the mechanism of stress-induced cellular adaptation1 and to compare this mechanism with the stress-induced cellular adaptation of mammalian cells summarized above and reviewed in more detail in [3].
Based on the recently published studies, the following experimental observations are common for bacterial and mammalian cells and support the proposed model:
(1) Epigenetic mechanisms play a leading role in the adjustment of cellular functions to meet the needs of the environment.
(2) Senescence, programmed cell death, and mutator phenotype are alternative strategies activated by the cells in response to a sustained stress.
(3) Stress-induced proliferative and survival signalling drives the activation of the mutator phenotype strategy in the cells.
(4) Activation of the mutator phenotype requires epigenetic reprogramming of the genome by silencing of cell cycle inhibitors, programmed cell death, DNA repair and by activation of repetitive DNA sequences.
(5) Mutator phenotype is implemented by error-prone cell divisions with participation of Y family polymerases.
These observations are discussed in more detail throughout this paper.
EPIGENETIC MECHANISMS PLAY A LEADING ROLE IN THE ADJUSTMENT OF CELLULAR FUNCTIONS TO MEET THE NEEDS OF THE ENVIRONMENT
Epigenetic alterations provide an important mechanism of transcriptional control regulating genes expression in cells of most living organisms. They are inherited through cell division, but revocable by continuous cell exposure to a new environment. In a stable environment, epigenetic alterations efficiently control a meta-stable state of gene expression in the genome by silencing or activating certain genes. This way a gene expression pattern is propagated in the cell tissue or in the cell culture. In response to environmental changes, epigenetic alterations non-mutationally modify the genome to produce a new expression pattern, and then propagate this new pattern to the progeny cells.
In eukaryotic cells, epigenetic alterations can be induced by (a) modifications of the DNA via methylation, particularly, of the cytosine residues at 5’ carbon in CpG dinucleotides located in the promoter and coding regions of genes [7,8]; (b) modifications of the chromatin via histone acetylation, phosphorylation, methylation, and ubiquitination [9]; and (c) RNA interference [10]. These epigenetic mechanisms are related and can affect each other by providing several layers of regulation of gene activity, from secure activation to secure suppression [11]. Modifications of certain histones, for example, may increase gene methylation.
In bacterial cells, epigenetic control of gene expression also exists and is implemented via methylated DNA, via his-tone-like proteins, and via small RNAs1 (sRNA). Below we describe these epigenetic mechanisms in bacteria in more detail (see the summary of the epigenetic factors in Table 2).
Table 2.
Characteristics of Proteins Implementing Epigenetic Mechanisms in Bacteria
| Epigenetic factor | Stress-related changes in the activity | Functions related to regulation of gene expression | Ref. | Specificity of binding |
|---|---|---|---|---|
| DNA Methyltransferases | ||||
| Dam (Dam MTase) | The abundance of Dam in the cell is tightly regulated at the transcriptional level. This regulation is growth-rate dependent and involves multiple promoters and one terminator. Transcription of the dam gene increases with growth rate. | i Modulates gene expression by GATS methylation. | [13–17] | Catalyzes the transfer of a methyl group from Sadenosylmethionine to the N6 position of adenine in a specific target sequence GATC. Most nonmethylated GATC sites are found within noncoding regions that likely have a regulatory function. |
| ii Coordinates chromosome replication in cooperation with SeqA , DnaA, oriC. DamMT and SeqA (a negative regulator of chromosome regulation and an important factor in forming and maintaining of chromosome structure) compete for GATC binding sites in hemimethylated DNA. | ||||
| iii Regulates mismatch repair. This repair in bacteria is methylation directed and the presence of hemimethylated GATC sites is crucial for initiation of mismatch repair. | ||||
| iv Plays a role of a virulence factor in bacterial pathogenesis. | ||||
| v Regulates transposition modulating the expression of transposase. | ||||
| CcrM (cell cycle-regulated MTase) | CcrM expression is cell cycle dependent. | Plays an essential role in the regulation of cell cycle in the alpha subdivision of proteobacteria. | [18–20] | Methylates the N-6 adenine of a specific target sequence GAnTC. |
| Histone-Like Proteins (HLPs) | ||||
| HU (heatunstable nucleoid protein) | HU is abundant in the exponential phase and then gradually decreases its abundance. | i Mediates and maintains supercoiling by forming a scaffold on which the DNA is wrapped (analogous to the eukaryotic nucleosome). | [21–26] | Non-specific DNA binding with preference to various distortions in DNA, nicks, gaps, cruciforms, and phased loops. |
| ii Disrupts H-NS-imposed repression. | ||||
| iii Plays a critical role in transpososome assembly. | ||||
| iv Regulates DNA repair by binding specifically to DNA that contains single-strand breaks or gaps (cells lacking HU are extremely sensitive to gamma irradiation). | ||||
| v Reduces formation of double strand breaks. | ||||
| vi Suppresses illegitimate recombination and SOS response through interaction with DNA gyrase. | ||||
| vii Regulates RpoS translation. | ||||
| H-NS (histonelike nucleoid structuring protein) | HU/H-NS ratio is about 2.5 in logarithmic phase and only 1 in stationary phase. | Contributes to organization of transcriptionally inactive DNA and to global repression of gene expression (in many cases environmental signals, such as temperature, osmolarity, oxidative conditions lead to disruption of the repressive complexes). Regulates virulence gene expression, multidrug resistance, motility, and acid tolerance. | [27–35] | Binding preference for AT-rich curved DNA regions (commonly found at gene promoters) and for actively bending DNA. H-NS has the ability to bridge adjacent tracts of DNA and, thus, induces a higher-order compaction. |
| ii Mediates suppression of DNA repair after UV irradiation. | ||||
| iii Provides efficient transposition of IS903, Tn10 and Tn552 (Plays a role in target capture, either by providing regions of DNA more accessible to transposition or by stabilizing transposome binding to captured targets). | ||||
| iv Regulates DNA replication. | ||||
| v Regulates RpoS expression post-transcriptionally. | ||||
| DNA Methyltransferases | ||||
| Fis (factor for inversion stimulation) | Fis is abundant in the exponential phase and then decreases its abundance to an undetectable level. | i Disrupts H-NS-imposed repression. | [36–40] | A relaxed target specificity including the binding of SIDD regions of gene promoters. DNA binding results in a high degree of bending of the DNA and may stabilize the "loopdomain" structure of the bacterial chromosome. |
| ii Regulates site-specific DNA recombination. | ||||
| iii Regulates transcription of the growth-related genes (like rRNA and tRNA acting as an antagonist to H-NS and Lrp. | ||||
| iv Regulates transposition (transposon Tn5 and insertion sequence IS50). | ||||
| v Inhibits DNA replication. | ||||
| vi Represses RpoS during exponential growth. | ||||
| Dps (DNA binding protein from starved cells) | Dps exists at low level in the exponential phase and is the most abundant protein in the stationary phase. Dps expression is controlled by RpoS, OxyR, and IHF. | i Protects cells from oxidative stress during exponentialphase growth. | [41–43] | Non-specific DNA-binding activity. |
| ii Organizes the chromosome into a highly ordered, stable nucleoprotein complex, called the biocrystal, during the stationary phase. | ||||
| Integration host factor (IHF) | IHF becomes the second-mostabundant protein in the transition from the growth phase to the stationary phase. IHF expression is controlled by RpoS, ppGpp and by autoregulation. | i Regulates DNA replication via interplay with Fis and DnaA. | [26, 30, 36, 44, 45] | Binding to cognate sites represented by the consensus WATCARXXXXTTR (W is A or T; X is A, T, C or G; R is A or G). These sites may be situated in the supercoiling-induced duplex destabilized (SIDD) regions of gene promoters. |
| ii Regulates transposition in the stationary state in combination with RpoS and provides transposition efficiency (combination of IHF with RpoS maintains a bend in the IS10 ends, governs the choice of transposition pathway, and stimulates transposase binding to Tn1000 ends). | ||||
| iii Regulates site-specific recombination. | ||||
| Leucineresponsive regulatory protein (Lrp) | For cells in a rich medium, Lrp levels drop during the lag phase, and then rise to starting levels upon entry into the stationary phase. For cells in a minimal medium, there was not much variation in levels as a function of growth phase. Lrp expression is induced by ppGpp. | i Modulates a variety of metabolic functions on entry into stationary phase by inducing a DNA methylation pattern that matches gene activity and environmental signals.. | [39, 46–50] | Lrp competes with Dam for the same binding site in certain promoters and thus operates as a methylation-blocking factor. |
| ii Binds to the regulatory region of bacterial rRNA promoters and this way may contribute in combination with H-NS to the control of rRNA synthesis. | ||||
| ii Regulates Off-to-On switching of cell surface structures (pap-pili switch). | ||||
| iii Affects selectivity of sigma factors binding to many promoters. | ||||
| Small RNAs (sRNAs) | ||||
| sRNAs (DsrA; RprA; OxyS) | The sRNAs may be induced under specific environmental conditions and act on regulator genes (DsrA is temperature sensitive; OxyS is induced by oxidative stress; RprA activation is related to cell surface stress and biofilm formation). | i DsrA and RprA positively regulate RpoS; OxyS represses RpoS translation. | [51–54] | Base pairing with target mRNAs. DsrA and OxyS modify mRNA translation using an RNA chaperone protein Hfq. |
| ii DsrA negatively regulates translation of HNS by pairing just beyond the translation initiation codon. | ||||
DNA methylation in bacterial cells primarily serves as a mechanism of defence against the invasion of a foreign DNA into the cells. It functions as a part of restriction-modification systems1 [12] that are comprised of genes encoding a restriction enzyme and DNA methyltransferases (DNA MTases). Some bacterial DNA MTases, however, do not have associated restriction enzymes, and are involved in the epigenetic control of DNA replication and transcription. The Echerichia coli Dam and Caulobacter crescentus CcrM enzymes are the best studied DNA MTases involved in epigenetic alterations. Both enzymes catalyze the transfer of a methyl group from S-adenosylmethionine to the N6 position of adenine at a specific target sequence (GATC for Dam and GANTC for CcrM). GATC-methylation by Dam can influence gene expression by two different mechanisms. First, methylated GATC sequences in gene promoter regions can affect transcription initiation. Second, Dam and regulatory proteins, including Cap, Lrp or OxyR, compete for overlapping sites in, or near a gene promoter [17]. Binding (detaching) of such regulatory proteins to (from) the DNA controls DNA methylation by Dam, and thus determines a DNA methylation pattern in response to changes in environmental conditions. Well-known examples of this regulation are the control of the glucitol operon (gut) [55] and the reversible off-to-on switching of cell surface structures such as the regulation of pyelonephritis-associated pili (pap) [49] in bacteria.
