

Abstract
Mice were subjected to gastrectomy (GX) or food deprivation (24 h). The release of insulin and glucagon in response to different secretagogues was monitored in vivo and in isolated islets 3–4 weeks after surgery.
GX animals responded to glucose with an impaired glucose tolerance and a poor increase in plasma insulin. Islets from GX or food-deprived mice displayed impaired insulin release to high glucose and enhanced glucagon release at low glucose.
After GX the insulinogenic index, Δ insulin (μU ml−1)/Δ glucose (mg ml−1), was suppressed by 65% after oral glucose and by 59% after i.v. glucose. The integrated insulin response after oral glucose was reduced by 90% in GX mice. After i.v. glucose the reduction was 67%.
Carbachol-induced insulin release in vivo was reduced after food deprivation and exaggerated after GX. Carbachol-stimulated glucagon secretion was suppressed after GX and after food deprivation. A similar pattern was found in vitro.
Cyclic AMP activation (by the phosphodiesterase inhibitor isobutylmethylxanthine or the adenylate cyclase stimulator forskolin) induced a greater insulin response in GX or food-deprived mice than in sham-operated, fed mice. A similar pattern was found in vitro. The glucagon response was enhanced in vitro but not in vivo.
Crude extracts of rat oxyntic mucosa enhanced basal as well as glucose-induced insulin release from isolated islets, whereas glucagon release was markedly inhibited. The effects were dose dependent, the inhibition of glucagon release being achieved at lower concentrations than the potentiation of glucose-induced insulin release. The active principle was inactivated by incubation with trypsin or leucine aminopeptidase.
The data suggest that a circulating agent, probably a peptide, from gastric oxyntic mucosa stimulates glucose-induced insulin secretion. It also suppresses glucagon secretion. The GX-evoked impairment of the insulin (and glucagon) response to glucose is partly compensated for by an enhanced insulin response to cholinergic and/or cyclic AMP activation.
Hormones released from the digestive tract have been suggested to contribute to the metabolic disposal of ingested glucose. Thus, an oral glucose load is metabolized more rapidly than an intravenous glucose load, and more insulin is secreted after oral glucose than after an equivalent intravenous glucose load (McIntyre et al. 1964; Dupré, 1991; Creutzfeldt & Nauck, 1992). Several intestinal hormones, including glucagon-like peptide-1-(7-36)-amide (GLP-1) and glucose-dependent insulinotrophic peptide (GIP), are thought to affect blood glucose by modulating the secretion of insulin and glucagon (Dupré, 1991; Creutzfeldt & Nauck, 1992; Holst, 1994). However, the dysfunction of such intestinal hormone systems does not explain the alimentary hyperglycaemia often noted in patients who have undergone gastric surgery (Muir, 1949; Tobe et al. 1967). The possible existence of a gastro-insular axis prompted us to study the effect of gastrectomy in mice on the insulin and glucagon responses in vivo and in vitro to the three major types of regulators of islet hormone release, i.e. glucose (nutrient regulator), carbachol (phospholipase C regulator) and isobutylmethylxanthine (IBMX) or forskolin (cyclic AMP regulators). In addition, we tested the effect of a crude oxyntic mucosal extract on islet hormone release. We also monitored blood lipids since recent studies have indicated that free fatty acids may impair insulin release (Prentki & Corkey, 1996).
METHODS
Drugs and chemicals
Collagenase (CLS-4) was purchased from Worthington Biochemicals (Freehold, NJ, USA). Bovine serum albumin (BSA) was from ICN Biomedicals (High Wycombe, UK). Leucine aminopeptidase (EC 3.4.11.2., microsomal, type IV-S, porcine kidney; 40 units (mg protein)−1) and trypsin (EC 3.4.21.4., bovine pancreas; 9060 units (mg protein)−1) were obtained from Sigma. All other drugs and chemicals were from British Drug Houses (Poole, UK) or Merck (Darmstadt, Germany). Radioimmunoassay kits for determination of insulin were obtained from Novo Nordisk (Bagsværd, Denmark) or Diagnostica (Falkenberg, Sweden) and those for glucagon determination from Eurodiagnostica (Malmö, Sweden). The antiserum used in the glucagon assay recognizes pancreatic glucagon but not gut glucagon-like peptides. It was also ascertained that the glucagon antiserum did not cross-react with any constituent of oxyntic mucosal extract.
Animals
Female mice of the NMRI strain (B & K, Sollentuna, Sweden), weighing 25-30 g, were used. They were given a standard pellet diet (B & K) and tap water ad libitum unless otherwise stated. Before surgery, the mice were anaesthetized with mebumal (25 mg per mouse, i.p.). Gastrectomy was performed by resecting the stomach and by anastomosing the oesophagus and the duodenum end-to-end. Sham operation consisted of an abdominal mid-line incision and manipulation of the viscera (laparatomy). Vagotomy was performed by cutting both vagal trunks just below the diaphragm. At the same time a pyloroplasty was performed in order to prevent post-vagotomy gastric dilatation. Pyloroplasty alone was performed as a control to the vagotomy. The animals were allowed to recuperate for at least 3 weeks before they were subjected to experiments. During this time period suitable after-care was given to the animals to ensure that they suffered no pain or distress. Before the experiments one group of age-matched intact mice was deprived of food for 24 h but were allowed tap water ad libitum. The animal experiments were approved by the local animal welfare committee (Lund, Sweden).
Experimental protocol
In vivo studies
Glucose, carbachol and IBMX were dissolved in 0.9 % NaCl and injected intravenously into a tail vein (5 μl (g mouse)−1) or administered via an oro-gastric tube (glucose only) (5 μl (g mouse)−1). The doses chosen are known to give both an approximately half-maximal response and also a response of similar magnitude for the different secretagogues with regard to insulin release in mice (Lundquist, 1982; Lundquist & Panagiotidis, 1992; Panagiotidis et al. 1994). Controls received saline. Blood sampling was performed as described previously (Rerup & Lundquist, 1966). The mice were then killed by cervical dislocation. The concentrations of insulin and glucagon in plasma were determined by radioimmunoassay (Heding, 1966; Ahrén & Lundquist, 1982; Panagiotidis et al. 1992). Plasma glucose concentrations were determined enzymatically (Bruss & Black, 1978). Concentrations of FFA, triglycerides without free glycerol (TG) and cholesterol in serum were determined enzymatically with kits from Wako Chemicals and Boehringer Mannheim.
