

Abstract
The effects of different calcium channel blockers (ω-agatoxin IVA (ω-Aga IVA), ω-conotoxin GVIA (ω-CgTx GVIA) and dihydropyridines) were tested on spontaneous and evoked transmitter release at embryonic and newborn rat neuromuscular junctions (NMJs).
The nerve-evoked transmitter release quantal content (m) was strongly reduced by the P/Q-type voltage-dependent calcium channel (VDCC) blocker ω-Aga IVA (100 nM) at newly formed endplates of embryos and 0- to 11-day-old rats, in agreement with the effect of this blocker on transmitter release at mature and reinnervating muscles.
ω-CgTx GVIA (1–5 μm), the N-type VDCC blocker, also caused a significant reduction in m at newly formed NMJs early in development (embryos and 0- to 4-day-old rats), while it was ineffective in more mature animals (5- to 11-day-old rats).
L-type channel blockers, nitrendipine (1 μm) and nifedipine (1 μm), did not significantly affect neurally evoked release at developing NMJs. However, nifedipine (10 μm) was able to increase m significantly at 0- to 4-day-old rat NMJs.
At developing NMJs, K+-evoked transmitter release was dependent on Ca2+ entry through VDCCs of the P/Q-type family (100 nmω-Aga IVA reduced 70% of the K+-evoked miniature endplate potential frequency). N- and L-type VDCC blockers did not affect this type of release.
We conclude that at rat neuromuscular junctions the presynaptic calcium channel types involved in transmitter release undergo developmental changes during the early postnatal period.
The influx of Ca2+ through voltage-dependent calcium channels (VDCCs) at nerve terminals is the link between action potential and transmitter release (Katz & Miledi, 1970; Augustine et al. 1987). At mature mammalian neuromuscular junctions, neurotransmitter release is mediated by VDCCs of the P/Q-type family based on the strong inhibitory effect of funnel-web spider toxin (FTX), ω-agatoxin IVA (ω-Aga IVA) and ω-conotoxin MVIIC (ω-CTX MVIIC) on nerve-evoked transmission and on calcium presynaptic currents (Uchitel et al. 1992; Protti & Uchitel, 1993; Bowersox et al. 1995; Katz et al. 1997). In contrast, several studies have shown the lack of effects of dihydropyridines (L-type calcium channel blockers) (Penner & Dreyer, 1986; Katz et al. 1996; but see Atchison, 1989) and of the N-type calcium channel blocker ω-conotoxin GVIA (ω-CgTx GVIA) (Sano et al. 1987; Protti et al. 1991; but see Rossoni et al. 1994).
This pharmacological evidence has been reinforced using immunocytochemical techniques with labelled antibodies against different types of VDCC α1-subunits, revealing the nerve terminal localization of the P/Q-type calcium channels in human and rat neuromuscular junctions. Furthermore, VDCC α1-subunits from N- and R-type channels have not been identified at nerve terminals but have been found at the neighbouring structures of the neuromuscular junction (Day et al. 1997).
Many physiological and pharmacological changes have been described during the course of neuromuscular junction maturation (Redfern, 1970; Bennett & Pettigrew, 1974; Okamoto et al. 1992). Muscle fibres are multiply innervated, and the endplate potentials have distinct peaks and a lower mean quantal content of transmitter release compared with that found in adult muscles (Dennis et al. 1981). Changes in the pharmacology of the VDCCs coupled to neurotransmitter release were also reported in avian (Gray et al. 1992) and amphibian neuromuscular junctions (Fu & Huang, 1994), in cultured rat hippocampal neurons (Scholz & Miller, 1995) and in rat superior olivary complex preparations (Iwasaki & Takahashi, 1998). However, limited information exists about the identity of the VDCCs that mediate synaptic transmission at neonatal mammalian neuromuscular junctions (Sugiura & Ko, 1997).
The principal aim of this work was to evaluate changes in the subtype/s of VDCCs coupled to the release process during neuromuscular junction maturation.
We found that ω-Aga IVA, a P/Q-type VDCC blocker, produced a strong inhibition of the nerve- and K+-evoked neurotransmitter release during all the studied stages of development. On the other hand, the nerve-evoked transmitter release was inhibited by ω-CgTx GVIA only at early stages of neuromuscular junction development, and this effect became negligible during maturation. Therefore we conclude that VDCCs involved in synaptic transmission at rat NMJs undergo developmental changes during the early postnatal period.
