

Abstract
Using the conventional whole-cell clamp method, the electrical responses of individual smooth muscle and endothelial cells to acetylcholine (ACh) were observed in multicellular preparations where the two types of cells remained in close apposition.
In both types of cells, ACh induced similar hyperpolarizing responses which, when recorded in current clamp mode, had two phases (an initial fast and a second slower phase).
After blocking gap junctions, including myoendothelial junctions, with 18β-glycyrrhetinic acid, ACh induced an outward current with two phases in voltage-clamped endothelial cells. The outward current appeared around −90 mV and increased linearly with the membrane depolarization.
In smooth muscle cells, ACh failed to induce a membrane current after gap junctions had been blocked with 18β-glycyrrhetinic acid. The inhibition of ACh-induced response by 18β-glycyrrhetinic acid was observed using either sharp or patch electrodes.
Nominally Ca2+-free solution reduced the initial phase and abolished the second phase of ACh-induced responses of endothelial cells. Both phases were also reduced by charybdotoxin (CTX).
Our results indicate that in guinea-pig mesenteric arterioles, ACh hyperpolarizes endothelial cells by activating Ca2+-activated K+ channels which are sensitive to CTX. On the other hand, hyperpolarizing responses detected in smooth muscle cells seem to originate in endothelial cells and conduct to the muscle layer via myoendothelial gap junctions.
Vascular smooth muscle cells hyperpolarize in response to various vasoactive substances such as acetylcholine (ACh) and substance P in an endothelium-dependent manner; this phenomenon is thought to be due to the release of an unknown humoral substance from vascular endothelium (Chen et al. 1988). Thus, the term endothelium-derived hyperpolarizing factor or EDHF was introduced. In endothelium-dependent relaxation induced by ACh, EDHF seems to be a major factor in small arteries while nitric oxide (NO) is the major factor in large arteries (Hwa et al. 1994). EDHF may be a cytochrome P450-derived arachidonic acid metabolite (Hecker et al. 1994; Campbell et al. 1996; Popp et al. 1996), but evidence against this has been reported (Edwards et al. 1996; Fukao et al. 1997; Vanheel & Van de Voorde, 1997).
Since isolated vascular endothelial cells were found to hyperpolarize in response to ACh (Busse et al. 1988; Sakai, 1990), the existence of EDHF has been challenged by the suggestion that the hyperpolarization in smooth muscle cells is merely conducted from the endothelial cells through myoendothelial gap junctions, because smooth muscle and endothelial cells are coupled (von der Weid & Bény, 1993; Little et al. 1995; Bény, 1997; Yamamoto et al. 1998). The importance of gap junctions has often been assessed by applying substances such as heptanol, octanol and halothane which clearly block gap junctions in cardiac muscle (for review, see Spray & Burt, 1990). Although heptanol at 1 mM was found to effectively block dye transfer between smooth muscle and endothelial cells in arteriolar preparations (Little et al. 1995), its effectiveness in blocking electrical communication has been questioned (Hashitani & Suzuki, 1997; Yamamoto et al. 1998).
18α- or 18β-glycyrrhetinic acid has been found to be an effective blocker of gap junctions in human fibroblast cell lines (Davidson et al. 1986), in liver epithelial cells (Guan et al. 1996) and in guinea-pig mesenteric arterioles (Yamamoto et al. 1998). In the present experiments, electrical responses of individual smooth muscle and endothelial cells to ACh were observed in multicellular preparations where the two types of cells remained closely located before and after gap junctions had been blocked by 18β-glycyrrhetinic acid. Our results indicate that at least in guinea-pig mesenteric arterioles, ACh-induced hyperpolarization of smooth muscle cells seems to be conducted from endothelial cells through myoendothelial gap junctions.
METHODS
Tissue preparation
Male guinea-pigs, weighing 250-400 g, were killed by a rising concentration of CO2 and exsanguination. For the patch-clamp experiments, the ileum was excised and the first-order arterioles (diameter, 50-100 μm) were dissected free. Two kinds of preparations were made as described previously (Yamamoto et al. 1998). Briefly, for the experiments where recordings were made from the smooth muscle cells, the adventitial layer was removed to reveal the smooth muscle layer after a short incubation in nominally Ca2+-free solution containing 0.5 mg ml−1 collagenase (Wako Pure Chemical Industries, Ltd, Osaka, Japan). When recording from the endothelial cells, the arteriole segment was inverted and no enzymatic treatment was used. One millimetre segments of these preparations were pinned out in a small (0.8 mm × 1.5 mm) recording chamber. Preparations were superfused with preheated (35°C) bath solution at a constant rate (1 ml min−1).
For the microelectrode experiments, the ileum was slit open along the mesenteric border and pinned out in a dissecting chamber with the mucosa uppermost. The mucosal layer was removed and the sheet of submucosal connective tissue containing arterioles was separated from the underlying circular smooth muscle layer. The sheet of connective tissue was pinned out in a small recording chamber (volume approximately 1 ml) and superfused at a constant flow rate (about 4 ml min−1) with preheated (35°C) bath solution.
