

Abstract
We have reported previously that in mouse pancreatic β-cells H2O2 hyperpolarizes the membrane and increases the ATP-sensitive K+ current recorded in the perforated patch configuration of the patch-clamp technique. The present study was undertaken to elucidate the underlying mechanisms.
The intracellular ATP concentration measured by chemoluminescence was reduced by H2O2. The ADP concentration increased in parallel during the first 10 min, resulting in a pronounced decrease in the ATP/ADP ratio.
Consistent with these results, glucose-stimulated insulin secretion from isolated islets was inhibited by H2O2.
Membrane hyperpolarization measured with intracellular microelectrodes in intact islets and inhibition of insulin secretion were counteracted by tolbutamide, indicating that the channels are still responsive to inhibitors and that the ATP concentration is not too low to trigger exocytosis. However, the sensitivity of the β-cells to tolbutamide was reduced after treatment with H2O2.
H2O2 increased the intracellular Ca2+ activity ([Ca2+]i) in a biphasic manner. A first transient rise in [Ca2+]i due to mobilization of Ca2+ from intracellular stores was followed by a sustained increase, which was at least partly dependent on Ca2+ influx. The first phase seems to reflect Ca2+ mobilization from mitochondria.
Our results demonstrate that H2O2 interferes with glucose metabolism, which influences the membrane potential and ATP-sensitive K+ current via the intracellular concentration of ATP. These events finally lead to an inhibition of insulin secretion despite an increase in [Ca2+]i.
Several studies have shown that the infiltration of macrophages into the islets of Langerhans during the early stages of insulitis is one of the first steps in the development of insulin-dependent diabetes mellitus (Kolb-Bachofen et al. 1988; Lee et al. 1988; Hanenberg et al. 1989). It is generally believed that macrophages produce large amounts of reactive oxygen species such as hydrogen peroxide (H2O2), resulting in an undesired cytotoxic attack against β-cells. Alloxan and streptocotocin, two agents widely used to induce diabetes in animals, also lead to the formation of H2O2, which may thus contribute to the manifestation of diabetes (Takasu et al. 1991). Pancreatic β-cells are extremely sensitive to oxidative stress because of the low expression and activity of the enzymes defending cells against an assault by oxidants (Malaisse et al. 1982; Lenzen et al. 1996). Moreover, the GSH/GSSG (reduced/oxidized glutathione) ratio in islets is low compared with other tissues (Ammon et al. 1983). It is well known that reactive oxygen species alter β-cell function and finally lead to the destruction of β-cells. They interfere with enzymes involved in glucose metabolism (Welsh et al. 1991; Dimmeler et al. 1993; Nakazaki et al. 1995), inhibit DNA synthesis, and lead to DNA fragmentation, and probably via the activation of the poly(ADP-ribose) polymerase to a decrease of the cellular NAD+ content (Yamamoto et al. 1981; Takasu et al. 1991; Fehsel et al. 1993; Radons et al. 1994). We were interested in investigating whether H2O2 interferes with an early step in stimulus-secretion coupling, i.e. at the level of the plasma membrane. In a previous paper we showed that H2O2 hyperpolarizes the cell membrane and we assumed that an intracellular factor is involved in this effect (Krippeit-Drews et al. 1994b). In the present paper we have further elucidated the events underlying membrane hyperpolarization and we have studied how [Ca2+]i and insulin secretion are influenced by H2O2.
METHODS
Cell and islet preparation
The experiments were performed on islets or single cells of fed female NMRI mice (25-30 g), killed by cervical dislocation. For cell membrane potential measurements a piece of pancreas was fixed in a perifusion chamber and islets were micro-dissected by hand. The other experiments were performed on islets isolated by collagenase digestion of the pancreas. For patch-clamp experiments and measurements of [Ca2+]i, islet cells were dispersed in Ca2+-free medium and cultured for up to 4 days in RPMI 1640 medium supplemented with 10 % fetal calf serum, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Plant, 1988). Insulin secretion was determined with freshly prepared islets.
Solutions and chemicals
The extracellular fluid for cell membrane potential measurements was composed of (mM): 120 NaCl, 5 KCl, 2.5 CaCl2, 1.2 MgCl2, 24 NaHCO3, 15 glucose, gassed with 95 % O2 and 5 % CO2 to maintain a pH of 7.4 at 37°C.
Whole-cell ATP-sensitive K+ current recordings were performed at 32°C with nystatin in the pipette solution (150-250 μm). The pipette solution also contained (mM): 10 KCl, 10 NaCl, 70 K2SO4, 4 MgCl2, 2 CaCl2, 10 EGTA, 20 Hepes, pH adjusted to 7.15 with KOH. The bath solution was composed of (mM): 140 NaCl, 5 KCl, 1.2 MgCl2, 2.5 CaCl2, 15 glucose, 10 Hepes, pH adjusted to 7.4 with NaOH. The same bath solution was used for the determination of [Ca2+]i at 37°C. In these experiments β-cells were identified by the glucose-evoked rise in [Ca2+]i induced by changing the bath glucose concentration from 0.5 to 15 mM. Insulin secretion and the ATP/ADP ratio were determined at 37°C in a bath solution with the following composition (mM): 122 NaCl, 4.7 KCl, 1.1 MgCl2, 2.5 CaCl2, 10 Hepes, 15 glucose, supplemented with 0.5 % bovine serum albumin. The insulin secretion measurements were started in a bath solution containing 3 mM glucose, and in each experiment the biphasic increase in insulin release following the augmentation of the glucose concentration to 15 mM was registered as a control for the quality of the islets.
Fura-2 AM was obtained from Molecular Probes, FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone), fura-salt, flufenamic acid, ionomycin, gadolinium oxide, thapsigargin and tolbutamide were from Sigma, ATP and ADP were from Boehringer, the BioOrbit 1243-102 ATP-monitoring-kit was from Merlin Diagnostika GmbH (Bornheim-Fersel, Germany) and H2O2 was from Hedinger (Stuttgart, Germany). D600 was kindly provided by Knoll AG (Ludwigshafen, Germany). All other chemicals were purchased from Merck in the purest form available.
Recording methods
Membrane potential measurements
. The potential difference across the cell membrane was determined using high resistance microelectrodes (Meissner & Schmelz, 1974). The β-cells were identified by the characteristic oscillations of cell membrane potential which they display in the presence of 15 mM glucose at 37°C.