METHODS
Experiments were carried out on the phrenic-diaphragm preparation of embryonic (17-21 days) and newborn (0-11 days) Wistar rats. At all stages the animals were cared for in accordance with national guidelines for the humane treatment of laboratory animals, which are as protective as those of the National Institutes of Health, USA. The date of fertilization was determined by the appearance of sperm plugs on the cage floors in the morning and this was designated as day 0 in our reckoning of embryonic age. After the desired length of gestation, the mother was anaesthetized with 2 % tribromoethanol (0.15 ml (10 g body wt)−1, i.p.) and two to three embryos were removed to a bath of oxygenated saline solution and immediately dissected (see below). The newborn rats were anaesthetized as indicated above and immediately exsanguinated. The mothers were killed with an overdose of anaesthetic.
The muscle with its nerve supply was excised and dissected on a Sylgard-coated Petri dish containing a physiological saline solution (dissecting solution) of the following composition (mM): NaCl, 137; KCl, 5; CaCl2, 2; MgSO4, 1; NaHCO3, 12; Na2HPO4, 1 and glucose, 11; continuously bubbled with 95 % O2-5 % CO2. The preparation was then transferred to the recording chamber to which the different working solutions and drugs were applied. Experiments were performed at room temperature (19-24°C).
Evoked endplate potentials (EPPs) and miniature endplate potentials (MEPPs) were recorded intracellularly with conventional glass microelectrodes filled with 3 M KCl (20-30 MΩ resistance). In order to evaluate the transmitter release in low Ca2+-high Mg2+ solution the muscles were incubated in a saline solution, which differed from the dissecting solution in CaCl2 (0.7-1 mM) and MgSO4 (5-8 mM) concentrations. Control and toxin-treated fibres were assayed in the presence of 0.01 % bovine serum albumin (BSA) in the recording solution.
The mean quantal content (m) was estimated by the failure method:
where N is the total number of successive trials (100 at 0.5 Hz) and n0 is the number of trials in which the response fails (absence of EPP). Nerve action potentials were recorded using a microelectrode inserted, under visual control, into the perineural sheath of the nerve.
The nerves were stimulated with a supramaximal stimulus by using a suction electrode coupled to a Grass S88 stimulator with an associated stimulus isolation unit. The recording electrodes were connected to an I-E 201 amplifier (Warner Instrument Corp.). A distant Ag-AgCl electrode connected to the bath solution via an agar bridge (agar 3.5 % in 137 mM NaCl) was used as a reference. The signals were digitized (DigiData 1200 interface; Axon Instruments, Inc.), stored and computer analysed (Axoscope 1.0, Axon Instruments, Inc.).
K+-evoked release was evaluated after incubating the muscles for 30 min with high K+ saline solution of the following composition (mM): NaCl, 122; KCl, 20; CaCl2, 2; MgSO4, 1; NaHCO3, 12; Na2HPO4, 1 and glucose, 11; continuously bubbled with 95 % O2-5 % CO2. MEPPs were recorded and stored on tape for further analysis. The mean amplitude per fibre was calculated and corrected assuming a membrane potential of -70 mV. Their frequency of appearance was counted over 1 min periods.
The animals were separated into two experimental groups (i.e. embryos and 0- to 4-day-old rats vs. 5- to 11-day-old rats) based on the differential effect of ω-CgTx GVIA on nerve-evoked transmitter release during the neuromuscular junction maturation (see Fig. 4A). Values are expressed as means ±s.e.m. The statistical significance (P values in figure legends) was evaluated by the two-tailed Welch's t test (for unpaired values and not assuming equal variances).
Figure 4. Correlation between the effect of different VDCC blockers and the maturation of the neuromuscular junctions.
