

Abstract
Given at the Meeting of the Physiological Society held at the University of Southampton on 10 September 1998
Inflammation in the brain
Interest in neuroimmunology and the actions of cytokines in the brain has grown exponentially over the last decade or so, from a variety of different biological disciplines. Scientists and clinicians studying inflammatory diseases of the CNS, such as multiple sclerosis, began to look to proteins associated with the immune system as potential mediators of these conditions. In parallel, those scientists interested in fever recognized the importance of cytokines as endogenous pyrogens, while others, trying to understand the ‘sickness behaviour’ which is common to most diseases and injury, also began to study cytokines. My own interest in cytokines also arose from unexpected beginnings. In 1978, my first presentation to The Physiological Society described experiments on body weight regulation (Rothwell & Stock, 1978). Over the next decade our research identified the role of diet-induced thermogenesis and sympathetically mediated thermogenesis in the regulation of energy balance and body weight (Rothwell & Stock, 1984, 1986). The field of energy balance regulation was then, as now, dominated by the goal of understanding the causes of excess fat deposition and obesity and developing possible treatments.
Obesity is a problem of positive energy balance and excess fat deposition. Its antithesis is cachexia, a condition of wasting and weight loss which is usually associated with chronic diseases such as cancer, arthritis and AIDS, and acute disease such as severe injury or sepsis (Tisdale, 1997). In all of these conditions, cachexia contributes to morbidity and mortality and has been proposed as the major cause of death in cancer (Garrattini et al. 1980). Cytokines are recognized as primary mediators of cachexia through actions in the periphery and the CNS. Indeed, one of the first cytokines identified was initially named ‘cachectin’ (now known as tumour necrosis factor, TNFα) because it was believed to be an important mediator of cachexia (Beutler & Cerami, 1989).
During the late 1980s my interest turned towards cachexia and responses to injury, and therefore to cytokines. After studying the involvement of these proteins in CNS and immune responses to peripheral inflammation and tissue injury (Rothwell & Luheshi, 1994; Rothwell & Hopkins, 1995; Rothwell, 1997), we began to address the question of whether pro-inflammatory cytokines such as interleukin-1 (IL-1) might also be involved in host responses (including fever and hypermetabolism) to brain injury, and indeed whether such proteins might actually mediate neuronal damage and death.
Early studies on the actions of cytokines in the brain were conducted in the face of a widespread belief that these proteins were associated mainly with immune activation and peripheral inflammation, and therefore unlikely to influence the CNS, and that the brain was an ‘immune privileged organ’, which failed to exhibit clinical inflammatory or immune responses.
It is now recognized that cytokines have diverse actions in the brain, which modulate and mediate both systemic host responses to disease and local changes caused by CNS inflammation, infection and injury (see Rothwell & Luheshi, 1994; Rothwell & Hopkins, 1995; Rothwell, 1997).
Cytokines
This large and rapidly growing group of polypeptides comprises the interleukins, chemokines, tumour necrosis factors, interferons, and growth and cell stimulating factors; neurotrophins have also been included in this category. Cytokines have diverse actions on cell growth and differentiation, immune and inflammatory responses, and on a number of physiological systems particularly in disease.
Although most cytokines are expressed at low or undetectable levels in the healthy adult brain, many are induced in response to injury or infection (see Beneviste, 1992; Hopkins & Rothwell, 1995). For example, expression in the brain of interleukins (IL) 1, 2, 3, 4, 6, 8 and 10, several chemokines, TNFα, interferons and numerous growth factors is induced rapidly by experimental and clinical insults to the CNS (Table 1). The functions and actions of many of these cytokines in the brain remain to be elucidated, but probably include both beneficial and detrimental effects. However, there is now evidence that IL-1, TNFα, several chemokines and interferon-γ may contribute directly to neurodegeneration or impaired neuronal function. This review will focus on IL-1 and its involvement and mechanisms of action in neurodegeneration.
Table 1.
Factors which induce cytokine expression in the brain
| Systemic or brain infection or inflammation |
| Brain injury |
| Stroke |
| Excitotoxic brain damage |
| Multiple sclerosis and experimental allergic encephalomyelitis |
| Scrapie and Creutzfeldt–Jakob Disease |
| Down's syndrome |
| Alzheimer's disease |
| Parkinson's disease |
Interleukin-1
The IL-1 family comprises at least three proteins, IL-1α, IL-1β and IL-1ra - the products of separate genes which share a significant homology and probably derived from a common gene ancestor about 350 million years ago (Fig. 1). IL-1α and IL-1β are both agonists which, at least in the periphery, are believed to exert identical actions (Dinarello, 1991, 1998). IL-1 receptor antagonist (IL-1ra) is probably the only current example of a selective and specific endogenous receptor antagonist, which blocks the actions of IL-1α and β but has no known agonist activity (Dinarello & Thompson, 1991; Dinarello, 1996, 1998). IL-1ra appears to function as a naturally occurring inhibitor of IL-1 actions in the periphery (Dinarello, 1996, 1998) and in the brain (see below), and also provides a very useful pharmacological tool to study IL-1 functions.
Figure 1. The interleukin-1 (IL-1) family.
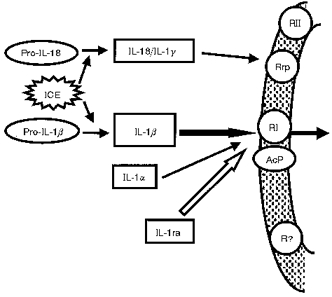
The IL-1 family comprises the two agonists IL-1α and IL-1β and the IL-1 receptor antagonist IL-1ra. Pro-IL-1β is inactive and must be cleaved by the enzyme interleukin-1β converting enzyme (ICE; also known as caspase 1). Actions of IL-1 are believed to be mediated through an 80 kDa type 1 (RI) receptor which requires an accessory protein (AcP) for signal transduction. IL-18 (also known as interferon gamma-inducing factor or IL-1γ) shares structural homology with the IL-1 family, is activated through cleavage by ICE and acts on an IL-1 receptor-related protein (Rrp), but is not confirmed as a member of the IL-1 family.
The IL-1 proteins are formed as precursors which lack leader sequences, and are enzymatically cleaved to the mature form by specific cellular proteases. Pro-IL-1α is biologically active but remains mostly within the cell, whereas the IL-1β precursor is inactive, so cleavage is an important regulatory step. Pro-IL-1β is cleaved to release mature, active IL-1β by an enzyme called IL-1β converting enzyme (ICE) (Cerretti et al. 1992; Thornberry et al. 1992), which has recently gained notoriety because of its potential role in apoptosis (Yuan et al. 1993). ICE shares homology with the gene ced-3, one of the death genes in the nematode worm Caenorhabditis elegans, and was the first identified member of the caspases - a family of cysteine proteases believed to execute apoptosis (Miura et al. 1993; Nicholson & Thornberry, 1997).
A third potential member of the IL-1 family has been identified recently. Interferon gamma-inducing factor (IGIF), also known as IL-18, was cloned from mouse liver (Okamura et al. 1995) but because of its homology with the IL-1 family, the name IL-1γ was proposed (Bazan et al. 1996). Interestingly pro-IL-18, like pro-IL-1β, is cleaved by ICE (Gu et al. 1997; Rano et al. 1997) and is in fact the preferred substrate for the enzyme (Rano et al. 1997). We have cloned rat IL-18/IL-1γ from rat brain, shown that it is also cleaved by ICE, and that, in contrast to IL-1α or β, it appears to be expressed constitutively in the rat brain (Culhane et al. 1998). The issue of whether IL-18 is a true member of the IL-1 family remains to be resolved, but similarities in its structure, cleavage, receptors and signalling mechanisms suggest a close relationship with IL-1 (Dinarello et al. 1998).
Several putative IL-1 receptors have been characterized, but all actions of IL-1 are believed to be effected through binding to the type 1 (80 kDa) receptor (IL-1RI) (Bomsztyk et al. 1989; Sims et al. 1993; O'Neill, 1996; Martin & Falk, 1997; Loddick et al. 1998), which requires an accessory protein (AcP) for signal transduction (Greenfeder et al. 1995). The type II (68 kDa) receptor (RII) has a short intracellular domain and is not thought to initiate signalling, but rather acts as a decoy which is shed from the membrane and binds IL-1 (Colotta et al. 1993). However, this receptor may mediate responses to IL-1 in the brain because an antibody to RII inhibits febrile responses to IL-1β, but not IL-1α (Luheshi et al. 1993; Rothwell et al. 1996b). There has been much speculation about the presence of novel or atypical IL-1 receptors in the brain, and an IL-1 receptor-related protein (IL-1RrP) was identified on the basis of its homology with known IL-1 receptors (Lovenberg et al. 1996; Parnet et al. 1996). Although RrP does not bind IL-1α or β it has recently been proposed as a receptor for IL-18 (Torigoe et al. 1997).
