

Abstract
We determined the effect of cortisol (200 nm for 48 h) on the intracellular Ca2+ concentration ([Ca2+]i) and parameters of Ca2+i signalling in 19 lymphoblastoid cell lines (LCLs).
Using the fluorescent dye fura-2, the basal [Ca2+]i in Ca2+-containing medium was 63.5 ± 2.4 nm in vehicle (ethanol)-treated LCLs and 55.7 ± 2.6 nm (mean ±s.e.m.) in cortisol-treated LCLs.
Ca2+i signalling following platelet-activating factor (PAF, 100 nm) addition was enhanced by cortisol treatment, with LCLs having small PAF responses showing the largest percentage increase after cortisol treatment. Mean peak [Ca2+]i responses to PAF were enhanced 67.0% and 55.7% in Ca2+-free and Ca2+-containing medium, respectively.
The endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin (100 nm) caused a transient increase in [Ca2+]i in Ca2+-free medium in which the peak change was increased in cortisol-treated cells (98.5 ± 5.8 vs. 79.8 ± 4.5 nm). Peak changes in the freely exchangeable Ca2+ in response to 5 μm ionomycin were also enhanced in cortisol-treated cells (923.7 ± 113.9 vs. 652.2 ± 64.5 nm) and correlated to the PAF-evoked [Ca2+]i response.
Cortisol-treated LCLs exposed to thapsigargin to empty intracellular Ca2+ stores (10 min treatment in Ca2+-free medium) and exposed to CaCl2 or MnCl2 had a greater rate of Ca2+ entry (18.6 ± 1.8 vs. 13.8 ± 1.5 nm s−1) and higher rate constant for Mn2+ entry (0.0345 ± 0.0029 vs. 0.0217 ± 0.0020) than vehicle-treated cells. Peak [Ca2+]i in cells exposed to CaCl2 was also enhanced (869.4 ± 114.7 vs. 562.6 ± 61.7 nm). Parameters of divalent cation influx were highly correlated to the peak [Ca2+]i elicited by thapsigargin or ionomycin.
Inclusion of RU 486 (a glucocorticoid antagonist) with cortisol prevented the decrease in basal [Ca2+]i and stimulatory actions of cortisol on all Ca2+i parameters. RU 486 alone had no apparent effects on basal [Ca2+]i or signalling.
Based on data obtained over a wide range of responses (in the presence and/or absence of cortisol or RU 486), the results show that cortisol stimulation of glucocorticoid receptors decreases basal [Ca2+]i and enhances PAF-evoked [Ca2+]i signalling, most probably through its effects on intracellular Ca2+ stores. In turn, the extent of Ca2+ entry via store-operated plasma membrane Ca2+ channels is closely linked to the size of the Ca2+ stores.
Stimulation of B lymphocytes leads to diverse cellular functions including secretion, proliferation, differentiation or programmed cell death (apoptosis). Inflammatory agents binding or cross-linking surface membrane receptors increase protein kinase/phosphatase activities and activate phospholipase C, generating inositol 1,4,5-trisphosphate (IP3). IP3 mobilizes Ca2+ from intracellular Ca2+ stores and produces a transient rise in cytosolic Ca2+ (Ca2+1). The emptying of Ca2+ from IP3-sensitive Ca2+ stores also results in enhanced Ca2+ influx across the plasma membrane, which is manifested as an enhancement of the Ca2+1 transient, oscillatory increases in Ca2+1, or a secondary, sustained increase in [Ca2+]i. This Ca2+ entry pathway, termed store-operated Ca2+ entry (SOCE), is responsible for refilling intracellular Ca2+ stores (Putney, 1990) and is an important regulator of Ca2+-dependent gene expression (Negulescu et al. 1994; Dolmetsch et al. 1998; for review, see Putney & Bird, 1993). In B lymphocytes, elevation of [Ca2+]i serves as a differentiation signal towards antibody-secreting cells (Clevers et al. 1985; Huang et al. 1995) and enables interleukin-2 (IL-2) synthesis (Taira et al. 1987) and cell proliferation (Ambrus et al. 1991). The inhibition of Ca2+ influx via the SOCE pathway results in marked decreases in [Ca2+]i and IL-2 (Diegel et al. 1994), supporting the hypothesis that the B cell response is related to the degree of [Ca2+]i elevation (Clark & Lane, 1991). Additional changes in Ca2+1 regulation in anti-IgM-stimulated malignant B-1 cells suggest that dysfunctional Ca2+1 signalling may increase the susceptibility for these cells to undergo apoptosis (Dang et al. 1995). Thus, the regulation of [Ca2+]i represents a key factor in the control of B lymphocyte development and function.
Cortisol and synthetic glucocorticoids such as dexamethasone are important modulators of the immune system (Cupps & Fauci, 1982), and exert both inhibitory and stimulatory actions on lymphocyte development and cellular responses to inflammatory stimuli (Guyre et al. 1988; Wilckens & De Rijk, 1997). In murine precursor B cells, slight elevation of the circulating level of glucocorticoids decreased the number of cycling cells by inducing apoptosis (Garvey et al. 1993). In B cells undergoing lymphopoiesis, negative regulatory effects of glucocorticoids were countered by the presence of (T) lymphocyte-supportive stromal cells (Borghesi et al. 1997). Glucocorticoids produce variable responses in cell surface Ia (sIa) expression, the response being dependent on the duration of exposure and the state (i.e. resting or activated) of the B cell (McMillan et al. 1988). In a report examining cell activation of resting B cells, cortisol acted as an inhibitor of anti-Ig-antibody-mediated IP3 synthesis, Ca2+1 signalling and entry into the cell cycle (Dennis et al. 1987). These results suggest that, similar to T lymphocytes, cortisol effects on B cell function are dependent on the extent of glucocorticoid exposure and the context in which inflammatory mediators are presented. In light of the significant influence of Ca2+ homeostasis on B cell proliferation and differentiation, widely different effects of glucocorticoids on Ca2+1 signalling and B cell physiology could be expected at different stages of B lymphocyte development.
To examine the glucocorticoid regulation of Ca2+ homeostasis in activated B lymphocytes, we studied cortisol-induced alterations of Ca2+ signalling in Epstein- Barr virus (EBV)-transformed B lymphoblasts. In vitro transformation of peripheral blood cells by EBV leads to partial differentiation and immortalization of cells of the B lymphocyte lineage, lending to their characterization as a model of activated B cells. EBV-generated lymphoblastoid cell lines (LCLs) are often used where stable, proliferating cultures of pure B cells are required. Their growth in tissue culture surmounts obstacles often associated with freshly isolated cell populations, including low cell number, deleterious effects of cell isolation chemicals or procedures, and exposure to leucocyte-derived cytokines or hormones. Several groups have suggested that LCLs exhibit signalling and membrane ion transport properties common to non-transformed cells (Schulam et al. 1990; Kuruvilla et al. 1993; Kojima et al. 1994; Siffert et al. 1995), supporting the idea that independent of viral transformation, LCLs retain membrane signalling properties representative of B lymphocytes. Thus, to study the role of cortisol on Ca2+ homeostasis in activated B cells, we examined parameters of receptor-mediated [Ca2+]i signalling, intracellular Ca2+ stores and Ca2+ influx across the plasma membrane (SOCE) in LCLs.
METHODS
Chemicals
Unless otherwise specified, the chemicals used in this study were obtained from Sigma. Fura-2-acetoxymethylester (fura-2 AM) was from Molecular Probes; ionomycin and platelet-activating factor (PAF) were from Calbiochem; thapsigargin (TG) was from Alexis Corporation; mifepristone (RU 486) was from Biomol Research Laboratories, Inc.
Culture of B lymphoblasts
LCLs from the Centre d'Etude du Polymorphisme Humain (CEPH) collection (Coriell Institute for Medical Research, Camden, NJ, USA) were cultured as previously described (Brzustowicz et al. 1997) in RPMI 1640 culture medium containing 2 mM L-glutamine, 100 u ml−1 penicillin, 100 μg l−1 streptomycin and 15 % heat-inactivated fetal bovine serum (Irvine Scientific). Cells were passaged 2-3 times per week, and cell density was maintained between 0.5 × 106 and 1 × 106 cells ml−1. Cells used for experiments were derived from fresh cultures thawed from frozen stocks at 3-4 month intervals. Cells were routinely monitored for mycoplasma contamination with the Mycotrim TC Triphasic Culture System (Irvine Scientific).