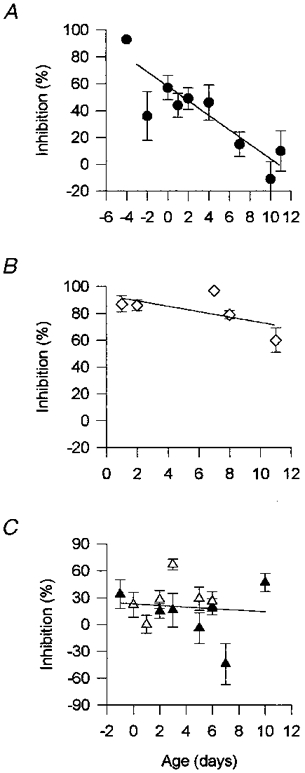
The plots show the correlation between the percentage of inhibition (of quantal content) and the time of development (age of the animals) in ω-CgTx GVIA (1 μm; A); ω-Aga IVA (100 nM; B) and 1 μm nitrendipine (▴) and 1 μm nifedipine (▵;C) treated preparations. Each value represents the mean ±s.e.m. of the data pooled from a nerve-muscle preparation (at least 12 endplates per muscle). The lines represent the best linear fit of the data of ω-CgTx GVIA (r2= 0.8, P < 0.01), ω-Aga IVA (r2= 0.37, P= 0.27), nitrendipine (r2= 0.01, P= 0.79) and nifedipine (r2= 0.01, P= 0.74).
Chemicals
BSA, dimethyl sulphoxide (DMSO) and all other salts and reagents used were of analytical grade and purchased from Sigma. Tribromoethanol was purchased from Aldrich.
Dihydropyridines (nifedipine and nitrendipine) were purchased from RBI. These drugs were dissolved in DMSO, stored at 4°C and protected from light. In both control and treated fibres, the final total DMSO concentration did not exceed 0.3 % in the bath medium. All the experiments with these drugs were carried out avoiding a direct illumination of the preparations.
The synthetic polypeptide ω-conotoxin GVIA (ω-CgTx GVIA) was purchased from Alomone Laboratories (Jerusalem, Israel). The synthetic polypeptide ω-agatoxin IVA (ω-Aga IVA) was a generous gift of Dr N. Saccomano (Pfizer Inc., Groton, CT, USA).
RESULTS
Nerve-evoked endplate potentials (EPPs) were recorded at the rat neuromuscular junctions (NMJs) during the first two weeks after birth. EPPs are relatively slower in their rise and decay rates and they appear after a longer latency period compared with the ones recorded in mature muscles, as reported by Dennis et al. (1981). At early stages, EPPs showed a high variance in their amplitude, which diminished at later stages indicating an increase in the quantal content of transmitter release during maturation (data not shown).
Effect of Ca2+ channel blockers on nerve-evoked neurotransmitter release in neonatal rats
Studies in embryonic and 0- to 4-day-old rats
In order to investigate the pharmacological profile of the VDCCs coupled to the release process during the maturation of NMJs, we studied the effects of specific P/Q-, L- and N-type calcium channel blockers on the estimated mean quantal content (m) of transmitter release.
As shown in Fig. 1A, ω-CgTx GVIA reduced transmitter release (percentage of blockade ∼50 %) at a newly formed endplate (2-day-old rat). EPPs were recorded from the same NMJ before (Control) and after treatment with the toxin (1 μmω-CgTx GVIA). The toxin increased the number of failures of transmitter release and decreased EPP amplitude without affecting the compound nerve action potential (Fig. 1B).
Figure 1. Effect of ω-conotoxin GVIA (ω-CgTx GVIA) on synaptic transmission at a 2-day-old rat neuromuscular junction.

A, representative recordings from the same neuromuscular junction of 100 successive stimuli delivered at 0.5 Hz recorded before (Control) and after treatment with 1 μmω-CgTx GVIA. Note that the toxin reduced the amplitude of EPPs and significantly inhibited the quantal content (increasing the number of failures) to 50 % of the control value. Stimulation artifacts were reduced for clarity. B, extracellular recordings of compound nerve action potential before (Control) and after application of the toxin. This blocker showed no effect on the conduction of the nerve impulse. Each trace represents an average of 30 successive traces. C, temporal course plot of the effect of ω-CgTx GVIA on quantal content. The inhibition began to be apparent 10-20 min after bath application of this blocker (indicated with the bar) reaching a plateau within 35 min. This effect lasted after washout of the preparation with toxin-free saline solution (data not shown).
Figure 1C shows a time course of the effect of ω-CgTx GVIA on quantal content. Typically, 10-20 min after the application of the toxin, the quantal content value began to decrease, reaching a plateau within 35-40 min. This effect was persistent after washout of the preparation with toxin-free saline solution (data not shown). A similar blockade was obtained when we evaluated the quantal content of 46 NMJs (5 muscle preparations) after the application of 1-5 μm of the toxin (Fig. 3A).