Histone-like proteins (HLPs) in bacterial cells, also refered as nucleoid-associated proteins, organize the structure of the bacterial nucleoid and serve as global regulators of gene expression in response to environmental changes [24, 37, 42, 56]. The nucleoid, however, does not have such a complex and heterogeneous structure as does the chromatin in a eukaryotic genome. Therefore, a sophisticated regulation of transcriptional activity by modification of histones is absent in bacteria. Related epigenetic regulations are implemented by direct interaction of bacterial HLPs with DNA.
Among the major known HLPs are HU, H-NS, Fis, Dps, IHF, and Lrp. Most HLPs regulate gene expression by non-specific DNA-binding and by changing DNA supercoiling. A gene promoter often includes a supercoiling-induced duplex destabilized (SIDD) region. It is the site where negative superhelicity can destabilize the DNA duplex and facilitate transcription of the gene. SIDD sites have 80% chance of containing a promoter [57]. SIDD regions are usually A+T-rich and contain HLP binding sites. Binding of HLP changes negative superhelicity in the region and affects transcriptional activity of the gene. Such a mechanism of the transcriptional regulation by IHF and Fis has been studied in depth in [56]. Table 2 presents some specific DNA binding properties of the HLPs and the effect of this binding on gene activity.
Small RNAs (sRNAs) in bacteria are involved in epige-netic alterations that trigger transcriptional gene silencing [10]. The mechanisms underlying these epigenetic effects include synthesis of small RNAs that subsequently recognize the RNA or DNA target sequences by base-pairing. This binding of sRNAs changes the activity of the target gene, and this change is transferred to progenies. There are some differences in how sRNAs are processed in bacteria and eu-karyotes to implement the epigenetic effect [53]. More than 60 sRNAs are identified in E. coli; most of them function as regulatory molecules. They modify translation and stability of mRNAs by base-pairing in a manner similar to eukaryotic miRNAs and siRNAs. A growing body of data suggests that sRNAs are crucial coordinators of adaptive processes in bacteria and have profound effects on cell physiology [58]. Properties of three bacterial sRNAs involved in stress response regulation are considered in Table 2.
In summary, comparison of the aforementioned epigenetic mechanisms show their importance in gene silencing or activating and thus in adjusting cellular functions in bacteria and mammals to changes in the environment. Specific implementation of epigenetic regulation in mammals is more advanced than in bacteria. Also, epigenetic reprogramming of cellular functions is not as fast as a transcriptional regulation of gene activities, because it involves modifications of DNA supercoiling in bacteria and modifications of histones and chromatin structure in mammals. Considering a slow rate of the modifications, a sustained signaling from the environment becomes an important condition for epigenetic reprogramming. This signaling is also important for defining what cellular functions are active and, therefore, what gene expression patterns are propagated under different environmental conditions. We refer to a set of cellular functions epigenetically fixed in the genome as the cellular strategy1.
SENESCENCE, PROGRAMMED CELL DEATH, AND MUTATOR PHENOTYPE ARE ALTERNATIVE STRATEGIES ACTIVATED BY CELLS IN RESPONSE TO A SUSTAINED STRESS
According to the proposed model (see Introduction and Fig. (1)), the mutator phenotype of mammalian tumour cells is their survival strategy activated in a subpopulation of normal cells under sustained stress. This strategy is an alternative to cellular senescence, programmed cell death (apop-tosis), and cellular differentiation. Each of these strategies requires certain signals from the environment followed by epigenetic activation and suppression of specific cellular processes and involved genes [3]. Bacterial cells can activate similar survival strategies in a stressful environment: stationary-state mutagenesis, programmed cell death, “conditional” senescence, or cellular differentiation. Each of them is considered below in more detail.
Stationary-state mutagenesis, or the generation of mutations in stationary phase bacteria in response to growth-limiting stress, is well-documented [59–67]: (a) mutator phenotype is activated in a small subpopulation of cells in response to stress [65–67] and (b) different molecular mechanisms are involved in generation of adaptive mutations. Well-documented examples include adaptive mutations in E. coli Lac system (compensatory frameshift mutations and amplifications) [64].
Programmed cell death1 was also reported in bacterial cells in response to damage and to nutritional stress [68–69], though this response is not as sophisticated as the apoptosis in mammalian cells. In Pseudomonas putida, increased death of bacterial cells takes place under the conditions of prolonged starvation; and this phenomenon might be important for the generation of stationary phase mutations in the bacteria [70]. Programmed cell death may be considered as a survival strategy, because it allows bacteria to eliminate defective cells in the population, and thus to release deficient nutrients and signalling molecules to support survivors. In addition, programmed cell death provides a mechanism for natural selection of cells with beneficial mutations in a stressful environment [71].
Stress-induced senescence is another important survival strategy of the cell. Cellular senescence usually refers to the state of the cell in which the cell irreversibly stops its replication. Cellular (also referred as replicative) senescence of mammalian cells is their general stress response program [72, 73]. Important biological functions of the replicative senescence include the onset of arrest on proliferation and on apoptosis. Although replicative senescence is absent from bacteria, a sustained stress environment may induce bacteria to enter a non-proliferative state (stationary phase). In this state bacterial cells fail to grow normally (at an exponential rate) and to increase their biomass. In many aspects the bacterial stationary state is similar to senescence in mammalian cells [74] and has even been called “conditional” senescence. Senescent mammalian cells remain viable, but they cannot divide, because cell cycle inhibitors are activated and arrest replication. The bacterial stationary state is also a form of growth arrest. When bacteria enter a stationary phase, viable cells appear that can survive for long periods without division and without life-supporting activities. In conclusion, the replicative senescence in mammalian cells and “conditional” senescence in bacterial cells represents an important survival strategy triggered by sustained stress. In both cases the active state of genes involved in the onset of arrest on proliferation is indispensable for the implementation of the senescence strategy.
Competence development is a specific form of cellular differentiation evolved in Bacillus subtilis and some other bacteria in response to stress. This survival strategy involves cellular differentiation of normal cells into naturally transformable competent cells [75]. These cells are able to synthesize specific proteins to take up DNA from the environment. The acquired DNA is used within the cell for recombination with the genomic DNA. Competence development is induced in stationary state bacterial cells growing at a high cell density by the competence transcription factor ComK. As in stationary-state mutagenesis, only a subpopulation of cells (1 to 10% of cell culture) becomes competent. In a way this mechanism is reminiscent of a primitive sexual life-cycle, because there is a striking genetic polymorphism in quorum-sensing systems mediating competence development in different gram-positive bacteria. This polymorphism provides a sort of sexual isolation mechanism, namely the import of DNA into the genome of competent cells occurs only from closely related strains [76].
“Persister cells” represent another form of cellular differentiation in response to a sustained stress [77, 78]. The proportion of such cells in wild-type E .coli is approximately 10−6 of the total number of cells, but this quantity may be increased by “gain-of-function” mutations in the hipA gene that inhibits cellular proliferation. Differentiation into “persister cells” allows bacteria to resist environmental stresses such as high antibiotic concentration.
A specific pattern of up- or down-regulation of the decisive set of cellular processes and involved genes may determine which of the aforementioned cellular survival strategies takes place in response to the prolonged stress (Table 3). The set includes cell-cycle arrest, stress-related proliferative signalling, and differentiation pathways. These strategies are likely mutually exclusive, because one survival strategy may require the activation of specific genes and pathways, but others require their suppression. Thus, the survival strategies are not executed simultaneously in a single cell. Specifically, cell cycle inhibitors must be activated in differentiated cells, senescent cells or apoptotic cells. But the inhibitors must be suppressed in proliferating cells activating the mutator phenotype. Stress-related proliferative signalling, however, is absent from senescent cells and differentiating cells, but this signalling is important in inducing the mutator phenotype strategy or driving the cell cycle arrested cells to programmed cell death. A differentiation program is activated in differentiating cells.
Table 3.
Activation of Cellular Survival Strategies in Sustained Stress Environment. Activity of Genes Involved in the Onset of Cell Cycle Arrest, in the Stress-Induced Proliferation, and in the Differentiation Pathways Determines the Choice of the Survival Strategy
| Activity of the decisive cellular processes (“+”activation; “–” suppression) | Activating survival strategy | |||
|---|---|---|---|---|
| Cell cycle arrest | Stress-related proliferative signal | Differentiation pathways | Mammalian cells | Bacterial cells arrest |
| + | – | – | Replicative senescence | Conditional senescence in the stationary phase |
| + | + | – | Apoptosis | Programmed cell death |
| – | + | – | Mutator phenotype (tumorigenic transformations) | Mutator phenotype (stationary-state mutagenesis) |
| + | – | + | Differentiation | Competence development “Persister cells” |
Thus, each strategy is implemented by a subpopulation of stressed cells, and the choice of a particular strategy is regulated in the cells by signalling from the environment. This signalling is crucial for epigenetic reprogramming of the cells. The reprogramming leads to sustained activation of genes involved in implementation of the target strategy and to sustained suppression of genes activating alternative survival strategies. The sustained change in activity of the genes leads to fixation of the gene expression pattern epigenetically in the genome and then to the accomplishment of the target strategy. The next section reviews in more detail some of the signalling pathways leading to activation of the mutator phenotype strategy.
STRESS-INDUCED PROLIFERATIVE AND SURVIVAL SIGNALLING DRIVES THE ACTIVATION OF THE MUTATOR PHENOTYPE STRATEGY IN THE CELLS
Two main pathways, which are strongly involved in tu-morigenic transformation of different mammalian cells, implement stress-related survival and proliferative signalling. They are Ras/MAP kinase pathway and phosphatidylinositol-3-kinase (PI3K)-mediated pathway. The Ras/MAP kinase pathway is a well-known indicator of stress in mammalian cells. Sustained activation of this pathway induces senescence of the cell by cell cycle arrest on proliferation [79]. In human cells, cell cycle inhibitors, p53 and p16, are two independent barriers to proliferation activated in normal cellular senescence and in apoptosis. Both inhibitors activate Rb and this way they suppress the initiation of proliferation.
The PI3K-mediated pathway promotes proliferation and survival of mammalian cells. Sustained activity of the pathway may epigenetically silence the crucial genes involved in cell cycle arrest (like p53, p16, p21 and pRb), and, therefore, inhibit the activation of senescence and apoptosis in stressed cells.