Expression of IL-1 in the brain
IL-1β seems to be the main IL-1 agonist induced in the brain in response to systemic (e.g. injury, infection) or local (e.g. injury, stroke) insults (see Rothwell & Luheshi, 1994; Hopkins & Rothwell, 1995; Rothwell et al. 1997). In rodents, IL-1β mRNA expression is increased within 15–30 min, and protein within 1 h of experimental cerebral ischaemia (stroke), brain injury or infusion of excitotoxins (Minami et al. 1992; Liu et al. 1993; Buttini et al. 1994; Yabuuchi et al. 1994), all of which lead to neuronal death. Several groups have also reported increased expression of IL-1β protein after experimental brain damage (e.g. Ianotti et al. 1993; Taupin et al. 1993).
We have demonstrated by immunocytochemistry using anti-rat IL-1β antibodies and cell-specific markers that in rat brain after ischaemic or excitotoxic insults, early expression (up to 24 h) of IL-1β is predominantly by microglia and meningeal macrophages around and within the emerging infarct (Davies et al. 1998; Pearson et al. 1998). In contrast, delayed expression (between 24 h and 7 days) is seen, often at distant sites, by astrocytes and invading immune cells (Davies et al. 1998). IL-1β expression appears to be associated with increased expression of ICE (Bhat et al. 1996) and cleavage of pro-IL-1β, since bioactive IL-1 is also markedly increased and is usually maximal 6–8 h after the insult (Fig. 2).
Figure 2. Bioactive IL-1 levels are increased after cerebral ischaemia.
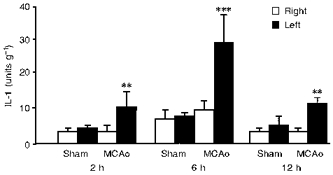
The figure shows bioactivity of IL-1 in the left and right cerebral hemispheres of rats subjected to sham surgery or unilateral focal cerebral ischaemia (middle cerebral artery occlusion (MCAo) in the left hemisphere). IL-1 was measured using a D10 thymocyte proliferation assay. Mean values ±s.e.m. are shown (n= 6). S. Loddick, S. Hopkins & N. Rothwell, unpublished data.
IL-1α expression is increased by stimuli similar to those increasing expression of IL-1β but at lower levels, and slightly after IL-1β in the brain. Expression of IL-1ra, the endogenous inhibitor of IL-1, is also increased by brain damage in rodents, usually slightly (30–60 min) after IL-1β and in different cells, mainly neurones (Toulmond & Rothwell, 1995a; Loddick et al. 1997).
Actions of IL-1 in the brain
Research into the actions of IL-1 in the brain initially focused on its role in host defence responses to systemic disease. Thus, it has been shown that IL-1 injected into the cerebral ventricles or the brain parenchyma, at doses as little as picomoles, induces responses which mimic those that occur during disease and injury, i.e. fever, anorexia, sickness behaviour, slow-wave sleep, and alterations in neuroendocrine (e.g. activation of the hypothalamic pituitary adrenal axis), cardiovascular and immune system function (see Rothwell & Luheshi, 1994; Rothwell & Hopkins, 1995; Rothwell, 1997). IL-1 appears to act as an endogenous mediator of these diverse host defence responses mainly through actions in the hypothalamus, since localized injections of IL-1ra or antibodies to IL-1β attenuate such responses to systemic stimuli (see Rothwell & Luheshi, 1994; Rothwell & Hopkins, 1995; Rothwell, 1997).
IL-1 can also elicit an array of responses which could either inhibit, exacerbate or induce neuronal damage and death. These include induction of fever and oedema, damage to the cerebral vasculature, activation of glia, induction of neurotrophins, growth factors/phospholipase A2, cyclooxygenase (COX-2), β-amyloid precursor protein, adhesion molecules and corticotrophin-releasing factor (CRF), release of nitric oxide and other free radicals, activation of complement, and modification of calcium homeostasis (see Beneviste, 1992; Giulian et al. 1993; Betz et al. 1996; Martin et al. 1996; Rothwell et al. 1996a,c; Rothwell, 1997; Rothwell et al. 1997).
The infusion of IL-1α or β into the brain of normal rodents or the application to neuronal cultures, even at doses in the high nanomolar range, does not result in overt damage or death. However, injection of recombinant IL-1β at picomolar doses into the cerebral ventricles (i.c.v.) or into specific brain regions (see below) of the rat markedly exacerbates neuronal damage induced by cerebral ischaemia, traumatic injury or excitotoxins (Relton & Rothwell, 1992; Loddick & Rothwell, 1996; Lawrence et al. 1998; and S. Toulmond, S. M. Allan, R. Grundy & N. J. Rothwell, unpublished data), and IL-1β can enhance cell death in cultures exposed to apoptotic stimuli (Friedlander et al. 1996; Troy et al. 1996). These data indicate either that IL-1 causes (or exacerbates) damage only to threatened cells, or that it synergizes with, or depends on, other factor(s) or responses present in the damaged brain.
Functional role of IL-1 in neurodegeneration
The most direct and substantial evidence indicating that endogenous IL-1 plays a functional role in neurodegeneration derives from in vivo studies in which its release or action has been blocked. The majority of such experiments have used recombinant IL-1ra - a highly effective and selective receptor antagonist for IL-1 (see above).
The first report that inhibition of IL-1 reduces brain damage in vivo (Relton & Rothwell, 1992) demonstrated that i.c.v. injection of IL-1ra at the time of induction of permanent focal cerebral ischaemia (middle cerebral artery occlusion (MCAo)) in the rat inhibited subsequent brain damage (infarct volume) by almost 70 % (Fig. 3). Numerous studies have subsequently verified and extended this observation (e.g. Betz et al. 1995, 1996; Garcia et al. 1995; Loddick & Rothwell, 1996; Relton et al. 1996; Rothwell et al. 1996a, 1997; Stroemer & Rothwell, 1997). It is now known that even systemic injection of IL-1ra, at considerably higher doses (50–100 mg kg−1), also inhibits ischaemic brain damage (Garcia et al. 1995; Relton et al. 1996), and indeed both IL-1 and IL-1ra are actively transported into the brain from the circulation (Gutierrez et al. 1994). IL-1ra is neuroprotective when administered 30–60 min after focal cerebral ischaemia and reduces not only infarct volume but also oedema, glial activation and neuronal loss, and largely reverses neurological impairment caused by MCAo (Garcia et al. 1995; Relton et al. 1996). In addition to these effects on MCAo in the adult rat or mouse, IL-1ra also markedly reduces brain damage caused by hypoxia/ischaemia in neonatal rats (Martin et al. 1995), global cerebral ischaemia in gerbils (Martin et al. 1996), lateral, cortical fluid percussion injury in the rat (Toulmond & Rothwell, 1995a) and heat stroke damage in rabbits (Lin et al. 1995), and reduces the clinical symptoms of experimental allergic encephalomyelitis (EAE, a rodent model of multiple sclerosis) (Martin & Near, 1995). In several of these paradigms, IL-1ra is effective when administered up to 4 h after the insult, and these actions of IL-1ra are not associated with any changes in body temperature or cardiovascular parameters in normal or brain-damaged rats.
Figure 3. Effect of injection of IL-1ra (10 μg, i.c.v.) or vehicle on ischaemic brain damage.
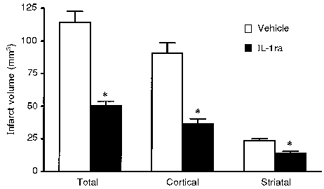
Treatments were given immediately after MCAo and infarct volumes were determined histologically 24 h later. Adapted from Loddick & Rothwell (1996). *P < 0.01vs. vehicle.
Since IL-1ra blocks all known actions of both IL-1α and IL-1β, the results described above do not distinguish the relative importance of each of these proteins. There is, however, some evidence to suggest that IL-1β is the primary mediator of neurodegeneration: IL-1β is the predominant form of IL-1 induced by brain insults (see above), and central administration of a neutralizing IL-1β antibody inhibits damage caused by reversible cerebral ischaemia in the rat (Yamasaki et al. 1992, 1994, 1995).
We have also demonstrated that injection (i.c.v.) of an irreversible peptide inhibitor of ICE activity (z-VAD), the enzyme required to cleave pro-IL-1β, reduces ischaemic brain damage in a pattern very similar to that of IL-1ra (Loddick et al. 1996) (Fig. 4) and this has now been verified by others in focal (Hara et al. 1997; Endres et al. 1998) and global ischaemia (Cheng et al. 1998). However, ICE and related members of the caspase family have also been implicated in apoptosis, and available ICE inhibitors are not selective for ICE (or caspase 1). Nevertheless, several recent studies support the involvement of ICE itself (caspase 1) in ischaemic brain damage. Mice in which the ICE (caspase 1) gene has been deleted (knocked out) or disabled (by over-expression of a dominant negative mutant form of the enzyme) exhibit reduced brain damage in response to MCAo (Friedlander et al. 1997; Schielke et al. 1998).
Figure 4. Pattern of brain damage (shown as dark areas) in coronal brain sections of rats measured histologically 24 h after MCAo.