Electrophysiological techniques
The tight-seal patch-clamp method was used in conventional whole-cell clamp configuration. Current or voltage signals were acquired using an EPC-7 patch-clamp system (List-Medical), filtered with an 8-pole low-pass Bessel filter (900C/9L8L; Frequency Devices, Haverhill, MA, USA), and then digitized with a Digidata 1200A data acquisition system (Axon Instruments). Voltage clamp protocols were controlled with pCLAMP software (Axon Instruments). In current clamp experiments, voltage signals were stored on a DAT tape with a bandwidth of DC -10 kHz (PC-204, Sony Magnescale Inc., Tokyo) before digitization.
In some experiments, conventional high resistance microelectrodes were used to record membrane potential changes from intact preparations of arterioles (diameter, 30-80 μm). Smooth muscle cells were impaled with glass capillary microelectrodes filled with 1 M KCl (tip resistance, 100-250 MΩ). Membrane potentials were recorded using a high input impedance amplifier (Axoclamp-2B, Axon Instruments). The voltage signals were digitized and stored with a Digidata 1200A data acquisition system.
Solutions and chemicals
The composition of standard bath solution for the patch-clamp experiments was (mM): NaCl, 141.5; KCl, 5.4; CaCl2, 1.8; MgCl2, 1; Hepes, 10; glucose, 5. The pH was adjusted to 7.3 with NaOH. The solution was aerated with O2. Nominally Ca2+-free solution was prepared by simply omitting CaCl2. The pipette solution contained (mM): KCl, 143; MgCl2, 1; EGTA, 1; Hepes, 10; glucose, 5. The pH was adjusted to 7.3 with KOH. The bath solution for the microelectrode experiments contained (mM): NaCl, 120.7; KCl, 5.9; NaHCO3, 15.5; NaH2PO4, 1.2; CaCl2, 2.5; MgCl2, 1.2; glucose, 11.5. The solution was aerated with O2 containing 5 % CO2 to maintain the pH of the solution at 7.2-7.3. ACh and 18β-glycyrrhetinic acid were obtained from Sigma; charybdotoxin (CTX) and Nω-nitro-L-arginine (nitroarginine) from Peptide Institute (Osaka, Japan); diclofenac sodium salt from Research Biochemicals International (Natick, MA, USA). 18β-Glycyrrhetinic acid was dissolved in DMSO to make a stock solution of 100 mM.
Statistics
Numerical data are represented as the mean ± standard deviation, with n referring to the number of measurements. Statistical evaluation was performed by Student's paired t test with a P value < 0.05 considered statistically significant.
RESULTS
In guinea-pig mesenteric arterioles, smooth muscle and endothelial cells are all electrically coupled together, and these cells are thought to be isopotential (Yamamoto et al. 1998). The mean resting membrane potentials of smooth muscle and endothelial cells measured with the conventional whole-cell clamp method were -45.1 ± 6.9 mV (n= 21) and -39.6 ± 6.3 mV (n= 44), respectively. The apparent difference in the membrane potentials between the two types of cells was probably due to the presence of an additional population of uncoupled endothelial cells (Yamamoto et al. 1998). Membrane potentials were recorded from smooth muscle or endothelial cells in current-clamp mode and 3 μm ACh was applied (Fig. 1). In both types of cell, ACh induced hyperpolarizing responses with two phases, an initial fast phase followed by a slower second phase. During the second phase, small fluctuations of membrane potential sometimes occurred (for example, see Figs 5 and 6). The mean peak amplitudes of the initial and second phases were 13.6 ± 6.2 mV (n= 21) and 12.2 ± 8.5 mV (n= 21), respectively, in smooth muscle cells, and 11.8 ± 5.8 mV (n= 44) and 13.3 ± 7.7 mV (n= 44), respectively, in endothelial cells. The question to be answered was which type of cell played a dominant role in producing hyperpolarization, the smooth muscle or the endothelial cell. To answer this, the effect of ACh on the two types of cells was examined after blocking myoendothelial gap junctions with 18β-glycyrrhetinic acid.
Figure 1. Effect of ACh on membrane potentials recorded from smooth muscle and endothelial cells.