A stressful environment leads to a sustained production of stress-related survival and proliferative signalling in mammalian cells through these pathways [3, 71]. This signalling results in a continuing death of some cells in the environment and to the emergence of a subpopulation of proliferating cells. The proliferating cells epigenetically reprogram their genome by activation or suppression of processes required to implement the mutator phenotype survival strategy. Eventually, the activated mutator phenotype leads to tumorigenic transformation of the epigenetically reprogrammed cells.
Signalling inducing the stress-related cell-cycle arrest and the proliferation is also activated in stressed bacterial cells. Below we describe this signaling in more detail.
Sensing stress in bacteria occurs at the onset of the stationary phase by Ras-like GTP binding proteins and by the GTP-to-GDP ratio. Particularly, bacterial cells growing under optimal nutritional conditions contain a high concentration of GTP and a small concentration of GDP. In response to different types of nutritional stress, bacteria synthesize the alarmone molecule, guanosine tetraphosphate ppGpp, at the expense of GTP, the level of which decreases [80]. A number of essential GTP-binding proteins in the bacterial cell respond to the change in the GTP-to-GDP ratio. Activity of these proteins is controlled by the conformational state. Specifically, they are turned “on” or “off” depending on whether they form a complex with GTP or GDP. After sensing the intracellular GTP-to-GDP ratio, the GTP binding proteins signal to downstream proteins about the nutritional state of the cell.
In B. subtilis, the activation of Ras-related GTPase Obg during the entry of the bacterium into the stationary state is well documented [81]. The stationary state in B. subtilis is controlled by the activity of the alternative sigma factor sigma(B). Stresses activate sigma(B) by means of a signalling pathway leading to the release of sigma(B) from an anti-sigma factor inhibitor. Ras-related GTPase Obg is an important regulator in this signal transduction. B. subtilis depleted of Obg does not activate sigma(B) in response to environmental stresses.
In E. coli, the transition to stationary state is regulated by the guanosine tetraphosphate ppGpp pool [82, 83]. Accumulation of ppGpp in response to stress changes the competition between two sigma factors (RpoS and σ70) in the structure of RNA polymerase in favour of the equipment of this enzyme with RpoS instead of σ70 factor. RpoS, an E. coli general stress response sigma factor, is nearly absent in rapidly growing cells, but it is strongly induced during entry into stationary phase and causes rapid changes in the pattern of gene expression. Though specific mechanisms by which Ras-signaling may regulate the induction of the stationary phase in this bacterium are not known, the involvement of E. coli Ras-related GTP-biding genes Obg and Era in this regulation has been reported in [84, 85]. In addition, the Ras-like Obg protein regulates the onset of cell cycle arrest and controls the initiation of replication by sensing changes of the intracellular GDP-to-GTP ratio in E. coli and other bacteria [86–88]. These findings provide a link between the Ras-like signalling, cell cycle arrest and subsequent senescence of stressed bacterial cells. This link also exists in mammalian cells, as has been discussed previously.
Proliferative signalling in bacteria may occur with participation of phospholipids in their membrane [89] and is not as complicated as in mammalian cells. In mammals the proliferative signalling is mediated by activity of a phospholipid second messenger in the PI3K-mediated pathway that is initiated at the cell membrane by growth factors. The PI3K-mediated pathway activates proliferation and also mediates SREBPs (sterol regulatory element binding proteins), which are transcription factors regulating cellular lipid homeostasis in eukaryotic cells. In bacteria, DnaA, a sequence-specific DNA-binding protein, plays a key role in the initiation of chromosomal DNA replication in vivo and in vitro [90]. Accumulating evidence, both in vitro and in vivo, suggests that acid phospholipids in the cell membrane play an important role in the regulation of DnaA reactivation and in the increasing fluidity of the membrane [89, 91, 92]. Both effects relate to bacterial survival and are well documented for acidic phospholipids such as phosphatidylinositol, phos-phatidylglycerol, and cardiolipin. Specifically, an increased level of acidic phospholipids is observed in the exponentially growing cells, while stationary phase bacteria have an increased level of basic phospholipids [91]. Extracts of total cell lipids from stationary phase cells failed to cause nucleotide release from DnaA (to reactivate DnaA), in contrast to lipids extracted from exponentially growing cells. A study of adaptation of E. coli to trisodium phosphate (an antimicrobial agent) also showed that the increased membrane fluidity imposed by this adaptation was associated with a significant increase in the acid resistance and survival of the bacteria [92]. The above studies suggest that although most bacterial cells in the stationary phase do not divide because of the cell cycle arrest initiating by Ras-related GTP-biding proteins, a small subpopulation of the stressed cells may proliferate maintaining a high concentration of acid phospholipids. The acid phospholipids act in a similar way to the PI3K-mediated pathway promoting proliferation and survival of mammalian cells.
In summary, Ras-like GTP binding proteins sense a sustained stress environment in both mammalian and bacterial cells. Sustained activation of this signalling can induce senescence in the mammalian and bacterial cells. In the mammalian cells a stressful environment also activates a proliferative signalling by phospholipid second messengers. This signalling may lead to a continuing proliferation of a sub-population of stressed mammalian cells, their reprogramming and, ultimately, activation of the mutator phenotype. The relationship between the abundance of acidic phosphol-ipids in the cellular membrane and survival of the bacterial stressed cells has also been reported. This observation suggests that survival of the bacterial proliferating subpopulation in the stationary phase and subsequent activation of the mutator phenotype may relate to an increased level of acidic phospholipids in cellular membranes of this subpopulation.
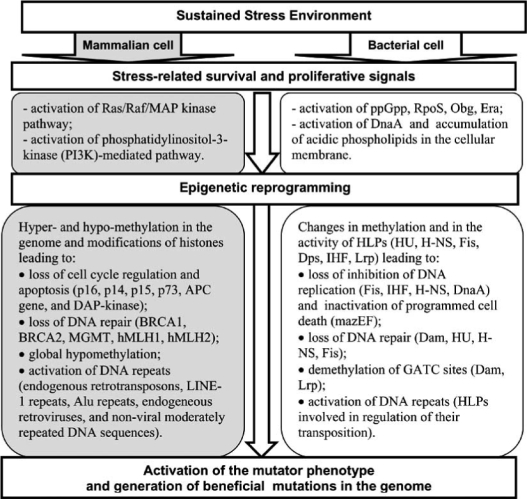
ACTIVATION OF THE MUTATOR PHENOTYPE REQUIRES EPIGENETIC REPROGRAMMING OF THE GENOME BY SILENCING OF CELL CYCLE INHIBITORS, PROGRAMMED CELL DEATH, DNA REPAIR AND BY ACTIVATION OF REPETITIVE DNA SEQUENCES
Reprogramming of the mammalian cell resulting from sustained stress-related survival and proliferative signalling includes hypermethylation of genes involved in cell cycle regulation, apoptosis, cellular differentiation, DNA repair genes, and activation of DNA repeats by global demethylation of the genome. Epigenetic silencing and activation of the cellular processes also occurs in the bacterial stationary phase and will be considered below.
Bacterial HLPs are Working Horses in the Stress-Induced Epigenetic Reprogramming of the Bacterial Genome
The most studied mammalian epigenetic alterations induced by stresses are changes in cytosine methylation. These alterations are also found at premalignant and malignant stages of cancer. Although changes in methylation related to activity of E. coli Dam are also well-documented in stationary state bacteria, much more information on reprogramming of gene activity in the stationary phase is associated with bacterial HLPs.
There are several common HLPs’ features implying that HLPs act as working horses in stress-induced epigenetic reprogramming of the genome (Table 2). First, HLPs change their abundance during the transition of bacteria from exponential growth to stationary phase growth. The abundance of HU and Fis proteins decreases and the abudnance of H-NS, Lrp, INF, and Dps increases as the stationary phase is being reached. These changes in protein abundance indicate that cellular processes involved in the epigenetic reprogramming during the stationary phase are highly complex and intense.
Second, the activity of HLPs relates to the activity of rpoS, a major regulator of general stress response in E. coli (Table 2). Specifically, HU, H-NS and Fis are involved in the transcriptional or post-transcriptional regulation of RpoS activity. Dps, IHF and Lrp are under the control of RpoS. Considering the key role of RpoS in the bacterial general stress response, the relationship between this sigma factor and HLPs indicates that these proteins play an important, if not crucial, functional role in the survival of stationary phase bacteria.
Third, interplay between IHF, Fis, H-NS and Dam meth-yltransferase regulates bacterial replication (Table 2) showing a dependence of this cellular process on epigenetic reprogramming. Studies supporting the role of epigenetic mechanisms in the reprogramming of the bacterial genome in stationary phase are considered below.
Loss of Cell Cycle Inhibitors in the Stationary Phase
Epigenetic alterations play an important role in the regulation of replication in both mammalian and bacterial cells. In mammalian cells the activity of cell cycle inhibitors are crucial for normal cellular replication. They are activated by the transcription factor c-myc, a protooncogene and a crucial mediator of intracellular proliferative signaling. Several cell cycle inhibitors, like p16, p14, p15, p73, APC gene, and DAP-kinase are silenced by hypermethylation at the initial stages of tumorigenic transformations. A loss of these cell cycle inhibitors is a hallmark of tumor cells.
In bacteria, the binding of DnaA (a key initiator of chromosomal DNA replication) to oriC (the replication origin) is the first conservative step in the assembly of the initiation complex for replication [90]. In addition to recognition sequences for DnaA binding, oriC have binding sites for histone-like proteins IHF, Fis, H-NS, and also several recognition sequences for the Dam methyltransferase. This indicates a tight epigenetic regulation of replication in bacteria. Histone-like protein Fis functions as a cell cycle inhibitor in this regulation. It inhibits DNA replication from oriC by suppressing DnaA, IHF binding, and oriC unwinding [93, 94]. Fis is abundant in the exponential phase and its abundance decreases to an undetectable level at the stationary phase (Table 2). Synthesis of Fis may be induced only after stationary cells are subcultured into a rich medium [95]. Unlike this pattern of Fis activity, the pattern of DnaA activity has two peaks in expression [30]. The first peak corresponds to the rapidly growing cells (the mid-exponential-growth phase). The second peak represents the late stationary phase, when Fis is undetectable. The second rise of DnaA in the stationary phase represents the resumption of cellular growth in a subpopulation of cells. This subpopulation may represent the outgrowing reprogrammed cells. Because of the lack of cell cycle inhibition by Fis, replication of these cells may be not normal. Similar to tumor cells, they may not arrest their cycle and, therefore, have a growth advantage.