In vitro studies
Isolated pancreatic islets were prepared by injection of a collagenase solution into the bile-pancreatic duct of the mouse (Gotoh et al. 1985) directly after being killed by cervical dislocation. These mice had not previously been used in the in vivo experiments. Freshly isolated islets were preincubated for 30 min at 37°C in Krebs-Ringer bicarbonate buffer (KRB), pH 7.4, supplemented with 10 mM Hepes, 0.1 % bovine serum albumin and 7 mM glucose. Each incubation vial was gassed with 95 % O2 and 5 % CO2 to obtain constant pH and oxygenation. After pre-incubation the buffer was changed to a medium containing the agents to be tested and the islets (10 islets per millilitre medium) were incubated for 60 min. All incubations were performed at 37°C in a metabolic shaker (30 cycles per minute). Immediately after incubation, aliquots (400 μl) of the medium were removed for assay of insulin and glucagon (Heding, 1966; Ahrén & Lundquist, 1982; Panagiotidis et al. 1992).
Extract of rat oxyntic mucosa was prepared as follows: 60 male Sprague-Dawley rats (250-300 g body wt) were killed by exsanguination via the abdominal aorta under anaesthesia (chloral hydrate, 300 mg kg−1, i.p.) and each stomach was opened along the major curvature, rinsed with ice-cold 0.9 % NaCl and flattened on a glass board with the mucosa upwards. The oxyntic mucosa was scraped off the gastric wall with a scalpel, weighed and homogenized in 0.01 M phosphate buffer, pH 7.0, at a concentration of 100 mg tissue (ml buffer)−1. The homogenate was boiled for 30 min in a water bath and centrifuged at 1000 g. The supernatant was freeze-dried, weighed and stored. On the day of the experiment the freeze-dried material was dissolved in the incubation medium. An equivalent amount of albumin was added to the control medium. In one series of experiments trypsin (5 μg; 50 units) or leucine aminopeptidase (10 units) was dissolved in 500 μl of the Hepes-buffered KRB (pH 8.0) and incubated with 32.4 mg of the freeze-dried material (115 μg protein) for 12 h at 37°C. For control purposes 32.4 mg of the material was incubated under identical conditions but without trypsin or leucine aminopeptidase, and the peptidases were also incubated without freeze-dried material. The digestion was terminated by boiling for 1 h. Protein was assayed according to Bradford with BSA as standard (Bradford, 1976).
Statistics
Results were expressed as means ±s.e.m. The level of significance for the difference between sets of data was assessed using Student's unpaired t test or analysis of variance followed by Tukey-Kramer's test whenever appropriate. P < 0.05 was considered statistically significant.
RESULTS
Body weights and plasma concentrations of glucose, insulin and glucagon
Table 1 shows an overview of some characteristics of the different groups of mice 3-4 weeks after the various operations. An age-matched group of intact mice deprived of food (24 h) was also included. There were no apparent differences between the groups with respect to body weight or basal plasma levels of glucose, insulin or glucagon (Table 1), except for the expected decrease in body weight and plasma glucose and insulin concentrations after food deprivation.
Table 1.
Body weights and plasma concentrations of glucose, insulin and glucagon in mice subjected to different surgical procedures or food deprivation
| Body weight (g) | Plasma glucose (mmol l−1) | Plasma insulin (pmol l−1) | Plasma glucagon (ng l−1) | |
|---|---|---|---|---|
| Sham operation | 32.1 ± 1.1 | 9.1 ± 0.5 | 82.2 ± 5.0 | 204 ± 12 |
| Gastrectomy | 29.4 ± 1.6 | 10.2 ± 0.6 | 70.4 ± 12.6 | 221 ± 21 |
| Food deprivation (24 h) | 27.5 ± 0.8 ** | 5.2 ± 0.3 *** | 25.0 ± 8.8 *** | 238 ± 21 |
| Pyloroplasty | 31.4 ± 0.9 | 11.2 ± 0.3 | 101.4 ± 21.0 | 193 ± 21 |
| Vagotomy | 30.0 ± 0.6 | 10.5 ± 0.8 | 62.4 ± 15.0 | 222 ± 2.6 |
Mean values ± S.E.M. for 6–11 animals in each group.
Significant difference versus sham-operated mice.
P < 0.01
P < 0.001.
Plasma insulin, glucagon and glucose in response to oral or intravenous glucose in sham-operated or gastrectomized mice
Gastrectomized and sham-operated mice received glucose (5.6 mmol kg−1) or an equivalent volume of 0.9 % NaCl injected via an oro-gastric tube. Gastrectomized mice displayed a defective insulin response and an impaired glucose tolerance. The insulinogenic index, Δ insulin (μU ml−1)/Δ glucose (mg ml−1), at 10 min (insulin peak level) was 6.2 (gastrectomy) versus 17.8 (sham operation), i.e. a suppression by 65 % in gastrectomized versus sham-operated mice. Moreover, glucose lowered plasma glucagon in sham-operated but not in gastrectomized mice (Fig. 1). Figure 2 shows a comparison between gastrectomized and sham-operated mice, given the same glucose load as above but now by the intravenous route. Glucose failed to stimulate insulin secretion appropriately and to suppress glucagon secretion in the gastrectomized mice. This resulted in impaired glucose tolerance (Fig. 2). The insulinogenic index at 2 min (insulin peak level) was 14.1 (gastrectomy) versus 34.1 (sham operation) and thus reduced by 59 % in gastrectomized mice, i.e. a difference of the same magnitude as after oral glucose. The integrated (120 min) insulin response (area under the curve as calculated from Fig. 1) to the oral glucose load amounted to 4370 pmol of insulin (mean value) in sham-operated mice versus 421 pmol in gastrectomized mice. The corresponding values (60 min) in response to intravenous glucose (Fig. 2) were 2787 pmol versus 915 pmol. Hence, the total insulin response was greatly reduced by gastrectomy, more so after oral than after intravenous glucose.
Figure 1. Effect of an oro-gastric load on plasma levels of insulin, glucagon and glucose.
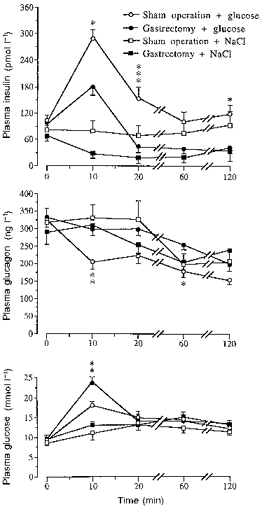
Effect of an oro-gastric glucose load (5.6 mmol kg−1) on plasma insulin, glucagon and glucose levels in sham-operated and gastrectomized mice. Mice given 0.9 % NaCl are included. Values are means ±s.e.m. (vertical bars) for 10-16 (controls) or 21-23 (glucose load) mice in each group. When no s.e.m. is indicated the value was smaller than the size of the symbol. Significant differences between sham operation and gastrectomy are denoted by *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 2. Effect of an i.v. glucose load on plasma levels of insulin, glucagon and glucose.