Patch-clamp recordings
. Patch pipettes were pulled from borosilicate glass capillaries (Clark Electromedical, Pangbourne, UK). They had resistances between 3 and 5 MΩ when filled with pipette solution. Membrane currents were recorded with an EPC-9 patch-clamp amplifier and ‘Pulse’ software (HEKA, Lambrecht, Germany). For off-line analysis, data were also stored on video tape, played back by means of a MacLab4S interface with ‘Chart’ software (WissTech, Spechbach, Germany) and evaluated with ‘Igor’ software (WaveMetrics, Lake Oswego, Oregon, USA). Patch-clamp recordings were performed at 32°C. Whole-cell ATP-sensitive K+ currents were measured in the perforated patch configuration at a holding potential of -70 mV and during 300 ms pulses to -80 and -60 mV at 15 s intervals. Perforation was monitored by the decrease in series resistance (Gs). Perforation was usually adequate for voltage-clamping (Gs < 30 MΩ) within 10 min of seal formation.
Measurement of insulin release
. After isolation, the islets were placed in batches of 50 in two parallel perifusion chambers (control and test solution). The islets were pre-incubated for 15 min in a medium containing 3 mM glucose at 37°C. The perifusion flow rate was 0.65 ml min−1. Effluent fractions were sampled at 2 min intervals and insulin was measured by a double-antibody radioimmunoassay with rat insulin as the standard (Linco Research, St Louis, MO, USA).
Measurement of the ATP/ADP ratio
. The ATP/ADP ratio was determined by measuring ATP and ADP in the same batch of eight islets. At the end of the incubation period extracellular fluid was sucked off and islets were disintegrated with NaOH-cysteine solution (40 mM-0.5 mM) and stored at -20°C. For luminescence measurements aliquots of each sample were dissolved with a buffer containing (mM): 20 creatine phosphate, 100 glycine, 1 MgSO4 at pH 9.0, with or without creatine kinase (20 pg.ml−1). Aliquots were either neutralized with HCl to pH 7.65 immediately or after an incubation with creatine kinase for 10 min at room temperature (20-25°C) to convert all ADP to ATP. The ATP concentration was measured in a luciferin/luciferase assay using the ATP-monitoring kit with a luminescence biometer 1253 (Bio-orbit, Merlin, Bornheim-Mersel, Germany).
Measurement of [Ca2+]i
. Intracellular Ca2+ activity ([Ca2+]i) was measured by the fura-2 method according to Grynkiewicz et al. (1985). Single cells or small clusters of cells were loaded with fura-2 AM (5 μm) for 30 min. Intracellular fura-2 was excited with light at 340, 360 and 380 nm wavelengths, generated by directing light from a monochromator light source through appropriate filters in a computer-controlled wheel. The excitation light was then directed through the objective lens (NPlanL × 40, Leica, Bensheim, Germany) by means of a glass fibre light guide and a dichroic mirror. The emitted light was filtered (512 nm) and measured by an ICCD camera. The ratio of the emitted light intensity at 340 nm/380 nm excitation wavelength (F340/F380) was used to calculate [Ca2+]i according to an in vitro calibration with fura-2 salt. The microscope type DMIRB was from Leica. All other hardware and the software needed for fluorescence measurements were from IonOptix (Milton, MA, USA). Alternatively, [Ca2+]i was measured with equipment and software from TILL photonics (Planegg, Germany) where the light wavelength is adjusted by means of a diffractive grating. The microscope was a Zeiss Axiovert 100 equipped with a PlanNeofluar × 40 objective lens.
Presentation of results
Electrophysiological experiments and Ca2+ measurements are illustrated by recordings that are representative of the indicated number of experiments performed with different cells. Cells of at least three different cell preparations have been used for each series of experiments. If possible, means ±s.e.m. are given in the text for the indicated number of experiments. For the other experiments data are presented as means ±s.e.m. in the figures. The statistical significance of differences between means was assessed by a Student's one-sample t test or a t test for paired values when two samples were compared. Multiple comparisons were made by ANOVA followed by Student-Newman-Keuls test. P≤ 0.05 was considered as indicating a significant difference.
RESULTS
Effects of H2O2 on membrane potential and ATP-sensitive K+ current
Figure 1A shows that the addition of 1 mM H2O2 hyperpolarized the cell membrane and suppressed the oscillations of the cell membrane potential observed in the presence of 15 mM glucose. The membrane potential measurements were performed with intracellular microelectrodes in intact islets. Previously, we have reported that the hyperpolarization was sustained after removal of H2O2 from the bath solution (Krippeit-Drews et al. 1994b), suggesting that glucose has lost the ability to depolarize the β-cells. Figure 1A demonstrates that the addition of 0.1 mM tolbutamide depolarizes the membrane again and small spikes were observed. However, 1 mM of the sulfonylurea further depolarized the membrane and induced spikes of large amplitude. On average, the fraction of plateau phase (percentage of time with spike activity) was 55 ± 6 % under control conditions in 15 mM glucose (calculated for the last 4 min under control conditions) (n= 4). Treatment of the cells with 1 mM H2O2 hyperpolarized the membrane by -25 ± 5 mV from the plateau potential (n= 4). The addition of 0.1 mM tolbutamide for 5-7 min after wash-out of H2O2 depolarized the membrane by 18 ± 3 mV, and in three out of four experiments small spikes appeared. Increasing the tolbutamide concentration to 1 mM depolarized the cells further by 10 ± 2 mV, and large spikes could be detected in all four experiments. The small oscillations of the cell membrane potential which can be seen in Fig. 1A after addition of H2O2 have been observed in 11 out of 19 experiments in which H2O2 was tested.
Figure 1. H2O2 hyperpolarizes the membrane potential by increasing the ATP-sensitive K+ current.
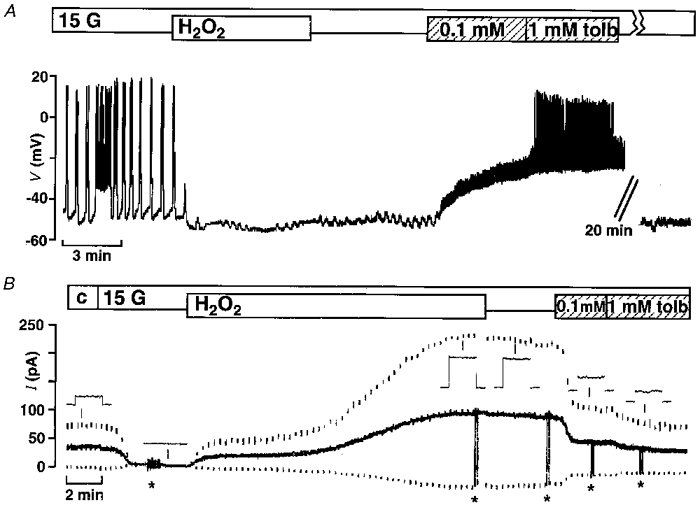
A, effects of 1 mM H2O2 and 0.1 and 1 mM tolbutamide (tolb) on the membrane potential of mouse pancreatic β-cells in the presence of 15 mM glucose (15 G). The H2O2-induced hyperpolarization was irreversible but could be counteracted by inhibiting the ATP-sensitive K+ current with tolbutamide. The record is representative of four experiments with similar results. B, ATP-sensitive K+ current monitored in the perforated-patch mode in the presence of 15 mM glucose (15 G). The holding potential was -70 mV (continuous trace) and every 15 s 300 ms voltage steps to -80 and -60 mV (lower and upper dashed traces, respectively) were applied. H2O2 dramatically increased the current. Again the irreversible effect of H2O2 was at least partly counteracted by tolbutamide. The insets show the corresponding currents on an extended time scale. At the points marked by asterisks the amplifier was switched from voltage-clamp to current-clamp mode and the membrane potential was registered. The record is representative of five experiments with similar results.