Silencing of the Programmed Cell Death
An important feature of mammalian reprogramming is silencing of the cellular apoptotic machinery. Programmed cell death in bacteria is caused by stress-induced toxin-antitoxin modules. They are comprised of mRNA-cleaving enzymes activated by different environmental stresses. Free-living bacteria have many toxin-antitoxin modules, and they play an important role in the cellular stress response [69]. In E. coli, mazEF loci encode for the stable toxin MazF and the unstable antitoxin MazE. A continuing synthesis of the antitoxin is essential for the cellular survival to prevent the lethal action of the toxin. The mazEF module is under the control of ppGpp [96]. Various stressful conditions in the exponential phase of growth lead to overproduction of ppGpp and to inhibition of mazE expression and, ultimately, to cell death. Bacterial mazEF-mediated death is activated during the exponential growth, and may be inactive in the stationary phase [97]. This stress-related suppression of cell death is similar to silencing of the apoptotic system in tumour cells.
Decreased Efficiency of DNA Repair
Early epigenetic silencing of genes involved in DNA repair by hypermethylation, like BRCA1, BRCA2, MGMT, hMLH1, and hMLH2, is well documented in different tumours. In addition to direct epigenetic silencing of DNA repair pathways, their inactivation may follow persistent suppression of genes involved in cell cycle control (like APC gene and p53), because these genes mediate DNA repair in mammalian cells. A loss or a decrease of the efficiency of DNA repair and a related increase in the rate of adaptive mutations in the bacterial stationary phase is also well documented [70, 98–101]. A recent study of adaptive mutagenesis in E. coli showed that the bacterial general-stress-response sigma factor RpoS may control a switch between a high-fidelity DNA repair and an error-prone repair in the case of double-strand breaks [102]. The study also provided evidence that, though error-prone repair increased the rate of mutations, the mutations were not stochastic and were localized near double-strand breaks.
Epigenetic mechanisms, including methylation and activity of some HLPs, play an important role in the regulation of bacterial DNA repair. Table 2 gives examples of this regulation including mismatch repair by Dam, mediation of the base excision repair, and recombinational DNA repair by HU, H-NS and Fis. Because the activity of these HLPs depends on bacterial growth stage, they may be responsible for growth stage dependent changes in the efficiency of DNA repair.
Activation of Repetitive DNA Sequences
Hypomethylation of repetitive DNA elements including transposoms (DNA sequences that are able to move from an original site on a DNA molecule to a new site on the same or a new molecule) and subsequent activation of these elements is an important component of the epigenetic reprogramming of mammalian cells in a sustained stress. It is known that hypomethylation of DNA repeats predisposes cells to chromosomal instability and to reorganization and rebuilding of mammalian genome by transpositions of the activated transposable elements. The effect of a sustained stress environment on mobility of transposable elements and instability of simple DNA repeats is also well-documented in bacteria. Transcriptional activity of transposoms can be induced in this organism by a variety of stresses, including UV radiation [103], limited nutrient availability [104], and reduced oxygen [105]. Instability of short-sequence DNA repeats was also demonstrated in pathogenic and nonpathogenic bacteria in response to different stresses [106–110].
Epigenetic mechanisms play a role in the adjustment of DNA repeats activity to changing environmental and physiological conditions [111–115]. In addition to direct regulation of transposition by RpoS [111], many transposons use histone-like proteins. Such histone-like proteins as IHF, HU, H-NS and Fis modulate activity of transposons (Table 2). Since the abundance of these proteins depends on the growth phase, the proteins adjust transpositions to physiological conditions of the bacteria. Though details of the regulation of transposition are not well understood, it is proposed that an increase in IHF concentration and a decline in supercoiling may couple the rate of transposition to the physiological state of the cell [112]. DNA methylation is also involved in regulation of the transposition rate by modulating the expression of transposase (a protein that performs the insertion of a DNA sequence into a new location). Several insertion elements carry GATC methylation sites in their transposase promoter regions. Absence of the GATC methylation sites increases activity of the transposase promoters [113–115].
Consideration of the aforementioned evidence suggests that stationary phase bacterial cells may be characterised by reprogramming of their functions. This reprogramming may include (i) loss of cell cycle regulators, (ii) silencing of the programmed cell death, (iii) decreased efficiency of DNA repair, and (iv) activation of repetitive DNA sequences. These processes are similar to cellular processes in mammalian cells at premalignant stages of tumour development. In both organisms epigenetic mechanisms play an important role in the stress-induced reprogramming of the cellular processes.
MUTATOR PHENOTYPE IS IMPLEMENTED BY ERROR-PRONE CELL DIVISIONS WITH PARTICIPATION OF Y FAMILY POLYMERASES
Mutator phenotype of mammalian cells is characterised by increased rate of mutations through error-prone cell divisions with participation of Y family polymerases. These po-lymerases belong to low fidelity translesion synthesis DNA polymerases that implement a bypass of DNA lesions when high fidelity replicative polymerases (normally replicate undamaged chromosomal DNA) are stalled at a lesion [116, 117]. Members of Y family polymerases do not have a proofreading activity and, therefore, can replicate the lesion with high fidelity. In mammalian cells, cell cycle inhibitors p53 and p21 are global regulators of error-free and error-prone DNA repair. Activity of the inhibitors is important to arrest cell cycle and to prevent mutations at the price of reduced repair efficiency and increased apoptosis [118,119]. Inactivation of the cell cycle inhibitors decreases fidelity of DNA repair and causes an overall increase in mutation rate.
The observations that mutator phenotype is induced in stationary phase bacteria and that Y family polymerases are important in the process have strong experimental support. This is reviewed below.
Mutator Phenotype in Bacterial Cells
An elevated rate of mutations (the production of a higher number of mutations per cell per generation) is a well-established feature of bacterial stationary phase [120, 121]. It may be an order of magnitude higher than when growth rate is exponential [122]. It is believed that this increase allows cells to adapt to a new environment. Different kinds of mutations may be accumulated in the genome of bacterial cells in the stationary phase including deletions [123], transpositions [124], genomic rearrangements [125], single gene mutations [126], and loss [123] or acquisition [127] of genetic material. A mutator phenotype was also demonstrated in biofilms. Specifically, the opportunistic pathogen Pseudomonas aeru-ginosa undergoes extensive genetic diversification during short-term growth in biofilms [128]. Mutational modifications in the genome of the bacterium lead to subpopulations with specialized functions in the biofilm. Molecular mechanisms that trigger the mutator phenotype in bacterial cells are not clear, but the importance of a sustained stress environment for generation of adaptive mutations is well-documented. The role of stress is demonstrated by the involvement of stress-induced sigma factor RpoS in the adaptive mutations in E. coli and other bacteria [129–131].
Y Family Polymerases
The involvement of error-prone DNA polymerases (or Y family polymerases) in stationary-phase mutagenesis is also well-documented for different bacterial species [132–136]. E. coli has five DNA polymerases. Pol I, Pol II and Pol III have essentially higher fidelity than Y-family polymerases Pol IV and Pol V. The high fidelity DNA polymerases play an important role in normal DNA replication and in SOS-induced DNA repair [116, 137, 138]. Although error-prone Pol IV is also involved in the DNA repair activated by SOS response, several studies also suggest that Pol IV functions in implementation of the mutator phenotype. This function is independent of SOS-response and involves production of mutations instead of DNA repair. Specifically, E. coli Pol IV was found to be controlled by RpoS, and the expression level of Pol IV in the stationary phase significantly decreased when the rpoS gene was mutated [139]. Thus, Pol IV is regulated not only by the SOS response but also in response to sustained stress including starvation and other stresses, even if DNA damage is absent. The abundance of Pol IV in this study increased in late stationary phase and stayed elevated for several days of continuous incubation, whereas in rpoS defective cells Pol IV was not induced and declined during prolonged incubation. Another study of Pol IV in E. coli showed that in the exponential phase, Pol IV had only a small effect on mutation rates, but it had a substantial effect on mutations in stationary phase bacteria. E. coli mutants lacking Y family polymerases suffered considerable fitness reduction when competing with a wild-type strain under starvation conditions [140]. The involvement of error-prone po-lymerase Pol IV in the generation of 1-bp deletions was also shown in starving Pseudomonas putida cells. This study demonstrated that involvement of Pol IV in stationary phase mutagenesis became essential only in long-term-starved populations of P. putida and this mutagenesis was induced by a mechanism that was different from SOS [141]. A SOS-independent increase in the transcription of dinB gene encoding Pol IV and a consequent increase in the reversion of a +1 Lac frameshift mutation were also demonstrated in a recent study of antibiotic-induced inhibition of cell wall synthesis in E. coli. The antibiotic-induced stress did not affect DNA (damage or replication) or translation, and the dinB induction was independent of the SOS regulators LexA and RecA [135].
The above studies indirectly support the proposed mechanism for activation of the mutator phenotype in a subpopulation of epigenetically reprogrammed proliferating bacterial cells Fig. (3). It is likely that similar to mammalian cells, reprogrammed bacterial cells do not arrest their cycle and do not activate the SOS response. Instead they replicate the lesion using Y-family polymerases.
Fig. (3).
Adaptive mutagenesis in bacteria and tumorigenesis in mammals as processes of stress-induced cellular adaptation.
DISCUSSION AND CONCLUSION
Bacterial mutator phenotype was recently explained by “environmental tuning of mutation rates” [6, 142]. According to this hypothesis any bacterial population is expected to harbour a subpopulation of DNA repair deficient mutants. Environmental conditions may favour their outgrowth leading to activation of the mutator phenotype. This hypothesis is based solely on a natural selection of the mutants with the increased mutation rates; it takes no account of cellular processes in stressed cells leading to the emergence of the mutator phenotype. The hypothesis is also inconsistent with a study of mutations conferring resistance to antibiotics in bacteria [143]. This study shows that to evolve resistance bacteria require postexposure mutations. Preexposure mutaitons cannot explain clinically significant levels of resistance.