Figure 3. Effect of calcium channel blockers on nerve-evoked release at neuromuscular junctions of neonatal rats.

The bar diagrams show the effect of the drugs on quantal content (m) expressed as a percentage of the control value. A, effect of calcium channel blockers in embryonic and 0- to 4-day-old rats. The toxins ω-Aga IVA (□) and ω-CgTx GVIA (▪) significantly reduced the evoked response while the dihydropyridines nifedipine ( ) and nitrendipine (
) and nitrendipine ( ) lacked any effect. Nifedipine (10 μm), however, significantly increased quantal content. In control fibres, m= 0.85± 0.08 (mean ±s.e.m., n= 64 endplates from 16 muscles). B, effect of calcium channel blockers in 5- to 11-day-old rats. The toxin ω-Aga IVA (□) maintained its strong effect on the evoked response shown at early stages while ω-CgTx GVIA (▪) and the dihydropyridines nifedipine () and nitrendipine () lacked any effect. In control fibres, m= 1.27± 0.16 (mean ±s.e.m., n= 43 endplates from 13 muscles). Control fibres were assayed in a low Ca2+/high Mg2+ (0.7-1 mM/5-8 mM) saline solution. Treated fibres were assayed in the same muscles after a 1 h incubation with the respective drug. Each column represents the mean ±s.e.m. of data pooled from 2-5 nerve-muscle preparations (at least 12 endplates per muscle). m was calculated by the failure method. Stimulation frequency, 0.5 Hz. **P < 0.0001, *P < 0.05 compared with the values obtained in the same muscles before addition of the calcium channel blockers.
) lacked any effect. Nifedipine (10 μm), however, significantly increased quantal content. In control fibres, m= 0.85± 0.08 (mean ±s.e.m., n= 64 endplates from 16 muscles). B, effect of calcium channel blockers in 5- to 11-day-old rats. The toxin ω-Aga IVA (□) maintained its strong effect on the evoked response shown at early stages while ω-CgTx GVIA (▪) and the dihydropyridines nifedipine () and nitrendipine () lacked any effect. In control fibres, m= 1.27± 0.16 (mean ±s.e.m., n= 43 endplates from 13 muscles). Control fibres were assayed in a low Ca2+/high Mg2+ (0.7-1 mM/5-8 mM) saline solution. Treated fibres were assayed in the same muscles after a 1 h incubation with the respective drug. Each column represents the mean ±s.e.m. of data pooled from 2-5 nerve-muscle preparations (at least 12 endplates per muscle). m was calculated by the failure method. Stimulation frequency, 0.5 Hz. **P < 0.0001, *P < 0.05 compared with the values obtained in the same muscles before addition of the calcium channel blockers.
The action of the other VDCC blockers, at this stage of development, is summarized in the bar diagram of Fig. 3A. The P/Q-type VDCC blocker ω-Aga IVA (100 nM) strongly reduced (percentage of blockade > 80 %) transmitter release. This effect is in agreement with the effect of this blocker on neurotransmitter release at mature mammalian neuromuscular junctions (Protti & Uchitel, 1993; Katz et al. 1997) and at newly formed endplates on reinnervating muscles (Katz et al. 1996).
On the other hand, low concentrations (1 μm) of the L-type channel blockers nitrendipine and nifedipine did not significantly affect evoked release at developing endplates. However, high concentrations of nifedipine (10 μm) produced a significant increase in quantal content (P < 0.05). Control experiments using the vehicle alone (0.2 % DMSO) did not produce any effect (data not shown).
Studies in 5- to 11-day-old rats
In contrast to the effect of ω-CgTx GVIA on transmitter release at 0- to 4-day-old rat NMJs, the application of this toxin had no effect at this later stage of development. Figure 2A shows representative recordings of EPPs made from the same muscle fibre (11-day-old rat) before (Control) and after treatment with this toxin (ω-CgTx GVIA, 1 μm). As shown in Fig. 2B, ω-CgTx GVIA had no effect on quantal content within the 70 min studied.
Figure 2. Effect of ω-conotoxin GVIA (ω-CgTx GVIA) on synaptic transmission at an 11-day-old rat neuromuscular junction.