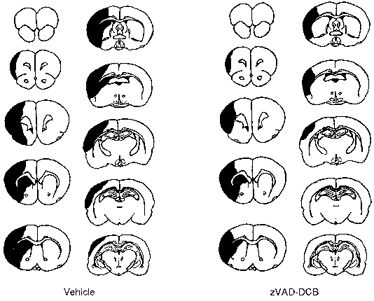
Animals were treated with either vehicle (left) or the ICE (caspase) inhibitor zVAD-DCB (right) (i.c.v.). Adapted from Loddick et al. (1996).
These observations further add to the current debate about the importance of apoptosis in neurodegeneration (e.g. Linnick, 1995; Henkart, 1996; Chalmers-Redman et al. 1997; Holtzman & Deshmukh, 1997; Barinaga, 1998) and the relationship between IL-1β, ICE and apoptosis. It had been believed that any involvement of ICE in apoptosis was independent of the release or actions of IL-1β. However, at least two studies have now suggested that actions of IL-1βitself are required for apoptosis in a variety of cell types, including neurones in vitro (Friedlander et al. 1996, 1997; Troy et al. 1996). Most notably, IL-1ra, or an antibody to IL-1RI, blocks apoptotic cell death (Friedlander et al. 1996; 1997). Indeed, the involvement of apoptosis in ischaemic brain damage is now generally accepted, though its quantitative importance is still uncertain (see Linnick, 1995; Henkart, 1996; Barinaga, 1998).
IL-18 is also cleaved by ICE (and signals through an IL-1-like receptor, see above), so caspase inhibitors will block release of active IL-18. However, the involvement of IL-18 in neurodegeneration has not yet been addressed, and it remains possible that further substrates for ICE and/or additional members of the IL-1 family remain to be discovered.
Mechanisms of action of IL-1 in neurodegeneration
Most forms of neuronal death in the adult brain have been ascribed to or associated with excitotoxicity, i.e. excessive release of excitotoxic amino acids (EAAs) such as glutamate, and subsequent activation of NMDA and AMPA receptors (see Doble, 1995; Boxer & Bigge, 1997). Thus, it is likely that IL-1 interacts in some way with this cascade to modify glutamate release, reuptake or actions.
In vitro studies indicate that neither IL-1 nor IL-1ra influences glutamate release or reuptake, at least in vitro (e.g. Allan et al. 1998; Lawrence et al. 1998). Experiments conducted in vivo indicate that expression of IL-1β (by immunohistochemistry) is stimulated by pharmacological activation of EAA receptors (Pearson et al. 1998), and that IL-1 acts at a point beyond EAA receptor activation to mediate neurodegeneration (Lawrence et al. 1998). Excitotoxins such as kainate or selective NMDA receptor agonists induce rapid expression of IL-1β mRNA and protein in the brain (see above), and the distribution and cell source of IL-1β protein induced by NMDA receptor activation in rat brain in vivo is remarkably similar to responses which follow cerebral ischaemia or brain trauma (Pearson et al. 1998). Co-infusion of IL-1ra with these excitotoxins markedly inhibits brain damage induced by pharmacological activation of either NMDA or AMPA receptors or infusion of kainate in the rat striatum in vivo (Lawrence, 1996; Lawrence et al. 1998; Panegyres & Hughes, 1998) (Fig. 5), suggesting that IL-1 mediates both of these forms of neuronal death.
Figure 5. Effect of IL-1ra on excitotoxic brain damage.
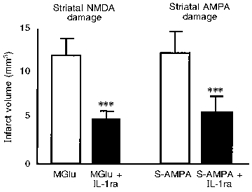
Infarct volume was measured histologically 24 h after infusion into the striatum of methanoglutamate (MGlu) or S-AMPA. Rats were either co-infused with vehicle (□) or IL-1ra (▪; 5 μg). Adapted from Lawrence et al. (1998). ***P < 0.001vs. MGlu or S-AMPA alone.
An obvious potential mechanism of IL-1 action on neurodegeneration is the induction of fever. IL-1 is a potent pyrogen (see Rothwell & Luheshi, 1994) and raised body temperature exacerbates some forms of neurodegeneration (Ridenour et al. 1992). However, IL-1ra does not affect body temperature in normal or ischaemic rats (Loddick & Rothwell, 1996). Furthermore we have found that the sites of action of IL-1 in the brain in fever differ from those involved in neurodegeneration, that cyclo-oxygenase inhibitors block IL-1-induced fever but do not affect ischaemic or excitotoxic brain damage (Relton & Rothwell, 1992; R. Grundy, N. Rothwell & S. M. Allan, unpublished data), and that the cytokine IL-6, which is a potent pyrogen, inhibits rather than enhances neural damage (see below).
Investigation of the mechanisms of neurodegeneration and IL-1 action should be facilitated by the use of in vitro approaches such as neuronal cell cultures. However, such approaches have yielded conflicting data. In primary neuronal cultures from the rat cortex or striatum, IL-1ra does not inhibit, and IL-1 does not enhance, excitotoxic cell death induced by glutamate or by selective NMDA or AMPA receptor agonists (Strijbos & Rothwell, 1995). Indeed, IL-1 applied in low (picomolar) concentrations (i.e. within the uncertain range which activates known IL-1 receptors) protects against excitotoxic damage, probably via induction of nerve growth factor (NGF) (Strijbos & Rothwell, 1995). Much higher concentrations (5–50 nM) of IL-1 are neurotoxic, but this far exceeds the amounts of IL-1 found even in injured brain. Similarly in neuronal cell lines, IL-1 fails to induce cell death, though it has been reported to exacerbate apoptosis (see above). Thus it has not been possible to mimic effects obtained reproducibly in vivo in cell culture systems.
In contrast, in mixed cultures of glia and neurones, IL-1 can be toxic even in the absence of other insults at low (picomolar) concentrations, and this has been ascribed to the release of secondary mediators, including nitric oxide and superoxides (e.g. Araujo, 1992; Beneviste, 1992; Giulian et al. 1993; Banati et al. 1993; Chao et al. 1995, 1996; Hu et al. 1995; and J. Relton, personal communication). Nevertheless, some caution must be exercised in interpreting these results because, unlike in the mixed cultures, IL-1 is not toxic to the normal adult brain in vivo (see above). Furthermore, these cultures comprise immature neurones without complex synaptic connections, and IL-1 actions may involve the activation of multiple neuronal pathways and several distinct brain regions (see below).
Studies both in vivo and in vitro have revealed diverse activities of IL-1, which could be either neuroprotective - e.g. through induction of NGF, inhibition of calcium concentrations and calcium entry into neurones and enhanced GABA activity (Spranger et al. 1990; Plata-Salaman & Ffrench-Mullen, 1991; Coogan & O'Connor, 1997) or neurotoxic - such as enhanced cell calcium entry, induction of other pro-inflammatory cytokines, eicosanoids, adhesion molecules, corticotrophin-releasing factor (CRF), acute proteins such as β-amyloid precursor protein, release of nitric oxide and other free radicals, activation of complement and neutrophins and blood-brain barrier damage (see Hartung et al. 1989; Moser et al. 1989; Sullivan et al. 1989; Quagliarello et al. 1991; Lee et al. 1993; Simmons & Murphy, 1993; Shrikant et al. 1994; Rothwell et al. 1997). Any number of these responses could contribute to the overall effects of endogenous and exogenous IL-1, which may depend on the concentration of the cytokine, the environment, and other factors present at specific sites within the brain.
Site-specific effects of IL-1
In vivo studies described above have revealed that IL-1 enhances and IL-1ra inhibits various forms of neuronal damage when injected into rodents either i.c.v. or peripherally, but both of these molecules appear to have highly site-specific effects in the brain on neurodegeneration.
We have found that injection of IL-1β (2 ng) into the striatum of rats exacerbates both striatal and cortical damage caused by cerebral ischaemia (MCAo), but is ineffective when administered into the cortex, even at a higher dose (Stroemer & Rothwell, 1998) (Fig. 6). Injection of low doses of IL-1ra into the striatum of rats subjected to MCAo inhibits subsequent damage in both the striatum and the cortex, whereas injection of the same, or higher, doses into the cortex fails to protect either region (Stroemer & Rothwell, 1997) (Fig. 7). Surprisingly, IL-1ra injected into the contralateral striatum also offers some protection against MCAo damage (Stroemer & Rothwell, 1997).
Figure 6. Local effects of IL-1 on ischaemic brain damage.
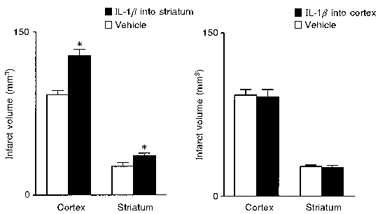
Effects of local infusion of either vehicle or IL-1β (5 ng) into the cortex or striatum on ischaemic brain damage induced by MCAo in the rat, measured as infarct volume. Adapted from Stroemer & Rothwell (1998). *P < 0.05vs. vehicle.
Figure 7. Local effects of IL-1ra on ischaemic brain damage.