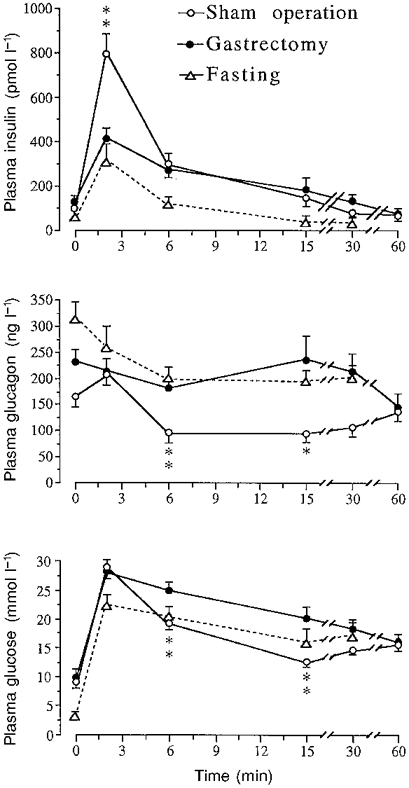
Effect of an i.v. injection of glucose (5.6 mmol kg−1) on plasma insulin, glucagon and glucose in sham-operated, fasted, and gastrectomized mice. Values are means ±s.e.m. for 8 mice in each group. When no s.e.m. is indicated the value was smaller than the size of the symbol. Significant differences between sham operation and gastrectomy are denoted by *P < 0.05, **P < 0.01.
Analysis of the insulin secretory capacity obtained during intravenous versus oral glucose in normal mice was performed by calculating the total increase in circulating insulin in relation to the total increase in plasma glucose (area under the curves) in sham-operated mice. We found an insulinogenic index of 5.0 after intravenous glucose versus 11.4 after oral glucose, i.e. the insulin secretory capacity after intravenous glucose was only 43 % of that after oral glucose. For comparison a group of food-deprived (‘gastric rest’) mice received glucose by the intravenous route. These animals displayed normal suppression of the glucagon levels and a marked impairment of the insulin response (Fig. 2).
Insulin and glucagon responses following an intravenous challenge of glucose, carbachol or IBMX in mice subjected to gastrectomy, food deprivation or vagotomy
Figure 3 shows the insulin response 2 min (peak level) after the intravenous injection of a half-maximal dose of glucose (3.3 mmol kg−1) (Lundquist & Panagiotidis, 1992). The insulin response to glucose was greatly impaired in gastrectomized mice (-50 % of that in sham-operated animals) (P < 0.001). For comparison, a group of intact fasted mice was again included. The insulin response was even more reduced in these animals (-80 % of control values) (P < 0.001) than in the gastrectomized mice. Since total gastrectomy could not be performed without cutting the vagi, experiments were conducted on mice subjected to vagotomy (VTPP) and/or pyloroplasty (PP). The insulin response to glucose was only slightly reduced in vagotomized mice (-10 %) (P < 0.05) and the plasma glucagon levels were unaffected within this short time period (2 min) (Fig. 3).
Figure 3. Acute effects of an i.v. glucose load on the insulin and glucagon responses in the different categories of mice.
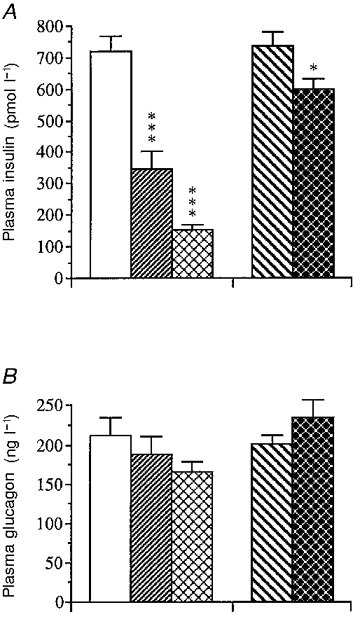
Acute insulin (A) and glucagon (B) secretory responses following an i.v. injection of glucose (3.3 mmol kg−1) in mice subjected to sham operation (□), gastrectomy ( ), fasting (
), fasting ( ), pyloroplasty (PP;
), pyloroplasty (PP;  ) or vagotomy (VTPP;
) or vagotomy (VTPP;  ). Values are means ±s.e.m. of 15-17 animals in each group except for the fasted group, which consisted of 8 animals. Significant differences versus sham operation are denoted by *P < 0.05, ***P < 0.001.
). Values are means ±s.e.m. of 15-17 animals in each group except for the fasted group, which consisted of 8 animals. Significant differences versus sham operation are denoted by *P < 0.05, ***P < 0.001.
The hormone responses (peak levels) to the muscarinic agonist carbachol (0.16 μmol kg−1) are illustrated in Fig. 4. When compared with sham-operated controls the carbachol-induced insulin response in gastrectomized mice was increased by +45 % (P < 0.05). An increase was also noted after vagotomy (+30 %) (P < 0.05). In contrast, the glucagon response was markedly reduced in gastrectomized mice (-45 %) (P < 0.001) versus sham-operated mice while being unaffected by vagotomy. Fasted mice displayed a marked impairment of the insulin response (-45 %) (P < 0.001) and a modest reduction of the glucagon response (-30 %) (P < 0.05) to carbachol.
Figure 4. Acute effect of an i.v. carbachol injection on the insulin and glucagon responses in the different categories of mice.
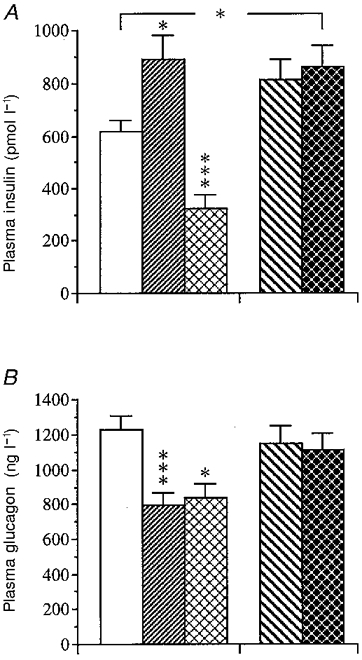
Acute insulin (A) and glucagon (B) secretory responses following an i.v. injection of the cholinergic muscarinic agonist carbachol (0.16 μmol kg−1) in mice subjected to sham operation (□), gastrectomy (), fasting (), pyloroplasty (PP; ) or vagotomy (VTPP; ). Values are means ±s.e.m. of 21-26 animals in each group except for the fasted group which consisted of 8 animals. Significant differences versus sham operation are denoted by *P < 0.05, ***P < 0.001.