A, representative recordings from the same neuromuscular junction of 100 successive stimuli delivered at 0.5 Hz recorded before (Control) and after treatment with 1 μmω-CgTx GVIA. The toxin reduced neither the amplitude of EPPs nor the quantal content of the synaptic transmission. Stimulation artifacts were reduced for clarity. B, temporal course plot of the effect of ω-CgTx GVIA on quantal content. At this age of development, the N-type VDCC blocker showed no effect on quantal content within 70 min after its bath application (indicated with the bar).
Figure 3B shows the effect of VDCC blockers studied at population level. The P/Q-type VDCC blocker ω-Aga IVA (100 nM) produced, as at previous stages, a strong blockade of neurotransmitter release. However, the blockade of synaptic transmission exerted by ω-CgTx GVIA (1 μm) at early stages of development disappeared at this age. L-type channel blockers nitrendipine (1 μm) and nifedipine (1-10 μm) were also ineffective in altering nerve-evoked transmitter release at these ages.
Age-dependent effect of VDCC blockers
Temporal course plots of the effects exerted by the different VDCC blockers during the studied stages are shown in Fig. 4.
Consistent with the disappearance of sensitivity to the N-type VDCC blocker during development, the degree of blockade of neurotransmitter release observed with 1 μmω-CgTx GVIA was inversely correlated with the age of the animals (Fig. 4A). A weak and non-significant correlation (r2= 0.37, P= 0.27), however, was found between the effect of 100 nM ω-Aga IVA on synaptic transmission and the time of development. The blockade caused by this toxin did not change during neuromuscular junction maturation, showing an elevated percentage of inhibition of neurotransmitter release (∼80-90 %) during the studied period of development (Fig. 4B). The effects of the L-type VDCC blockers, nitrendipine (1 μm) and nifedipine (1 μm), also did not show any correlation (r2= 0.01) with the time of the endplate maturation (Fig. 4C).
Effect of Ca2+ channel blockers on K+-evoked neurotransmitter release in neonatal rats
Application of high K+ concentrations to induce transmitter release is also a convenient way to study the identity of the VDCCs present at synaptic terminals without using a nerve-stimulation protocol. The K+-evoked depolarization increased MEPP frequency which is dependent on Ca2+ entry from the extracellular space (del Castillo & Katz, 1954).
As already reported by Dennis et al. (1981), spontaneous MEPPs at immature NMJs appear at very low frequency and their amplitudes vary over a wide range (0.5-5 mV) compared with mature endplates.
In 0- to 4-day-old rat muscle preparations, 100 nM ω-Aga IVA was able to reduce strongly the K+-evoked frequency of MEPPs (Fig. 5Aa) without affecting their amplitudes (Fig. 5Ab). Nifedipine (10 μm) and ω-CgTx GVIA (1 μm), however, were ineffective at inhibiting either MEPP frequency or MEPP amplitude (Fig. 5Aa and Ab, respectively) evoked by this type of stimulation.
Figure 5. Effect of calcium channel blockers on K+-evoked release at neuromuscular junctions of neonatal rats.

A, 0- to 4-day-old rat NMJs. a, effect of the calcium channel blockers on MEPP frequency (min−1). ω-Aga IVA (100 nM; ▪) strongly inhibited the frequency of MEPPs but not the other blockers, ω-CgTx GVIA (1 μm; ) and nifedipine (10 μm; ). b, effect of the calcium channel blockers on MEPP amplitudes. None of the blockers showed any significant effect on MEPP amplitude. B, 5- to 11-day-old rat NMJs. a, at this stage of development a similar effect of these blockers was observed on MEPP frequency (min−1). ω-Aga IVA (100 nM; ▪), but not ω-CgTx GVIA (1 μm; ) or nifedipine (10 μm; ), significantly inhibited MEPP frequency. b, calcium channel blocker action on MEPP amplitudes. Similar to 0- to 4-day-old rat NMJs, none of the blockers showed any effect on MEPP amplitude. In A and B, control fibres (□) were assayed in high K+ (20 mM) saline solution and treated fibres were assayed in the same muscles after 1 h incubation with the respective drug. Mean amplitude per fibre was calculated and corrected assuming a membrane potential of -70 mV. Each column represents the mean ±s.e.m. of data pooled from 2 nerve- muscle preparations (at least 12 endplates per muscle). *P < 0.001 compared with its control value.