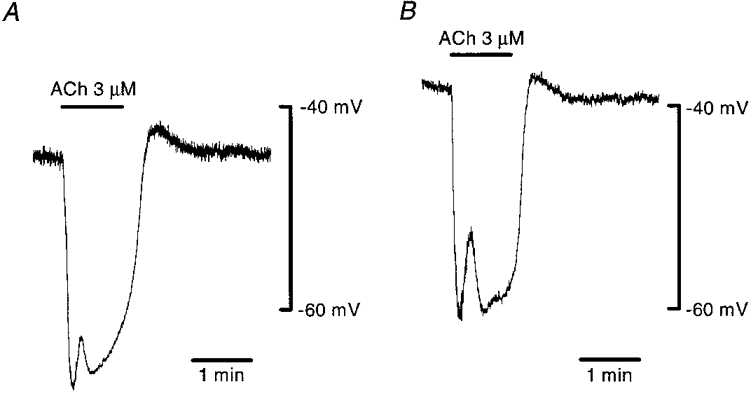
In current clamp mode of the conventional whole-cell clamp method, membrane potentials in a smooth muscle (A) and in an endothelial cell (B) were recorded. ACh (3 μm) was applied during periods indicated by bars at the top. Signals were filtered at a cut-off frequency (-3 dB) of 10 Hz. A and B show recordings made from different preparations.
Figure 5. Effect of Ca2+ removal on ACh-induced hyperpolarization in endothelial cells.
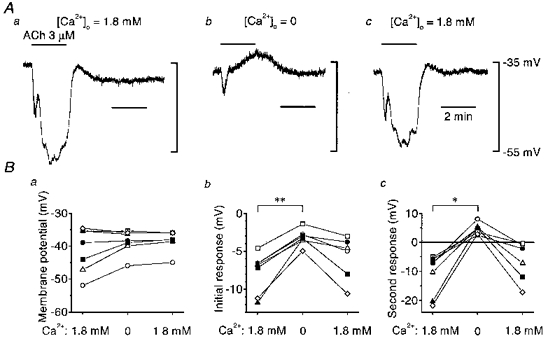
A, in current clamp mode of the conventional whole-cell clamp method, 3 μm ACh was applied during periods indicated by the bars at the top in control solution (1.8 mM Ca2+; a), 1 min after applying nominally Ca2+-free solution (b) and 9 min after reintroduction of the control solution (c). Signals were filtered at a cut-off frequency of 10 Hz. B, summaries of membrane potentials (a), the amplitude of the initial responses (b) and the amplitude of the second responses (c) before and after removing external Ca2+, and after reintroduction of control solution. Each symbol represents data from a separate experiment. In b and c, negative values represent hyperpolarizations and positive values depolarizations. *P < 0.002, **P < 0.001.
Figure 6. Effect of CTX on ACh-induced hyperpolarization in endothelial cells.
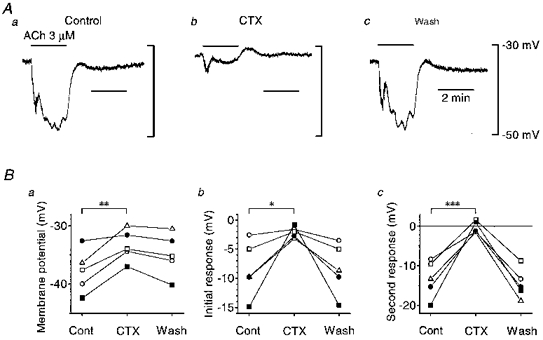
A, in the current clamp mode of the conventional whole-cell clamp method, 3 μm ACh was applied during periods indicated by bars at the top before (a), 5 min after application of 50 nM CTX (b) and 30 min after washing out CTX (c). Signals were filtered at a cut-off frequency of 10 Hz. B, summaries of membrane potentials (a), amplitude of the initial responses (b) and amplitude of the second responses (c) before and after application of CTX, and after washing it out. Each symbol represents data from a separate experiment. In b and c, negative values represent hyperpolarizations and positive values depolarizations. *P < 0.05, **P < 0.02, ***P < 0.005.