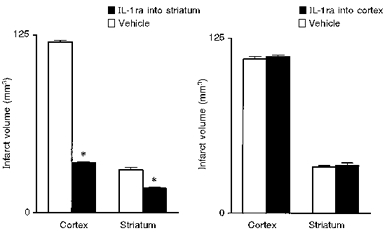
Effects of local infusion of vehicle or IL-1ra (5 μg) into the cortex or striatum on ischaemic brain damage induced by MCAo in the rat, measured as infarct volume. Adapted from Stroemer & Rothwell (1997). *P < 0.01vs. vehicle.
Similarly in excitotoxic damage, IL-1ra infused into the striatum reduces the volume of lesions caused by NMDA or AMPA agonists administered into the striatum, but infusion of IL-1ra together with these toxins into the cortex does not inhibit damage (Lawrence, 1996; Lawrence et al. 1998) (Fig. 8). Recombinant IL-1 also has surprising and dramatic effects on excitotoxic damage, since again it has no effect when administered into the cortex with an excitotoxin, but when co-infused into the striatum with S-AMPA, it causes extensive damage throughout the cortex, which cannot simply be ascribed to the diffusion of IL-1 (Lawrence et al. 1998) (Fig. 9). The resulting initial infarct (which is more than five times greater than the primary striatal damage) is associated with marked oedema, which resembles the effects of a major stroke.
Figure 8. Effects of IL-1ra on excitotoxic damage induced by methanoglutamate or S-AMPA in the striatum or cortex.
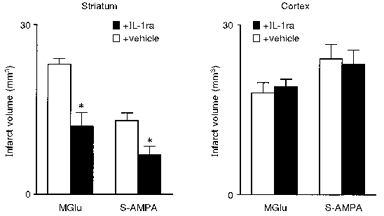
Infarct volume was measured 72 h after co-infusion of either methanoglutamate (MGlu) or S-AMPA plus vehicle or IL-1ra into the striatum (left) or cortex (right). Adapted from Lawrence (1996), Lawrence et al. (1998). *P < 0.01vs. vehicle.
Figure 9. Effects of IL-1 on striatal and cortical damage caused by S-AMPA.
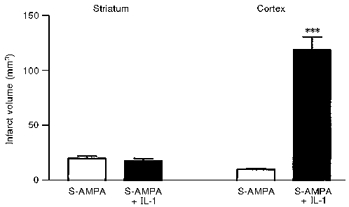
Effects of co-infusion of either vehicle or IL-1 (5 ng) with S-AMPA into the striatum on infarct volume in the striatum and cortex. Adapted from Lawrence et al. (1998). ***P < 0.001vs. S-AMPA.
These effects of IL-1 in the striatum do not correlate with its effects on body temperature (see above), and the sites of action are not consistent with the reported distribution of the IL-1 type I receptor, which is believed to mediate all actions of IL-1 (Sims et al. 1993). Published reports indicate that IL-1RI mRNA is localized mainly in the hippocampus, with low levels of expression in the cortex but little or none reported in the striatum (Cunningham et al. 1992; Ericsson et al. 1995), although the accessory protein, required for full binding of the ligand and signal transduction (Huang et al. 1997), is found in this region (see Loddick et al. 1998). These data might suggest that the effects of exogenous and endogenous IL-1 in the striatum may be dependent on novel or atypical IL-1 receptor(s). However, we have detected low levels of IL-1RI in the rat striatum by reverse transcriptase-polymerase chain reaction (RT-PCR) (L. Parker, G. N. Luheshi & N. Rothwell, unpublished data) which could mediate these actions of IL-1.
The effects described above indicate that IL-1 or IL-1ra infused into the striatum can have effects at distant brain sites to cause or inhibit damage in the cortex (see Fig. 10). The mechanisms of these distant effects are unknown, but there are several possibilities. IL-1 could induce expression or release of molecule(s) in the striatum (but not the cortex), which can then diffuse to, or indirectly influence, the cortex to cause neuronal death. Secondly, IL-1 could influence the cortex via activation of neuronal pathways. Since few, if any, striato-cortical afferent pathways have been identified, IL-1 would have to induce damage via retrograde neuronal pathways or by stimulating complex multi-synaptic pathways involving several other brain regions. It seems that glutamatergic pathways and NMDA receptors are involved in the distant cortical damage caused by striatal IL-1, because AMPA-induced damage in the striatum is not affected by NMDA antagonists, but cortical damage resulting from co-infusion of IL-1 and AMPA in the striatum appears to be inhibited by NMDA antagonists (S. M. Allan & N. Rothwell, unpublished data).
Figure 10. Site-specific actions of IL-1/IL-1ra.
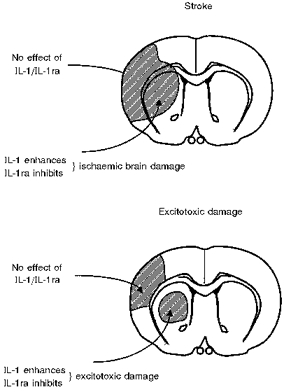
Diagram depicting site-specific effects of IL-1 and IL-1ra on ischaemic (stroke, top panel) and excitotoxic brain damage (bottom panel). For details, see text and Figs 7–9.
It also seems likely that other brain regions are associated with these distant effects of IL-1, since damage often occurs in the thalamus as well as the cortex (Lawrence, 1996). A possible mediator of these complex actions of IL-1 is the neuropeptide, corticotrophin-releasing factor (CRF), best known for its involvement in stress and activation of the hypothalamic pituitary adrenal axis. Several years ago it was reported that a CRF receptor antagonist reduced the damage caused by global ischaemia in the gerbil (Lyons et al. 1991). We have now shown that CRF mRNA is increased by cerebral ischaemia or traumatic brain damage in the rat cortex associated with the primary injury, but more specifically in the amygdala unilateral to the insult, where no neuronal loss occurs in response to damage in the rat (Wong et al. 1995). Injection of a CRF receptor antagonist (i.c.v.) significantly reduces subsequent damage in the cortex (Wong et al. 1995; Roe et al. 1998) (Fig. 11). Our preliminary data suggest that unilateral lesions of the amygdala reduce subsequent cortical injury (S. Roe & N. Rothwell, unpublished data). The mechanisms of action of CRF on neuronal death are not known, and indeed it remains to be proven that CRF mediates directly by the actions of IL-1.
Figure 11. Inhibition of corticotrophin-releasing factor (CRF) reduces brain damage.
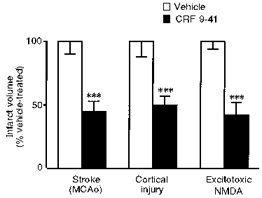
Effects of injection of a CRF receptor antagonist (α-helical CRF 9–41; 25 μg i.c.v.) on neuronal damage induced by cerebral ischaemia (left), lateral fluid percussion injury (centre) or striatal infusion of an NMDA agonist (right). Open bars show values for vehicle-treated rats depicted as 100 %. ***P < 0.001.
At present the mechanism(s) of IL-1 actions on neurodegeneration remain unknown. It is likely that IL-1 exerts diverse effects on a variety of cell types, including neurones, glia and endothelial cells (Fig. 12) to influence neurodegeneration. However, as yet the precise sites, receptors, signalling pathways and mediators of IL-1 actions have not been identified.
Figure 12. Scheme depicting potential actions of IL-1 in neurodegeneration.
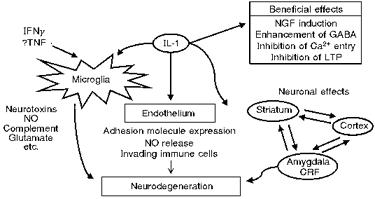
IFNγ, interferon γ. TNF, tumour necrosis factor.
Endogenous inhibitors of cytokines
A number of endogenous inhibitors of cytokine synthesis or action have been identified in the periphery, and several appear to be functionally active in the brain (Fig. 13). For IL-1, the most notable of these is IL-1ra, the naturally occurring receptor antagonist (Dinarello, 1996, 1998). IL-1ra mRNA and protein are induced in the brain by the same stimuli which upregulate IL-1 expression, i.e. excitotoxic, ischaemic and traumatic brain injury (Toulmond & Rothwell, 1995b; Loddick et al. 1997; Wang et al. 1997). Although the spatial patterns of expression of IL-1ra and IL-1 in the brain after injury are similar, these molecules are expressed by different cells (IL-1ra by neurones, IL-1 by glia) and IL-1ra is upregulated slightly later than IL-1 (Toulmond & Rothwell, 1995b; Loddick et al. 1997). We have suggested that IL-1ra is a functional inhibitor of IL-1 action and of neuronal death, since administration of anti-IL-1ra antibodies (injected i.c.v. into the rat) markedly exacerbates ischaemic and brain damage (Toulmond & Rothwell, 1995b; Loddick et al. 1997) (Fig. 14).
Figure 13. Regulation of IL-1 expression and action.

The factors shown may regulate expression and/or release of IL-1 and therefore modify neurodegeneration.
Figure 14. Effects of inhibition of endogenous IL-1ra.