The phosphodiesterase inhibitor IBMX (45 μmol kg−1) brought about a much greater insulin response (peak level) in gastrectomized mice than in sham-operated controls (Fig. 5). Vagotomy did not affect the insulin response to IBMX. The glucagon response to IBMX was not affected by either gastrectomy or vagotomy, and fasting, finally, did not influence the hormone responses to IBMX.
Figure 5. Acute effects of an i.v. injection of IBMX on the insulin and glucagon responses in the different categories of mice.
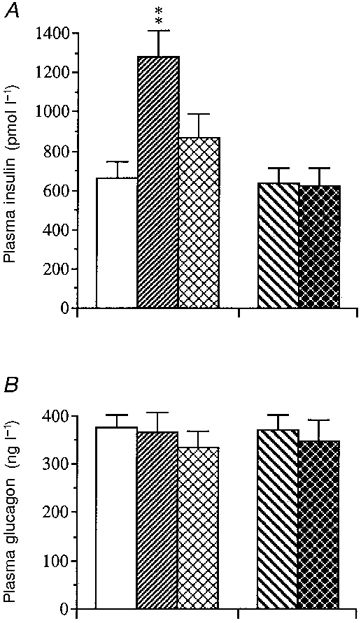
Acute insulin (A) and glucagon (B) secretory response following an i.v. injection of the phosphodiesterase inhibitor IBMX (45 μmol kg−1) in mice subjected to sham operation (□), gastrectomy (), fasting (), pyloroplasty (PP; ) or vagotomy (VTPP; ). Values are means ±s.e.m. of 13-18 animals in each group except for the fasted group which consisted of 8 animals. Significant differences versus sham operation are denoted by **P < 0.01.
Insulin and glucagon release from isolated islets in the basal state and in response to glucose, carbachol, IBMX or forskolin
Basal insulin release in response to low glucose (1 mmol l−1) (Fig. 6A and C) was the same in islets from gastrectomized, vagotomized or fasted mice. In contrast, glucagon release from isolated islets was markedly enhanced after gastrectomy, modestly increased after fasting, and slightly reduced after vagotomy (Fig. 6B and D).
Figure 6. Influence of glucose on hormone release from isolated islets.
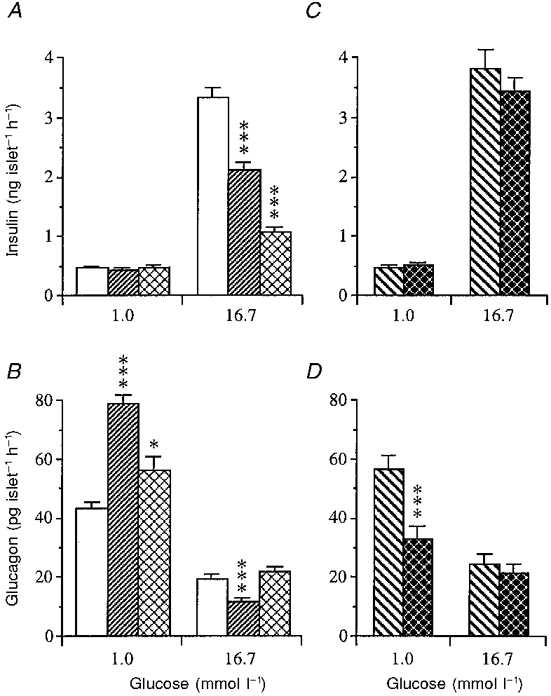
Influence of low (1 mmol l−1) and high (16.7 mmol l−1) glucose concentrations on insulin (A) and glucagon (B) release from islets isolated from mice subjected to sham operation (controls; □), gastrectomy () or fasting (). Hormone release following pyloroplasty (PP; ) or vagotomy (VTPP; ) is illustrated in C for insulin and D for glucagon. Values are means ±s.e.m. for 8-12 batches of islets in each group. Each batch contained 10 islets. Significant differences versus sham operation (A and B) or pyloroplasty (C and D) are denoted by *P < 0.05, ***P < 0.001.
At a high glucose concentration (16.7 mmol l−1) the insulin response was greatly impaired after gastrectomy (Fig. 6A). Food deprivation induced a similar but much more pronounced inhibition of insulin secretion at high glucose than was noted after gastrectomy (Fig. 6A), while the glucagon response was increased rather than suppressed (Fig. 6B). Vagotomy did not alter the insulin or glucagon secretion from isolated islets at high glucose (Fig. 6C and D).
At a physiological concentration of glucose (7 mmol l−1) (Fig. 7A and C) basal insulin release was slightly suppressed after gastrectomy but unaffected after vagotomy or fasting. Basal glucagon release at 7 mmol l−1 glucose was unaffected after gastrectomy or fasting and slightly reduced after vagotomy (Fig. 7B). Exactly the same pattern of basal hormone release can be seen in a later series of experiments (Fig. 8A-D).
Figure 7. Influence of carbachol on hormone release from isolated islets.
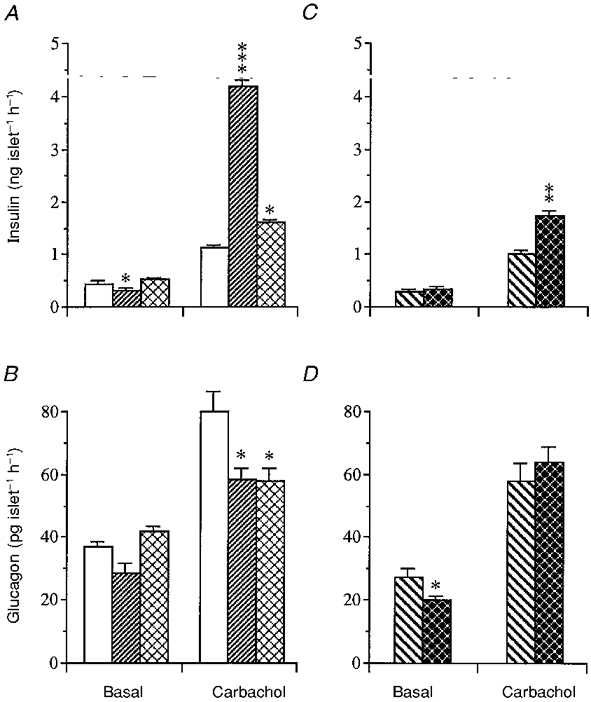
Insulin (A) and glucagon (B) release in response to carbachol (20 μmol l−1) in the presence of glucose (7 mmol l−1) from islets isolated from mice subjected to sham operation (controls; □), gastrectomy () or fasting (). Hormone release following pyloroplasty (PP; ) or vagotomy (VTPP; ) is illustrated in C for insulin and D for glucagon. Basal hormone release in the absence of carbachol (but in the presence of 7 mmol l−1 glucose) is also shown. Values are means ±s.e.m. for 8-12 batches of islets in each group. Each batch contained 10 islets. Significant differences versus sham operation (A and B) or pyloroplasty (C and D) are denoted by *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 8. Influence of IBMX or forskolin on hormone release from isolated islets.