In three experiments (not shown) the hyperpolarization induced by H2O2 was compared in the same cell to the hyperpolarization provoked by 100 μm diazoxide, which maximally repolarizes the membrane to close to the K+ reversal potential by opening the ATP-sensitive K+ channels. Addition of diazoxide led to a hyperpolarization of -32 ± 2 mV from the plateau potential while H2O2 evoked a hyperpolarization of -18 ± 3 mV.
To elucidate the mechanism underlying the hyperpolarization, the effect of H2O2 on the ATP-sensitive K+ current was investigated in the perforated patch configuration with dissociated cells. Figure 1B shows a representative recording from this series of experiments, demonstrating that H2O2 markedly increased the current. For quantification, the current elicited by a 10 mV depolarizing voltage step from the holding potential of -70 mV was used. In four out of the five experiments in this series the membrane potential was measured simultaneously (switch from voltage-clamp to current-clamp mode is marked by asterisks in the experiment shown in Fig. 1B). On average, the current under control conditions (c) in 0.5 mM glucose was 15 ± 6 pA and the membrane potential was -80 ± 2 mV. The current was completely abolished in all experiments when the glucose concentration was raised to 15 mM, the membrane potential depolarized and spikes of up to -25 mV were observed. Subsequent addition of 1 mM H2O2 in the presence of 15 mM glucose significantly increased the current to 73 ± 16 pA and hyperpolarized the membrane potential to -80 ± 1 mV. Thus, in contrast to intact islets in single cells, H2O2 is able to hyperpolarize the membrane potential to the K+ reversal potential. After removal of H2O2, the current and the membrane potential did not change significantly (69 ± 17 pA and -80 ± 1 mV, respectively). The addition of 0.1 mM tolbutamide did not abolish the current but reduced it to 25 ± 9 pA. The membrane potential was -79 ± 2 mV under these conditions. At 1 mM, the sulfonylurea induced a significant further decrease in the current, to 19 ± 7 pA, and slightly depolarized the membrane to -76 ± 1 mV, but no spikes occurred.
Effect of H2O2 on the intracellular ATP and ADP concentration
Since the increase in the ATP-sensitive K+ current was only observed when the metabolism of the cells was intact (Krippeit-Drews et al. 1994), we tested whether 1 mM H2O2 influences the ATP/ADP ratio, which is the main intracellular regulator of channel activity (Ashcroft & Rorsman, 1989). Figure 2 shows the effect of 1 mM H2O2 on the ATP/ADP ratio (panel A) and the ATP (panel B) and ADP (panel C) concentration in the islets. H2O2 reduced the ATP concentration in the presence of 15 mM glucose from 10.2 ± 0.4 to 4.8 ± 0.5 pM islet−1 within 30 min, with the main decrease occurring within the first 10 min. Under control conditions, the ATP concentration did not change within 30 min (11.0 ± 1.3 pM islet−1). The ADP concentration first increased after addition of H2O2 from 3.7 ± 0.2 to 5.1 ± 0.4 pM islet−1 within 5 min. Afterwards it decreased again, reaching a value of 3.6 ± 0.3 pM islet−1 after 30 min. The changes in the ATP and ADP concentration resulted in a decrease of the ATP/ADP ratio to about half of the control values. The maximum reduction of the ATP/ADP ratio was reached within 10 min.
Figure 2. H2O2 diminishes intracellular ATP content.
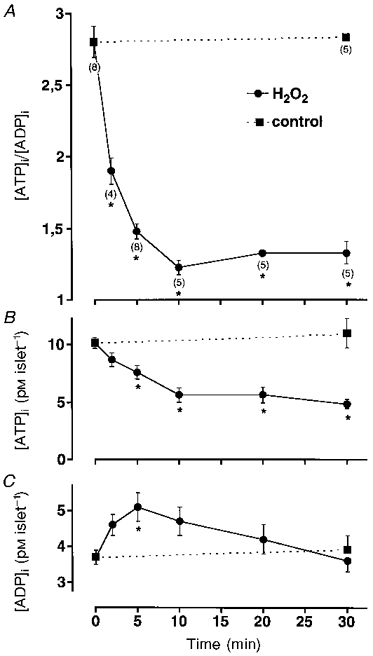
A, mean changes in the ATP/ADP ratio monitored with isolated islets after addition of 1 mM H2O2in the presence of 15 mM glucose. Numbers of experiments (n) given in parentheses below the symbols in A also apply to corresponding symbols in B and C. *Values significantly different from controls. B and C, H2O2 induced changes in intracellular ATP (B) and ADP (C) content. Note that the H2O2 effect is biphasic: within the first 5 min the loss in ATP coincided with an increase in ADP whereas afterwards the concentration of both nucleotides decreased.
Influence of H2O2 on insulin secretion
The first part of Fig. 3 demonstrates the typical biphasic rise in the rate of insulin secretion in response to an increase of the extracellular glucose concentration from 3 to 15 mM. The addition of 1 mM H2O2 to glucose-stimulated islets caused a marked inhibition of insulin release which was not reversible after removal of H2O2 (n= 4). However, tolbutamide (1 mM) was still able to augment insulin secretion (n= 4). After wash-out of tolbutamide, insulin release decreased again, emphasizing that glucose has lost its ability to stimulate hormone secretion from β-cells.
Figure 3. H2O2 decreases the insulin secretion from isolated mouse islets.