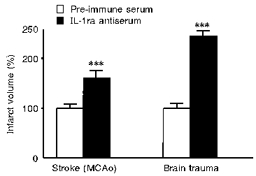
The effects of inhibiting endogenous IL-1ra by i.c.v. injection of an anti-rat IL-1ra antiserum or pre-immune serum on infarct volume (expressed as a percentage of pre-immune serum-treated groups) induced by cerebral ischaemia (left) or lateral fluid percussion injury (right). ***P < 0.001.
A number of other endogenous regulators of cytokine action have been identified including, for example, soluble IL-1 receptors, which are released and bind cytokines to inhibit their action - though the functions of these are not yet established in the brain.
Glucocorticoids are potent inhibitors of the synthesis and actions of pro-inflammatory cytokines such as IL-1 and TNFα, but have complex actions on neurodegeneration (Sapolsky & Pulsinelli, 1985; Reagan & McEwen, 1997). Lipocortin-1 (annexin-1) is a mediator of glucocorticoid action (Rothwell & Flower, 1992), that inhibits cytokine synthesis and actions and is a very potent neuroprotective agent. Lipocortin expression is upregulated in response to focal cerebral ischaemia in the rat brain (Relton et al. 1991). We have further shown that intracerebroventricular injection of recombinant lipocortin-1 markedly inhibits ischaemic and excitotoxic damage in the rat brain, while injection of a blocking antibody to lipocortin-1 enhances damage (Relton et al. 1991; Strijbos et al. 1994) (Fig. 15). The mechanisms by which lipocortin exerts its neuroprotective effects are unclear, but it has a number of actions which may contribute to these effects (see Rothwell & Flower, 1992).
Figure 15. Lipocortin-1 is an endogenous neuroprotectant.
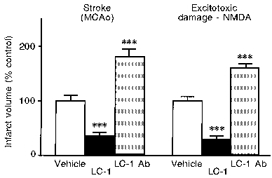
Effects of injection (i.c.v.) of vehicle, 1 μg recombinant lipocortin-1 (LC-1) or 3 μl anti-rat lipocortin-1 antibody (LC-1 Ab) on infarct volume (expressed as a percentage of vehicle-treated rats) induced by cerebral ischaemia (MCAo, left) or striatal infusion of an NMDA agonist (right). ***P < 0.001vs. vehicle.
Another, somewhat surprising inhibitor of ischaemic and excitotoxic brain damage is the cytokine IL-6, which shares many actions with IL-1, including induction of fever (Rothwell et al. 1991). IL-6 protects against excitotoxic (Toulmond et al. 1992) and ischaemic (Loddick et al. 1998) brain damage in the rat in vivo (Fig. 16). A recent report suggests that IL-6 induces lipocortin-1 expression and causes translocation to the cell surface (Solito et al. 1998), which may contribute to its neuroprotective effects.
Figure 16. Effect of IL-6 on ischaemic brain damage.
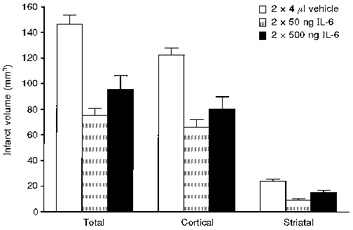
Effects of injection (i.c.v.) of saline vehicle or two doses of recombinant IL-6 (each given twice) on infarct volume induced by focal cerebral ischaemia (MCAo). Adapted from Loddick et al. (1998).
Several anti-inflammatory cytokines have been identified, such as IL-4, IL-10, IL-13 and transforming growth factor β (TGFβ), which can inhibit the release or actions of IL-1 and TNFα, and may induce IL-1ra (e.g. Vannier et al. 1992; Chao et al. 1993; Ohmori et al. 1996) (Fig. 17). Several of these molecules are induced by brain injury and are neuroprotective.
Figure 17. Diagram depicting cytokine modification of IL-1 and IL-1ra.
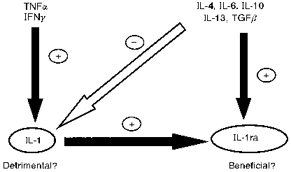
TNFα, tumour necrosis factor α; IFNγ, interferon γ; TGFβ, transforming growth factor β.
The pharmacological induction of these molecules which inhibit IL-1 activity, or the development of new agents which mimic their actions, may be of therapeutic value. It is likely that the balance between IL-1 and its inhibitors (particularly IL-1ra) determines the fate of injured neurones and inflammatory events in the brain.
Summary and implications
There is now considerable evidence to suggest that specific cytokines, particularly IL-1, are involved directly in neuronal death resulting from diverse insults and diseases. The mechanisms of these effects are not yet fully understood, but probably include complex actions on several types of brain cells and physiological systems. In addition to the involvement of IL-1 in acute neurodegeneration, circumstantial evidence also implicates this cytokine in chronic conditions such as Alzheimer's and Parkinson's disease (Royston et al. 1992; Sheng et al. 1996). Inhibition of the synthesis or actions of cytokines therefore provides an attractive therapeutic approach to the treatment of both acute and chronic neurodegenerative conditions.
References
- Allan SM, Lawrence CB, Rothwell NJ. Interleukin-1β and interleukin-1 receptor antagonist do not affect glutamate release or calcium entry in rat striatal synaptosomes. Molecular Psychiatry. 1998;3:178–182. doi: 10.1038/sj.mp.4000351. [DOI] [PubMed] [Google Scholar]
- Araujo DM. Contrasting effects of specific lymphokines on the survival of hippocampal neurons in culture. In: Meyer EM, editor. Treatment of Dementias. New York: Plenum Press; 1992. pp. 113–122. [Google Scholar]
- Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- Barinaga M. Stroke-damaged neurons may commit cellular suicide. Science. 1998;281:1302–1303. doi: 10.1126/science.281.5381.1302. [DOI] [PubMed] [Google Scholar]
- Bazan J, Timans JC, Kastlelein RA. A newly defined interleukin-1? Nature. 1996;379:591. doi: 10.1038/379591a0. [DOI] [PubMed] [Google Scholar]
- Beneviste EN. Cytokines: influence on glial cell gene expression and function. Neuroimmunoendocrinology. 1992;52:106–153. [PubMed] [Google Scholar]
- Betz K, Schielke GP, Yang G-Y. Interleukin-1 in cerebral ischaemia. Keio Journal of Medicine. 1996;45:230–238. doi: 10.2302/kjm.45.230. [DOI] [PubMed] [Google Scholar]
- Betz AL, Yang G-Y, Davidson BL. Attenuation of stroke size in rats using an adenoviral vector to induce overexpression of interleukin-1 receptor antagonist in brain. Journal of Cerebral Blood Flow and Metabolism. 1995;15:547–551. doi: 10.1038/jcbfm.1995.68. [DOI] [PubMed] [Google Scholar]
- Beutler B, Cerami A. The biology of cachectin/TNF-α: a primary mediator of the host response. Annual Review of Immunology. 1989;7:625–655. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- Bhat RV, DiRocco R, Marcy VR, Flood DG, Zhu Y, Dobrzanski P, Siman R, Scott R, Contreras PC, Miller M. Increased expression of IL-1beta converting enzyme in hippocampus after ischemia: selective localization in microglia. Journal of Neuroscience. 1996;16:4146–4154. doi: 10.1523/JNEUROSCI.16-13-04146.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsztyk K, Sims JE, Stanton TH, Slack J, McMahan CJ, Valentine MA, Dower SK. Evidence for different interleukin-1 receptors in murine B- and T-cell lines. Proceedings of the National Academy of Sciences of the USA. 1989;86:8034–8038. doi: 10.1073/pnas.86.20.8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer PA, Bigge CF. Mechanisms of neuronal cell injury/death and targets for drug intervention. Drug Discovery Today. 1997;2:219–228. [Google Scholar]
- Buttini M, Sauter A, Boddeke HWGM. Induction of interleukin-1 beta messenger RNA after focal cerebral ischaemia in the rat. Molecular Brain Research. 1994;23:126–134. doi: 10.1016/0169-328x(94)90218-6. [DOI] [PubMed] [Google Scholar]
- Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness KV, Greenstreet TA, March CJ, Kronheim SR, Druck T, Cannizzaro LA, Huebner K, Black RA. Molecular cloning of the interleukin-1β converting enzyme. Science. 1992;256:97–102. doi: 10.1126/science.1373520. [DOI] [PubMed] [Google Scholar]
- Chalmers-Redman RME, Fraser AD, Ju WYH, Wadia J, Tatton NA, Tatton WG. Mechanisms of nerve cell death: apoptosis or necrosis after cerebral ischaemia. In: Green AR, Cross AJ, editors. Neuroprotective Agents and Cerebral Ischaemia. New York: Academic Press; 1997. pp. 1–25. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumour necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behaviour and Immunology. 1995;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Sheng WS, Bu D, Bukrinsky MI, Peterson PK. Cytokine-stimulated astrocytes damage human neurones via a nitric oxide mechanism. Glia. 1996;16:276–284. doi: 10.1002/(SICI)1098-1136(199603)16:3<276::AID-GLIA10>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Chao CC, Molitor TW, Hu S. Neuroprotective role of IL-4 against activated microglia. Journal of Immunology. 1993;151:1473–1481. [PubMed] [Google Scholar]
- Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Holtzman DM. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. Journal of Clinical Investigation. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colotta F, Re F, Muzio M, Bertini B, Polentarutti N, Sironi M, Giri JG, Dower SK, Sims JE, Mantovani A. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science. 1993;261:472–475. doi: 10.1126/science.8332913. [DOI] [PubMed] [Google Scholar]
- Coogan A, O'Connor JJ. Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1β. NeuroReport. 1997;8:2107–2110. doi: 10.1097/00001756-199707070-00004. [DOI] [PubMed] [Google Scholar]
- Culhane AC, Hall MD, Rothwell NJ, Luheshi GN. Cloning of rat brain interleukin-18 cDNA. Molecular Psychiatry. 1998;3:362–366. doi: 10.1038/sj.mp.4000389. [DOI] [PubMed] [Google Scholar]
- Cunningham E, Wada E, Carter D, Tracey D, Battey J, De Souza E. In situ histochemical localization of type I interleukin-1 receptor messenger RNA in the central nervous system, pituitary and adrenal gland of the mouse. Journal of Neuroscience. 1992;12:1101–1114. doi: 10.1523/JNEUROSCI.12-03-01101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ. The progression and topographic distribution of interleukine-1β expression after permanent middle cerebral artery occlusion in the rat. Journal of Cerebral Blood Flow and Metabolism. 1998 doi: 10.1097/00004647-199901000-00010. (in the Press) [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. International Review of Immunology. 1998;16:457–499. doi: 10.3109/08830189809043005. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Novick D, Puren AJ, Fantuzzi G, Shapiro L, Muhl H, Yoon D-Y, Reznikov LL, Kim S-H, Rubinstein M. Overview of interleukin-18: more than an interferon-γ inducing factor. Journal of Leukocyte Biology. 1998;63:658–664. [PubMed] [Google Scholar]
- Dinarello CA, Thompson RC. Blocking IL-1: interleukin 1 receptor antagonist in vivo and in vitro. Immunology Today. 1991;12:404–410. doi: 10.1016/0167-5699(91)90142-G. [DOI] [PubMed] [Google Scholar]
- Doble A. Excitatory amino acid receptors and neurodegeneration. Therapie. 1995;50:319–337. [PubMed] [Google Scholar]
- Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischaemia in mice by inhibition of the caspase family. Journal of Cerebral Blood Flow and Metabolism. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- Ericsson A, Liu C, Hart RP, Sawchenko PE. Type 1 interleukin1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. Journal of Comparative Neurology. 1995;261:681–698. doi: 10.1002/cne.903610410. [DOI] [PubMed] [Google Scholar]
- Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, Macdonald G, Fishman MC, Greenberry AH, Moskowitz MA, Yan Y. Expression of a dominant negative mutant of interleukin-1β converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischaemic brain injury. Journal of Experimental Medicine. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander RM, Gagliardini V, Rotello RJ, Yuan J. Functional role of interleukin-1β (IL-1) in IL-1β converting enzyme mediated apoptosis. Journal of Experimental Medicine. 1996;184:717–724. doi: 10.1084/jem.184.2.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Liu K-F, Relton JK. Interleukin-1 receptor antagonist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. American Journal of Pathology. 1995;147:1477–1486. [PMC free article] [PubMed] [Google Scholar]
- Garrattini S, Bizz A, Donelli MG, Guaitani A, Samanin R, Spreafico F. Anorexia and cancer in animals and man. Cancer Treatment Reviews. 1980;7:115–139. doi: 10.1016/s0305-7372(80)80027-2. [DOI] [PubMed] [Google Scholar]
- Giulian D, Vaca K, Corpuz M. Brain glia release factors with opposing actions upon neuronal survival. Journal of Neuroscience. 1993;13:29–37. doi: 10.1523/JNEUROSCI.13-01-00029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfeder SA, Nunes P, Kwee L, Labow M, Chizzonite RA, Ju G. Molecular cloning and characterisation of a second subunit of the interleukin-1 receptor complex. Journal of Biological Chemistry. 1995;270:13757–13765. doi: 10.1074/jbc.270.23.13757. [DOI] [PubMed] [Google Scholar]
- Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakaniski K, Kurimoto M, Tanimoto T, Flavell RA, Sato V, Harding MW, Livingstone DJ, Su MSS. Activation of interferon-γ inducing factor mediated by interleukin-1β converting enzyme. Science. 1997;275:206–209. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- Gutierrez EG, Banks WA, Kastin AJ. Blood-borne interleukin-1 receptor antagonist crosses the blood brain barrier. Journal of Neuroimmunology. 1994;55:153–160. doi: 10.1016/0165-5728(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proceedings of the National Academy of Sciences of the USA. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung H-P, Schafer B, Heininger K, Toyka KV. Recombinant interleukin-1β stimulates eicosanoid production in rat primary culture astrocytes. Brain Research. 1989;489:113–118. doi: 10.1016/0006-8993(89)90013-9. [DOI] [PubMed] [Google Scholar]
- Henkart PA. ICE family proteases: mediators of apoptotic cell death? Immunity. 1996;4:195–201. doi: 10.1016/s1074-7613(00)80428-8. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Deshmukh M. Caspases: A treatment target for neurodegenerative diseases? Nature Medicine. 1997;3:954–955. doi: 10.1038/nm0997-954. [DOI] [PubMed] [Google Scholar]
- Hopkins SJ, Rothwell NJ. Cytokines in the nervous system I: Expression and recognition. Trends in Neurosciences. 1995;18:83–88. [PubMed] [Google Scholar]
- Hu S, Sheng WS, Peterson PK, Chao CC. Cytokine modulation of murine microglial cell superoxide production. Glia. 1995;13:45–50. doi: 10.1002/glia.440130106. [DOI] [PubMed] [Google Scholar]
- Huang J, Gao X, Li S, Cao Z. Recruitment of IRAK to the interleukin 1 receptor complex requires interleukin 1 receptor accessory protein. Proceedings of the National Academy of Sciences of the USA. 1997;94:12829–12832. doi: 10.1073/pnas.94.24.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianotti F, Kida S, Weller R, Buhagier G, Hillhouse E. Interleukin-1β in focal cerebral ischaemia in rats. Journal of Cerebral Blood Flow and Metabolism. 1993;13:s125. [Google Scholar]
- Lawrence CB. 1996. Involvement of interleukin-1 (IL-1) in excitotoxic brain damage. PhD Thesis, Manchester University. [Google Scholar]
- Lawrence CB, Allan SM, Rothwell NJ. Interleukin-1β and the interleukin-1 receptor antagonist act in the striatum to modify excitotoxic brain damage in the rat. European Journal of Neuroscience. 1998;10:1188–1195. doi: 10.1046/j.1460-9568.1998.00136.x. [DOI] [PubMed] [Google Scholar]
- Lee SC, Dickson DC, Liu W, Brosnan CF. Induction of nitric oxide synthase activity in human astrocytes by interleukin-1α and interferon-γ. Journal of Neuroimmunology. 1993;46:19–23. doi: 10.1016/0165-5728(93)90229-r. [DOI] [PubMed] [Google Scholar]
- Lin MT, Kao TY, Jin YT, Chen CF. Interleukin-1 receptor antagonist attenuates the heat stroke-induced neuronal damage by reducing the cerebral ischemia in rats. Brain Research. 1995;37:595–598. doi: 10.1016/0361-9230(95)00046-h. [DOI] [PubMed] [Google Scholar]
- Linnick MD. Programmed cell death in cerebral ischaemia. CNS Drugs. 1995;3:239–244. [Google Scholar]
- Liu T, McDonnell PC, Young PR, White RF, Siren AL, Hallenbeck JM, Barone FC, Feuerstein GZ. Interleukin-1 beta mRNA expression in ischaemic rat cortex. Stroke. 1993;24:1746–1751. doi: 10.1161/01.str.24.11.1746. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Liu C, Takao T, Hashimoto K, De Souza EB. Interleukin-1 receptors: cloning studies and role in central nervous system disorders. Brain Research Reviews. 1998;26:306–319. doi: 10.1016/s0165-0173(97)00037-4. [DOI] [PubMed] [Google Scholar]
- Loddick SA, MacKenzie A, Rothwell NJ. An ICE inhibitor, z-VAD-DCB attenuates ischaemic brain damage in the rat. NeuroReport. 1996;7:1465–1468. doi: 10.1097/00001756-199606170-00004. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Rothwell NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. Journal of Cerebral Blood Flow and Metabolism. 1996;16:932–940. doi: 10.1097/00004647-199609000-00017. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Turnbull AV, Rothwell NJ. Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischaemia in the rat. Journal of Cerebral Blood Flow and Metabolism. 1998;18:176–179. doi: 10.1097/00004647-199802000-00008. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Wong M-L, Bongiorno PB, Gold PW, Licinio J, Rothwell NJ. Endogenous interleukin-1 receptor antagonist is neuroprotective. Biochemical and Biophysical Research Communications. 1997;234:211–215. doi: 10.1006/bbrc.1997.6436. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Crowe PD, Liu C, Chalmers DT, Liu XJ, Liaw C, Clevenger W, Oltersdorf T, De Souza EB, Maki RA. Cloning of a cDNA encoding a novel interleukin-1 receptor related protein (IL-1R-rp2) Journal of Neuroimmunology. 1996;70:113–122. doi: 10.1016/s0165-5728(96)00047-1. [DOI] [PubMed] [Google Scholar]