The comparison of cellular processes leading to activation of the mutator phenotype in bacterial and mammalian cells suggests that activation of this phenotype may represent a conservative cellular survival strategy of stress-induced adaptation. This strategy is similar in these diverse organisms at the cellular level of organisation. In both organisms activation of the strategy requires sustained stress environment characterised by continuing survival and proliferative signalling. This environment leads to appearance of cells activating programmed cell death, stress-induced senescence, and the mutator phenotype. Each of these strategies is implemented in a subpopulation of the stressed cells, and epigenetic mechanisms are employed to reprogram cellular functions according to the requirements of each strategy.
A sequence of cellular events leading to activation of the mutator phenotype strategy in mammalian and bacterial cells is presented in Fig. (3). A combination of signals that indicate a sustained cellular stress, on the one hand, and promote cellular proliferation and survival, on the other hand, is essential for the activation of the mutator phenotype strategy in cells of both organisms. A lack of proliferative signalling will activate cellular senescence of a cell or programmed cell death instead of the mutator phenotype in the stressful environment. Stress-induced proliferation of a cell is an alternative option to senescence and programmed cell death in this environment. The proliferation induces epigenetic alterations in the genome leading to loss of programmed cell death, inhibition of DNA replication and DNA repair, and to activation of DNA repeats. These changes in cellular processes may be necessary to implement the mutator phenotype, and they occur in both mammalian and bacterial cells.
Although similar epigenetic mechanisms are involved in cellular reprogramming, pathways and activated or suppressed genes are specific to the organism. In the bacterial genome the stress-induced epigenetic reprogramming of gene expression involves changes in methylation of GATC sites by Dam and direct modifications of gene promoters by changes in nonspecific binding of HLPs, including HU, H-NS, Fis, Dps, IHF, and Lrp. In mammalian cells the stress-induced reprogramming occurs by hyper- and hypo-methylation of CpG dinucleotides in the genome and by modification of histones. In both organisms the epigenetic alterations drive cells for activation of the mutator phenotype.
The presented hypothesis is only a general overview of the processes that may underlie cellular adaptation to stresses in diverse organisms. Though this overview does not provide direct evidence, it may improve understanding of several aspects of molecular and cellular evolution and predict some unknown phenomena in bacterial adaptation. For example, the effect of composition of the bacterial membrane, and in particular, a prevalence of acidic phospholipids (signals to proliferation, which are akin to mammalian growth factors), on the rate of adaptive mutations has not been reported in literature. A parallel between mammalian and bacterial cells in signalling for activation of the mutator phenotype shows that this effect likely exists. An increase of acidic phospholipids in the cellular membrane of stationary state bacteria likely increases the rate of adaptive mutations and facilitates bacterial adjustment to a new environment. Conversely, an increase of basic phospholipids likely suppresses adaptive mutations. Therefore, basic phospholipids may be efficient at preventing the development of antibiotic resistance in bacteria. Further studies are needed to test the proposed effect of membrane composition on bacterial adaptation.
The review provides only indirect support to the proposed hypothesis of stress-induced cellular adaptation. Direct experimental confirmation of the hypothesis may be complicated by the presence of different subpopulations in the sustained stress environment. As demonstrated in Table 3, the bacterial subpopulations in the stationary phase represent survival strategies with different regulation (up-regulation versus down-regulation) of the common cellular processes and genes. Therefore, initial separation of the sub-populations is very important for measuring gene activities or protein abundances that are specific for each strategy.
In conclusion, the review provides support for the model of activation of the mutator phenotype by stress-induced cellular adaptation. According to the model, stress-related survival and proliferative signals in a sustained stress environment lead to the emergence of a subpopulation of epige-netically reprogrammed proliferating cells. The reprogrammed subpopulation, in turn, activates the mutator phenotype as an alternative survival strategy to senescence and programmed cell death. The mutator phenotype strategy and conditions activating it may share many common characteristics at the cellular level across different organisms.
Fig. (2).
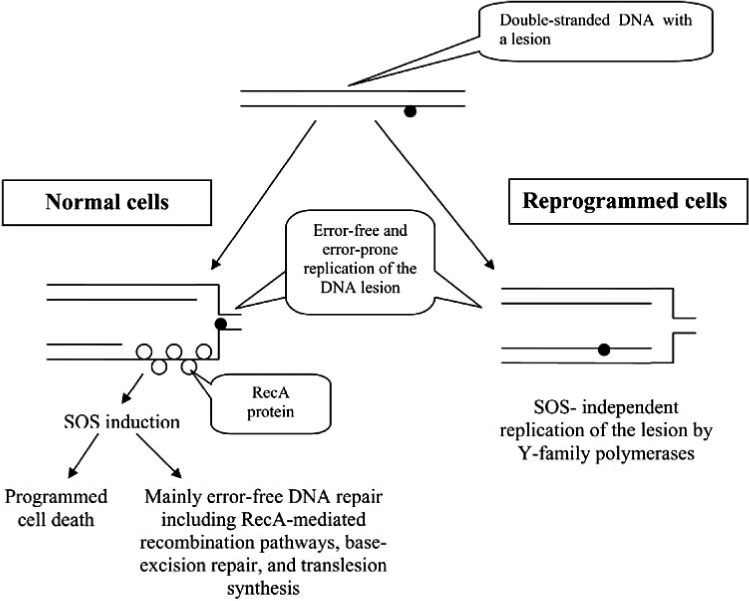
Replication of DNA lesion in normal bacterial cells and in epigenetically reprogrammed proliferating cells. In a normal cell the formation of single-stranded DNA downstream from the DNA lesion arrests DNA replication. The gap is covered with RecA proteins and SOS response is activated to repair the lesion. If the lesion cannot be repaired, the cell activates the programmed cell death. In the reprogrammed proliferating cells, cell cycle arrest cannot be activated because of epigenetic silencing of cell cycle inhibitors. DNA lesion is replicated by Y-family polymerases without SOS induction.
Table 1.
Glossary of Terms Used in the Paper
| Epigenetic alterations | Inherited but revocable changes in the genome (without modification of DNA sequences) that lead to metastable changes in gene expression. |
| Restriction–modification system | Bacterial mechanism of defence against invasion by foreign DNA (for example, viruses). It is comprised of genes that encode a restriction enzyme and a modification methylase. |
| Histone Like Proteins (HLPs) | Proteins with non-specific DNA-binding activity that regulate gene expression in the bacterial genome by changes in DNA supercoiling and that play a role in a structural organization of the bacterial DNA. |
| Small RNAs (sRNA) | Small non-coding RNAs regulating gene expression at the level of transcription, translation, chromatin organisation by base-pairing with a target mRNA or DNA. In mammalian cells they include small nuclear RNAs (snoRNA), microRNAs (miRNA), and short interfering RNAs (siRNA/iRNA). |
| Cellular strategy | A set of cellular functions that are epigenetically fixed in the genome. |
| Stationary phase/state | A steady-state equilibrium at which the continuing cell division is balanced by the continuing cell death. |
| Sustained stress environment | Cellular environment leading to continuing cell death and, therefore, sustained generation of stress-induced survival and proliferative signaling in a subpopulation of the cells. |
| Epigenetic reprogramming | A set of epigenetic alterations in the genome that are induced by sustained changes in the cellular environment and that allow to implement a cellular strategy. |
| Stationary-state mutagenesis | The acquisition of beneficial mutations by bacteria in a growth-limited state. |
| Mutator phenotype | A cellular survival strategy induced by sustained stress environment and characterized by increased rate of mutations. |
| Stress-induced cellular adaptation | Epigenetic reprogramming of a subpopulation of cells in a sustained stress environment to implement the mutator phenotype. |
Footnotes
See Table 1 for definitions of underlined terms.