In preparation for experimental protocols, 107 cells (0.5 × 106 ml−1) were resuspended for 48 h in fresh culture medium containing glucocorticoid (200 nM cortisol), glucocorticoid receptor (GR) antagonist (RU 486: Gagne et al. 1985) or vehicle (ethanol, final concentration of 0.04 %). Cell numbers were determined with a Coulter Counter ZM.
Measurements of [Ca2+]i
Lymphoblasts were washed at 37°C with Hepes-buffered solution (HBS) comprising (mM): 140 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 20 N-2-hydroxyethyl-piperazine-N′-2-ethanesulfonic acid (Hepes), 10 glucose (pH 7.4), and 0.1 % fatty acid-free bovine serum albumin. Cells (2 × 107 ml−1) were then incubated for 30 min at 37°C with 2 μm fura-2 AM and 0.125 mM sulfinpyrazone (an anionic transport inhibitor used to retard fura-2 extrusion: Di Virgilio et al. 1988) as previously described (Dang et al. 1995; Brzustowicz et al. 1997). Aliquots of lymphoblasts (2 × 106 (100 μl)−1) were centrifuged to remove extracellular fura-2, washed once with HBS and resuspended in a cuvette containing 3 ml of either Ca2+-free HBS (0.3 mM ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) substituted for CaCl2) or HBS. [Ca2+]i measurements were performed in a CM3 spectrophotometer equipped with stirring and temperature control (Instruments SA, Inc.). Excitation wavelengths were set at 340 nm/380 nm and emission wavelength at 505 nm. [Ca2+]i was calculated from the 340 nm/380 nm fluorescence ratio (R) according to the equation described by Grynkiewicz et al. (1985). Calibration was achieved by treating cells with 100 μm digitonin in the presence of 1 mM CaCl2 (Rmax), followed by 15 mM EGTA (pH 8.0; Rmin). In some experiments, TG, a sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) inhibitor (Lytton et al. 1991), was used to inhibit Ca2+ reuptake by intracellular Ca2+ stores. Autofluorescence of unloaded cells in HBS was subtracted from the cellular fluorescence of the dye prior to all calculations for [Ca2+]i.
Mn2+ uptake assay
Mn2+ was used as a Ca2+ surrogate to study divalent cation entry (Hallam & Rink, 1985). Fura-2-loaded cells were centrifuged, resuspended in 500 μl microfuge tubes in Ca2+-free HBS and treated with vehicle (DMSO; 0.5 % final volume) or TG (500 nM) for 10 min. Cells were recentrifuged and suspended in cuvettes containing Ca2+-free medium; after 10 s, 0.35 mM MnCl2 was added. Fluorescence was monitored at excitation and emission wavelengths of 360 nm and 505 nm. Data were normalized using 360 nm values obtained immediately prior to addition of MnCl2. Autofluorescence of Mn2+-treated (1 mM), digitonin-permeabilized cells and buffer for each experiment was subtracted from the fluorescence record prior to normalization.
Data analysis and statistics
Mn2+ quenching of fura-2 (percentage fluorescence quenching at times 0, 10, 30 and 60 s compiled from individual experiments of vehicle- and TG-treated cells) was best-fitted to the monoexponential function:
where A is the cellular pool at time t, a1 is the fura-2 fluorescence before treatment with Mn2+, and k1 is the rate constant. To obtain an estimate of the TG-sensitive rate of Mn2+ influx, Mn2+ quenching in DMSO-treated cells was subtracted from quenching in TG-treated cells and fitted to the equation:
in which parameters were defined as above and a constraint on the maximal difference between DMSO- and TG-treated cells (45 %, cf. Fig. 4B) was incorporated.
Figure 4. Cortisol treatment increases Mn2+ entry in TG-treated cells.
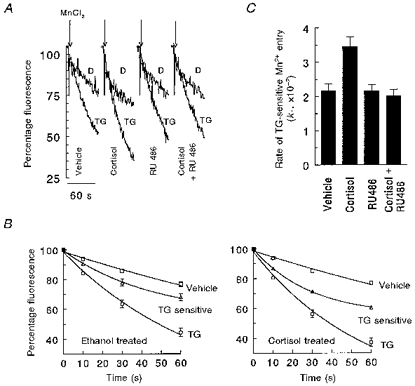
A, representative experiments are depicted in which fura-2-loaded cells were centrifuged and pretreated with 500 nM TG (TG-labelled traces) or vehicle (D-labelled traces, for 0.5 % DMSO) in Ca2+-free medium for 10 min. Cells were then centrifuged and the pellet resuspended in 3 ml Ca2+-free medium; traces show the normalized response of fluorescence decline immediately after addition of 0.35 mM MnCl2 (arrows). B, data used to calculate the rate constant for TG-sensitive quenching of fura-2 in vehicle (left panel) and cortisol-treated (right panel) LCLs (n= 18) as described in Methods and legend to Table 1. Shown are means ±s.e.m. of LCLs evaluated for Mn2+ entry; s.e.m.s smaller than the symbol are not shown. Mn2+ entry in vehicle- (□, DMSO) and TG-treated cells (○); TG-sensitive Mn2+ influx is given by (▵), and curve-fitted lines describing the exponential decay of fluorescence are given for each condition. C, results of weighted least-squares analysis of k1 (rate constant, see also Table 1) indicated a P value < 0.01 between vehicle- and cortisol-treated LCLs.
Repeated measures analysis of variance (ANOVA) was performed with an IBM-compatible PC using the general linear model procedure (PROC GLM, Statistical Analysis Systems (SAS) Institute, Cary, NC, USA). This analysis controlled between-cell line variability, which, as shown for PAF-evoked Ca2+1 transients and other parameters of Ca2+ signalling, varies widely between LCLs from different individuals. Duncan's multiple range test was used to assess differences between treatment groups. Correlation analysis was performed using SAS PROC CORR. Non-linear regression analysis for computations of parameters of Mn2+ quenching of fura-2 quench experiments utilized SAS REG. Statistical analyses of the rate constants derived from curve-fitting the Mn2+ data were performed using weighted least squares according to the method described by Johnson & Milliken (1983). P values < 0.05 were taken as statistically significant. Data are reported as mean values ±s.e.m. for n LCLs.
RESULTS
Effects of cortisol on basal [Ca2+]i and receptor-mediated [Ca2+]i transient
To examine Ca2+1 signalling in LCLs, we used platelet-activating factor (PAF), a potent cytokine that has diverse actions throughout the immune system (Snyder, 1990). In B cells, this lipid mediator alters several immunomodulatory functions including nuclear-factor-κ B-mediated immunoglobulin synthesis and secretion (Smith & Schearer, 1994; Rosskopf et al. 1995), cytokine expression (e.g. TNFα and IL-2; Smith et al. 1994) and Ca2+-dependent expression of cell cycle genes and proliferation (Mazer et al. 1991, 1994; Rosskopf et al. 1995). The PAF receptor is expressed in both resting and activated cells (Nguer et al. 1992, 1996); its activation results in phosphatidylinositol bisphosphate hydrolysis, IP3 generation and a transient Ca2+1 response (Mazer et al. 1991). Previous work by the present authors and Siffert's group showed that the PAF-evoked [Ca2+]i response in lymphoblasts displays marked variability between cell lines established from different individuals (Siffert et al. 1995; Brzustowicz et al. 1997). Based on widely differing [Ca2+]i responses to PAF, we selected a panel of 19 LCLs from the CEPH collection to study the effects of cortisol on Ca2+1 homeostasis. For these experiments, the effects of cortisol on the PAF-evoked [Ca2+]i response, intracellular Ca2+ store size and SOCE were assessed. In addition, we determined whether modulation of Ca2+1 occurred through the glucocorticoid-GR complex by performing the study in the presence of RU 486.