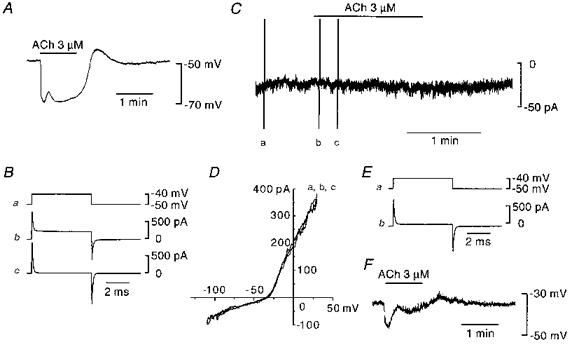
Changes in time course of the current required to impose a 10 mV voltage clamp step were used to determine the extent of coupling between cells (de Roos et al. 1996). As shown in Fig. 2Ab, the current produced by a 10 mV voltage step recorded from an endothelial cell relaxed slowly in a multi-exponential manner, indicating the existence of electrical coupling to neighbouring cells. The input resistance of this cell was calculated to be 1.7 MΩ according to the method described by Yamamoto et al. (1998). The mean input resistance of sixteen cells examined was 3.4 ± 1.8 MΩ. The application of 18β-glycyrrhetinic acid (20 μm) almost abolished the current in 6 min leaving only a rapid capacitive current, the relaxation of which could be described by a single exponential function, followed by a sustained current (Fig. 2Ac). In the presence of 18β-glycyrrhetinic acid, the input resistance and capacitance of this cell were 0.5 GΩ and 10.5 pF, respectively. The mean input resistance and capacitance were 0.3 ± 0.2 GΩ and 13.0 ± 3.6 pF (n= 16), respectively. Once such a current was obtained, we considered that most of the electrical communications, including myoendothelial couplings had been blocked, and it was possible to control the membrane potential of the patched cells in the intact vessel segment (see Yamamoto et al. 1998). As the ACh-induced hyperpolarization of endothelial cells persisted after disrupting gap junctions with 18β-glycyrrhetinic acid (Yamamoto et al. 1998), we expected that an ACh-induced whole-cell current would be observed in voltage-clamped endothelial cells. To test this, the membrane potential of the endothelial cell was clamped at -60 mV and 3 μm ACh was applied 9 min after the application of 18β-glycyrrhetinic acid (Fig. 2B). ACh induced an outward current which again had two phases with similar temporal characteristics to the hyperpolarizations shown in Fig. 1. The current-voltage relationships of the ACh-induced currents (Fig. 2D) obtained both at the peak of the initial phase (b - a) and during the second phase (d - a) were qualitatively the same in that the outward current appeared around -90 mV and increased linearly with the membrane depolarization. From linear regressions calculated by the least-squares method, the conductance and null potential of ACh-induced current in this particular cell were 0.87 nS and -89.3 mV, respectively during the initial phase, and 0.28 nS and -89.9 mV, respectively, during the second phase. Current-voltage relationships of ACh-induced currents were recorded in five endothelial cells and the mean null potential during the initial and second phases were -91.3 ± 3.9 mV and -88.7 ± 5.3 mV, respectively. Although the magnitudes of the currents varied among preparations, ACh induced similar responses to those shown in Fig. 2 in each of the sixteen endothelial cells examined.
Figure 2. Effect of ACh on membrane current recorded from an endothelial cell after blocking gap junctions.
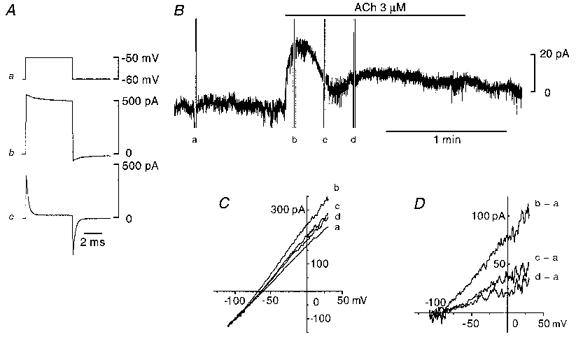
Conventional whole-cell clamp method. A, membrane current produced by a 10 mV voltage step (a) recorded from an endothelial cell before (b) and 6 min after application of 20 μm 18β-glycyrrhetinic acid (c). B, in the presence of 18β-glycyrrhetinic acid, membrane current was recorded while the membrane potential was clamped at -60 mV. ACh (3 μm) was applied during the period indicated by the upper bar. C, current-voltage relationships at the points marked by a-d in B obtained with a ramp protocol. The actual duration of the ramp pulse was 0.8 s. D, current-voltage relationships of ACh-induced current were obtained by subtracting a from b, c and d in C. Signals were filtered at a cut-off frequency of 10 kHz (A) or 100 Hz (B-D). All traces were recorded from the same cell.
The effect of ACh on membrane current was also examined in several smooth muscle cells (Fig. 3). First, the ability of 3 μm ACh to induce a membrane hyperpolarization was checked in current clamp mode (Fig. 3A). The mode was switched to voltage clamp and the current recorded. Again the current produced by a 10 mV voltage step relaxed in a multi-exponential manner (Fig. 3Bb); the input resistance of this cell was found to be 31.0 MΩ. The mean input resistance of six cells examined was 55.0 ± 23.0 MΩ. Six minutes after application of 18β-glycyrrhetinic acid (20 μm), the relaxation of the capacitive current could be well fitted with a single exponential function (Fig. 3Bc), indicating that most of the electrical communications with neighbouring cells had been blocked. The input resistance and capacitance of this cell were 1.1 GΩ and 7.5 pF, respectively. The mean input resistance and capacitance were 0.7 ± 0.4 GΩ and 9.7 ± 2.4 pF (n= 6), respectively. Seven minutes after introduction of 18β-glycyrrhetinic acid, 3 μm ACh was applied while the membrane potential of the smooth muscle cell was clamped at -50 mV (Fig. 3C). ACh did not induce a change in membrane current. The current-voltage relationships obtained before and after application of ACh completely overlapped (Fig. 3D). After washing out 18β-glycyrrhetinic acid, the current produced by a 10 mV voltage step gradually returned towards its control, indicating some recovery of syncytial behaviour (Fig. 3Eb). The input resistance decreased to 125.8 MΩ 9 min after the washout. Although the magnitudes of the responses were small, ACh-induced hyperpolarizations could be detected (Fig. 3F). In the presence of 18β-glycyrrhetinic acid, ACh did not induce a membrane current in each of the six smooth muscle cells examined.