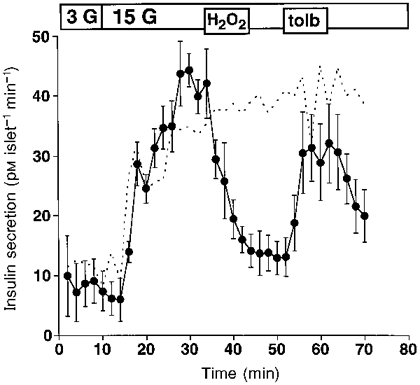
Effects of 1 mM H2O2 and 1 mM tolbutamide (tolb) on insulin release from perifused mouse islets. Switching from 3 to 15 mM glucose (3 G, 15 G) elicited the response of the islets to glucose. H2O2 irreversibly inhibited glucose-stimulated insulin secretion, an effect which was blunted by tolbutamide. Values are means ±s.e.m. of four experiments. Control experiments without H2O2 and tolbutamide are represented by the dashed line.
Effect of H2O2 on [Ca2+]i
Figure 4 shows one example for the biphasic effect of H2O2 on [Ca2+]i. Under control conditions (c) the bath solution contained 0.5 mM glucose. Increasing the glucose concentration to 15 mM resulted first in a small decrease in [Ca2+]i as described previously (Gylfe, 1989). Thereafter, [Ca2+]i markedly increased, and often this rise was followed by small oscillations of [Ca2+]i. On average, [Ca2+]i was 141 ± 22 nM under control conditions and increased to a maximum of 443 ± 22 nM with 15 mM glucose (n= 10). The addition of H2O2 first led to a drop in [Ca2+]i as a result of the closure of L-type Ca2+ channels induced by the hyperpolarization. Thereafter, a transient increase in [Ca2+]i (marked by the arrowhead in Fig. 4) was observed, which was followed by a marked sustained increase. In the experiment shown in Fig. 4, which is representative of five similar experiments, the dependency of the second steep rise on extracellular Ca2+ has been studied. During the rising phase, [Ca2+]i decreased after removal of extracellular Ca2+, but not to basal levels. The effect of extracellular Ca2+ withdrawal diminished with increasing [Ca2+]i and was almost abolished in the steady state. Ca2+ ionophores like ionomycin or A23187 (1 μm) still markedly decreased [Ca2+]i in the absence of extracellular Ca2+. An unspecific leakage of fura-2 out of the cells or an increase in the F340/F380 ratio due to an augmentation of reduced pyridine nucleotides (Pralong et al. 1990; Gilon & Henquin, 1992) can be excluded because H2O2 did not change the fura-2 signal when the excitation wavelength was 360 nm (n= 5). In addition, H2O2 did not induce any change in the fluorescence signals of fura-salt (5 μm) when tested in vitro in the presence of 300 nM free Ca2+ (n= 3, not shown).
Figure 4. Effect of H2O2 on [Ca2+]i.
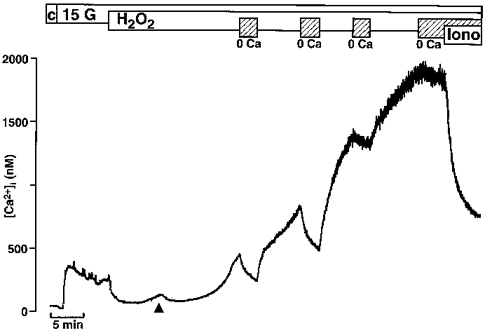
Switching the extracellular glucose concentration from 0.5 mM (c = control) to 15 mM (15 G) led to an increase in [Ca2+]i. The subsequent addition of 1 mM H2O2 led to a marked drop in [Ca2+]i followed by a biphasic increase in [Ca2+]i, a first transient phase (see arrowhead) was followed by a large sustained increase. At the time intervals marked by the hatched bars Ca2+ was removed from the extracellular bath solution and 1 mM EGTA was added. After a steady-state level of [Ca2+]i had been reached, the Ca2+ ionophore ionomycin (1 μm) was applied in the absence of extracellular Ca2+. The experiment shown is representative of five with similar results.
Figure 5 shows that the second increase in [Ca2+]i induced by H2O2 is not blockable by either 100 μm D600 (Fig. 5A, n= 4), an inhibitor of L-type Ca2+ channels, or by 100 μm Gd3+ (Fig. 5B, n= 4) or flufenamate (Fig. 5C, n= 4). Gd3+ and flufenamate are blockers of non-selective cation channels and of the Ca2+ release-activated Ca2+ current (Icrac).
Figure 5. Influence of D600, Gd3+ and flufenamate on H2O2-induced changes in [Ca2+]i.
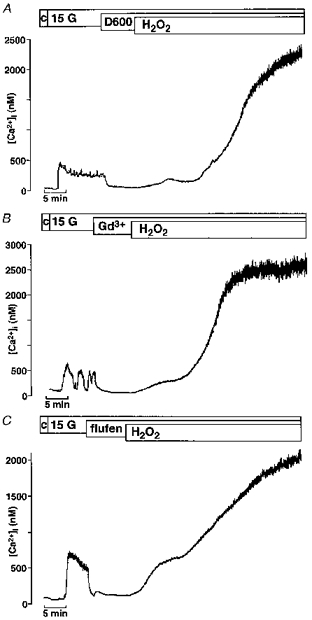
In all experiments [Ca2+]i was first increased by switching from 0.5 mM glucose (c = control) to 15 mM glucose (15 G) in the bath solution. Neither the L-type Ca2+ channel blocker D600 (100 μm; A) nor the inhibitors of non-selective cation channels and the Ca2+ release-activated Ca2+ current Icrac, Gd3+ (100 μm; B) or flufenamate (flufen, 100 μm; C), suppressed the second marked rise in [Ca2+]i induced by 1 mM H2O2. Each experiment presented in this figure is representative of four experiments with similar results.
Figure 6 demonstrates that the first transient rise was also observed in a Ca2+-free bath solution. It was not different from the effect observed in Ca2+-containing extracellular medium. On average, the first increase in [Ca2+]i induced by H2O2, expressed as the area under the curve, was 425 ± 45 nM min in the bath solution with Ca2+ (n= 10) and 475 ± 70 nM min in the Ca2+-free bath solution (n= 9).
Figure 6. Effect of H2O2 on [Ca2+]i in Ca2+-free solution.
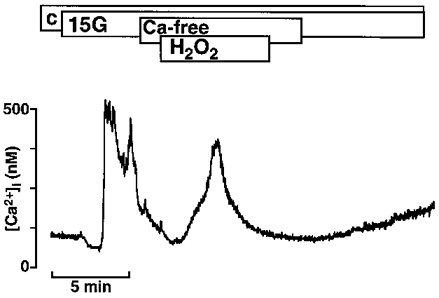
Raising the glucose concentration from 0.5 mM (c = control) to 15 mM (15 G) increased [Ca2+]i. A subsequent switch to Ca2+-free solution (1 mM EGTA) restored [Ca2+]i to control values. Under these conditions the addition of 1 mM H2O2 led to a transient increase in [Ca2+]i, pointing to Ca2+ release from intracellular sites. The experiment shown is representative of nine with similar results.