Preliminary experiments showed that 200 nM cortisol added 1 or 15 min prior to 100 nM PAF did not alter basal [Ca2+]i or the PAF-evoked Ca2+1 response. Furthermore, enhancement of the PAF responses was observed only after 12-24 h treatment with this concentration of cortisol (data not shown). We therefore performed all experiments after 48 h of exposure of cells to 200 nM glucocorticoid or inhibitor. Figure 1A shows examples of the PAF-evoked Ca2+1 response in cortisol- or vehicle-treated cells suspended in HBS. Cells treated with glucocorticoid exhibited larger Ca2+1 transients over the entire range of agonist concentration, with no apparent alteration of sensitivity or time course of response. As peak Ca2+1 signals at 100 nM PAF are maximal and highly reproducible for a given LCL (Brzustowicz et al. 1997), this concentration of agonist was used to evaluate the influence of cortisol on Ca2+1 signalling. In Ca2+-containing medium, basal [Ca2+]i in cortisol-treated cells was slightly decreased (55.7 ± 2.6 nM) compared with vehicle-treated cells (63.5 ± 2.4 nM, P < 0.05, n= 19). LCLs treated with RU 486, or the combination of RU 486 and cortisol, exhibited a basal [Ca2+]i that was not significantly different from that of ethanol-treated cells.
Figure 1. Cortisol treatment enhances PAF-evoked signalling.
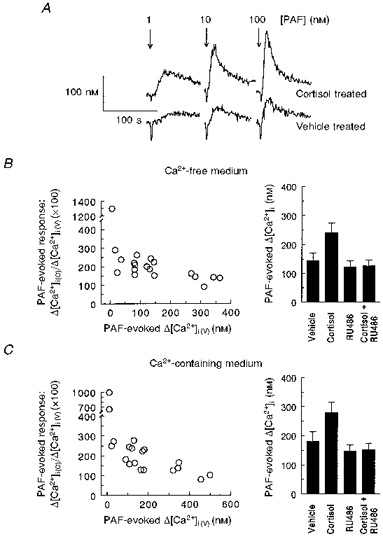
A, example of transients in an LCL exposed to 1, 10 or 100 nM PAF in HBS. 10 × 106 cells were treated with vehicle (ethanol) or cortisol (200 nM) for 48 h and processed for measurements as described in Methods. Basal [Ca2+]i was determined as the mean [Ca2+]i value immediately preceding addition of agonist, and the PAF-evoked [Ca2+]i response was calculated as the difference between basal [Ca2+]i and peak [Ca2+]i after PAF addition. For this LCL, PAF addition (arrow) to cortisol-treated cells resulted in peak [Ca2+]i increases of 123 % (1 nM), 163 % (10 nM) and 158 % (100 nM) compared with vehicle-treated cells. B and C, PAF-evoked [Ca2+]i responses in LCLs suspended in Ca2+-free and Ca2+-containing HBS. Left panels: percentage stimulation of PAF-evoked [Ca2+]i response in cortisol-treated cells (Δ[Ca2+]i(C)) to vehicle-treated cells (Δ[Ca2+]i(V)) as a function of control response. Right panels: mean changes in PAF-evoked [Ca2+]i in cells treated for 48 h with vehicle, cortisol (200 nM), RU 486 (200 nM), or cortisol (200 nM) and RU 486 (200 nM) (n= 19). ANOVA indicated overall P values < 0.0001 for both Ca2+-free and Ca2+-containing HBS, with significant differences between cortisol and all other treatments.
Figure 1B and C shows the extent of stimulation following cortisol treatment on peak PAF-evoked [Ca2+]i responses, in Ca2+-free and Ca2+-containing HBS. In the left panels, the range of responses in vehicle-treated cells is graphed as the dependent variable, and the percentage stimulation following cortisol treatment is shown on the ordinate. LCLs exhibiting small PAF responses demonstrated the greatest percentage stimulation following glucocorticoid treatment. This relation was maintained for individual LCLs in Ca2+-free or Ca2+-containing HBS, as analysis of the percentile stimulation of the cortisol-enhanced PAF responses in the two conditions showed a high degree of correlation (r= 0.8890, P < 0.0001, n= 19). Additional experiments showed the lack of cortisol responsiveness in LCLs with inherently large responses was not a result of the high dose of agonist used; one cell line that showed no enhancement following cortisol treatment similarly failed to show increased [Ca2+]i responses at lower (1-10 nM) PAF concentrations (data not shown). The right panels of Fig. 1B and C show that the mean PAF-evoked [Ca2+]i response for 19 LCLs was significantly increased by cortisol treatment (by 67.0 and 55.7 % in Ca2+-free and Ca2+-containing HBS, respectively). RU 486 had no discernible effect on the PAF response, and the stimulatory actions of cortisol were absent in cells treated with equimolar concentrations of RU 486 and glucocorticoid.
Effects of cortisol on intracellular Ca2+ stores
PAF responses in cortisol-treated cells were increased in the absence of extracellular Ca2+ influx (Fig. 1B). This result suggests that the size of the IP3-releasable Ca2+ pool(s) was altered by glucocorticoid treatment. We examined whether cortisol increased intracellular Ca2+ stores by challenging LCLs with TG and ionomycin, two agents that mobilize Ca2+ from Ca2+ stores independently of IP3. TG, a sarco(endo)-plasmic reticulum (SER) Ca2+-ATPase (SERCA) inhibitor (Lytton et al. 1991), inhibits SERCA-mediated Ca2+ uptake into the SER. Its addition to lymphoblasts in Ca2+-free HBS results in a transient increase in Ca2+1, owing to an endogenous leak of Ca2+ from these stores and subsequent extrusion of Ca2+ across the plasma membrane. Alternatively, addition of the Ca2+ ionophore ionomycin to cells results in its insertion into intracellular membranes and the electroneutral exchange of Ca2+ for H+. Addition of 100 nM ionomycin to cells in Ca2+-free HBS results in a rapid increase in [Ca2+]i that serves as an estimate of the freely exchangeable Ca2+ (FEC). Cortisol treatment (48 h, 200 nM) resulted in significant increases in the TG-mediated rise in [Ca2+]i (Fig. 2A) and the FEC (Fig. 2B). The left panels illustrate the protocols used to obtain these parameters, and the right panels indicate the mean values for the four treatment groups. Treatment with the glucocorticoid antagonist had no effects of its own (on either parameter), and treatment of cells with RU 486 eliminated cortisol-induced increases in the TG-mediated rise in [Ca2+]i and FEC. The TG-evoked rise in [Ca2+]i appeared to reflect the FEC measurement, as comparison of these two parameters gave a correlation coefficient (r) of 0.5622 (P < 0.0001, n= 71). These data suggest that cortisol enhances SERCA-regulated and freely exchangeable intracellular Ca2+ stores in LCLs.
Figure 2. Cortisol treatment enhances intracellular Ca2+ stores in LCLs.
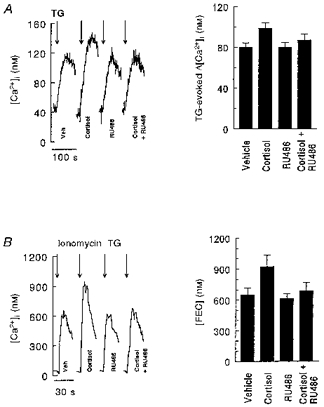
The left panels show representative examples of the TG-evoked [Ca2+]i response (A) and the ionomycin-TG-evoked rise in [Ca2+]i (FEC) (B) in LCLs treated as described in the legend to Fig. 1. Arrows indicate addition of 100 nM TG (A) or 5 μm ionomycin-100 nM TG (B) to cells suspended in Ca2+-free medium; TG was added to prevent reuptake of Ca2+ and diminution of the [Ca2+]i transient. The peak [Ca2+]i response minus the basal [Ca2+]i was used to estimate TG-releasable Ca2+ pools defined by the SER and FEC. The right panels show the results from 19 (A) and 17 (B) LCLs. ANOVA indicated overall P values < 0.0001 (TG-evoked [Ca2+]i) and < 0.0025 (ionomycin-TG-evoked [Ca2+]i), with significant differences between cortisol and all other treatment conditions.