Similarly, K+-evoked transmitter release showed the same pharmacological profile at 5- to 11-day-old rat NMJs. Thus MEPP frequency was inhibited only by ω-Aga IVA (100 nM) without affecting the amplitude of the MEPPs (Fig. 5B a and Bb). On the other hand, nifedipine (10 μm) and ω-CgTx GVIA (1 μm) showed no effect on MEPP frequency or MEPP amplitude (Fig. 5B a and Bb).
DISCUSSION
In this work we investigated the identity of calcium channel/s involved in nerve- and K+-evoked transmitter release at developing NMJs. We have found that ω-CgTx GVIA, the N-type VDCC blocker, significantly and irreversibly inhibited nerve-evoked release at newly formed endplates in the early stages of development (embryos and 0- to 4-day-old rats) without affecting the compound nerve action potential. However, this toxin was ineffective in 5- to 11-day-old rats. In neuromuscular junctions, during the first postnatal weeks, nerve terminals withdraw from multiply innervated muscle fibres. This process occurs concomitantly with the reduction in sensitivity to the N-type calcium channel blocker. Thus, this loss of sensitivity to ω-CgTx GVIA might be associated with the elimination of aberrant multiple synapses at developing muscles. A similar change in the pharmacological profile of VDCCs was found at the brainstem auditory synapse of neonatal rats where the contribution of ω-CgTx GVIA-sensitive channels to synaptic transmission diminishes during postnatal development (Iwasaki & Takahashi, 1998).
At newly formed NMJs of embryos and 0- to 11-day-old rats, the polypeptide toxin ω-Aga IVA was capable of inhibiting nerve-evoked transmitter release indicating that, during all stages of development, the P/Q-type calcium channels mediate synaptic transmission. The sensitivity of transmitter release to the effect of this toxin is in the same range (100 nM caused 90 % blockade) as that found in normal and reinnervated rodent muscles (Katz et al. 1996, 1997). These results also agree with those of Sugiura & Ko (1997) who blocked the transmitter release at newborn rat NMJs using the toxin ω-CTX MVIIC, which is able to block both P/Q- and N-type calcium channels (Olivera et al. 1994).
Our pharmacological studies showed that a low concentration (1 μm) of the dihydropyridines (DHPs) nitrendipine and nifedipine did not significantly affect the neurally evoked release at any stage of rat neuromuscular junction development. However, 10 μm nifedipine increased quantal content at early stages of development.
Sugiura & Ko (1997) have reported an increase in EPP amplitude after the application of DHPs (1-10 μm) in neonatal neuromuscular junctions. They postulated that DHPs decrease the release of a neuromodulator, which exerts a tonic inhibition on transmitter release. We tested the effect of 10 μm nifedipine using their experimental conditions ([Ca2+]o= 10 mM) and also observed a significant increase of EPP amplitude to ∼300 %, in agreement with their results. Therefore, the small increase (∼40 %) in m produced by 10 μm nifedipine and the lack of effect of 1 μm of DHPs in our experimental conditions ([Ca2+]o= 0.7- 1 mM), might result from an effect of this drug on a diminished L-type-dependent release of the neuromodulator. Nevertheless, a careful interpretation of the effect of these blockers must be done because there is evidence that DHPs can affect other types of ionic channels (Yatani & Brown, 1985; Kamath et al. 1995).
An overlapping of the inhibitory effect of ω-Aga IVA and ω-CgTx GVIA at early stages of neonatal development was observed. This effect cannot simply be accounted for by an overlapping action of these blockers on calcium currents because there is a non-linear relationship between [Ca2+]o and transmitter release (Dodge & Rahamimoff, 1967; Takahashi & Momiyama, 1993). Also, at these new synapses, Ca2+ entering through more than one type of channel might be necessary to trigger secretion at each single release site (Mintz et al. 1995). Our results suggest that both P/Q- and N-type calcium channels are involved in controlling nerve-evoked transmitter release at newly formed endplates during the early stages of development. Nevertheless, we cannot rule out the possibility that an embryonic VDCC with non-standard properties might exist (i.e. sensitive to both ω-Aga IVA and ω-CgTx GVIA).