As shown in Fig. 7A in one representative experiment out of four, pretreatment of the β-cells with 1 μm thapsigargin for 30 min did not suppress the first transient increase in [Ca2+]i induced by H2O2 (right arrowhead). Due to the irreversible inhibition of the Ca2+ pumps by thapsigargin, the initial decrease in [Ca2+]i is abolished (left arrowhead). In contrast, 100 μm of FCCP, a mitochondrial uncoupler which reduces the mitochondrial membrane potential and Ca2+ uptake into the mitochondria driven by the membrane potential, clearly blunted the first phase of the H2O2 effect (Fig. 7B, n= 5). The mean value of the area under the curves after addition of H2O2 in the presence of FCCP was 217 ± 49 nM min compared with 425 ± 45 nM min with H2O2 alone.
Figure 7. Effect of the Ca2+ pump inhibitor thapsigargin and the mitochondria uncoupler FCCP on the first phase increase in [Ca2+]i induced by H2O2.
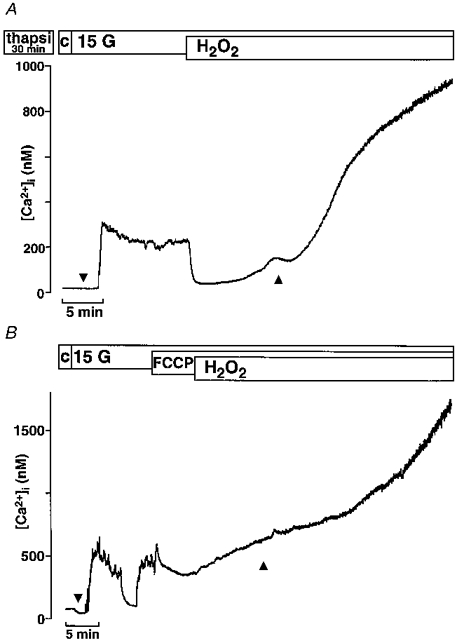
A, the cells were pre-incubated for 30 min with 1 μm thapsigargin. The effectiveness of the treatment is demonstrated by the complete suppression of the first initial decrease in [Ca2+]i after increasing the glucose concentration from 0.5 to 15 mM (compare traces at ▾ in A and B). Ca2+ store depletion by thapsigargin did not prevent the first rise in [Ca2+]i induced by H2O2 (▴). The experiment shown is representative of four with similar results. B, addition of 100 μm FCCP in the presence of 15 mM glucose suppressed the first phase of the H2O2-evoked rise in [Ca2+]i (compare traces at ▴ in A and B). The experiment shown is representative of five with similar results.
DISCUSSION
H2O2 plays an important role as a cytotoxic molecule attacking pancreatic β-cells and leading to alteration of function and eventually β-cell destruction. It is released from activated macrophages, infiltrating the islets during early stages of insulitis, and it is also produced by agents which chemically induce diabetes like alloxan and streptozotocin (Takasu et al. 1991). However, relatively little information is available on the mechanism by which H2O2 interferes with insulin secretion in β-cells. In the present paper we have shown that H2O2 inhibits insulin secretion from pancreatic islets by decreasing the ATP/ADP ratio, an effect leading to the opening of ATP-sensitive K+ channels and membrane hyperpolarization. Moreover, we observed that H2O2 biphasically increased the intracellular Ca2+ activity. Thus, H2O2 may induce a deviation from the strong coupling between [Ca2+]i and insulin secretion normally found in β-cells.
We observed an irreversible inhibition of glucose-induced insulin secretion by 1 mM H2O2. An inhibition of glucose-stimulated insulin release by H2O2 at concentrations between 0.1 and 0.5 mM has also been reported for rat (Kim et al. 1994) and human (Jahr et al. 1995) islets. We propose that the rapid inhibition of insulin secretion induced by H2O2 is due to membrane hyperpolarization which is caused by the opening of ATP-sensitive K+ channels. The activation of ATP-sensitive K+ current after addition of H2O2 was only observed in the perforated patch configuration with intact cell metabolism but, as previously reported, not in the standard whole-cell configuration (Krippeit-Drews et al. 1994b). Similar results were obtained by Nakazaki et al. (1995) who investigated the effect of H2O2 on single ATP-sensitive K+ channels. They found an activation of the channels by H2O2 in the cell-attached mode but not in inside-out membrane patches.
Comparison of the action of H2O2 and diazoxide revealed that in intact islets 1 mM H2O2 did not hyperpolarize the membrane potential to the K+ reversal potential and even small oscillations of the membrane potential persisted in some experiments. In contrast, in single cells, H2O2 hyperpolarized the membrane potential to -80 mV and accordingly spike activity was completely suppressed in all experiments. In an intact islet, an average membrane potential is registered from all coupled cells and we assume that, compared with single cells in intact islets, H2O2 does not have unhampered access to all cells. Consistent with this assumption is the observation that the effectiveness of tolbutamide in reversing the hyperpolarizing effect of H2O2 is more pronounced in intact islets, where the membrane potential stayed above the K+ equilibrium potential after additon of H2O2. Microelectrode and patch-clamp experiments clearly show that the effectiveness of tolbutamide is strongly reduced after addition of H2O2 in the presence of 15 mM glucose. In cells not treated with H2O2, 100 μm of the sulfonylurea in the presence of 15 mM glucose is sufficient to induce continuous large spikes (Debuyser et al. 1991).
It seems reasonable to speculate that the intracellular factor mediating the activating effect of H2O2 on the ATP-sensitive K+ current is a reduction of ATP, and we have demonstrated that H2O2 indeed lowered the ATP concentration in pancreatic β-cells. It has been shown for a variety of other cells such as endothelial cells, platelets and P388D1 cells that H2O2 diminishes the intracellular ATP concentration (Holmsen & Robkin, 1977; Spragg et al. 1985; Hyslop et al. 1988). Thus, it is likely that H2O2 also influences ATP-sensitive K+ channels in other tissues. It has been speculated that the relaxation induced in rabbit tracheal smooth muscles by H2O2 is mediated by ATP-sensitive K+ channels (Gupta & Prasad, 1992). Opening of ATP-sensitive K+ channels by H2O2 has been detected in patch-clamp experiments in ventricular myocytes (Goldhaber et al. 1989) and in the renal epithelial cell line LLC-PK1 (Filipovic & Reeves, 1997). In the latter study a concomitant reduction in the cellular ATP concentration has been shown. Several studies have shown that H2O2 interferes with enzymes of the glycolytic pathway (Brodie & Reed, 1987; Hyslop et al. 1988; Chatham et al. 1989; Goldhaber et al. 1989) and of oxidative phosphorylation (Hyslop et al. 1988; Goldhaber et al. 1989; Zhang et al. 1990). Nakazaki et al. (1995) have used various secretagogues to antagonize the ATP-sensitive K+ channel activation induced by H2O2 in rat β-cells. From the relative potencies of 2-ketoisocaproic acid, glyceraldehyde and glucose in counteracting the H2O2 effect, they concluded that the enzymes in mitochondria are less sensitive to H2O2 than the glycolytic enzymes.