- Luheshi G, Hopkins SJ, Lefeuvre RA, Dascombe MJ, Ghiara P, Rothwell NJ. Importance of brain IL-1 type II receptors in fever and thermogenesis in the rat. American Journal of Physiology. 1993;265:585–591. doi: 10.1152/ajpendo.1993.265.4.E585. [DOI] [PubMed] [Google Scholar]
- Lyons MK, Anderson RE, Meyer FB. Corticotrophin-releasing factor antagonist reduces ischaemic hippocampal neuronal injury. Brain Research. 1991;545:339–342. doi: 10.1016/0006-8993(91)91310-w. [DOI] [PubMed] [Google Scholar]
- Martin D, Chinookoswong N, Miller G. The interleukin-1 receptor antagonist (rhIL-1ra) protects against cerebral infarction in a rat model of hypoxia-ischaemia. Experimental Neurology. 1995;130:362–367. doi: 10.1006/exnr.1994.1215. [DOI] [PubMed] [Google Scholar]
- Martin MU, Falk W. The interleukin-1 receptor complex and interleukin-1 signal transduction. European Cytokine Network. 1997;8:5–17. [PubMed] [Google Scholar]
- Martin D, Near SL. Protective effect of the interleukin-1 receptor antagonist (IL-1ra) on experimental allergic encephalomyelitis in rats. Journal of Neuroimmunology. 1995;61:241–245. doi: 10.1016/0165-5728(95)00108-e. [DOI] [PubMed] [Google Scholar]
- Martin D, Relton JK, Muller G, Bendele A, Fischer N, Russell D. Cytokines as therapeutic agents in neurological disorders. In: Rothwell NJ, editor. Cytokines in the Nervous System. Boston: Chapman & Hall; 1996. pp. 162–178. [Google Scholar]
- Minami M, Kuraishi K, Yabuuchi K, Yamazaki K, Satoh M. Induction of interleukin-1β mRNA in rat brain transient forebrain ischaemia. Journal of Neurochemistry. 1992;58:390–392. doi: 10.1111/j.1471-4159.1992.tb09324.x. [DOI] [PubMed] [Google Scholar]
- Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- Moser R, Schleiffenbaum B, Groscurth P, Fehr J. Interleukin-1 and tumour necrosis factor stimulate vascular endothelial cells to promote transendothelial neutrophil passage. Journal of Clinical Investigation. 1989;83:444–451. doi: 10.1172/JCI113903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry N. Caspases: killer proteases. Trends in Biochemical Sciences. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Ohmori Y, Smith MF, Hamilton TA. IL-4 induced expression of the IL-1 receptor antagonist gene is mediated by STAT6. Journal of Immunology. 1996;57:2058–2065. [PubMed] [Google Scholar]
- Okamura H, Tsutsui H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, Okura T, Nukada YL, Hattori K, Akita K, Namba M, Tanabe F, Konishi K, Fukuda S, Kurimoto M. Cloning of a new cytokine that induces IFN-gamma production by T-cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- O'Neill LAJ. Pharmacological targets in the immune response. Interleukin-1 receptors and signal transduction. Biochemical Society Transactions. 1996;24:207–211. doi: 10.1042/bst0240207. [DOI] [PubMed] [Google Scholar]
- Panegyres PK, Hughes J. The neuroprotective effects of the recombinant interleukin-1 receptor antagonist rhIL-1ra after excitotoxic stimulation with kainic acid and its relationship to the amyloid precursor protein gene. Journal of the Neurological Sciences. 1998;154:123–132. doi: 10.1016/s0022-510x(97)00214-1. [DOI] [PubMed] [Google Scholar]
- Parnet P, Garka KE, Bonnert TP, Dower SK, Sims JE. IL-1Rrp is a novel receptor like molecule similar to the type I interleukin-1 receptor and its homologues T1/ST2 and IL-1R AcP. Journal of Biological Chemistry. 1996;271:3967–3970. doi: 10.1074/jbc.271.8.3967. [DOI] [PubMed] [Google Scholar]
- Pearson V, Rothwell NJ, Toulmond S. Excitotoxic brain damage in the rat induces IL-1 protein in microglia and astrocytes: correlation with the progression of cell death. Glia. 1998. in the Press. [PubMed]
- Plata-Salaman CR, Ffrench-Mullen JMH. Interleukin-1β depresses calcium currents in CA1 hippocampal, neurones at pathophysiological concentrations. Brain Research Bulletin. 1991;29:221–225. doi: 10.1016/0361-9230(92)90029-w. [DOI] [PubMed] [Google Scholar]
- Quagliarello VJ, Wispelwey B, Long WJ, Jr, Scheld WM. Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in the rat. Characterization and comparison with tumor necrosis factor. Journal of Clinical Investigation. 1991;87:1360–1366. doi: 10.1172/JCI115140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rano TA, Timkey T, Peterson EP, Rotonda J, Nicholson DW, Becker JW, Chapman KT, Thornberry NA. A combinatorial approach for determining protease specificities; application to interleukin-1β converting enzyme (ICE) Chemistry and Biology. 1997;4:149–155. doi: 10.1016/s1074-5521(97)90258-1. [DOI] [PubMed] [Google Scholar]
- Reagan LP, McEwen BS. Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. Journal of Chemical Neuroanatomy. 1997;13:149–167. doi: 10.1016/s0891-0618(97)00031-8. [DOI] [PubMed] [Google Scholar]
- Relton JK, Martin D, Thompson RC, Russell DA. Peripheral administration of interleukin-1 receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Experimental Neurology. 1996;138:206–213. doi: 10.1006/exnr.1996.0059. [DOI] [PubMed] [Google Scholar]
- Relton JK, Rothwell NJ. Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Research Bulletin. 1992;29:243–246. doi: 10.1016/0361-9230(92)90033-t. [DOI] [PubMed] [Google Scholar]
- Relton JK, Strijbos PJLM, O'Shaughnessy CT, Carey F, Forder RA, Tilders FJH, Rothwell NJ. Lipocortin-1 is an endogenous inhibitor of ischemic damage in the rat brain. Journal of Experimental Medicine. 1991;174:305–310. doi: 10.1084/jem.174.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridenour TR, Warner DS, Todd MM, McAllister AC. Mild hypothermia reduces infarct size resulting from temporary but not permanent focal ischemia in rats. Stroke. 1992;23:733–738. doi: 10.1161/01.str.23.5.733. [DOI] [PubMed] [Google Scholar]
- Roe SY, McGowan EM, Rothwell NJ. Evidence for the involvement of corticotrophin releasing hormone (CRH) in the pathogenesis of traumatic brain injury. European Journal of Neuroscience. 1998;10:553–559. doi: 10.1046/j.1460-9568.1998.00064.x. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ. Neuroimmune interactions: the role of cytokines. British Journal of Pharmacology. 1997;121:841–847. doi: 10.1038/sj.bjp.0701248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell NJ, Allan S, Toulmond S. Cytokines and the brain: the role of interleukin-1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. Journal of Clinical Investigation. 1997;100:2648–2652. doi: 10.1172/JCI119808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell NJ, Busbridge NJ, Lefeuvre RA, Hardwick AJ, Gauldie J, Hopkins SJ. Interleukin-6 is a centrally acting endogenous pyrogen in the rat. Canadian Journal of Physiology and Pharmacology. 1991;69:1465–1469. doi: 10.1139/y91-219. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Flower R. Lipocortin-1 exhibits novel actions providing clinical opportunities. Trends in Pharmacological Sciences. 1992;13:45–46. doi: 10.1016/0165-6147(92)90019-3. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Hopkins SJ. Cytokines and the nervous system II: Actions and mechanisms of action. Trends in Neurosciences. 1995;18:130–136. doi: 10.1016/0166-2236(95)93890-a. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Loddick SA, Stroemer P. Interleukins and cerebral ischaemia. In: Green AR, Cross AJ, editors. Neuroprotective Agents and Cerebral Ischaemia. New York: Academic Press; 1996a. pp. 281–298. [Google Scholar]
- Rothwell NJ, Luheshi GN. Pharmacology of interleukin-1 actions in the brain. Advances in Pharmacology. 1994;25:1–20. doi: 10.1016/s1054-3589(08)60428-7. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Luheshi G, Toulmond S. Cytokines and their receptors in the central nervous system: Physiology, pharmacology and pathology. Pharmacological Therapeutics. 1996b;69:85–95. doi: 10.1016/0163-7258(95)02033-0. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Stock MJ. Mechanisms of weight gain and loss in reversible obesity in the rat. The Journal of Physiology. 1978;276:60–61P. [PubMed] [Google Scholar]