We evaluated the effect of these VDCC blockers on K+-evoked transmitter release and observed that none of them inhibited MEPP amplitude, indicating that the effect of these blockers on synaptic transmission could not be accounted for by a postsynaptic effect. ω-Aga IVA (100 nM) is capable of inhibiting MEPP frequency evoked by high extracellular potassium, as has been shown in reinnervated muscle (Katz et al. 1996) and in adult rodent neuromuscular junctions (Protti & Uchitel, 1993; Losavio & Muchnik, 1997). However, in spite of its effect on evoked release, ω-CgTx GVIA (1 μm) was ineffective on K+-evoked transmitter release. This pharmacological difference between both protocols of stimulation may indicate that the VDCCs involved in K+-induced release are different from those operating during nerve-evoked depolarization (Momiyama & Takahashi, 1994), or that the tonic calcium influx, induced by [K+]o= 20 mM, could be masking the participation of other VDCCs in this type of release.
In conclusion, we have demonstrated that there is a change in the pharmacological profile of the VDCCs that mediate nerve-evoked neurotransmitter release at neonatal rat neuromuscular junctions. VDCCs of the N-type are involved in synaptic transmission early in development while P/Q-type VDCCs play a major role in this process at all stages of development. The participation of the N-type channel in the synapse elimination process at newly formed NMJs remains to be investigated.
Acknowledgments
We wish to thank Dr Tomoyuki Takahashi, Dr F. Urbano and Dr E. Katz for reading the manuscript and for useful discussions. We would also like to thank Dr N. Saccomano of Pfizer, Inc. for generously providing the toxin ω-Aga IVA. This work was supported by the Muscular Dystrophy Association, UBA (ME 064) and CONICET (PICT 0310).
References
- Augustine GJ, Charlton MP, Smith SJ. Calcium action in synaptic transmitter release. Annual Review of Neuroscience. 1987;10:633–693. doi: 10.1146/annurev.ne.10.030187.003221. 10.1146/annurev.ne.10.030187.003221. [DOI] [PubMed] [Google Scholar]
- Atchison WD. Dihydropiridine-sensitive and -insensitive components of acetylcholine release from rat motor nerve terminals. Journal of Pharmacology and Experimental Therapeutics. 1989;251:672–678. [PubMed] [Google Scholar]
- Bennett MR, Pettigrew AG. The formation of synapses in striated muscle during development. The Journal of Physiology. 1974;241:515–545. doi: 10.1113/jphysiol.1974.sp010670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowersox SS, Miljanich GP, Sugiura Y, Li C, Nadasdi L, Hoffman BB, Ramachandran J, Ko C. Differential blockade of voltage-sensitive calcium channels at the mouse neuromuscular junction by novel ω-conopeptides and ω-agatoxin-IVA. Journal of Pharmacology and Experimental Therapeutics. 1995;273:248–256. [PubMed] [Google Scholar]
- Day NC, Wood SJ, Ince PG, Volsen SG, Smith W, Slater CR, Shaw PJ. Differential localisation of voltage-dependent calcium channel α1 subunits at the human and rat neuromuscular junction. Journal of Neuroscience. 1997;17:6226–6235. doi: 10.1523/JNEUROSCI.17-16-06226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B. Changes in end-plate activity produced by presynaptic polarization. The Journal of Physiology. 1954;124:586–604. doi: 10.1113/jphysiol.1954.sp005131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MJ, Ziskind-Conhaim L, Harris AJ. Development of neuromuscular junctions in rat embryos. Developmental Biology. 1981;81:266–279. doi: 10.1016/0012-1606(81)90290-6. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Rahamimoff R. Co-operative action of calcium ions in transmitter release at the neuromuscular junction. The Journal of Physiology. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu WM, Huang FL. L-type Ca2+ channel is involved in the regulation of spontaneous transmitter release at developing neuromuscular synapses. Neuroscience. 1994;58:131–140. doi: 10.1016/0306-4522(94)90160-0. 10.1016/0306-4522(94)90160-0. [DOI] [PubMed] [Google Scholar]
- Gray DB, Brusés JL, Pilar GR. Developmental switch in the pharmacology of Ca2+ channels coupled to acetylcholine release. Neuron. 1992;8:1–20. doi: 10.1016/0896-6273(92)90092-r. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. The Journal of Physiology. 1998;509:419–423. doi: 10.1111/j.1469-7793.1998.419bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath A, Larson K, Shibata EF, Hoshi T. Mechanisms of dihydropyridine block of Shaker potassium channels. Society for Neuroscience Abstracts. 1995;21 720.8. [Google Scholar]