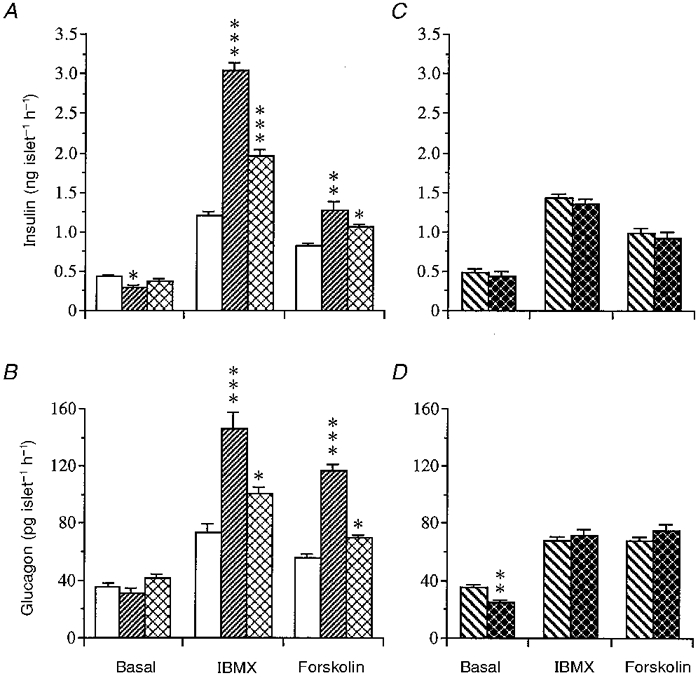
Insulin (A) and glucagon (B) release in response to IBMX (1 mmol l−1) or forskolin (20 μmol l−1) in the presence of basal glucose (7 mmol l−1) from islets isolated from mice subjected to sham operation (controls; □), gastrectomy () or fasting (). Hormone release following pyloroplasty (PP; ) or vagotomy (VTPP; ) is illustrated for insulin in C and for glucagon in D. Basal hormone release is also shown. Values are means ±s.e.m. for 8-12 batches of islets in each group. Each batch contained 10 islets. Significant differences versus sham operation (A and B) or pyloroplasty (C and D) are denoted by *P < 0.05, **P < 0.01, ***P < 0.001.
In the presence of a basal glucose concentration of 7 mmol l−1, carbachol (20 μmol l−1) stimulated the secretion of both insulin and glucagon from islets of sham-operated controls (Fig. 7A and B). Islets from gastrectomized mice responded to carbachol with a markedly exaggerated insulin secretion and a modest suppression of glucagon secretion compared with controls. In islets from fasted mice the insulin response to carbachol was slightly greater (P < 0.05) than in islets from fed mice (Fig. 7A); glucagon secretion, on the other hand, was slightly suppressed (Fig. 7B). Vagotomy brought about a somewhat greater insulin response to carbachol in isolated islets than food deprivation. The glucagon response, however, was unaffected (Fig. 7C and D).
IBMX (1 mmol l−1) and forskolin (20 μmol l−1) mobilized both insulin and glucagon from isolated islets at 7 mmol l−1 glucose. Islets from gastrectomized mice displayed a 2-fold greater insulin and glucagon response compared with islets from sham-operated mice (Fig. 8A and B). A similar response pattern, although less pronounced, was observed in islets from fasted mice. Vagotomy did not influence islet hormone secretion in response to IBMX or forskolin (Fig. 8C and D).
Effects of extract of rat oxyntic mucosa on hormone release from isolated islets
An aqueous extract of rat oxyntic mucosa was added to the incubation medium containing either 7 or 12 mmol l−1 glucose. Figure 9A and B shows that the freeze-dried extract (32.0 μg protein ml−1) brought about a marked increase in insulin secretion and a strong suppression of glucagon secretion from isolated islets at both glucose concentrations. To elucidate whether the active principle in the extract would act in a dose-dependent manner we performed a series of dilutions of the freeze-dried material (from 256.0 μg protein ml−1 to 1.0 μg protein ml−1) and tested the effects of the different dilutions on islet hormone release in the presence of 12 mmol l−1 glucose. As can be seen from Fig. 9C the potentiating action of the extract on glucose-induced insulin release displayed a linear dose- response relationship between 4.0 and 64.0 μg protein ml−1. In contrast, the inhibiting effect on glucagon release started at much lower concentrations (2.0-4.0 μg protein ml−1) (Fig. 9D). Finally, we tested the sensitivity of the active principle to peptidases by incubating the freeze-dried material with trypsin or leucine aminopeptidase and then testing the activity of the treated extracts (19 μg protein ml−1) on islet hormone release in the presence of 12 mmol l−1 glucose. The results are illustrated in Fig. 9E and F and show that both the potentiating action on glucose-induced insulin release as well as the inhibiting action on glucagon release were abolished by treatment with peptidases.
Figure 9. Effect of extract of rat oxyntic mucosa on islet hormone release.