- Rothwell NJ, Stock MJ. Brown adipose tissue. In: Baker PF, editor. Recent Advances in Physiology. Vol. 10. Edinburgh: Churchill Livingstone; 1984. pp. 349–384. [Google Scholar]
- Rothwell NJ, Stock NJ. Brown adipose tissue and diet-induced thermogenesis. In: Trayhurn P, Nicholls DG, editors. Brown Adipose Tissue. London: Edward Arnold; 1986. pp. 269–298. [Google Scholar]
- Rothwell NJ, Stroemer P, Lawrence C, Davies C. Actions of IL-1 in neurodegeneration. In: Kriegelstein J, editor. Pharmacology of Cerebral Ischaemia. Stuttgart: Medpharm; 1996c. pp. 125–129. [Google Scholar]
- Royston MC, Rothwell NJ, Roberts GW. Alzheimer's disease: pathology to potential treatments. Trends in Pharmacological Sciences. 1992;13:131–133. doi: 10.1016/0165-6147(92)90047-a. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Pulsinelli WA. Glucocorticoids potentiate ischaemic injury to neurons: therapeutic implications. Science. 1985;229:1397–1399. doi: 10.1126/science.4035356. [DOI] [PubMed] [Google Scholar]
- Schielke GP, Yang GY, Shivers BD, Betz AL. Reduced ischaemic brain injury in interleukin-1 beta converting enzyme-deficient mice. Journal of Cerebral Blood Flow and Metabolism. 1998;18:180–185. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Ito K, Skinner RD, Mrak RE, Rovnaghi CR, Van Eldik L, Griffin WST. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiology of Ageing. 1996;7:761–766. doi: 10.1016/0197-4580(96)00104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrikant P, Chung IY, Ballestas ME, Benveniste EN. Regulation of intercellular adhesion molecule-1 gene expression by tumor necrosis factor-alpha, interleukin-1 beta, and interferon-gamma in astrocytes. Journal of Neuroimmunology. 1994;51:209–220. doi: 10.1016/0165-5728(94)90083-3. [DOI] [PubMed] [Google Scholar]
- Simmons ML, Murphy S. Cytokines regulate L-arginine-dependent cyclic GMP production in rat glial cells. European Journal of Neuroscience. 1993;5:825–832. doi: 10.1111/j.1460-9568.1993.tb00934.x. [DOI] [PubMed] [Google Scholar]
- Sims J, Gayle MA, Slack JL, Alderson MR, Bird TA, Giri JG, Colotta F, Re F, Mantovani A, Shanebeck K, Grabstein KH, Dower SK. Interleukin-1 signalling occurs exclusively via the type I receptor. Proceedings of the National Academy of Sciences of the USA. 1993;90:6155–6159. doi: 10.1073/pnas.90.13.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solito E, De Coupade C, Parente L, Flower RJ, Russo-Marie F. IL-6 stimulates annexin-1 expression and translocation, and suggests a new biological role as class II acute phase protein. Cytokine. 1998. in the Press. [DOI] [PubMed]
- Spranger M, Lindholm D, Bandtlow C, Heumann R, Ghahn H, Näher-Noé M, Thoenen H. Regulation of nerve growth factor (NGF) synthesis in the rat central nervous system: comparison between the effects of interleukin-1 and various growth factors in astrocyte cultures and in vivo. European Journal of Neuroscience. 1990;2:69–76. doi: 10.1111/j.1460-9568.1990.tb00382.x. [DOI] [PubMed] [Google Scholar]
- Strijbos PJLM, Relton JK, Rothwell NJ. Corticotrophin-releasing factor antagonist inhibits neuronal damage induced by focal cerebal ischaemia or activation of NMDA receptors in the rat brain. Brain Research. 1994;656:405–408. doi: 10.1016/0006-8993(94)91485-0. [DOI] [PubMed] [Google Scholar]
- Strijbos PJLM, Rothwell NJ. Interleukin-1β attenuates excitatory amino acid induced neurodegeneration in vitro: involvement of nerve growth factor. Journal of Neuroscience. 1995;15:3468–3474. doi: 10.1523/JNEUROSCI.15-05-03468.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroemer RP, Rothwell NJ. Cortical protection by localised striatal injection of IL-1ra following cerebral ischaemia in the rat. Journal of Cerebral Blood Flow and Metabolism. 1997;17:597–604. doi: 10.1097/00004647-199706000-00001. [DOI] [PubMed] [Google Scholar]
- Stroemer RP, Rothwell NJ. Exacerbation of ischaemic brain damage by localised striatal injection of IL-1β in the rat. Journal of Cerebral Blood Flow and Metabolism. 1998 doi: 10.1097/00004647-199808000-00003. in the Press. [DOI] [PubMed] [Google Scholar]
- Sullivan GW, Carper HT, Sullivan JA, Murata T, Mandell GL. Both recombinant interleukin-1 (beta) and purified human monocyte interleukin-1 prime human neutrophils for increased oxidative activity and promote neutrophil spreading. Journal of Leukocyte Biology. 1989;45:389–395. doi: 10.1002/jlb.45.5.389. [DOI] [PubMed] [Google Scholar]
- Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with R05 4864 a peripheral-type (p site) benzodiazepine ligand. Journal of Neuroimmunology. 1993;42:177–185. doi: 10.1016/0165-5728(93)90008-m. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Ellistan KO, Ayala JM, Casano FJ, Chin J, Ding G J-F, Egger LA, Gaffney EP, Limjuco G, Palyha OC, Raju SM, Rolando AM, Salley JP, Yanin T-T, Lee TD, Shively JE, Maccross M, Mumford RA, Schmidt JA, Tocci M. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Tisdale MJ. Cancer cachexia: metabolic alterations and clinical manifestations. Nutrition. 1997;13:1–7. doi: 10.1016/s0899-9007(96)00313-9. [DOI] [PubMed] [Google Scholar]
- Torigoe K, Ushio S, Okura T, Kobayashi S, Taniai M, Kunikata T, Murakami T, Sanou O, Kojima H, Fujii M, Ohta T, Ikeda M, Ikegami H, Kurimoto M. Purification and characterisation of the human interleukin-18 receptor. Journal of Biological Chemistry. 1997;272:25737–25742. doi: 10.1074/jbc.272.41.25737. [DOI] [PubMed] [Google Scholar]
- Toulmond S, Rothwell NJ. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Research. 1995a;671:261–266. doi: 10.1016/0006-8993(94)01343-g. [DOI] [PubMed] [Google Scholar]
- Toulmond S, Rothwell NJ. Time course of IL-1 receptor antagonist (IL-1ra) expression after brain trauma in the rat. Society for Neuroscience Abstracts. 1995b;21:200.2. [Google Scholar]
- Toulmond S, Vige X, Fage D, Benavides J. Local infusion of interleukin-6 attenuates the neurotoxic effects of NMDA on rat striatal cholinergic neurons. Neuroscience Letters. 1992;144:49–52. doi: 10.1016/0304-3940(92)90713-h. [DOI] [PubMed] [Google Scholar]
- Troy CC, Stefanis L, Prochiantz A, Greene LA, Shelanski ML. The contrasting roles of ICE family proteases and interleukin-1β in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase downregulation. Proceedings of the National Academy of Sciences of the USA. 1996;93:5635–5640. doi: 10.1073/pnas.93.11.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannier E, Miller LC, Dinarello CA. Coordinated antiinflammatory effects of interleukin 4: interleukin 4 suppresses interleukin 1 production but up-regulates gene expression and synthesis of interleukin 1 receptor antagonist. Proceedings of the National Academy of Sciences of the USA. 1992;89:4076–4080. doi: 10.1073/pnas.89.9.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Barone F, Aiyar NV, Feuerstein GZ. Interleukin-1 receptor and receptor antagonist gene expression after focal stroke in rats. Stroke. 1997;28:155–162. doi: 10.1161/01.str.28.1.155. [DOI] [PubMed] [Google Scholar]
- Wong ML, Loddick SA, Bongiorno PB, Gold PW, Licinio J, Rothwell NJ. Focal cerebral ischaemia induces CRH mRNA in rat cerebral cortex and amygdala. NeuroReport. 1995;6:1785–1788. doi: 10.1097/00001756-199509000-00019. [DOI] [PubMed] [Google Scholar]
- Yabuuchi K, Minami M, Katsumata S, Yamasaki A, Satoh MA. An in situ hybridisation study of interleukin-1 beta induced by transient forebrain ischaemia in the rat brain. Molecular Brain Research. 1994;26:135–142. doi: 10.1016/0169-328x(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Matsuura N, Shozuhara H, Onodera H, Itoyama Y, Kogure K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 1995;26:676–681. doi: 10.1161/01.str.26.4.676. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Shozuhara H, Onodera H, Kogure K. Blocking of interleukin-1 activity is a beneficial approach to ischemia brain edema formation. Acta Neurochirugica Supplement. 1994;60:300–302. doi: 10.1007/978-3-7091-9334-1_80. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Suzuki T, Yamaya H, Matsuura N, Onodera H, Kogure K. Possible involvement of interleukin-1 in ischemic brain edema formation. Neuroscience Letters. 1992;142:45–47. doi: 10.1016/0304-3940(92)90616-f. [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]