Effects of cortisol on parameters of SOCE
The size, or capacity, of intracellular Ca2+ stores is tightly linked to the control of Ca2+ entry from the external medium. As cortisol enhanced intracellular Ca2+ stores, we examined whether a similar upregulation of store-operated Ca2+ entry (SOCE) was present in cortisol-treated cells. For these experiments, we measured both the rate of Ca2+ entry (change in [Ca2+]i during the first 10 s) and peak change in [Ca2+]i following Ca2+ reintroduction to cells with depleted Ca2+ stores (10 min of TG treatment in Ca2+-free medium). Figure 3A shows an example of an experiment used to determine the rate and peak [Ca2+]i after CaCl2 was introduced into the medium (final extracellular [Ca2+]= 0.15 mM). Parameters of Ca2+ influx following Ca2+ readdition to TG-treated cells showed considerable variability between LCLs with regard to rate of Ca2+ entry (3.1-39.3 nM s−1) and peak [Ca2+]i levels (233.1-2233 nM). Within individual experiments, the correlation coefficient for the rate of Ca2+ entry and the peak [Ca2+]i level was 0.7464 (P < 0.0001, n= 71), indicating a high degree of consistency between the two measurements. Figure 3 also shows that cortisol treatment significantly increased both the rate of Ca2+ entry (Fig. 3B) and peak [Ca2+]i (Fig. 3C). These results suggest glucocorticoids enhance the Ca2+ entry pathway activated by intracellular Ca2+ store depletion.
Figure 3. Cortisol enhancement of Ca2+ entry and peak [Ca2+]i following Ca2+ addition to TG-treated cells.
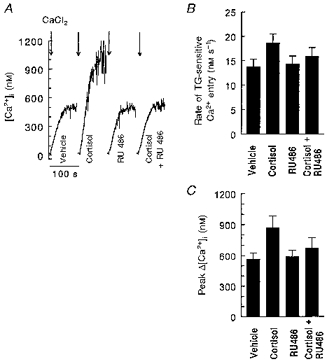
In representative experiments (A), cells challenged with 100 nM TG in Ca2+-free medium for 10 min (not shown) had 0.45 mM CaCl2 added to the cuvette (final [Ca2+]o= 0.15 mM). The first 10 s of the increase in [Ca2+]i were used to estimate the rate of Ca2+ influx (nM s−1, B), and peak changes in [Ca2+]i were determined as the difference between [Ca2+]i immediately prior to CaCl2 addition and the maximal [Ca2+]i (C). Control experiments with cells treated with DMSO (vehicle, 0.2 %) for 10 min had low (< 0.05 nM s−1) rates of Ca2+ influx after CaCl2 addition, hence no correction for non-TG-mediated increases in was performed. ANOVA indicated overall P values < 0.0001 for both analyses, with significant differences between cortisol and all other treatment conditions.
Estimates of rates of Ca2+ influx, or peak changes in [Ca2+]i following Ca2+ readdition to cells, may be affected by Ca2+-binding proteins and intracellular or plasma membrane Ca2+ transport mechanisms. We therefore assessed fura-2 quenching by Mn2+ as an indicator of store-operated divalent cation entry in TG-treated cells. Mn2+ has been used previously to characterize Ca2+ entry and SOCE in a variety of cells (Hallam & Rink, 1985; Missiaen et al. 1990). It has substantially greater affinity than Ca2+ to fura-2, and is a poor substrate for SERCA and other Ca2+ transport systems (Gomes da Costa & Madeira, 1986; Missiaen et al. 1990). In addition, quenching is monitored at the isosbestic point of fura-2, enabling changes in the rate of Mn2+ entry to be monitored independently of changes in [Ca2+]i. Figure 4A illustrates a representative experiment showing fluorescence records of TG- and vehicle-elicited Mn2+ influx for the four treatment conditions. To quantify the rate of Mn2+ entry, data obtained with cells treated with TG, vehicle or the difference between these curves (i.e. TG-sensitive Mn2+ entry) were best-fitted to a monoexponential function (Fig. 4B, and see Methods and Table 1 for additional details). Rates of Mn2+ quenching of intracellular fura-2 showed considerable variability between LCLs; e.g. from 0.11 to 1.79 % s−1 for vehicle-treated cells and 0.14 to 2.46 % s−1 for cortisol-treated cells. Rate constants derived for each treatment indicated cortisol-treated cells had a significantly greater mean rate constant for TG-sensitive Mn2+ entry than control or RU 486-treated cells (Fig. 4C and Table 1). The parameters describing rates of TG-sensitive Ca2+ entry and Mn2+ entry (quenching at 30 s) were highly correlated with each other (r= 0.5935, P < 0.0001, n= 69). These experiments, coupled with the findings of Fig. 3, suggest that measurements of rates of Ca2+ entry and peak changes in [Ca2+]i in TG-treated lymphoblasts are largely unaffected by Ca2+ transport or sequestration mechanisms. Importantly, the data indicate that LCLs treated with cortisol have enhanced SOCE.
Table 1.
Quenching of intracellular fura-2 by Mn2+: k1 (s−1)
| Treatment | Vehicle | TG | TG sensitive |
|---|---|---|---|
| Control | 0.446 ± 0.030 | 1.392 ± 0.077 | 2.169 ± 0.203 |
| Cortisol | 0.441 ± 0.047 | 1.731 ± 0.090* | 3.450 ± 0.293** |
| RU 486 | 0.495 ± 0.044 | 1.483 ± 0.074 | 2.169 ± 0.178 |
| Cortisol + RU486 | 0.489 ± 0.040 | 1.413 ± 0.079 | 2.005 ± 0.198 |
Cells were treated with vehicle, cortisol, RU 486 or cortisol and RU 486 (as described in text and legend to Fig. 4) and assessed for Mn2+ influx (cf. Fig. 4). Values are means ± asymptotic S.E.M. of k1 (× 10−2), the rate constant. Percentage fluorescence quenching values at 0, 10, 30 and 60 s were compiled for vehicle- and thapsigargin (TG)-treated cells and were best fitted by the monoexponential function A=a1e−k1t, described in Methods. TG-sensitive quenching of fura-2 describes the exponential fit of Mn2+ quenching of TG-treated cells minus the quenching in vehicle-treated cells. a1 values ranged from 96.99 ± 1.44 to 99.18 ± 0.05 in vehicle- and TG-treated cells, and 44.37 ± 1.56 to 44.97 ± 4.53 for curve-fitted data describing TG-sensitive Mn2+ influx, and were not significantly different within treatments. n= 18 for each treatment. Significantly different at
P < 0.05
P < 0.01.
These results indicate that glucocorticoid treatment enhances Ca2+ homeostasis in LCLs. Correlation of the extent of cortisol stimulation of PAF-evoked [Ca2+]i signals (cf. Fig. 1B and C, left panels) with intracellular Ca2+ store or SOCE parameters could suggest potential determinants effecting the cortisol-induced stimulation. However, these analyses resulted in weak correlations showing high dependence on one or two data points. We therefore performed a correlation analysis of the entire data set of Ca2+ and Mn2+ parameters to examine the relation between PAF-evoked increases in [Ca2+]i and Ca2+ store size and parameters of SOCE. Although this analysis does not specifically examine the cortisol-induced enhancement of the PAF response, it examines parameters of Ca2+1 signalling throughout the range of cortisol-modulated Ca2+1 responses. The correlation coefficients for these comparisons are indicated in Table 2. In Ca2+-free medium, PAF-evoked [Ca2+]i responses were positively correlated to Ca2+ stores and parameters describing SOCE. Similar correlations were obtained when the PAF response in Ca2+-containing HBS was used. The strongest correlation occurred with FEC, and is shown in Fig. 5A. As intracellular Ca2+ stores can regulate Ca2+ entry, we also compared the relation between store size and parameters of SOCE (Table 3). In general, correlation coefficients for these comparisons were higher than those for PAF-mediated [Ca2+]i responses and intracellular Ca2+ store size or SOCE. Two examples of the strong association are given by the correlations between the peak [Ca2+]i in TG-treated cells and the peak [Ca2+]i achieved following TG addition (Fig. 5B), and TG-sensitive Mn2+ entry and FEC (Fig. 5C). These results suggest that the magnitude of the PAF-evoked Ca2+1 signal is dependent on the size of the intracellular Ca2+ stores, which in turn are related to Ca2+ entry through the SOCE pathway(s).
Table 2.