REFERENCES
- 1.Hall BG. Adaptive mutagenesis: a process that generates almost exclusively beneficial mutations. Genetica. 1998;102-103:109–125. [PubMed] [Google Scholar]
- 2.Janion C. A new look at adaptive mutations in bacteria. Acta Biochim Pol. 2001;47:451–457. [PubMed] [Google Scholar]
- 3.Karpinets TV, Foy BD. Tumorigenesis: the adaptation of mammalian cells to sustained stress environment by epigenetic alterations and succeeding matched mutations. Carcinogenesis. 2005;26:1323–1334. doi: 10.1093/carcin/bgi079. [DOI] [PubMed] [Google Scholar]
- 4.Tenaillon O, Denamur E, Matic I. Evolutionary significance of stress-induced mutagenesis in bacteria. Trends Microbiol. 2004;12:264–270. doi: 10.1016/j.tim.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Finkel SE. Long-term survival during stationary phase: evolution and the GASP phenotype. Nat Rev Microbiol. 2006;4:113–120. doi: 10.1038/nrmicro1340. [DOI] [PubMed] [Google Scholar]
- 6.Denamur E, Matic I. Evolution of mutation rates in bacteria. Mol Microbiol. 2006;60:820–827. doi: 10.1111/j.1365-2958.2006.05150.x. [DOI] [PubMed] [Google Scholar]
- 7.Nephew KP, Huang TH. Epigenetic gene silencing in cancer initiation and progression. Cancer Lett. 2003;190:125–133. doi: 10.1016/s0304-3835(02)00511-6. [DOI] [PubMed] [Google Scholar]
- 8.Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629–656. doi: 10.1146/annurev.pharmtox.45.120403.095832. [DOI] [PubMed] [Google Scholar]
- 9.Cheung P, Lau P. Epigenetic regulation by histone methylation and histone variants. Mol Endocrinol. 2005;19:563–573. doi: 10.1210/me.2004-0496. [DOI] [PubMed] [Google Scholar]
- 10.Mattick JS, Makunin IV. Small regulatory RNAs in mammals. Hum Mol Genet. 2005;14:R121–132. doi: 10.1093/hmg/ddi101. [DOI] [PubMed] [Google Scholar]
- 11.Talbert PB, Henikoff S. Spreading of silent chromatin: inaction at a distance. Nat Rev Genet. 2006;7:793–803. doi: 10.1038/nrg1920. [DOI] [PubMed] [Google Scholar]
- 12.Wion D, Casadesus J. N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat Rev Microbiol. 2006;4:183–192. doi: 10.1038/nrmicro1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin JCP, Krebs MP, Reznikoff WS. Effect of dam methylation on Tn5 transposition. J Mol Biol. 1988;199:35–45. doi: 10.1016/0022-2836(88)90377-4. [DOI] [PubMed] [Google Scholar]
- 14.Rasmussen LJ, Lobner-Olesen A, Marinus MG. Growth-rate-dependent transcription initiation from the dam P2 promoter. Gene. 1995;157:213–215. doi: 10.1016/0378-1119(94)00619-4. [DOI] [PubMed] [Google Scholar]
- 15.Low DA, Weyand NJ, Mahan MJ. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect Immun. 2001;69:7197–7204. doi: 10.1128/IAI.69.12.7197-7204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- 17.Lobner-Olesen A, Skovgaard O, Marinus MG. Dam methylation: coordinating cellular processes. Curr Opin Microbiol. 2005;8:154–160. doi: 10.1016/j.mib.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Stephens C, Reisenauer A, Wright R, Shapiro L. A cell cycle-regulated bacterial DNA methyltransferase is essential for viability. Proc Natl Acad Sci USA. 1997;93:1210–1214. doi: 10.1073/pnas.93.3.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahng LS, Shapiro L. The CcrM DNA methyltransferase of Agrobacterium tumefaciens is essential, and its activity is cell cycle regulated. J Bacteriol. 2001;183:3065–3075. doi: 10.1128/JB.183.10.3065-3075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kossykh VG, Lloyd RS. A DNA adenine methyltransferase of Escherichia coli that is cell cycle regulated and essential for viability. J Bacteriol. 2004;186:6340. doi: 10.1128/JB.186.18.6340.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shanado Y, Kato J, Ikeda H. Escherichia coli HU protein suppresses DNA-gyrase-mediated illegitimate recombination and SOS induction. Genes Cells. 1998;3:511–520. doi: 10.1046/j.1365-2443.1998.00208.x. [DOI] [PubMed] [Google Scholar]
- 22.Kobryn K, Lavoie BD, Chaconas GZ. Supercoiling-dependent site-specific binding of HU to naked Mu DNA. J Mol Biol. 1999;289:777–784. doi: 10.1006/jmbi.1999.2805. [DOI] [PubMed] [Google Scholar]
- 23.Balandina A, Claret L, Hengge-Aronis R, Rouviere-Yaniv J. The Escherichia coli histone-like protein HU regulates rpoS translation. Mol Microbiol. 2001;39:1069–1079. doi: 10.1046/j.1365-2958.2001.02305.x. [DOI] [PubMed] [Google Scholar]
- 24.Dorman CJ, Deighan P. Regulation of gene expression by histone-like proteins in bacteria. Curr Opin Genet Dev. 2003;13:179–184. doi: 10.1016/s0959-437x(03)00025-x. [DOI] [PubMed] [Google Scholar]
- 25.Sagi D, Friedman N, Vorgias C, Oppenheim AB, Stavans J. Modulation of DNA conformations through the formation of alternative high-order HU-DNA complexes. J Mol Biol. 2004;341:419–428. doi: 10.1016/j.jmb.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 26.Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Yamashino T, Ueguchi C, Mizuno T. Quantitative control of the stationary phase-specific sigma factor, sigma S, in Escherichia coli: involvement of the nucleoid protein H-NS. EMBO J. 1995;14:594–602. doi: 10.1002/j.1460-2075.1995.tb07035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atlung T, Ingmer H. H-NS: a modulator of environmentally regulated gene expression. Mol Microbiol. 1997;24:7–17. doi: 10.1046/j.1365-2958.1997.3151679.x. [DOI] [PubMed] [Google Scholar]
- 29.Palchaudhuri S, Tominna B, Leon MA. H-NS regulates DNA repair in Shigella. J Bacteriol. 1998;180:5260–5262. doi: 10.1128/jb.180.19.5260-5262.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol. 1999;181:6361–6370. doi: 10.1128/jb.181.20.6361-6370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hommais F, Krin E, Laurent-Winter C, Soutourina O, Malpertuy A, Le Caer JP, Danchin A, Bertin P. Large-scale monitoring of pleiotropic regulation of gene expression by the prokaryotic nucleoid-associated protein, H-NS. Mol Microbiol. 2001;40:20–36. doi: 10.1046/j.1365-2958.2001.02358.x. [DOI] [PubMed] [Google Scholar]
- 32.Schroder O, Wagner R. The bacterial regulatory protein H-NS-a versatile modulator of nucleic acid structures. Biol Chem. 2002;383:945–960. doi: 10.1515/BC.2002.101. [DOI] [PubMed] [Google Scholar]
- 33.Rimsky S. Structure of the histone-like protein H-NS and its role in regulation and genome superstructure. Curr Opin Microbiol. 2004;7:109–114. doi: 10.1016/j.mib.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 34.Nishino K, Yamaguchi A. Role of histone-like protein H-NS in multidrug resistance of Escherichia coli. J Bacteriol. 2004;186:1423–1429. doi: 10.1128/JB.186.5.1423-1429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dame RT, Luijsterburg MS, Krin E, Bertin PN, Wagner R, Wuite GJ. DNA bridging: a property shared among H-NS-like proteins. J Bacteriol. 2005;187:1845–1848. doi: 10.1128/JB.187.5.1845-1848.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Opel ML, Aeling KA, Holmes WM, Johnson RC, Benham CJ, Hatfield GWZ. Activation of transcription initiation from a stable RNA promoter by a Fis protein-mediated DNA structural transmission mechanism. Mol Microbiol. 2004;53:665–674. doi: 10.1111/j.1365-2958.2004.04147.x. [DOI] [PubMed] [Google Scholar]
- 37.Luijsterburg MS, Noom MC, Wuite GJ, Dame RT. The architectural role of nucleoid-associated proteins in the organization of bacterial chromatin: A molecular perspective. J Struct Biol. 2006;156:262–272. doi: 10.1016/j.jsb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 38.Hirsch M, Elliott T. Fis regulates transcriptional induction of RpoS in Salmonella enterica. J Bacteriol. 2005;187:1568–1580. doi: 10.1128/JB.187.5.1568-1580.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pul U, Wurm R, Lux B, Meltzer M, Menzel A, Wagner R. LRP and H-NS--cooperative partners for transcription regulation at Escherichia coli rRNA promoters. Mol Microbiol. 2005;58:864–876. doi: 10.1111/j.1365-2958.2005.04873.x. [DOI] [PubMed] [Google Scholar]
- 40.Skoko D, Yan J, Johnson RC, Marko JF. Low-force DNA condensation and discontinuous high-force decondensation reveal a loop-stabilizing function of the protein Fis. Phys Rev Lett. 2005;95:208101. doi: 10.1103/PhysRevLett.95.208101. [DOI] [PubMed] [Google Scholar]
- 41.Nair S, Finkel SE. Dps protects cells against multiple stresses during stationary phase. J Bacteriol. 2004;186:4192–4198. doi: 10.1128/JB.186.13.4192-4198.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frenkiel-Krispin D, Ben-Avraham I, Englander J, Shimoni E, Wolf SG, Minsky A. Nucleoid restructuring in stationary-state bacteria. Mol Microbiol. 2004;51:395–405. doi: 10.1046/j.1365-2958.2003.03855.x. [DOI] [PubMed] [Google Scholar]
- 43.Su M, Cavallo S, Stefanini S, Chiancone E, Chasteen ND. The so-called Listeria innocua ferritin is a Dps protein. Iron incorporation, detoxification, and DNA protection properties. Biochemistry. 2005;44:5572–5578. doi: 10.1021/bi0472705. [DOI] [PubMed] [Google Scholar]
- 44.Murtin C, Engelhorn M, Geiselmann J, Boccard F. A quantitative UV laser footprinting analysis of the interaction of IHF with specific binding sites: reevaluation of the effective concentration of IHF in the cell. J Mol Biol. 1998;284:949–961. doi: 10.1006/jmbi.1998.2256. [DOI] [PubMed] [Google Scholar]
- 45.Ilves H, Horak R, Teras R, Kivisaar M. IHF is the limiting host factor in transposition of Pseudomonas putida transposon Tn4652 in stationary phase. Mol Microbiol. 2004;51:1773–1785. doi: 10.1111/j.1365-2958.2003.03948.x. [DOI] [PubMed] [Google Scholar]
- 46.Landgraf JR, Wu J, Calvo JM. Effects of nutrition and growth rate on Lrp levels in Escherichia coli. J Bacteriol. 1997;178:6930–6936. doi: 10.1128/jb.178.23.6930-6936.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tani TH, Khodursky A, Blumenthal RM, Brown PO, Matthews RG. Adaptation to famine: a family of stationary-phase genes revealed by microarray analysis. Proc Natl Acad Sci USA. 2002;99:13471–13476. doi: 10.1073/pnas.212510999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brinkman AB, Ettema TJ, de Vos WM, van der Oost J. The Lrp family of transcriptional regulators. Mol Microbiol. 2003;48:287–294. doi: 10.1046/j.1365-2958.2003.03442.x. [DOI] [PubMed] [Google Scholar]
- 49.Hernday A, Braaten B, Low D. The intricate workings of a bacterial epigenetic switch. Adv Exp Med Biol. 2004;547:83–89. doi: 10.1007/978-1-4419-8861-4_7. [DOI] [PubMed] [Google Scholar]