Recently, Herson & Ashford (1997) have shown that H2O2 depolarizes the membrane of the rat insulin-secreting cell line CRI-G1. They suggest that this depolarization is due to the activation of a non-selective cation channel. Although we did not observe the activation of an inward current by H2O2 in our preparation, we cannot entirely rule out that it exists in normal pancreatic β-cells. However, according to our experiments with normal mouse β-cells and those of Nakazaki et al. (1995) with normal rat β-cells, the predominant effect of H2O2 is activation of ATP-sensitive K+ channels, since the cell membrane hyperpolarizes after addition of H2O2 and insulin secretion is inhibited.
Membrane hyperpolarization would close voltage-dependent Ca2+ channels and thus reduce [Ca2+]i. However, after an initial drop, H2O2 led to a biphasic increase in [Ca2+]i. The initial transient increase was also observed in Ca2+-free extracellular solution. Since the areas under the curve, calculated in order to quantify and compare the effect of H2O2 on [Ca2+]i in Ca2+-free and Ca2+-containing solutions, were equal, we assume that the elevation of [Ca2+]i during this phase is exclusively due to mobilization of Ca2+ from intracellular sites. The observation that this initial increase is clearly reduced by the mitochondrial uncoupler FCCP but not by the Ca2+ pump inhibitor thapsigargin strongly suggests that it is caused by mobilization of Ca2+ from mitochondria. The second dramatic increase in [Ca2+]i provoked by H2O2 seems to be, at least in part, due to Ca2+ influx. At high [Ca2+]i the effect of the removal of extracellular Ca2+ was diminished or even abolished, suggesting that Ca2+ efflux pathways are insufficient or even inhibited under these conditions. The strong decrease in [Ca2+]i observed after the addition of an ionophore supports this assumption. It seems unlikely that the second increase in [Ca2+]i induced by H2O2 is due to a direct activation of L-type Ca2+ channels, since H2O2 did not alter the activity of the voltage-dependent Ca2+ current in mouse β-cells (Krippeit-Drews et al. 1994a) and the second rise in [Ca2+]i was not blocked by D600. One has to assume that other Ca2+ influx pathways are activated by H2O2. However, the second increase in [Ca2+]i evoked by H2O2 is not blocked by Gd3+ or flufenamate, two inhibitors of unspecific cation channels and Icrac. Moreover, since we did not observe a current corresponding to the increase in [Ca2+]i, either the current conductance may be very small or the influx pathway may be electroneutral. An elevation of [Ca2+]i induced by H2O2 has been observed in a variety of other cells such as smooth muscle cells (Roveri et al. 1992; Krippeit-Drews et al. 1995), endothelial cells (Doan et al. 1994), mesangial cells (Shaw et al. 1995; Meyer et al. 1996), a B-cell line (Qin et al. 1996), neuronal cells (Whittemore et al. 1995), myocytes (Nakamura et al. 1993) and renal tubular cells (Ueda & Shah, 1992), and the effect has been associated with H2O2-induced cell injury in, for example, renal tubular cells (Ueda & Shah, 1992), myocytes (Nakamura et al. 1993), glomerular mesangial cells (Shaw et al. 1995) and neuronal cells (Whittemore et al. 1995). The augmentation of [Ca2+]i has been attributed to influx across the plasma membrane (Meyer et al. 1996), to mobilization from intracellular stores (Ueda & Shah, 1992) or to both mechanisms (Roveri et al. 1992; Doan et al. 1994; Krippeit-Drews et al. 1995; Shaw et al. 1995; Qin et al. 1996).
The observation that H2O2 decreased the ATP concentration raises the question of whether H2O2 also influences an ATP-dependent step in stimulus-secretion coupling distal to the membrane potential. Experiments carried out on permeabilized insulin-secreting cells have indicated that withdrawal of ATP from the cytoplasm resulted in a dramatic reduction in exocytosis (Regazzi et al. 1995). It has been suggested that the readily releasable pool of granules (i.e. releasable without consumption of ATP) is small in pancreatic β-cells and that ATP hydrolysis is required to translocate granules from the reserve pool to the readily releasable pool (Rorsman, 1997). However, the suggestion that the ATP concentration is too low to trigger exocytosis normally after addition of H2O2 is not supported by our experiments with tolbutamide, since the sulfonylurea is, at least at high concentrations, able to reverse the H2O2-induced inhibition of insulin secretion. Alternatively, the compartmentation of the cells could account for the discrepancy between the Ca2+ signal and insulin secretion. Recently, it has been suggested that the Ca2+ concentration in the vicinity of the secretory sites may principally control the secretory machinery (Augustine & Neher, 1992; Bokvist et al. 1995), indicating a different connection between Ca2+ mobilization and various Ca2+ influx pathways and secretion. In pancreatic β-cells, L-type Ca2+ channels are clustered in the part of the cell containing the secretory granules (Bokvist et al. 1995). However, the H2O2-induced increase in [Ca2+]i is mediated by a pathway different from that leading to opening of L-type Ca2+ channels and thus the Ca2+ activity in the direct vicinity of the granules may not be sufficient.
The present paper reveals that a reduction in ATP concentration plays a central role in the suppression of insulin release by H2O2, since it influences secretion at the level of the plasma membrane.
Acknowledgments
We acknowledge the excellent technical support of Mrs K. Stögerer, Mrs I. Hagenloh and Mr M. Dröge. This work was supported by the Deutsche Forschungsgemeinschaft (Dr 225/3-1) and the Deutsche Diabetesgesellschaft.