Correlation analysis of PAF-evoked [Ca2+]i response with intracellular Ca2+ store size and parameters of SOCE
| Parameter | r | n | P | |
|---|---|---|---|---|
| PAF-evoked [Ca2+]i in Ca2+-free HBS | TG-evoked [Ca2+]i | 0.2713 | 74 | 0.0194 |
| FEC | 0.3544 | 72 | 0.0023 | |
| Rate of Ca2+ entry | 0.2701 | 74 | 0.0199 | |
| Peak [Ca2+]i | 0.2425 | 72 | 0.0401 | |
| Mn2+ quenching | 0.2703 | 70 | 0.0236 |
Analyses were performed for the PAF-evoked [Ca2+]i response obtained in Ca2+-free HBS vs. parameters of intracellular Ca2+ store size (TG-evoked [Ca2+]i and FEC), rate of Ca2+ entry and and peak [Ca2+]i in TG-treated cells, and the degree of TG-sensitive Mn2+ quenching at 30 s (obtained as described in legends to Figs 2–4).
Figure 5. Correlations between PAF-evoked [Ca2+]i response and intracellular Ca2+ store size (A) and intracellular Ca2+ store size and parameters of SOCE (B and C).
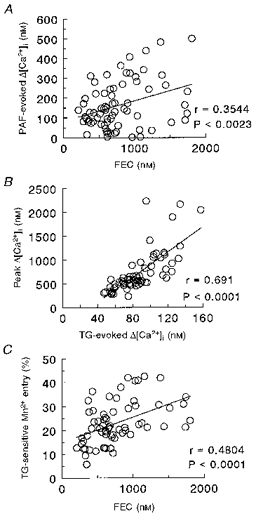
A, correlation of PAF-evoked [Ca2+]i responses (in Ca2+-free HBS) and FEC; parameters were obtained as described in legends to Figs 1 and 2. B, correlation of TG-evoked [Ca2+]i and peak [Ca2+]i levels achieved after 10 min TG treatment and readdition of CaCl2. TG-evoked [Ca2+]i was obtained as described in legend to Fig. 2, and peak [Ca2+]i values were obtained as in legend to Fig. 3. C, intracellular Ca2+ store size (FEC) and TG-sensitive Mn2+ entry. FEC was determined as in the legend to Fig. 2, and Mn2+ entry was obtained from TG-sensitive decreases in fluorescence at 30 s after addition of MnCl2 (cf. Fig. 4). Correlations were obtained with data from the four treatment conditions for 17-19 LCLs (for A, n= 72; for B, n= 71; for C, n= 67).
Table 3.
Correlation analysis of measurements of intracellular Ca2+ store size and parameters of SOCE
| Parameter | r | n | P | |
|---|---|---|---|---|
| TG-evoked [Ca2+]i | Rate of Ca2+ entry | 0.4433 | 73 | 0.0001 |
| Peak [Ca2+]i | 0.6905 | 71 | 0.0001 | |
| Mn2+ quenching | 0.2969 | 69 | 0.0132 | |
| FEC | Rate of Ca2+ entry | 0.3551 | 71 | 0.0024 |
| Peak [Ca2+]i | 0.4134 | 72 | 0.0003 | |
| Mn2+ quenching | 0.4804 | 67 | 0.0001 |
DISCUSSION
This report characterizes several novel aspects of glucocorticoid action on cellular Ca2+ homeostasis in human B lymphoblasts. Forty-eight hour treatment of cells with cortisol decreased basal [Ca2+]i and increased the PAF-evoked [Ca2+]i transient. Cortisol increased the size of intracellular Ca2+ stores and increased the rate of TG-sensitive Ca2+ and Mn2+ entry, indicating enhanced SOCE. These results identify links among the size of the Ca2+ stores, Ca2+ mobilization from these stores, and the pathways that refill the Ca2+ stores. As the stimulatory effects of cortisol on Ca2+1 signalling occur in conjunction with increases in intracellular Ca2+ stores and SOCE, the results indicate a major regulatory role of glucocorticoids on lymphoblast Ca2+ homeostasis.
Cortisol-induced changes in Ca2+ homeostasis could result via modulation of transcriptional events, or through a non-genomic mode of action initiated at the plasma membrane. In the former, more traditional, mechanism of steroid action, the triggering of genomic events is mediated by cortisol binding to a glucocorticoid receptor (GR) and subsequent modulation of nuclear transcription through the glucocorticoid-GR complex (Boumpas et al. 1993). Alternatively, a non-genomic mechanism of action is characterized as a rapid (< 2 min) onset, membrane-mediated response that occurs independently of DNA transcription or protein synthesis inhibition (for review, see Wehling, 1997). Non-genomic responses usually involve ion transport systems and are presumed to occur through a membrane-resident receptor. For example, cortisol both acutely and chronically affects cAMP concentrations and 45Ca2+ accumulation in cichlid fish pituitary cells (Borski et al. 1991), and acutely hyperpolarizes guinea-pig neurons, an effect inhibited by GR antagonists (Hua & Chen, 1989). However, we failed to observe rapid effects of cortisol on Ca2+1 signalling. Furthermore, the long (12 h) latency for enhancement of the Ca2+1 signal is more consistent with a transcriptional/translational mechanism of action. As LCLs contain GRs (Tomita et al. 1985; Sinclair et al. 1994), we performed experiments with RU 486, a competitive antagonist of the cytosolic GR (Agarwal et al. 1987). The results of these experiments showed complete inhibition of cortisol-induced changes in all measured parameters of Ca2+ homeostasis and Mn2+ influx, suggesting activation of endogenous GR is critical for the modulatory effects of cortisol. Taken together, these results do not support a non-genomic action of cortisol and instead suggest the primary effect of cortisol occurs through the classically described, genomic mechanism of glucocorticoid action.
The present observations showing glucocorticoid-stimulated decreases in basal [Ca2+]i and enhanced Ca2+ signalling in a model of activated lymphocytes are, with one exception, quite different from those reported in the literature for lymphocytes or lymphoid cells. That is, in response to increased cortisol, activated T cells and resting B cells exhibit reduced Ca2+1 signalling (Dennis et al. 1987; Baus et al. 1996) whereas thymocytes demonstrate an increase in basal [Ca2+]i (McConkey et al. 1989). S49 lymphoma cells exhibit increased basal [Ca2+]i and decreased intracellular Ca2+ stores (TG-evoked [Ca2+]i changes in Ca2+-free medium: Lam et al. 1993; Bian et al. 1997) in response to dexamethasone (18-24 h) treatment. The discrepancies between glucocorticoid effects in these cells and immortalized lymphoblasts suggest that Ca2+1 signalling pathways downstream of glucocorticoid-GR activation differ between resting and activated lymphocytes, subsets of lymphocytes, and lymphoma cells. These differences are likely to be key factors responsible for some of the well-described effects of glucocorticoids on lymphocyte function, i.e. inhibition of cell proliferation (Bowen & Fauci, 1984) and induction of apoptosis (McConkey et al. 1989; Lam et al. 1993; Bian et al. 1997). Interestingly, one recent report showed dexamethasone enhanced receptor-mediated [Ca2+]i transients and other parameters of Ca2+ signalling in U937 and erythroleukaemia cells (Willmott et al. 1997). Although these investigators were unable to associate alterations in Ca2+ homeostasis with an increase in a glucocorticoid-regulated protein (lipocortin 1), their work lends support to the finding that glucocorticoids enhance Ca2+-signalling pathways through modulation of intracellular Ca2+ stores. As this phenomenon is observed in monocytic, erythroid and lymphocytic cell lines, and occurs independently of viral transformation, glucocorticoid modulation of Ca2+ homeostasis may reflect differentiation-specific responses of haematopoietic cells to elevated levels of cortisol.
In cortisol-treated LCLs, the increase in the PAF-evoked [Ca2+]i response was observed in Ca2+-free medium and was strongly correlated to the FEC. The TG-evoked [Ca2+]i response and FEC, two markers of the size of intracellular Ca2+ stores, were similarly enhanced by cortisol treatment. As PAF-evoked responses in Ca2+-free HBS are approximately 70 % of the response in Ca2+-containing medium (Brzustowicz et al. 1997), it is apparent that not only the PAF-induced [Ca2+]i signal is highly dependent on the size of the Ca2+ stores, but cortisol alters the size of these Ca2+ stores. Enhanced Ca2+ sequestration capabilities of these compartments could also explain the decrease in basal [Ca2+]i. The simplest explanation of the cortisol-induced enhancement of Ca2+1 signalling would postulate an upregulation of SERCA Ca2+ pumps, SER Ca2+ binding proteins (e.g. calreticulin), or modifiers of these systems, assuming glucocorticoid-GR signalling events exist for enhancing the expression or activity of these proteins. However, assigning the involvement of a particular Ca2+ system as a target of glucocorticoid action, in the absence of specific experiments, is highly speculative. For instance, overexpression of a plasma membrane Ca2+ pump in endothelial cells downregulated SERCA Ca2+ pump and IP3-channel expression, and enhanced store-dependent Ca2+ entry (Liu et al. 1996). This example illustrates the highly interdependent nature of Ca2+ signalling on multiple Ca2+ transport and buffering systems. We have ruled out the potential involvement of the Na+-Ca2+ exchanger as a modifier of enhanced Ca2+ responses, as LCLs fail to exhibit measurable Na+-Ca2+ exchange activity under conditions optimized to detect its activity (Balasubramanyam et al. 1996; Gardner & Balasubramanyam, 1996).