- 50.Weber H, Polen T, Heuveling J, Wendisch VF, Hengge R. Genome-wide analysis of the general stress response network in Escherichia coli: sigmaS-dependent genes, promoters, and sigma factor selectivity. J Bacteriol. 2005;187:1591–1603. doi: 10.1128/JB.187.5.1591-1603.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masse E, Majdalani N, Gottesman S. Regulatory roles for small RNAs in bacteria. Curr Opin Microbiol. 2003;6:120–124. doi: 10.1016/s1369-5274(03)00027-4. [DOI] [PubMed] [Google Scholar]
- 52.Repoila F, Majdalani N, Gottesman S. Small non-coding RNAs, co-ordinators of adaptation processes in Escherichia coli: the RpoS paradigm. Mol Microbiol. 2003;48:855–861. doi: 10.1046/j.1365-2958.2003.03454.x. [DOI] [PubMed] [Google Scholar]
- 53.Gottesman S. The small RNA regulators of Escherichia coli: roles and mechanisms. Annu Rev Microbiol. 2004;58:303–328. doi: 10.1146/annurev.micro.58.030603.123841. [DOI] [PubMed] [Google Scholar]
- 54.Majdalani N, Vanderpool CK, Gottesman S. Bacterial small RNA regulators. Crit Rev Biochem Mol Biol. 2005;40:93–113. doi: 10.1080/10409230590918702. [DOI] [PubMed] [Google Scholar]
- 55.van der Woude M, Hale WB, Low DA. Formation of DNA methylation patterns: nonmethylated GATC sequences in gut and pap operons. J Bacteriol. 1998;180:5913–5920. doi: 10.1128/jb.180.22.5913-5920.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Travers A, Muskhelishvili G. Bacterial chromatin. Curr Opin Genet Dev. 2005;15:507–514. doi: 10.1016/j.gde.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Noordewier M, Benham CJ. Stress-induced DNA duplex destabilization (SIDD) in the E. coli genome: SIDD sites are closely associated with promoters. Genome Res. 2004;14:1575–1584. doi: 10.1101/gr.2080004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Altuvia S. Regulatory small RNAs: the key to coordinating global regulatory circuits. J Bacteriol. 2004;186:6679–6680. doi: 10.1128/JB.186.20.6679-6680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kasak L, Horak R, Kivisaar M. Promoter-creating mutations in Pseudomonas putida: a model system for the study of mutation in starving bacteria. Proc Natl Acad Sci USA. 1997;94:3134–3139. doi: 10.1073/pnas.94.7.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sung HM, Yasbin RE. Adaptive, or stationary-phase, mutagenesis, a component of bacterial differentiation in Bacillus subtilis. J Bacteriol. 2002;84:5641–53. doi: 10.1128/JB.184.20.5641-5653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bjedov I, Tenaillon O, Gerard B, Souza V, Denamur E, Radman M, Taddei F, Matic I. Stress-induced mutagenesis in bacteria. Science. 2003;300:1404–1409. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- 62.Rosenberg SM, Hastings PJ. Adaptive Point Mutation and Adaptive Amplification Pathways in the Escherichia coli Lac System: Stress Responses Producing Genetic Change. J Bacteriol. 2004;186:4838–4843. doi: 10.1128/JB.186.15.4838-4843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hersh MN, Ponder RG, Hastings PJ, Rosenberg SM. Adaptive mutation and amplification in Escherichia coli: two pathways of genome adaptation under stress. Res Microbiol. 2004;155:352–359. doi: 10.1016/j.resmic.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 64.Roth JR, Andersson DI. Amplification-mutagenesis--how growth under selection contributes to the origin of genetic diversity and explains the phenomenon of adaptive mutation. Res Microbiol. 2004;155:342–51. doi: 10.1016/j.resmic.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 65.Torkelson J, Harris RS, Lombardo MJ, Nagendran J, Thulin C, Rosenberg SM. Genome-wide hypermutation in a subpopulation of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO J. 1997;16:3303–3311. doi: 10.1093/emboj/16.11.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lombardo MJ, Torkelson J, Bull HJ, McKenzie GJ, Rosenberg SM. Mechanisms of genome-wide hypermutation in stationary phase. Ann N Y Acad Sci. 1999;870:275–289. doi: 10.1111/j.1749-6632.1999.tb08888.x. [DOI] [PubMed] [Google Scholar]
- 67.Bridges BA. Hypermutation under stress. Nature. 1997;387:557–558. doi: 10.1038/42370. [DOI] [PubMed] [Google Scholar]
- 68.Lewis K. Programmed death in bacteria. Microbiol Mol Biol Rev. 2000;64:503–14. doi: 10.1128/mmbr.64.3.503-514.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 70.Saumaa S, Tarassova K, Tark M, Tover A, Tegova R, Kivisaar M. Involvement of DNA mismatch repair in stationary-phase mutagenesis during prolonged starvation of Pseudomonas putida. DNA Repair (Amst) 2006;5:505–514. doi: 10.1016/j.dnarep.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 71.Karpinets TV, Foy BD. Model of the developing tumorigenic phenotype in mammalian cells and the role of sustained stress. J Theoret Biol. 2004;227:253–264. doi: 10.1016/j.jtbi.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 72.Ben-Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Invest. 2004;113:8–13. doi: 10.1172/JCI200420663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 74.Nystrom T. Conditional senescence in bacteria: death of the immortals. Mol Microbiol. 2003;48:17–23. doi: 10.1046/j.1365-2958.2003.03385.x. [DOI] [PubMed] [Google Scholar]
- 75.Hamoen LW, Venema G, Kuipers OP. Controlling competence in Bacillus subtilis: shared use of regulators. Microbiology. 2003;149:9–17. doi: 10.1099/mic.0.26003-0. [DOI] [PubMed] [Google Scholar]
- 76.Tortosa P, Dubnau D. Competence for transformation: a matter of taste. Curr Opin Microbiol. 1999;2:588–592. doi: 10.1016/s1369-5274(99)00026-0. [DOI] [PubMed] [Google Scholar]
- 77.Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol. 2005;3:371–82. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 78.Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 2006;6:53. doi: 10.1186/1471-2180-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bihani T, Mason DX, Jackson TJ, Chen SC, Boettner B, Lin AW. Differential oncogenic Ras signaling and senescence in tumor cells. Cell Cycle. 2004;3:1201–1207. [PubMed] [Google Scholar]
- 80.Murray HD, Schneider DA, Gourse RL. Control of rRNA expression by small molecules is dynamic and nonredundant. Mol Cell. 2003;12:125–34. doi: 10.1016/s1097-2765(03)00266-1. [DOI] [PubMed] [Google Scholar]
- 81.Scott JM, Haldenwang WG. Obg, an essential GTP binding protein of Bacillus subtilis, is necessary for stress activation of transcription factor sigma(B) J Bacteriol. 1999;181:4653–4660. doi: 10.1128/jb.181.15.4653-4660.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nystrom T. Growth versus maintenance: a trade-off dictated by RNA polymerase availability and sigma factor competition? Mol Microbiol. 2004;54:855–862. doi: 10.1111/j.1365-2958.2004.04342.x. [DOI] [PubMed] [Google Scholar]
- 83.Magnusson LU, Farewell A, Nystrom T. ppGpp: a global regulator in Escherichia coli. Trends Microbiol. 2005;13:236–242. doi: 10.1016/j.tim.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 84.Wout P, Pu K, Sullivan SM, Reese V, Zhou S, Lin B, Maddock JR. The Escherichia coli GTPase CgtAE cofractionates with the 50S ribosomal subunit and interacts with SpoT, a ppGpp synthetase/hydrolase. J Bacteriol. 2004;186:5249–5257. doi: 10.1128/JB.186.16.5249-5257.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lerner CG, Inouye M. Pleiotropic changes resulting from depletion of Era, an essential GTP-binding protein in Escherichia coli. Mol Microbiol. 1991;5:951–957. doi: 10.1111/j.1365-2958.1991.tb00770.x. [DOI] [PubMed] [Google Scholar]
- 86.Britton RA, Powell BS, Dasgupta S, Sun Q, Margolin W, Lupski JR, Court DL. Cell cycle arrest in Era GTPase mutants: a potential growth rate-regulated checkpoint in Escherichia coli. Mol Microbiol. 1998;30:739–750. doi: 10.1046/j.1365-2958.1998.00719.x. [DOI] [PubMed] [Google Scholar]
- 87.Foti JJ, Schienda J, Sutera VA, Jr, Lovett ST. A bacterial G protein-mediated response to replication arrest. Mol Cell. 2005;17:549–560. doi: 10.1016/j.molcel.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 88.Michel B. Obg/CtgA, a signaling protein that controls replication, translation, and morphological development? Dev Cell. 2005;8:300–301. doi: 10.1016/j.devcel.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 89.Boeneman K, Crooke E. Chromosomal replication and the cell membrane. Curr Opin Microbiol. 2005;8:143–148. doi: 10.1016/j.mib.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 90.Messer W. The bacterial replication initiator DnaA. DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiol Rev. 2002;26:355–74. doi: 10.1111/j.1574-6976.2002.tb00620.x. [DOI] [PubMed] [Google Scholar]
- 91.Ichihashi N, Kurokawa K, Matsuo M, Kaito C, Sekimizu K. Inhibitory effects of basic or neutral phospholipids on acidic phospholipid-mediated dissociation of adenine nucleotide bound to DnaA protein, the initiator of chromosomal DNA replication. J Biol Chem. 2003;278:28778–28786. doi: 10.1074/jbc.M212202200. [DOI] [PubMed] [Google Scholar]
- 92.Yuk HG, Marshall DL. Effect of trisodium phosphate adaptation on changes in membrane lipid composition, verotoxin secretion, and acid resistance of Escherichia coli O157: H7 in simulated gastric fluid. Int J Food Microbiol. 2006;106:39–44. doi: 10.1016/j.ijfoodmicro.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 93.Wold S, Crooke E, Skarstad K. The Escherichia coli Fis protein prevents initiation of DNA replication from oriC in vitro. Nucleic Acids Res. 1997;24:3527–3532. doi: 10.1093/nar/24.18.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ryan VT, Grimwade JE, Camara JE, Crooke E, Leonard AC. Escherichia coli prereplication complex assembly is regulated by dynamic interplay among Fis, IHF and DnaA. Mol Microbiol. 2004;51:1347–1359. doi: 10.1046/j.1365-2958.2003.03906.x. [DOI] [PubMed] [Google Scholar]
- 95.Ninnemann O, Koch C, Kahmann R. The E. coli fis promoter is subject to stringent control and autoregulation. EMBO J. 1992;11:13–20. doi: 10.1002/j.1460-2075.1992.tb05146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aizenman E, Engelberg-Kulka H, Glaser G. An Escherichia coli chromosomal "addiction module" regulated by guanosine 3’,5’-bispyrophosphate: a model for programmed bacterial cell death. Proc Natl Acad Sci USA. 1997;93:6059–6063. doi: 10.1073/pnas.93.12.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hazan R, Sat B, Engelberg-Kulka H. Escherichia coli mazEF-Mediated Cell Death Is Triggered by Various Stressful Conditions. J Bacteriol. 2004;186:3663–3669. doi: 10.1128/JB.186.11.3663-3669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harris RS, Feng G, Ross KJ, Sidhu R, Thulin C, Longerich S, Szigety SK, Winkler ME, Rosenberg SM. Mismatch repair protein MutL becomes limiting during stationary-phase mutation. Genes Dev. 1997;11:2426–37. doi: 10.1101/gad.11.18.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bridges BA, Timms A. Effect of endogenous carotenoids and defective RpoS sigma factor on spontaneous mutation under starvation conditions in Escherichia coli: evidence for the possible involvement of singlet oxygen. Mutat Res. 1998;403:21–28. doi: 10.1016/s0027-5107(98)00013-x. [DOI] [PubMed] [Google Scholar]