References
- Ammon HPT, Hägele R, Youssif N, Eujen R, El-Amri N. A possible role of intracellular and membrane thiols of rat pancreatic islets in calcium uptake and insulin release. Endocrinology. 1983;112:720–726. doi: 10.1210/endo-112-2-720. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Progress in Biophysics and Molecular Biology. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Neher E. Calcium requirement for secretion in bovine chromaffin cells. The Journal of Physiology. 1992;450:247–271. doi: 10.1113/jphysiol.1992.sp019126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokvist K, Eliasson L, Ämmälä C, Renström E, Rorsman P. Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis in mouse pancreatic B-cells. EMBO Journal. 1995;14:50–57. doi: 10.1002/j.1460-2075.1995.tb06974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie AE, Reed DJ. Reversible oxidation of glyceraldehyde 3-phosphate dehydrogenase thiols in human lung carcinoma cells by hydrogen peroxide. Biochemical and Biophysical Research Communications. 1987;148:120–125. doi: 10.1016/0006-291x(87)91084-9. [DOI] [PubMed] [Google Scholar]
- Chatham JC, Gilbert HF, Radda GK. The metabolic consequences of hydroperoxide perfusion on the isolated rat heart. European Journal of Biochemistry. 1989;184:657–662. doi: 10.1111/j.1432-1033.1989.tb15063.x. [DOI] [PubMed] [Google Scholar]
- Debuyser A, Drews G, Henquin J-C. Adrenaline inhibition of insulin release: role of the repolarization of the B cell membrane. Pflügers Archiv. 1991;419:131–137. doi: 10.1007/BF00372998. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Ankarcrona M, Nicotera P, Brüne B. Exogenous nitric oxide NO generation or IL-1β-induced intracellular NO production stimulates inhibitory auto-ADP-ribosylation of glyceraldehyde-3-phosphate dehydrogenase in RINm5F cells. Journal of Immunology. 1993;150:2964–2971. [PubMed] [Google Scholar]
- Doan TN, Gentry DL, Taylor AA, Elliott SJ. Hydrogen peroxide activates agonist-sensitive Ca2+-flux pathways in canine venous endothelial cells. Biochemical Journal. 1994;297:209–215. doi: 10.1042/bj2970209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehsel K, Jalowy A, Qi S, Burkart V, Hartmann B, Kolb H. Islet cell DNA is a target of inflammatory attack by nitric oxide. Diabetes. 1993;42:496–500. doi: 10.2337/diab.42.3.496. [DOI] [PubMed] [Google Scholar]
- Filipovic DM, Reeves B. Hydrogen peroxide activates glibenclamide-sensitive K+ channels in LLC-PK1 cells. American Journal of Physiology. 1997;272:C737–743. doi: 10.1152/ajpcell.1997.272.2.C737. [DOI] [PubMed] [Google Scholar]
- Gilon P, Henquin J-C. Influence of membrane potential changes on cytoplasmic Ca2+ concentration in an electrically excitable cell, the insulin-secreting pancreatic B-cell. Journal of Biological Chemistry. 1992;267:20713–20720. [PubMed] [Google Scholar]
- Goldhaber JI, Ji S, Lamp ST, Weiss JN. Effects of exogenous free radicals on electromechanical function and metabolism in isolated rabbit and guinea pig ventricle. Implications for ischemia and reperfusion injury. Journal of Clinical Investigation. 1989;83:1800–1809. doi: 10.1172/JCI114085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gupta JB, Prasad K. Mechanism of H2O2-induced modulation of airway smooth muscle. American Journal of Physiology. 1992;263:L714–722. doi: 10.1152/ajplung.1992.263.6.L714. [DOI] [PubMed] [Google Scholar]
- Gylfe E. Glucose-induced buffering of cytoplasmic Ca2+ in the pancreatic β-cell - An artefact or a physiological phenomenon? Biochemical and Biophysical Research Communications. 1989;159:907–912. doi: 10.1016/0006-291x(89)92194-3. [DOI] [PubMed] [Google Scholar]
- Hanenberg H, Kolb-Bachofen V, Kantwerk-Funke G, Kolb H. Macrophage infiltration precedes and is a prerequisite for lymphocytic insulitis in pancreatic islets of pre-diabetic BB rats. Diabetologia. 1989;32:126–134. doi: 10.1007/BF00505185. [DOI] [PubMed] [Google Scholar]
- Herson PS, Ashford MLJ. Activation of a novel non-selective cation channel by alloxan and H2O2 in the rat insulin-secreting cell line CRI-G1. The Journal of Physiology. 1997;501:59–66. doi: 10.1111/j.1469-7793.1997.059bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmsen H, Robkin L. Hydrogen peroxide lowers ATP levels in platelets without altering adenylate energy charge and platelet function. Journal of Biological Chemistry. 1977;252:1752–1757. [PubMed] [Google Scholar]
- Hyslop PA, Hinshaw DB, Halsey WA, Schraufstätter IU, Sauerheber RD, Spragg RG, Jackson JH, Cochrane CG. Mechanisms of oxidant-mediated cell injury. The glycolytic and mitochondrial pathways of ADP phosphorylation are major intracellular targets inactivated by hydrogen peroxide. Journal of Biological Chemistry. 1988;263:1665–1675. [PubMed] [Google Scholar]
- Jahr H, Bretzel RG, Wacker T, Weinand S, Brandhorst H, Brandhorst D, Lau D, Hering BJ, Federlin K. Toxic effects of superoxide, hydrogen peroxide, and nitric oxide on human and pig islets. Transplantation Proceedings. 1995;27:3220–3221. [PubMed] [Google Scholar]
- Kim H-R, Rho H-W, Park B-H, Park J-W, Kim J-S, Kim U-H, Chung M-Y. Role of Ca2+ in alloxan-induced pancreatic β-cell damage. Biochimica et Biophysica Acta. 1994;1227:87–91. doi: 10.1016/0925-4439(94)90111-2. [DOI] [PubMed] [Google Scholar]
- Kolb-Bachofen V, Epstein S, Kiesel U, Kolb H. Low-dose streptozocin-induced diabetes in mice. Electron microscopy reveals single-cell insulitis before diabetes onset. Diabetes. 1988;37:21–27. doi: 10.2337/diab.37.1.21. [DOI] [PubMed] [Google Scholar]
- Krippeit-Drews P, Britsch S, Lang F, Drews G. Effects of SH-group reagents on Ca2+ and K+ channel currents of pancreatic B-cells. Biochemical and Biophysical Research Communications. 1994a;200:860–866. doi: 10.1006/bbrc.1994.1530. [DOI] [PubMed] [Google Scholar]
- Krippeit-Drews P, Haberland C, Fingerle J, Drews G, Lang F. Effects of H2O2 on membrane potential and [Ca2+]i of cultured rat arterial smooth muscle cells. Biochemical and Biophysical Research Communications. 1995;209:139–145. doi: 10.1006/bbrc.1995.1481. [DOI] [PubMed] [Google Scholar]