PAF-evoked [Ca2+]i transients demonstrated cell line-specific responses with regard to the extent of stimulation by cortisol (Fig. 1B and C). The negative relation describing the percentage increase in the PAF response in cortisol- and vehicle-treated cells could be the result of several factors, including (but not limited to) a decreased ability of cortisol to effect GR activation, variable upregulation of GR in transformed LCLs, and/or a finite capacity of receptor-mediated Ca2+ signalling to evoke Ca2+ mobilization. With regard to the first possibility, direct measurements of GR levels and their extent of activation in the presence of cortisol in individual LCLs may address this issue. In addition, several cultured cell types retain the metabolic pathways that convert active glucocorticoid to inactive metabolite (and vice versa: Nath et al. 1993; Reeves, 1995; Rajan et al. 1996; Brem et al. 1998). We have observed that LCLs treated with carbenoxolone (an inhibitor of 11β-hydroxysteroid dehydrogenase; Edwards et al. 1996) exhibit enhanced PAF responses, suggesting LCLs may express the cortisol ‘inactivating’ form of this isoenzyme (authors’ unpublished observations). The second possibility, i.e. that certain LCLs are more responsive to cortisol because of increased GR levels, is supported by reports that EBV-transformed LCLs exhibit increased levels of GR protein compared with EBV-positive cells (e.g. Burkitt's lymphoma cells; Sinclair et al. 1994) or human mononuclear leucocytes (Tomita et al. 1985). This is in keeping with idea that LCLs are representative of cytokine-activated B cells, which also express enhanced glucocorticoid receptors (Falus et al. 1995). However, it is difficult to reconcile glucocorticoid responsiveness with the ability of an LCL to respond to an agonist such as PAF. Instead, it is more likely that LCLs exhibit a maximal capacity for receptor-evoked Ca2+1 transients (with or without cortisol stimulation). This conclusion is supported by our study with a larger panel of CEPH LCLs (Brzustowicz et al. 1997), in which the maximal PAF-evoked [Ca2+]i response in HBS was ∼400 nM. The limits of receptor-mediated Ca2+ mobilization could be bound by the size of the intracellular Ca2+ stores, although the influence of IP3 metabolic pathways would appear to be of major importance.
The PAF-evoked [Ca2+]i transient and its dependence on the intracellular Ca2+ store-SOCE axis is supported by several converging lines of evidence: the significant correlation of the PAF response with intracellular Ca2+ stores, the correlation between intracellular Ca2+ store size and SOCE, and the positive, modulatory events of cortisol on upregulation of the PAF response and the SOCE pathway and capacity of intracellular Ca2+ stores. Recently, Ca2+ entry via a store-operated channel (ICRAC) was reported to be tightly linked to the filling status of intracellular Ca2+ stores (Hofer et al. 1998). Although our data do not address the time course of the activation of Ca2+ entry with Ca2+ depletion from intracellular stores, the results obtained with this limited number of LCLs suggest that over a range of intracellular Ca2+ store sizes, the magnitude of the Ca2+ entry process is coupled to the size of the Ca2+ store.
We observed that cortisol enhances Ca2+1 signalling in a subset of minimally responsive cells. Physiological consequences of increased circulating glucocorticoid may thus serve to augment certain functions attributed to activated B cells, e.g. immunoglobulin secretion. This idea is derived from observations in B lymphocytes showing glucocorticoid-induced potentiation of IL-1 and IL-6 secretion (Emilie et al. 1988) and of IL-4-induced IgE formation (Nusslein et al. 1992). In this regard, immunoglobulin formation is highly dependent on endoplasmic reticulum (ER) synthesis, sorting and trafficking; these functions in turn are partially dependent on ER Ca2+ levels (Shachar et al. 1994). Furthermore, low-secreting variants of rat basal leukaemia cells have defective antigen-mediated [Ca2+]i signals (Bingham et al. 1994), and LCLs with enhanced IgM and IgG secretion in response to PAF also have increased PAF-evoked [Ca2+]i responses (Rosskopf et al. 1995). Based on this information and the findings in this report, we suggest intracellular Ca2+ stores and Ca2+1-signalling pathways could serve as a major physiological target for glucocorticoid action.
The major finding in this report is that glucocorticoids significantly affect parameters of Ca2+ homeostasis in B lymphoblasts. In addition to previously discussed Ca2+ transport or IP3 metabolic pathways, several other candidate systems could be invoked to explain the mechanisms through which glucocorticoid produce its effects. Besides describing the modulatory effects of cortisol, the present study characterizes parameters of Ca2+ homeostasis in LCLs representative of the general population. The possibility exists that lymphoblasts may be used to examine the intrinsic regulation of numerous Ca2+ parameters, including G-protein-coupled receptors, SOCE and intracellular Ca2+ stores. These studies may be accomplished through the comparison of known or postulated modulators of Ca2+ metabolism in LCLs exhibiting markedly different responses. Alternatively, they may be performed through phenotypic expressions of Ca2+1 signalling, which, when coupled with non-parametric methods of linkage analysis (Brzustowicz et al. 1997), can be used to identify genetic components that mediate or regulate a specific Ca2+ parameter.
Acknowledgments
We thank Dr Abraham Aviv for helpful discussions. L. Z. was supported by an American Heart Association (NJ Affiliate) fellowship. This work was supported by NIH R29 HL-44196.