Figure 3. Effect of ACh on membrane current recorded from a smooth muscle cell after blocking gap junctions.
Conventional whole-cell clamp method. A, in current clamp mode, the membrane potential in a smooth muscle cell was recorded and 3 μm ACh was applied during the period indicated by the bar at the top. B, in voltage clamp mode, membrane currents produced by a 10 mV voltage step (a) were recorded before (b) and 6 min after application of 20 μm 18β-glycyrrhetinic acid (c). C, in the presence of 18β-glycyrrhetinic acid, membrane current was recorded while membrane potential was clamped at -50 mV. ACh (3 μm) was applied during the period indicated by the bar at the top. D, current-voltage relationships taken at points marked by a-c in C obtained with a ramp protocol. Actual duration of the ramp pulse was 0.8 s. E, membrane current produced by a 10 mV voltage step (a) was recorded 9 min after washing out 18β-glycyrrhetinic acid (b). F, again in current clamp mode, membrane potential was recorded and 3 μm ACh was applied during the period indicated by the bar at the top. Signals were filtered at a cut-off frequency of 10 Hz (A and F), 10 kHz (B and E) or 100 Hz (C and D). All traces were recorded from the same cell.
As the endothelium-dependent hyperpolarization might be affected by intracellular dialysis which occurs during conventional whole-cell clamp recordings, the responses to ACh were examined using conventional microelectrodes (Fig. 4). The mean resting membrane potential of smooth muscle was -74.5 ± 4.5 mV (n= 7); this value was somewhat more negative than that recorded with patch-clamp electrodes, perhaps reflecting removal of the adventitia in the preparations for patch-clamp experiments. To detect the endothelium-dependent hyperpolarization, the preparations were depolarized, to a mean membrane potential of -44.2 ± 5.3 mV (n= 7), by blocking inwardly rectifying K+ channels with 0.5 mM Ba2+ (Hashitani & Suzuki, 1997). In this series of experiments inhibitors of NO synthase (nitroarginine, 100 μm) and of cyclo-oxygenase (diclofenac, 1 μm) were incorporated in the bath solution to block any effects of NO and prostanoids. Before applying 18β-glycyrrhetinic acid, the mean amplitudes of the ACh-induced initial and second hyperpolarizing phases were 26.3 ± 2.8 mV and 24.6 ± 2.7 mV (n= 7), respectively. In the presence of 20 μm 18β-glycyrrhetinic acid, the membrane depolarized to -38.2 ± 5.8 mV (n= 7) and the mean amplitudes of initial and second phases of ACh-induced hyperpolarization decreased to 12.2 ± 5.5 mV and to 10.8 ± 6.1 mV (n= 7), respectively. After washing out 18β-glycyrrhetinic acid, the mean membrane potential returned to -41.5 ± 8.7 mV (n= 3) and the mean amplitudes of the initial and second phases of ACh-induced hyperpolarization increased to 27.6 ± 4.1 mV and 25.4 ± 3.5 mV (n= 3), respectively.
Figure 4. Effect of ACh on the membrane potential of smooth muscle cells recorded with a microelectrode after blocking gap junctions.
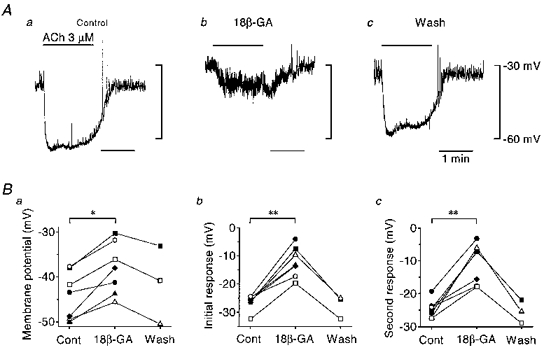
A, a smooth muscle cell was impaled with a conventional microelectrode and the membrane potential recorded. ACh (3 μm) was applied during periods indicated by bars at the top before (a), 10 min after application of 20 μm 18β-glycyrrhetinic acid (18β-GA; b) and 13 min after washing out 18β-glycyrrhetinic acid (c). Signals were filtered at a cut-off frequency of 40 Hz. B, summaries of membrane potentials (a), the amplitude of the initial responses (b) and the amplitude of the second responses (c) before and after application of 18β-glycyrrhetinic acid, and after washing it out. Each symbol represents data from a separate experiment. In b and c, negative values represent hyperpolarizations. *P < 0.002, **P < 0.001.