- 100.Harris RS, Feng G, Ross KJ, Sidhu R, Thulin C, Longerich S, Szigety SK, Hastings PJ, Winkler ME, Rosenberg SM. Mismatch repair is diminished during stationary-phase mutation. Mutat Res. 1999;437:51–60. [PubMed] [Google Scholar]
- 101.Pedraza-Reyes M, Yasbin RE. Contribution of the mismatch DNA repair system to the generation of stationary-phase-induced mutants of Bacillus subtilis. J Bacteriol. 2004;186:6485–6491. doi: 10.1128/JB.186.19.6485-6491.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ponder RG, Fonville NC, Rosenberg SM. Switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell. 2005;19:791–804. doi: 10.1016/j.molcel.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 103.Eichenbaum Z, Livneh Z. UV light induces IS10 transposition in Escherichia coli. Genetics. 1998;149:1173–1181. doi: 10.1093/genetics/149.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ilves H, Horak R, Kivisaar M. Involvement of sigma [S] in starvation-induced transposition of Pseudomonas putida transposon Tn4652. J Bacteriol. 2001;183:5445–5448. doi: 10.1128/JB.183.18.5445-5448.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ghanekar K, McBride A, Dellagostin O, Thorne S, Mooney R, McFadden J. Stimulation of transposition of the Mycobacterium tuberculosis insertion sequence IS6110 by exposure to a microaerobic environment. Mol Microbiol. 1999;33:982–993. doi: 10.1046/j.1365-2958.1999.01539.x. [DOI] [PubMed] [Google Scholar]
- 106.Jock S, Jacob T, Kim WS, Hildebrand M, Vosberg HP, Geider K. Instability of short-sequence DNA repeats of pear pathogenic Erwinia strains from Japan and Erwinia amylovora fruit tree and raspberry strains. Mol Genet Genomics. 2003;268:739–49. doi: 10.1007/s00438-003-0814-6. [DOI] [PubMed] [Google Scholar]
- 107.Jackson AL, Chen R, Loeb LA. Induction of microsatellite instability by oxidative DNA damage. Proc Natl Acad Sci USA. 1998;95:12468–12473. doi: 10.1073/pnas.95.21.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jackson AL, Loeb LA. Microsatellite instability induced by hydrogen peroxide in Escherichia coli. Mutat Res. 2000;447:187–198. doi: 10.1016/s0027-5107(99)00206-7. [DOI] [PubMed] [Google Scholar]
- 109.Yamamura E, Nunoshiba T, Nohmi T, Yamamoto K. Hydrogen peroxide-induced microsatellite instability in the Escherichia coli K-12 endogenous tonB gene. Biochem Biophys Res Commun. 2003;306:570–576. doi: 10.1016/s0006-291x(03)01027-1. [DOI] [PubMed] [Google Scholar]
- 110.Rocha EP, Matic I, Taddei F. Over-representation of repeats in stress response genes: a strategy to increase versatility under stressful conditions? Nucleic Acids Res. 2002;30:1886–1894. doi: 10.1093/nar/30.9.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ilves H, Horak R, Kivisaar M. Involvement of {sigma}S in Starvation-Induced Transposition of Pseudomonas putida Transposon Tn4652. J Bacteriol. 2001;183:5445–5448. doi: 10.1128/JB.183.18.5445-5448.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu D, Crellin P, Chalmers R. Cyclic changes in the affinity of protein-DNA interactions drive the progression and regulate the outcome of the Tn10 transposition reaction. Nucleic Acids Res. 2005;33:1982–1992. doi: 10.1093/nar/gki348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mahillon J, Chandler M. Insertion sequences. Microbiol Mol Biol Rev. 1998;62:725–774. doi: 10.1128/mmbr.62.3.725-774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roberts D, Hoopes BC, McClure WR, Kleckner N. IS10 transposition is regulated by DNA adenine methylation. Cell. 1985;43:117–130. doi: 10.1016/0092-8674(85)90017-0. [DOI] [PubMed] [Google Scholar]
- 115.Yin JCP, Krebs MP, Reznikoff WS. Effect of dam methylation on Tn5 transposition. J Mol Biol. 1988;199:35–45. doi: 10.1016/0022-2836(88)90377-4. [DOI] [PubMed] [Google Scholar]
- 116.Shcherbakova PV, Fijalkowska IJ. Translesion synthesis DNA polymerases and control of genome stability. Front Biosci. 2006;11:2496–2517. doi: 10.2741/1985. [DOI] [PubMed] [Google Scholar]
- 117.Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Annu Rev Microbiol. 2006;60:231–53. doi: 10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 118.Avkin S, Sevilyam Z, Toubem L, Geacintovm N, Chaneym SG, Orenm M, Livneh Z. p53 and p21 regulate error-prone DNA repair to yield a lower mutation load. MolCell. 2006;22:407–413. doi: 10.1016/j.molcel.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 119.Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440–450. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 120.Metzgar D, Wills C. Evidence for the adaptive evolution of mutation rates. Cell. 2000;101:581–584. doi: 10.1016/s0092-8674(00)80869-7. [DOI] [PubMed] [Google Scholar]
- 121.McKenzie GJ, Rosenberg SM. Adaptive mutations, mutator DNA polymerases and genetic change strategies of pathogens. Curr Opin Microbiol. 2001;4:586–594. doi: 10.1016/s1369-5274(00)00255-1. [DOI] [PubMed] [Google Scholar]
- 122.Loewe LL, Textor V, Scherer S. High Deleterious Genomic Mutation Rate in Stationary Phase of Escherichia coli. Science. 2003;302:1558–1560. doi: 10.1126/science.1087911. [DOI] [PubMed] [Google Scholar]
- 123.Faure D, Frederick R, Wloch D, Portier P, Blot M, Adams J. Genomic Changes Arising in Long-Term Stab Cultures of Escherichia coli. J Bacteriol. 2004;186:6437–6442. doi: 10.1128/JB.186.19.6437-6442.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Naas T, Blot M, Fitch WM, Arber W. Dynamics of IS-related genetic rearrangements in resting Escherichia coli K-12. Mol Biol Evol. 1995;12:198–207. doi: 10.1093/oxfordjournals.molbev.a040198. [DOI] [PubMed] [Google Scholar]
- 125.Zinser ER, Schneider D, Blot M, Kolter R. Bacterial evolution through the selective loss of beneficial genes: trade-offs in expression involving two loci. Genetics. 2003;164:1271–1277. doi: 10.1093/genetics/164.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zinser ER, Kolter R. Prolonged stationary-phase incubation selects for lrp mutation in Escherichia coli K-12. J Bacteriol. 2000;182:4361–4365. doi: 10.1128/jb.182.15.4361-4365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hamoen LW, Venema G, Kuipers OP. Controlling competence in Bacillus subtilis: shared use of regulators. Microbiology. 2003;149:9–17. doi: 10.1099/mic.0.26003-0. [DOI] [PubMed] [Google Scholar]
- 128.Boles BR, Thoendel M, Singh PK. Self-generated diversity produces "insurance effects" in biofilm communities. Proc Natl Acad Sci USA. 2004;101:16630–16635. doi: 10.1073/pnas.0407460101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bachman MA, Swanson MS. Genetic evidence that Legionella pneumophila RpoS modulates expression of the transmission phenotype in both the exponential phase and the stationary phase. Infect Immun. 2004;72:2468–2476. doi: 10.1128/IAI.72.5.2468-2476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lombardo MJ, Aponyi I, Rosenberg SM. General stress response regulator RpoS in adaptive mutation and amplification in Escherichia coli. Genetics. 2004;166:669–680. doi: 10.1534/genetics.166.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Madan R, Kolter R, Mahadevan S. Mutations that activate the silent bgl operon of Escherichia coli confer a growth advantage in stationary phase. J Bacteriol. 2005;187:7912–7917. doi: 10.1128/JB.187.23.7912-7917.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tompkins JD, Nelson JL, Hazel JC, Leugers SL, Stumpf JD, Foster PL. Error-prone polymerase, DNA polymerase IV, is responsible for transient hypermutation during adaptive mutation in Escherichia coli. J Bacteriol. 2003;185:3469–3472. doi: 10.1128/JB.185.11.3469-3472.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sung HM, Yeamans G, Ross CA, Yasbin RE. Roles of YqjH and YqjW, homologs of the Escherichia coli UmuC/DinB or Y superfamily of DNA polymerases, in stationary-phase mutagenesis and UV-induced mutagenesis of Bacillus subtilis. J Bacteriol. 2003;185:2153–2160. doi: 10.1128/JB.185.7.2153-2160.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2000;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 135.Perez-Capilla T, Baquero MR, Gomez-Gomez JM, Ionel A, Martin S, Blazquez J. SOS-independent induction of dinB transcription by β-lactam-mediated inhibition of cell wall synthesis in Escherichia coli. J Bacteriol. 2005;187:1515–1518. doi: 10.1128/JB.187.4.1515-1518.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Annu Rev Microbiol. 2006;60:231–53. doi: 10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 137.Camps M, Loeb LA. When pol I goes into high gear: processive DNA synthesis by pol I in the cell. Cell Cycle. 2004;3:116–118. [PubMed] [Google Scholar]
- 138.Banach-Orlowska M, Fijalkowska IJ, Schaaper RM, Jonczyk P. DNA polymerase II as a fidelity factor in chromosomal DNA synthesis in Escherichia coli. Mol Microbiol. 2005;58:61–70. doi: 10.1111/j.1365-2958.2005.04805.x. [DOI] [PubMed] [Google Scholar]
- 139.Layton JC, Foster PL. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol. 2003;50:549–61. doi: 10.1046/j.1365-2958.2003.03704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yeiser B, Pepper ED, Goodman MF, Finkel SE. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc Natl Acad Sci USA. 2002;99:8737–8741. doi: 10.1073/pnas.092269199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tegova R, Tover A, Tarassova K, Tark M, Kivisaar M. Involvement of error-prone DNA polymerase IV in stationary-phase mutagenesis in Pseudomonas putida. J Bacteriol. 2004;186:2735–2744. doi: 10.1128/JB.186.9.2735-2744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Saint-Ruf C, Matic I. Environmental tuning of mutation rates. Environment Microbiol. 2006;8:193–199. doi: 10.1111/j.1462-2920.2005.00968.x. [DOI] [PubMed] [Google Scholar]
- 143.Cirz RT, Romesberg FE. Induction and inhibition of ciprofloxacin resistance-conferring mutations in hypermutator bacteria. Antimicrob Agents Chemother. 2006;50:220–225. doi: 10.1128/AAC.50.1.220-225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]