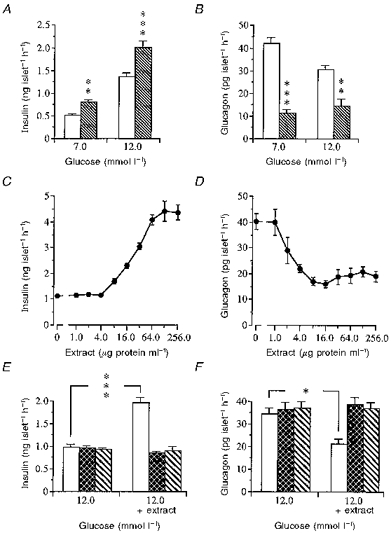
Insulin (A) and glucagon (B) release in response to extract (32 μg protein expressed as BSA equivalents ml−1) of rat oxyntic mucosa in the presence of either basal glucose (7 mmol l−1) or insulin stimulating glucose (12 mmol l−1) from islets isolated from normal freely fed mice. □, control; , + oxyntic mucosa. Values are means ±s.e.m. for 10-12 batches of islets in each group. Each batch contained 10 islets. They were isolated and pooled from a total of 8 mice. Significant differences versus controls are denoted by **P < 0.01, ***P < 0.001. C and D show dose-response relationships between different dilutions of the oxyntic mucosal extract and insulin (C) and glucagon (D) release in the presence of glucose at 12 mmol l−1. Values are means ±s.e.m. for 8-12 batches of islets on each point between 0 and 32 μg protein ml−1; n= 3-4 for 64, 128 and 256 μg protein ml−1. The islets were isolated and pooled from a total of 10 mice. E and F show the effects of oxyntic mucosal extract pre-incubated for 16 h in either the absence (□) or presence of trypsin () or leucine aminopeptidase () on insulin (E) and glucagon (F) release (right part of the panels). Islets (8-10) were then incubated in a medium containing 12 mmol l−1 glucose and an extract concentration of 19 μg protein ml−1. The peptidases were also pre-incubated without extract and then added to the islets (left part of the panels). Values are means ±s.e.m. for 6 batches of islets. The islets were isolated and pooled from a total of 6 mice in each group. *P < 0.05, **P < 0.01, ***P < 0.001.
Serum lipids after surgery
Table 2 shows the serum concentrations of free fatty acids, triglycerides and cholesterol. No differences were noted between the various groups of mice, except for the food-deprived group, where free fatty acids were markedly increased. Also triglycerides and cholesterol were elevated in these mice.
Table 2.
Concentrations of serum lipids in mice subjected to different surgical procedures or food deprivation
| Free fatty acids (mmol l−1) | Triglycerides (mmol l−1) | Cholesterol (mmol l−1) | |
|---|---|---|---|
| Sham operation (20) | 1.43 ± 0.12 | 0.94 ± 0.06 | 2.50 ± 0.08 |
| No operation, food deprivation (24 h) (10) | 2.44 ± 0.09 *** | 1.30 ± 0.12 ** | 3.20 ± 0.14 *** |
| Gastrectomy (17) | 1.27 ± 0.12 | 0.94 ± 0.09 | 2.73 ± 0.11 |
| Pyloroplasty (21) | 1.55 ± 0.15 | 1.10 ± 0.06 | 2.63 ± 0.08 |
| Vagotomy (25) | 1.39 ± 0.16 | 0.90 ± 0.03 | 2.56 ± 0.08 |
Mean values ± S.E.M. are presented; (n) indicates number of animals.
Significant difference versus Sham-operated mice.
P < 0.01
P < 0.001.
DISCUSSION
Oral glucose tolerance tests performed in 100 postgastrectomy patients revealed a reduced glucose tolerance (Tobe et al. 1967), and the insulin response relative to the plasma glucose concentration (insulinogenic index) was significantly lower in gastrectomized patients than in healthy, age-matched controls irrespective of whether glucose was given orally or intrajejunally (Breuer et al. 1972). An impaired insulin-glucose relationship in gastrectomized patients was observed also by Sudo et al. (1982) after an intravenous glucose load. Further, these authors described hyperglucagonaemia in response to orally administered glucose and suggested that the glucose intolerance in gastrectomized patients could be due to increased glucagon and reduced insulin levels in plasma (Sudo et al. 1982).
The recent recognition of the potential clinical usefulness of GLP-1 in the treatment of diabetes (Holst, 1994) has overshadowed the possible importance of a gastro-insular axis. In view of previous data we reasoned that the stomach might harbour an insulin-releasing factor, operating independently of or in addition to GIP (duodenum, jejunum) and GLP-1 (ileum, colon/rectum). In fact, from a physiological point of view, the stomach would be a more favourable site for a nutrient-mobilized insulinotrophic factor than the intestines. There are at least five types of endocrine cells in the mouse stomach, the ECL cells (oxyntic mucosa), the somatostatin cells (antral and oxyntic mucosa), the gastrin cells (antrum), the enterochromaffin cells (antrum) and the A-like cells (oxyntic mucosa) (cf. Sundler & Håkanson, 1991). Somatostatin, which is present also in somatostatin cells within the islets, inhibits both insulin and glucagon secretion (Creutzfeldt & Nauck, 1992). Gastrin is thought to stimulate insulin secretion but only at pharmacological concentrations (Ahrén & Lundquist, 1981; Creutzfeldt & Nauck, 1992). The islet cholecystokinin (CCK) receptor is of the CCKA-type (Monstein et al. 1996), i.e. it recognizes CCK quite well (Zawalich et al. 1988) but gastrin only poorly. As the peptide hormones produced by the ECL cells and the A-like cells are unknown, these two cell types represent possible candidates for producing and secreting messengers in an insulinotrophic gastro-insular axis.
Analogously to the situation in gastrectomized patients (Muir, 1949; Tobe et al. 1967; Breuer et al. 1972; Sudo et al. 1982), gastrectomized mice did not display any obvious impairment in their basal glucose homeostasis, and we did not observe any abnormalities in their serum lipid status. These results are pertinent because elevated free fatty acids may represent a hitherto overlooked cause of impaired glucose-induced insulin secretion (Sako & Grill, 1990; Zhou & Grill, 1994), and may also contribute to the poor insulin response in the fasted mice.
Insulin secretion
With regard to insulin secretion, the severely impaired insulin response to glucose in gastrectomized mice could not be ascribed to loss of vagal input, since vagotomy reduced the glucose-induced insulin secretion only modestly. The slight reduction of glucose-stimulated insulin secretion after vagotomy is in agreement with our earlier data obtained from studies of fasted vagotomized rats (Håkanson et al. 1971) and atropinized mice (Lundquist, 1982). Later studies have emphasized the importance of the vagus for glucose-induced insulin release (cf. Jansson & Hellerström, 1986). However, as suggested by the findings of the present study a disturbed vagal input to the gastro-pancreatic region can only partially explain the gastrectomy-evoked impairment of the insulin response to glucose.