- Krippeit-Drews P, Lang F, Häussinger D, Drews G. H2O2 induced hyperpolarization of pancreatic B-cells. Pflügers Archiv. 1994;426:552–554. doi: 10.1007/BF00378534. [DOI] [PubMed] [Google Scholar]
- Lee KU, Kim MK, Amano K, Pak CY, Jaworski MA, Mehta JG, Yoon J-W. Preferential infiltration of macrophages during early stages of insulitis in diabetes-prone BB rats. Diabetes. 1988;37:1053–1058. doi: 10.2337/diab.37.8.1053. [DOI] [PubMed] [Google Scholar]
- Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radical Biology & Medicine. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- Malaisse WJ, Malaisse-Lagae F, Sener A, Pipeleers DG. Determinants of the selective toxicity of alloxan to the pancreatic B cell. Proceedings of the National Academy of Sciences of the USA. 1982;79:927–930. doi: 10.1073/pnas.79.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner HP, Schmelz H. Membrane potential of beta-cells in pancreatic islets. Pflügers Archiv. 1974;351:195–206. doi: 10.1007/BF00586918. [DOI] [PubMed] [Google Scholar]
- Meyer TN, Gloy J, Hug MJ, Greger R, Schollmeyer P, Pavenstädt H. Hydrogen peroxide increases the intracellular calcium activity in rat mesangial cells in primary culture. Kidney International. 1996;49:388–395. doi: 10.1038/ki.1996.57. [DOI] [PubMed] [Google Scholar]
- Nakamura TY, Goda K, Okamoto T, Kishi T, Nakamura T, Goshima K. Contractile and morphological impairment of cultured fetal mouse myocytes induced by oxygen radicals and oxidants. Correlation with intracellular Ca2+ concentration. Circulation Research. 1993;73:758–770. doi: 10.1161/01.res.73.4.758. [DOI] [PubMed] [Google Scholar]
- Nakazaki M, Kakei M, Koriyama N, Tanaka H. Involvement of ATP sensitive K+ channels in free radical-mediated inhibition of insulin secretion in rat pancreatic β-cells. Diabetes. 1995;44:878–883. doi: 10.2337/diab.44.8.878. [DOI] [PubMed] [Google Scholar]
- Plant TD. Properties and calcium-dependent inactivation of calcium currents in cultered mouse pancreatic B-cells. The Journal of Physiology. 1988;404:731–747. doi: 10.1113/jphysiol.1988.sp017316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pralong W-F, Bartley C, Wollheim CB. Single islet β-cell stimulation by nutrients: relationship between pyridine nucleotides, cytosolic Ca2+ and secretion. EMBO Journal. 1990;9:53–60. doi: 10.1002/j.1460-2075.1990.tb08079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Inazu T, Takata M, Kurosaki T, Homma Y, Yamamura H. Cooperation of tyrosine kinases p72syk and p53/56lyn regulates calcium mobilization in chicken B cell oxidant stress signaling. European Journal of Biochemistry. 1996;236:443–449. doi: 10.1111/j.1432-1033.1996.00443.x. [DOI] [PubMed] [Google Scholar]
- Radons J, Heller B, Bürkle A, Hartmann B, Rodriguez M-L, Kröncke K-D, Burkart V, Kolb H. Nitric oxide toxicity in islets cells involves poly ADP-ribose polymerase activation and concomitant NAD+ depletion. Biochemical Biophysical Research Communications. 1994;199:1270–1277. doi: 10.1006/bbrc.1994.1368. [DOI] [PubMed] [Google Scholar]
- Regazzi R, Wollheim CB, Lang J, Theler J-M, Rossetto O, Montecucco C, Sadoul K, Weller U, Palmer M, Thorens B. VAMP-2 and cellubrevin are expressed in pancreatic β-cells and are essential for Ca2+- but not for GTPγS-induced insulin secretion. EMBO Journal. 1995;14:2723–2730. doi: 10.1002/j.1460-2075.1995.tb07273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P. The pancreatic beta-cell as a fuel sensor: an electrophysiologist's viewpoint. Diabetologia. 1997;40:487–495. doi: 10.1007/s001250050706. [DOI] [PubMed] [Google Scholar]
- Roveri A, Coassin M, Maiorino M, Zamburlini A, van Amsterdam FTM, Ratti E, Ursini F. Effect of hydrogen peroxide on calcium homeostasis in smooth muscle cells. Archives of Biochemistry and Biophysics. 1992;297:265–270. doi: 10.1016/0003-9861(92)90671-i. [DOI] [PubMed] [Google Scholar]
- Shaw S, Naegeli P, Etter JD, Weidmann P. Role of intracellular signalling pathways in hydrogen peroxide-induced injury to rat glomerular mesangial cells. Clinical and Experimental Pharmacology and Physiology. 1995;22:924–933. doi: 10.1111/j.1440-1681.1995.tb02328.x. [DOI] [PubMed] [Google Scholar]
- Spragg RG, Hinshaw DB, Hyslop PA, Schraufstätter IU, Cochrane CG. Alterations in adenosine triphosphate and energy charge in cultured endothelial and P388D1 cells after oxidant injury. Journal of Clinical Investigation. 1985;76:1471–147. doi: 10.1172/JCI112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasu N, Komiya I, Asawa T, Nagasawa Y, Yamada T. Streptozocin- and alloxan-induced H2O2 generation and DNA fragmentation in pancreatic islets. H2O2 as mediator for DNA fragmentation. Diabetes. 1991;40:1141–1145. doi: 10.2337/diab.40.9.1141. [DOI] [PubMed] [Google Scholar]
- Ueda N, Shah SV. Role of intracellular calcium in hydrogen peroxide-induced renal tubular cell injury. American Journal of Physiology. 1992;263:F214–221. doi: 10.1152/ajprenal.1992.263.2.F214. [DOI] [PubMed] [Google Scholar]
- Welsh N, Eizirik DL, Bendtzen K, Sandler S. Interleukin 1β-induced nitric oxide production in isolated rat pancreatic islets requires gene transcription and may lead to inhibition of the Krebs cycle enzyme aconitase. Endocrinology. 1991;129:3167–3173. doi: 10.1210/endo-129-6-3167. [DOI] [PubMed] [Google Scholar]
- Whittemore ER, Loo DT, Watt JA, Cotman CW. A detailed analysis of hydrogen peroxide-induced cell death in primary neuronal culture. Neuroscience. 1995;67:921–932. doi: 10.1016/0306-4522(95)00108-u. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Uchigata Y, Okamoto H. Streptozotocin and alloxan induce DNA strand breaks and poly(ADP-ribose) synthetase in pancreatic islets. Nature. 1981;294:284–286. doi: 10.1038/294284a0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. Journal of Biological Chemistry. 1990;265:16330–16336. [PubMed] [Google Scholar]