References
- Agarwal MK, Hainque B, Moustaid N, Lazer G. Glucocorticoid antagonists. FEBS Letters. 1987;217:221–226. doi: 10.1016/0014-5793(87)80667-1. 10.1016/0014-5793(87)80667-1. [DOI] [PubMed] [Google Scholar]
- Ambrus JL, Jr, Chesky L, McFarland P, Young KR, Jr, Mostowski H, August A, Chused TM. Induction of proliferation by high molecular weight B cell growth factor or low molecular weight B cell growth factor is associated with increases in intracellular calcium in different subpopulations of human B lymphocytes. Cellular Immunology. 1991;134:314–324. doi: 10.1016/0008-8749(91)90305-u. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam M, Condrescu M, Reeves JP, Gardner JP. Sodium/calcium exchange activities in cultured lymphocyte and monocyte cell lines. Biophysical Journal. 1996;70(A205) [Google Scholar]
- Baus E, Andris F, Dubois PM, Urbain J, Leo O. Dexamethasone inhibits the early steps of antigen receptor signaling in activated T lymphocytes. Journal of Immunology. 1996;156:4555–4561. [PubMed] [Google Scholar]
- Bian X, Hughes FM, Jr, Huang Y, Cidlowski JA, Putney JW., Jr Roles of cytoplasmic Ca2+ and intracellular Ca2+ stores in induction and suppression of apoptosis in S49 cells. American Journal of Physiology. 1997;272:C1241–1249. doi: 10.1152/ajpcell.1997.272.4.C1241. [DOI] [PubMed] [Google Scholar]
- Bingham BR, Monk PN, Helm BA. Defective protein phosphorylation and Ca2+ mobilization in a low secreting variant of the rat basophilic leukemia cell line. Journal of Biological Chemistry. 1994;269:19300–19306. [PubMed] [Google Scholar]
- Borghesi LA, Smithson G, Kincade PW. Stromal cell modulation of negative regulatory signals that influence apoptosis and proliferation of B lineage lymphocytes. Journal of Immunology. 1997;159:4171–4179. [PubMed] [Google Scholar]
- Borski RJ, Helms LMH, Richman NH, III, Grau EG. Cortisol rapidly reduced prolactin release and cAMP and 45Ca2+ accumulation in the cichlid fish pituitary in vitro. Proceedings of the National Academy of Sciences of the USA. 1991;88:2758–2762. doi: 10.1073/pnas.88.7.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumpas DT, Chrousos GP, Wilder RL, Cupps TR, Balow JE. Glucocorticoid therapy of immune-mediated diseases: basic and clinical correlates. Annals of Internal Medicine. 1993;119:1198–1208. doi: 10.7326/0003-4819-119-12-199312150-00007. [DOI] [PubMed] [Google Scholar]
- Bowen DL, Fauci AS. Selective suppressive effects of glucocorticoids on the early events in the human B cell activation process. Journal of Immunology. 1984;133:1885–1890. [PubMed] [Google Scholar]
- Brem AS, Bina RB, King TC, Morris DJ. Localization of 2 11β-OH steroid dehydrogenase isoforms in aortic endothelial cells. Hypertension. 1998;31:459–462. doi: 10.1161/01.hyp.31.1.459. [DOI] [PubMed] [Google Scholar]
- Brzustowicz LM, Gardner JP, Hopp L, Jeanclos E, Ott J, Yang XY, Fekete Z, Aviv A. Linkage analysis using platelet-activating factor Ca2+ response in transformed lymphoblasts. Hypertension. 1997;29:158–164. doi: 10.1161/01.hyp.29.1.158. [DOI] [PubMed] [Google Scholar]
- Clark E, Lane P. Regulation of human B-cell activation and adhesion. Annual Review of Immunology. 1991;9:97–127. doi: 10.1146/annurev.iy.09.040191.000525. [DOI] [PubMed] [Google Scholar]
- Clevers HC, Verstegen JM, Logtenberg TF, Gmelig-Meyling H, Ballieux RE. Synergistic action of A23187 and phorbol ester on human B cell activation. Journal of Immunology. 1985;135:3827–3830. [PubMed] [Google Scholar]
- Cupps TR, Fauci AS. Corticosteroid-mediated immunoregulation in man. Immunological Reviews. 1982;65:133–155. doi: 10.1111/j.1600-065x.1982.tb00431.x. [DOI] [PubMed] [Google Scholar]
- Dang A, Balasubramanyam M, Garcia Z, Raveche E, Gardner JP. Altered signal transduction in B-1 malignant clones. Immunology and Cell Biology. 1995;73:511–520. doi: 10.1038/icb.1995.81. [DOI] [PubMed] [Google Scholar]
- Dennis G, June CH, Mizuguchi J, Ohara J, Witherspoon K, Finkelman FD, McMillan V, Mond JJ. Glucocorticoids suppress calcium mobilization and phospholipid hydrolysis in anti-Ig antibody-stimulated B cells. Journal of Immunology. 1987;139:2516–2523. [PubMed] [Google Scholar]
- Diegel ML, Rankin BM, Bolen JB, Dubois PM, Kiener PA. Cross-linking of Fc gamma receptor to surface immunoglobulin on B cells provides an inhibitory signal that closes the plasma membrane calcium channel. Journal of Biological Chemistry. 1994;269:11409–11416. [PubMed] [Google Scholar]
- Di Virgilio F, Fasolato C, Steinberg TH. Inhibitors of membrane transport system for organic anions block fura-2 excretion from PC12 and N2A cells. Biochemical Journal. 1988;256:959–963. doi: 10.1042/bj2560959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- Edwards CR, Benediktsson R, Lindsay RS, Seckl JR. 11 beta-Hydroxysteroid dehydrogenases: key enzymes in determining tissue-specific glucocorticoid effects. Steroids. 1996;61:263–269. doi: 10.1016/0039-128x(96)00033-5. 10.1016/0039-128X(96)00033-5. [DOI] [PubMed] [Google Scholar]
- Emilie D, Crevon MC, Auffredou MT, Galanaud P. Glucocorticosteroid-dependent synergy between interleukin 1 and interleukin 6 for human B lymphocyte differentiation. European Journal of Immunology. 1988;18:2043–2047. doi: 10.1002/eji.1830181226. [DOI] [PubMed] [Google Scholar]
- Falus A, Biro J, Rakasz E. Cytokine networks and corticosteroid receptors. Annals of the New York Academy of Sciences. 1995;762:71–77. doi: 10.1111/j.1749-6632.1995.tb32315.x. [DOI] [PubMed] [Google Scholar]
- Gagne D, Pons M, Philibert D. RU 38486: a potent antiglucocorticoid in vitro and in vivo. Journal of Steroid Biochemistry. 1985;23:247–251. doi: 10.1016/0022-4731(85)90401-7. 10.1016/0022-4731(85)90401-7. [DOI] [PubMed] [Google Scholar]
- Gardner JP, Balasubramanyam M. Na/Ca exchange in circulating blood cells. Annals of the New York Academy of Sciences. 1996;779:502–514. doi: 10.1111/j.1749-6632.1996.tb44824.x. [DOI] [PubMed] [Google Scholar]
- Garvy BA, King LE, Telford WG, Morford LA, Fraker PJ. Chronic elevation of plasma corticosterone causes reductions in the number of cycling cells of the B lineage in murine bone marrow and induces apoptosis. Immunology. 1993;80:587–592. [PMC free article] [PubMed] [Google Scholar]
- Gomes da Costa A, Madeira VM. Magnesium and manganese ions modulate Ca2+ uptake and its energetic coupling in sarcoplasmic reticulum. Archives of Biochemistry and Biophysics. 1986;249:199–206. doi: 10.1016/0003-9861(86)90575-8. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guyre PM, Girard MT, Morganelli PM, Manganiello PD. Glucocorticoid effects on the production and actions of immune cytokines. Steroid Biochemistry. 1988;30:89–93. doi: 10.1016/0022-4731(88)90080-5. 10.1016/0022-4731(88)90080-5. [DOI] [PubMed] [Google Scholar]
- Hallam TJ, Rink TJ. Agonists stimulate divalent cation channels in the plasma membrane of human platelets. FEBS Letters. 1985;186:175–179. doi: 10.1016/0014-5793(85)80703-1. 10.1016/0014-5793(85)80703-1. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Fasolato C, Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: A study using simultaneous measurements of ICRAC and intraluminal [Ca2+] Journal of Cell Biology. 1998;140:325–334. doi: 10.1083/jcb.140.2.325. 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua SY, Chen YZ. Membrane receptor-mediated electrophysiological effects of glucocorticoid on mammalian neurons. Endocrinology. 1989;124:687–691. doi: 10.1210/endo-124-2-687. [DOI] [PubMed] [Google Scholar]
- Huang R, Cioffi J, Kimberly R, Edberg J, Mayer L. B cell differentiation factor-induced human B cell maturation: stimulation of intracellular calcium release. Cellular Immunology. 1995;164:227–233. doi: 10.1006/cimm.1995.1165. 10.1006/cimm.1995.1165. [DOI] [PubMed] [Google Scholar]
- Johnson P, Milliken GA. A simple procedure for testing linear hypotheses about the parameters of a nonlinear model using weighted least squares. Communications in Statistics - Simulation and Computation. 1983;12:135–145. [Google Scholar]