Fasting can induce starvation diabetes and is known to lower the sensitivity of the insulin cell to glucose, an effect being largely ascribed to impaired stimulus-secretion coupling in the insulin cell (Hedeskov, 1980; Lundquist, 1986; Prentki & Matschinsky, 1987). Islets isolated from gastrectomized or fasted mice responded to glucose in a manner similar to that of mice studied in vivo (compare Figs 2, 3 and 6). Hence, one should expect that gastrectomy would lead to a permanent diabetes-like condition. However, this was not the case in mice (present data) nor does it seem to occur very often in man (Muir, 1949; Tobe et al. 1967; Breuer et al. 1972; Sudo et al. 1982). Conceivably, the lack of a circulating gastric factor to promote glucose-induced insulin secretion is compensated for by an increase in secretory signals governed by non-glucose-activated transduction mechanisms (e.g. the phospholipase C and the cyclic AMP systems). Thus, while the insulin response to glucose was impaired, the responses to carbachol and IBMX were enhanced after gastrectomy. Also, direct stimulation with the adenylate cyclase activator forskolin resulted in exaggerated insulin secretion from islets of gastrectomized mice. Part of the exaggerated carbachol-induced insulin response in gastrectomized mice may be ascribed to the loss of vagal input, since vagotomy enhanced the insulin response to carbachol in contrast to the glucose-induced insulin release, which was modestly reduced after vagotomy. This enhanced response to carbachol may reflect denervation supersensitivity (see also Järhult et al. 1983). However, the insulin response to carbachol was greater in gastrectomized mice than in vagotomized mice, suggesting that mechanisms other than loss of vagal input operate after gastrectomy. In fasted mice, cholinergic stimulation of insulin secretion did not display the enhancement seen in gastrectomized mice, in fact it was modestly suppressed in vivo and marginally enhanced in vitro. Thus, gastrectomized mice responded better to stimulation of the phospholipase C system by carbachol than did fasted mice. A similar - although not identical - pattern of insulin secretion in gastrectomized versus fasted animals was seen after stimulation of the cyclic AMP system by IBMX or forskolin. Cyclic AMP-activating secretagogues greatly stimulated the insulin secretion after gastrectomy both in vivo and in vitro. Food deprivation, however, only slightly enhanced the insulin response to IBMX or forskolin. These results agree with previous reports showing that the insulin response to cyclic AMP-activating secretagogues, such as glucagon or the β-adrenoceptor agonists isopropylnoradrenaline and terbutaline, was unaffected by fasting (Lundquist & Lövdahl, 1983; Panagiotidis et al. 1993). Hence, the impaired insulin response to glucose after gastrectomy is associated with an enhanced responsiveness to secretagogues stimulating the phospholipase C and the cyclic AMP systems. This may prevent the development of a diabetes-like condition and, in addition, may have contributed to the fact that disturbances in glucose metabolism after gastrectomy in patients have received little attention in the literature.
The effects of gastrectomy can be explained by a gastric hormone deficiency and/or by deficient gastro-pancreatic neuronal signalling. In fact, Kirchgessner & Gershon (1991) demonstrated that injection of a retrograde tracer into rat pancreas labelled neurons in the myenteric plexus of the antrum of the stomach and the proximal part of the duodenum, suggesting the possibility that neurons in the upper digestive tract exercise control of the pancreas. However, our data from experiments with an extract of oxyntic mucosa seem to support the view that the putative gastric insulinotrophic and glucagon-suppressing effect is hormonally and not neurally mediated. It should be noted that our extract originated from the oxyntic mucosa and not from the antral part of the stomach. Furthermore, the active principle was found to potentiate glucose-induced insulin release in a dose-dependent manner and to be inactivated by trypsin and leucine-aminopeptidase. These data largely exclude any non-specific effects of the extracts and are suggestive of the active principle being of a peptide nature.
Glucagon secretion
Gastrectomy also induced marked disturbances in glucagon secretion. The well-known glucose-induced suppression of glucagon secretion was almost abolished by gastrectomy but not by food deprivation (cf. Figs 1 and 2). The gastrectomy-evoked failure of glucose to suppress glucagon secretion in vivo could not be reproduced in experiments with islets from gastrectomized mice. Rather, in fact, glucose evoked a stronger suppression of glucagon release in these islets than in islets from normal freely fed mice. This was especially notable since islets from gastrectomized mice released larger amounts of glucagon in response to low glucose (1 mmol l−1) (cf. Fig. 6) than the control islets. Such a pattern of glucagon release in islets isolated from gastrectomized mice is suggestive of the loss of a factor in these mice that normally restrains glucagon secretion. The absence of this factor might thus render the glucagon secretory machinery more sensitive to inhibition by glucose in analogy with the effect of its presence to increase the sensitivity of the insulin-releasing action of glucose. The mechanisms behind the gastrectomy-induced failure of glucose to suppress glucagon secretion in vivo is less clear. An increased activity in gastrectomized mice of adrenegic nerves innervating the glucagon-producing α-cells (Lundquist & Ericsson, 1978) might be a possible explanation, since catecholamines are known to potently stimulate glucagon secretion in mice (Skoglund et al. 1987). Moreover, insulin is reportedly a powerful inhibitor of glucagon release (Maruyama et al 1984) and it is possible that the difference between gastrectomized and intact mice with respect to the in vivo glucagon response can be explained by differences in the amount of insulin released by glucose, since there was a marked impairment, both in vivo and in vitro, of glucose-induced insulin release in gastrectomized animals. Furthermore, the difference is unlikely to be explained by vagal impairment since vagotomy did not alter the glucagon response to the different secretagogues. Gastrectomy, however, suppressed the glucagon response to carbachol both in vivo and in vitro. Since cholinergic stimulation after gastrectomy was associated with an enhanced insulin release, an inhibitory effect of insulin on glucagon release in this situation is not inconceivable. Although fasted mice also displayed a reduced glucagon response to cholinergic stimulation, there was no concomitant increase in insulin secretion in these animals in contrast to what was seen after gastrectomy. Hence, the extent of the insulin-evoked inhibition of glucagon secretion seems to vary with the experimental situation and cannot fully explain the disturbed glucagon secretion seen after gastrectomy. This was further emphasized in experiments conducted with cyclic AMP-activating secretagogues. Here an increased glucagon secretion in the different experimental groups was accompanied by increased insulin secretion, an effect more pronounced in islets isolated from gastrectomized mice than from sham-operated mice. Such a cyclic AMP-induced exaggeration of insulin and glucagon secretion in gastrectomized mice is compatible with an enhanced sensitivity to cyclic AMP-activating secretagogues in both insulin and glucagon cells. This may reflect the lack of a cyclic AMP-activating gastric factor, which normally exerts a ‘tonic’ influence on the islets. Such an assumption might also be inferred from our observation that the inhibitory action of the oxyntic mucosal extract on glucagon release was exerted at quite low concentrations and had a ‘GLP-like’ effect on islet hormone release (see below).