- Katz B, Miledi R. Further study of the role of calcium in synaptic transmission. The Journal of Physiology. 1970;207:789–801. doi: 10.1113/jphysiol.1970.sp009095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Ferro PA, Weisz G, Uchitel OD. Calcium channels involved in synaptic transmission at the mature and regenerating mouse neuromuscular junction. The Journal of Physiology. 1996;497:687–697. doi: 10.1113/jphysiol.1996.sp021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Protti DA, Ferro PA, Rosato Siri MD, Uchitel OD. Effects of Ca2+ channel blocker neurotoxins on transmitter release and presynaptic currents at the mouse neuromuscular junction. British Journal of Pharmacology. 1997;121:1531–1540. doi: 10.1038/sj.bjp.0701290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losavio A, Muchnik S. Spontaneous acetylcholine release in mammalian neuromuscular junctions. American Journal of Physiology. 1997;273:1835–1841. doi: 10.1152/ajpcell.1997.273.6.C1835. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Momiyama A, Takahashi T. Calcium channels responsible for potassium-induced transmitter release at rat cerebellar synapses. The Journal of Physiology. 1994;476:197–202. doi: 10.1113/jphysiol.1994.sp020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto M, Walewski JL, Artusio JF, Riker WF. Neuromuscular pharmacology in rat neonates: development of responsiveness to prototypic blocking and reversal drugs. Anesthesia and Analgesia. 1992;75:361–371. doi: 10.1213/00000539-199209000-00008. [DOI] [PubMed] [Google Scholar]
- Olivera BM, Miljanich GP, Ramachandran J, Adams ME. Calcium channel diversity and neurotransmitter release: The ω-conotoxins and ω-agatoxins. Annual Review of Biochemistry. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- Penner R, Dreyer F. Two different presynaptic calcium currents in mouse motor nerve terminals. Pflügers Archiv. 1986;406:190–197. doi: 10.1007/BF00586682. [DOI] [PubMed] [Google Scholar]
- Protti DA, Szczupak L, Scornik FS, Uchitel OD. Effect of ω-conotoxin GVIA on neurotransmitter release at the mouse neuromuscular junction. Brain Research. 1991;557:336–339. doi: 10.1016/0006-8993(91)90156-p. [DOI] [PubMed] [Google Scholar]
- Protti DA, Uchitel OD. Transmitter release and presynaptic Ca2+ currents blocked by the spider toxin ω-Aga-IVA. NeuroReport. 1993;5:333–336. doi: 10.1097/00001756-199312000-00039. [DOI] [PubMed] [Google Scholar]
- Redfern PA. Neuromuscular transmission in new-born rats. The Journal of Physiology. 1970;209:701–709. doi: 10.1113/jphysiol.1970.sp009187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossoni G, Berti F, La Maestra L, Clementi F. ω-Conotoxin GVIA binds to and blocks rat neuromuscular junction. Neuroscience Letters. 1994;176:185–188. doi: 10.1016/0304-3940(94)90078-7. [DOI] [PubMed] [Google Scholar]
- Sano K, Enomoto K, Maeno T. Effects of synthetic ω-conotoxin, a new type Ca2+ channel antagonist, on frog and mouse neuromuscular transmission. European Journal of Pharmacology. 1987;141:235–241. doi: 10.1016/0014-2999(87)90268-8. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Developmental changes in presynaptic calcium channels coupled to glutamate release in cultured rat hippocampal neurons. Journal of Neuroscience. 1995;15:4612–4617. doi: 10.1523/JNEUROSCI.15-06-04612.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, Ko C. Novel modulatory effect of L-type calcium channels at newly formed neuromuscular junctions. Journal of Neuroscience. 1997;17:1101–1111. doi: 10.1523/JNEUROSCI.17-03-01101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- Uchitel OD, Protti DA, Sánchez V, Cherksey BD, Sugimori M, Llinás R. P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proceedings of the National Academy of Sciences of the USA. 1992;89:3330–3333. doi: 10.1073/pnas.89.8.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatani A, Brown AM. The calcium channel blocker nitrendipine blocks sodium channels in neonatal rat cardiac myocytes. Circulation Research. 1985;57:868–875. doi: 10.1161/01.res.56.6.868. [DOI] [PubMed] [Google Scholar]