As Ca2+-activated K+ channels have been shown to be involved in ACh-induced hyperpolarization (Chen & Cheung, 1992; Marchenko & Sage, 1996), the effect of Ca2+ removal was examined in current-clamped endothelial cells (Fig. 5). In control solution containing 1.8 mM Ca2+, the mean membrane potential was -41.1 ± 6.8 mV (n= 7) and the mean amplitudes of the initial and second phases of ACh-induced hyperpolarization were 7.8 ± 2.6 mV and 11.1 ± 7.0 mV (n= 7), respectively. When external Ca2+ was removed, the membrane potential was not significantly changed (mean, -38.7 ± 3.6 mV; n= 7), but the mean amplitude of the initial phase of ACh-induced hyperpolarization was decreased to 3.2 ± 1.1 mV (n= 7) and it was no longer followed by a secondary hyperpolarization, rather a depolarization of 4.5 ± 1.9 mV (n= 7) was detected. The nature of this depolarization was not investigated in the present experiments. After reintroducing Ca2+, the mean membrane potential was -38.6 ± 3.3 mV (n= 6), the second component returned (6.7 ± 6.8 mV, n= 6) and the initial component increased in amplitude (5.8 ± 2.9 mV, n= 6).
In endothelial cells of the rat aorta, the Ca2+-activated K+ channels involved in ACh-induced hyperpolarization are blocked by CTX (Marchenko & Sage, 1996). The effects of CTX on ACh-induced hyperpolarization in current-clamped endothelial cells were examined (Fig. 6). Before application of CTX, the mean membrane potential was -37.8 ± 3.7 mV (n= 5), and the mean amplitudes of the initial and second phases of ACh-induced hyperpolarizations were 8.4 ± 4.8 mV and 13.4 ± 4.7 mV (n= 5), respectively. In the presence of 50 nM CTX, the membrane depolarized to -33.4 ± 2.7 mV (n= 5) and the mean amplitudes of the initial and second phases of ACh-induced hyperpolarization decreased to 2.1 ± 1.0 mV and to 0.3 ± 1.5 mV (n= 5), respectively. After washing out CTX, the mean membrane potential returned to -34.9 ± 3.7 mV (n= 5), and the mean amplitudes of the initial and second phases of ACh-induced hyperpolarization increased to 8.3 ± 4.4 mV and 14.5 ± 3.8 mV (n= 5), respectively.
DISCUSSION
Since Chen et al. (1988) reported endothelium-dependent hyperpolarization of vascular smooth muscle, this phenomenon has been thought to be produced by an unidentified humoral substance (EDHF) released from vascular endothelium (Chen et al. 1991). In the presence of 18β-glycyrrhetinic acid, electrical coupling between cells was effectively blocked and the membrane potential of individual cells could be well clamped while all cell types remained located within a multicellular preparation. If endothelial cells do indeed release EDHF, it should induce some outward current in voltage-clamped smooth muscle cells which had been electrically uncoupled, as the two types of cells were still closely located. In smooth muscle cells of guinea-pig mesenteric arteriole, however, when gap junctions had been blocked, ACh failed to induce a membrane current which would trigger membrane hyperpolarization. On the other hand, after gap junctions had been blocked, endothelial cells continued to respond to ACh and produced an outward current which was presumably responsible for the membrane hyperpolarization. These results suggest that ACh-induced hyperpolarization of smooth muscle cells is due to electrotonic conduction from endothelial cells through myoendothelial gap junctions.
In most of the experiments presented in this paper, we used the conventional whole-cell clamp method. With this method, the interior of the patched cell is eventually dialysed with the pipette solution. If soluble factors are involved in the action of EDHF, the dialysed cells may no longer respond to EDHF. When the cells were attached to neighbouring cells, responses generated in the neighbouring, non-dialysed, cells would conduct electrotonically to the dialysed cell. However after blocking gap junctions, the conducted responses would be abolished. To rule out the possibility that dialysis rather than blocking gap junctions abolished the responses to ACh, the experiments were repeated using conventional microelectrodes, which preserve the intracellular environment. In these experiments, 18β-glycyrrhetinic acid again inhibited ACh-induced hyperpolarization. While 18β-glycyrrhetinic acid completely blocked the ACh-induced response in the conventional whole-cell clamp experiments, small hyperpolarizations could still be observed in the presence of 18β-glycyrrhetinic acid when recordings were made using microelectrodes. This observation suggests that perhaps part of the response to EDHF had persisted and did indeed rely on intracellular soluble factors. Alternatively it could be that the preparations used with the microelectrode were embedded within the connective tissue and this limited the access of 18β-glycyrrhetinic acid to gap junctions.
In a large artery, changes in the membrane potential of smooth muscle cells can change the membrane potential of endothelial cells. However, changes in the membrane potential of endothelial cells do not change the membrane potential of smooth muscle cells and this has been attributed to the electrical mismatch between the large smooth muscle syncytium and the smaller endothelial syncytium (Bény & Pacicca, 1994). This is not the case in arterioles where the media consists of only one or two cell layers and both smooth muscle and endothelial cells have comparable cell membrane capacitance of around 10 pF (Yamamoto et al. 1998).