- Kojima H, Newton-Nash D, Weiss HJ, Zhao J, Sims PJ, Wiedmer T. Production and characterization of transformed B-lymphocytes expressing the membrane defect of Scott syndrome. Journal of Clinical Investigation. 1994;94:2237–2244. doi: 10.1172/JCI117586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla A, Putcha G, Poulos E, Shearer WT. Tyrosine phosphorylation of phospholipase C concomitant with its activation by platelet-activating factor in a human B cell line. Journal of Immunology. 1993;151:637–648. [PubMed] [Google Scholar]
- Lam M, Dubyack G, Distelhorst CW. Effect of glucocorticosteroid treatment on intracellular calcium homeostasis in mouse lymphoma cells. Molecular Endocrinology. 1993;7:686–693. doi: 10.1210/mend.7.5.8316252. 10.1210/me.7.5.686. [DOI] [PubMed] [Google Scholar]
- Liu BF, Xu X, Fridman R, Muallem S, Kuo TH. Consequences of functional expression of the plasma membrane Ca2+ pump isoform 1a. Journal of Biological Chemistry. 1996;271:5536–5544. doi: 10.1074/jbc.271.10.5536. 10.1074/jbc.271.10.5536. [DOI] [PubMed] [Google Scholar]
- Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. Journal of Biological Chemistry. 1991;266:17067–17071. [PubMed] [Google Scholar]
- McConkey DJ, Nicotera P, Hartzell P, Belloma G, Wyllie AH, Orrenius S. Glucocorticoids activate a suicide process in thymocytes through an elevation of cytosolic Ca2+ concentration. Archives of Biochemistry and Biophysics. 1989;269:365–370. doi: 10.1016/0003-9861(89)90119-7. [DOI] [PubMed] [Google Scholar]
- McMillan VM, Dennis GJ, Glimcher LH, Finkelman FD, Mond JJ. Corticosteroid induction of Ig+Ia− B cells in vitro is mediated via interaction with the glucocorticoid cytoplasmic receptor. Journal of Immunology. 1988;140:2549–2555. [PubMed] [Google Scholar]
- Mazer B, Domenico J, Sawami H, Gelfand EW. Platelet-activating factor induces an increase in intracellular calcium and expression of regulatory genes in human B lymphoblastoid cells. Journal of Immunology. 1991;146:1914–1920. [PubMed] [Google Scholar]
- Mazer BD, Domenico J, Szepesi A, Lucas JJ, Gelfand EW. Role for Ca2+ in expression of cell cycle regulated genes in PAF-stimulated cells. Journal of Lipid Mediators and Cell Signalling. 1994;10:269–281. [PubMed] [Google Scholar]
- Missiaen L, Declerck I, Droogmans G, Plessers L, De Smedt H, Raeymaekers L, Casteels R. Agonist-dependent Ca2+ and Mn2+ entry dependent on state of filling of Ca2+ stores in aortic smooth muscle cells of the rat. The Journal of Physiology. 1990;427:171–186. doi: 10.1113/jphysiol.1990.sp018166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath N, Lakshmi V, Rosenthal JC. Presence of 11 beta-hydroxysteroid dehydrogenase enzyme in the human prostate tumor cell line LNCaP. Prostate. 1993;23:225–233. doi: 10.1002/pros.2990230305. [DOI] [PubMed] [Google Scholar]
- Negulescu PA, Shastri N, Cahalan MD. Intracellular calcium dependence of gene expression in single T lymphocytes. Proceedings of the National Academy of Sciences of the USA. 1994;91:2873–2877. doi: 10.1073/pnas.91.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguer CM, Pellegrini O, Galanaud P, Benveniste J, Thomas Y, Richard Y. Regulation of paf-acether receptor expression in human B cells. Journal of Immunology. 1992;149:2742–2748. [PubMed] [Google Scholar]
- Nguer CM, Treton D, Rola-Pleszczynski M, Mishal Z, Thomas Y, Galanaud P, Richard Y. Regulation of platelet-activating factor receptor expression in human B cells and B cell lines. Lipids. 1996;31:1051–1058. doi: 10.1007/BF02522462. [DOI] [PubMed] [Google Scholar]
- Nusslein HG, Trag T, Winter M, Dietz A, Kalden JR. The role of T cells and the effect of hydrocortisone on interleukin-4-induced IgE synthesis by non-T cells. Clinical and Experimental Immunology. 1992;90:286–292. doi: 10.1111/j.1365-2249.1992.tb07944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. 10.1016/0143-4160(90)90016-N. [DOI] [PubMed] [Google Scholar]
- Putney JW, Jr, Bird GStJ. The inositol phosphate-calcium signalling system in non-excitable cells. Endocrine Research. 1993;14:610–631. doi: 10.1210/edrv-14-5-610. 10.1210/er.14.5.610. [DOI] [PubMed] [Google Scholar]
- Rajan V, Edwards CR, Seckl JR. 11 beta-Hydroxysteroid dehydrogenase in cultured hippocampal cells reactivates inert 11-dehydrocorticosterone, potentiating neurotoxicity. Journal of Neuroscience. 1996;16:65–70. doi: 10.1523/JNEUROSCI.16-01-00065.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves WB. NAD-dependent 11 beta-hydroxysteroid dehydrogenase in cultured human colonic epithelial cells. American Journal of Physiology. 1995;268:C1467–1473. doi: 10.1152/ajpcell.1995.268.6.C1467. [DOI] [PubMed] [Google Scholar]
- Rosskopf D, Hartung K, Hense J, Siffert W. Enhanced immunoglobulin formation of immortalized B cells from hypertensive patients. Hypertension. 1995;26:432–435. doi: 10.1161/01.hyp.26.3.432. [DOI] [PubMed] [Google Scholar]
- Schulam PG, Putcha G, Franklin-Johnson J, Shearer WT. Evidence for a platelet-activating factor receptor on human lymphoblastoid B cells: activation of the phosphatidylinositol cycle and induction of calcium mobilization. Biochemical and Biophysical Research Communications. 1990;166:1047–1052. doi: 10.1016/0006-291x(90)90916-b. [DOI] [PubMed] [Google Scholar]
- Shachar I, Rabinovich E, Kerem A, Bar-Nun S. Thiol-reducing agents and calcium perturbants alter intracellular sorting of immunoglobulin M. Journal of Biological Chemistry. 1994;269:27344–27350. [PubMed] [Google Scholar]
- Siffert W, Rosskopf D, Moritz A, Wieland T, Kaldenberg-Stasch S, Kettler N, Hartung K, Beckmann S, Jakobs KH. Enhanced G protein activation in immortalized lymphoblasts from patients with essential hypertension. Journal of Clinical Investigation. 1995;96:759–766. doi: 10.1172/JCI118120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair AJ, Jacquemin MG, Brooks L, Shanahan F, Brimmel M, Rowe M, Farrell PJ. Reduced signal transduction through glucocorticoid receptor in Burkitt's lymphoma cell lines. Virology. 1994;199:339–353. doi: 10.1006/viro.1994.1132. 10.1006/viro.1994.1132. [DOI] [PubMed] [Google Scholar]
- Smith CS, Parker L, Shearer WT. Cytokine regulation by platelet-activating factor in a human B cell line. Journal of Immunology. 1994;153:3997–4005. [PubMed] [Google Scholar]
- Smith CS, Shearer WT. Activation of NF-kappa B and immunoglobulin expression in response to platelet-activating factor in a human B cell line. Cellular Immunology. 1994;155:292–303. doi: 10.1006/cimm.1994.1123. 10.1006/cimm.1994.1123. [DOI] [PubMed] [Google Scholar]
- Snyder F. Platelet-activating factor and related acetylated lipids as potent biologically active cellular mediators. American Journal of Physiology. 1990;259:C697–708. doi: 10.1152/ajpcell.1990.259.5.C697. [DOI] [PubMed] [Google Scholar]
- Taira S, Matsui M, Hayakawa K, Yokoyama T, Nariuchi H. Interleukin secretion by B cell lines and splenic B cells stimulated with calcium ionophore and phorbol ester. Journal of Immunology. 1987;139:2957–2964. [PubMed] [Google Scholar]
- Tomita M, Chrousos GP, Brandon DD, Ben-Or S, Foster CM, De Vougn L, Taylor S, Loriaux DL, Lipsett MB. Glucocorticoid receptors in Epstein-Barr virus-transformed human lymphocytes. Hormone and Metabolic Research. 1985;17:674–678. doi: 10.1055/s-2007-1013641. [DOI] [PubMed] [Google Scholar]
- Wehling M. Specific, nongenomic actions of steroid hormones. Annual Review of Physiology. 1997;59:365–393. doi: 10.1146/annurev.physiol.59.1.365. 10.1146/annurev.physiol.59.1.365. [DOI] [PubMed] [Google Scholar]
- Wilckens T, De Rijk R. Glucocorticoids and immune function: unknown dimensions and new frontiers. Immunology Today. 1997;18:418–424. doi: 10.1016/s0167-5699(97)01111-0. 10.1016/S0167-5699(97)01111-0. [DOI] [PubMed] [Google Scholar]
- Willmott NJ, Choudhury Q, Flower R. Effects of dexamethasone and phorbol ester on P2 receptor-coupled Ca2+ signalling and lipocortin 1 presentation in U937 cells. British Journal of Pharmacology. 1997;122:1055–1060. doi: 10.1038/sj.bjp.0701490. [DOI] [PMC free article] [PubMed] [Google Scholar]