Ca2+-activated K+ channels appear to be involved in ACh-induced hyperpolarization in endothelial cells (Olesen et al. 1988; Chen & Cheung, 1992; Marchenko & Sage, 1996). In the present experiments, ACh induced a two-phase hyperpolarization, an initial rapid and a slower secondary phase. ACh-induced outward currents had similar phases. The current-voltage relationships taken during both the initial and second phases showed that the outward currents had null potentials of around -90 mV, close to the calculated K+ equilibrium potential of -87.0 mV. The currents both increased linearly with the membrane depolarization, indicating that both involve an increase of K+ conductance. Although somewhat diminished, the first phase of ACh-induced hyperpolarization persisted but the second phase was abolished by removing external Ca2+, suggesting that the first phase might be due to Ca2+ release from intracellular stores whereas the second phase required Ca2+ influx. Thus, in endothelial cells, ACh might increase intracellular Ca2+ first by releasing stored Ca2+, probably through an inositol (1,4,5)-trisphosphate-mediated mechanism (Marchenko & Sage, 1994), and then by following Ca2+ entry from the extracellular medium. Each increase in [Ca2+]i then activates K+ channels producing membrane hyperpolarization. As both phases of the ACh-induced responses were inhibited by CTX, Ca2+-activated K+ channels similar to those found in rat aortic endothelial cells (Marchenko & Sage, 1996) may be involved in guinea-pig mesenteric arterioles.
The existence of humoral EDHF was suggested by using sandwiched preparations in guinea-pig coronary artery (Chen et al. 1991), and the release of EDHF from either native or cultured porcine coronary endothelial cells has been bioassayed by monitoring the membrane potential in vascular smooth muscle cells located downstream (Popp et al. 1996). However, at least in guinea-pig mesenteric arterioles, the present experiments suggest that a humoral substance is not involved in ACh-induced hyperpolarization of smooth muscle cells. Rather, myoendothelial gap junctions seem to be crucial in this phenomenon. Although the significance of these gap junctions in large arteries was not assessed in the present experiments, recent studies show an important role of myoendothelial gap junctions in endothelium-dependent relaxation in rabbit thoracic aorta and superior mesenteric artery (Chaytor et al. 1998). When ACh is applied for a long period (10 min), endothelium-derived NO is responsible for a small sustained component of the ACh-induced hyperpolarization in the rat aorta (Vanheel et al. 1994). As neither nitroarginine nor diclofenac were used in the whole-cell clamp experiments, NO and prostanoids were probably released from the endothelium by ACh stimulation. Although we did not apply ACh for more than 2 min, these substances did not seem to induce any detectable membrane current in the presence of 18β-glycyrrhetinic acid.
In conclusion, endothelial cells control vascular smooth muscle cells not only by releasing vasoactive substances such as NO but also by affecting the membrane potential of nearby smooth muscle cells through myoendothelial gap junctions. It is also possible that smooth muscle cells control the release of vasoactive substances from endothelial cells by affecting their membrane potential and/or [Ca2+]i as suggested by Dora et al. (1997). It is unlikely that ACh-induced hyperpolarizations recorded from smooth muscle cells of guinea-pig mesenteric arterioles involve a humoral substance released from endothelial cells.
Acknowledgments
We thank Professor G. D. S. Hirst for his valuable comments on the manuscript.
References
- Bény J-L. Electrical coupling between smooth muscle cells and endothelial cells in pig coronary arteries. Pflügers Archiv. 1997;433:364–367. doi: 10.1007/s004240050289. [DOI] [PubMed] [Google Scholar]
- Bény J-L, Pacicca C. Bidirectional electrical communication between smooth muscle and endothelial cells in the pig coronary artery. American Journal of Physiology. 1994;266:H1465–1472. doi: 10.1152/ajpheart.1994.266.4.H1465. [DOI] [PubMed] [Google Scholar]
- Busse R, Fichtner H, Lückhoff A, Kohlhardt M. Hyperpolarization and increased free calcium in acetylcholine-stimulated endothelial cells. American Journal of Physiology. 1988;255:H965–969. doi: 10.1152/ajpheart.1988.255.4.H965. [DOI] [PubMed] [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circulation Research. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- Chaytor AT, Evans WH, Griffith TM. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. The Journal of Physiology. 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Cheung DW. Characterization of acetylcholine-induced membrane hyperpolarization in endothelial cells. Circulation Research. 1992;70:257–263. doi: 10.1161/01.res.70.2.257. [DOI] [PubMed] [Google Scholar]
- Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. British Journal of Pharmacology. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Yamamoto Y, Miwa K, Suzuki H. Hyperpolarization of arterial smooth muscle induced by endothelial humoral substances. American Journal of Physiology. 1991;260:H1888–1892. doi: 10.1152/ajpheart.1991.260.6.H1888. [DOI] [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM, Harley EH. Reversible inhibition of intercellular junctional communication by glycyrrhetinic acid. Biochemical and Biophysical Research Communications. 1986;134:29–36. doi: 10.1016/0006-291x(86)90522-x. [DOI] [PubMed] [Google Scholar]
- de Roos ADG, van Zoelen EJJ, Theuvenet APR. Determination of gap junctional intercellular communication by capacitance measurements. Pflügers Archiv. 1996;431:556–563. doi: 10.1007/BF02191903. [DOI] [PubMed] [Google Scholar]
- Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proceedings of the National Academy of Sciences of the USA. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Zygmunt PM, Hogestatt ED, Weston AH. Effects of cytochrome P450 inhibitors on potassium currents and mechanical activity in rat portal vein. British Journal of Pharmacology. 1996;119:691–701. doi: 10.1111/j.1476-5381.1996.tb15728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao M, Hattori Y, Kanno M, Sakuma I, Kitabatake A. Evidence against a role of cytochrome P450-derived arachidonic acid metabolites in endothelium-dependent hyperpolarization by acetylcholine in rat isolated mesenteric artery. British Journal of Pharmacology. 1997;120:439–446. doi: 10.1038/sj.bjp.0700932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Wilson S, Schlender KK, Ruch RJ. Gap-junction disassembly and connexin 43 dephosphorylation induced by 18β-glycyrrhetinic acid. Molecular Carcinogenesis. 1996;16:157–164. doi: 10.1002/(SICI)1098-2744(199607)16:3<157::AID-MC6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Hashitani H, Suzuki H. K+ channels which contribute to the acetylcholine-induced hyperpolarization in smooth muscle of the guinea-pig submucosal arteriole. The Journal of Physiology. 1997;501:319–329. doi: 10.1111/j.1469-7793.1997.319bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker M, Bara A, Bauersachs J, Busse R. Characterization of endothelium-dependent hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. The Journal of Physiology. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa JJ, Ghibaudi L, Williams P, Chatterjee M. Comparison of acetylcholine-dependent relaxation in large and small arteries of rat mesenteric vascular bed. American Journal of Physiology. 1994;266:H952–958. doi: 10.1152/ajpheart.1994.266.3.H952. [DOI] [PubMed] [Google Scholar]
- Little TL, Xia J, Duling BR. Dye tracers define differential endothelial and smooth muscle coupling patterns within the arteriolar wall. Circulation Research. 1995;76:498–504. doi: 10.1161/01.res.76.3.498. [DOI] [PubMed] [Google Scholar]
- Marchenko SM, Sage SO. Mechanism of acetylcholine action on membrane potential of endothelium of intact rat aorta. American Journal of Physiology. 1994;266:H2388–2395. doi: 10.1152/ajpheart.1994.266.6.H2388. [DOI] [PubMed] [Google Scholar]
- Marchenko SM, Sage SO. Calcium-activated potassium channels in the endothelium of intact rat aorta. The Journal of Physiology. 1996;492:53–60. doi: 10.1113/jphysiol.1996.sp021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen S-P, Davies PF, Clapham DE. Muscarinic-activated K+ current in bovine aortic endothelial cells. Circulation Research. 1988;62:1059–1064. doi: 10.1161/01.res.62.6.1059. [DOI] [PubMed] [Google Scholar]
- Popp R, Bauersachs J, Hecker M, Fleming I, Busse R. A transferable, β-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. The Journal of Physiology. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T. Acetylcholine induces Ca-dependent K currents in rabbit endothelial cells. Japanese Journal of Pharmacology. 1990;53:235–246. doi: 10.1254/jjp.53.235. [DOI] [PubMed] [Google Scholar]
- Spray DC, Burt JM. Structure-activity relations of the cardiac gap junction channel. American Journal of Physiology. 1990;258:C195–205. doi: 10.1152/ajpcell.1990.258.2.C195. [DOI] [PubMed] [Google Scholar]
- Vanheel B, Van de Voorde J. Evidence against the involvement of cytochrome P450 metabolites in endothelium-dependent hyperpolarization of the rat main mesenteric artery. The Journal of Physiology. 1997;501:331–341. doi: 10.1111/j.1469-7793.1997.331bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanheel B, Van de Voorde J, Leusen I. Contribution of nitric oxide to the endothelium-dependent hyperpolarization in rat aorta. The Journal of Physiology. 1994;475:277–284. doi: 10.1113/jphysiol.1994.sp020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Weid P-Y, Bény J-L. Simultaneous oscillations in the membrane potential of pig coronary artery endothelial and smooth muscle cells. The Journal of Physiology. 1993;471:13–24. doi: 10.1113/jphysiol.1993.sp019888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Fukuta H, Nakahira Y, Suzuki H. Blockade by 18β-glycyrrhetinic acid of intercellular electrical coupling in guinea-pig arterioles. The Journal of Physiology. 1998;511:501–508. doi: 10.1111/j.1469-7793.1998.501bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]