Conclusion
From the present data we propose that the stomach is the origin of a circulating factor (peptide hormone?) that enhances the glucose-induced insulin response and the glucose-induced suppression of glucagon secretion. Lack of this factor alters the way the insulin and the glucagon cells respond to stimuli. The pattern of the insulin and glucagon responses in mice lacking this gastric factor is such that the factor can be expected to act similarly to GLP-1, which is thought to enhance the insulin response to glucose and suppress the glucagon response (Creutzfeldt & Nauck, 1992; Holst, 1994). The two most likely sources of this putative gastric insulinotrophic agent are the ECL cells and the A-like cells (Sundler & Håkanson, 1988, 1991). There is some experimental evidence to suggest that the so-called gastrin- ECL cell axis is engaged in the control of bone metabolism (Persson & Håkanson, 1991). It is not inconceivable, therefore, that the putative hormone should originate in the A-like cells, which constitute the second largest endocrine cell population in the murine oxyntic mucosa. Also human oxyntic mucosa is known to harbour numerous A-like cells (cf. Bordi & D'Adda, 1991; Capella et al. 1991). Studies are in progress to explore the origin and detailed mechanisms of action of a gastro-insular axis.
Acknowledgments
The skilful assistance of Elsy Ling, Britt-Marie Nilsson and Anna Themner-Persson, and the secretarial help of Eva Björkbom are gratefully acknowledged. This study was supported by grants from the Stig and Ragna Gorthon Foundation, the Swedish Medical Research Council (14X-4286 and 04X-1007), the Swedish Diabetes Association and the Albert Påhlsson Foundation.
References
- Ahrén B, Lundquist I. Effects of vasoactive intestinal polypeptide (VIP), secretin and gastrin on insulin secretion in the mouse. Diabetologia. 1981;20:54–59. doi: 10.1007/BF00253818. [DOI] [PubMed] [Google Scholar]
- Ahrén B, Lundquist I. Glucagon immunoreactivity in plasma from normal and dystrophic mice. Diabetologia. 1982;22:258–263. doi: 10.1007/BF00281302. [DOI] [PubMed] [Google Scholar]
- Bordi C, D'Adda T. Ultrastructural morphometry of gastric endocrine cells. In: Håkanson R, Sundler F, editors. The Stomach as an Endocrine Organ. Amsterdam: Elsevier Science Publishers; 1991. pp. 53–69. Fernström Symposium No. 15. [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Breuer RI, Moses H, III, Hagen TC, Zuckerman L. Gastric operations and glucose homeostasis. Gastroenterology. 1972;62:1109–1119. [PubMed] [Google Scholar]
- Bruss ML, Black AL. Enzymatic microdetermination of glycogen. Analytical Biochemistry. 1978;84:309–312. doi: 10.1016/0003-2697(78)90514-6. [DOI] [PubMed] [Google Scholar]
- Capella C, Finzi G, Cornaggia M, Usellini L, Luinetti O, Buffa R, Solcia E. Ultrastructural typing of gastric endocrine cells. In: Håkanson R, Sundler F, editors. The Stomach as an Endocrine Organ. Amsterdam: Elsevier Science Publishers; 1991. pp. 27–51. Fernström Symposium No. 15. [Google Scholar]
- Creutzfeldt W, Nauck M. Gut hormones and diabetes mellitus. Diabetes Metabolism Reviews. 1992;8:149–177. doi: 10.1002/dmr.5610080206. [DOI] [PubMed] [Google Scholar]
- Dupré J. Influences of the gut on the endocrine pancreas. An overview of established and potential physiological mechanisms. In: Samols E, editor. The Endocrine Pancreas. Raven Press; 1991. pp. 253–281. [Google Scholar]
- Gotoh M, Maki T, Kiyoizumi T, Satomi S, Monaco AP. An improved method for isolation of mouse pancreatic islets. Transplantation. 1985;40:437–438. doi: 10.1097/00007890-198510000-00018. [DOI] [PubMed] [Google Scholar]
- Håkanson R, Liedberg G, Lundquist I. Effect of vagal denervation on insulin release after oral and intravenous glucose. Experientia. 1971;27:460–461. doi: 10.1007/BF02137312. [DOI] [PubMed] [Google Scholar]
- Hedeskov CJ. Mechanism of glucose-induced insulin secretion. Physiological Reviews. 1980;60:442–509. doi: 10.1152/physrev.1980.60.2.442. [DOI] [PubMed] [Google Scholar]
- Heding L. A simplified insulin radioimmunoassay method. In: Donato L, Milhaud G, Sirchis J, editors. Labelled Proteins in Tracer Studies. Brussels: Euratom; 1966. pp. 345–350. [Google Scholar]
- Holst JJ. Glucagonlike peptide 1: a newly discovered gastrointestinal hormone. Gastroenterology. 1994;107:1848–1855. doi: 10.1016/0016-5085(94)90831-1. [DOI] [PubMed] [Google Scholar]
- Jansson L, Hellerström C. Glucose-induced changes in pancreatic islets blood flow mediated by central nervous system. American Journal of Physiology. 1986;251:E644–647. doi: 10.1152/ajpendo.1986.251.6.E644. [DOI] [PubMed] [Google Scholar]
- Järhult J, Ahrén B, Erichsen C, Lundquist I. Improved insulin release to carbachol after vagotomy in rats: an effect due to cholinergic supersensitivity of the α-cells. Acta Physiologica Scandinavica. 1983;118:87–89. doi: 10.1111/j.1748-1716.1983.tb07246.x. [DOI] [PubMed] [Google Scholar]
- Kirchgessner AL, Gershon MD. Innervation and regulation of the pancreas by neurons in the gut. Zeitschrift für Gastroenterologie. 1991;26:230–233. [PubMed] [Google Scholar]
- Lundquist I. Cholinergic muscarinic effects on insulin release in mice. Pharmacology. 1982;25:338–347. doi: 10.1159/000137760. [DOI] [PubMed] [Google Scholar]
- Lundquist I. Islet amyloglucosidase activity: some characteristics, and its relation to insulin secretion stimulated by various secretagogues. Diabetes Research. 1986;3:31–41. [PubMed] [Google Scholar]
- Lundquist I, Ericson LE. β-Adrenergic insulin release and adrenergic innervation of mouse pancreatic islets. Cell Tissue Research. 1978;193:73–85. doi: 10.1007/BF00221602. [DOI] [PubMed] [Google Scholar]
- Lundquist I, Lövdahl R. Effect of fasting on islet lysosomal enzyme activities and the in vivo insulin response to different secretagogues. Hormone and Metabolic Research. 1983;15:11–14. doi: 10.1055/s-2007-1018615. [DOI] [PubMed] [Google Scholar]
- Lundquist I, Panagiotidis G. The relationship of islet amyloglucosidase activity and glucose-induced insulin secretion. Pancreas. 1992;7:352–357. doi: 10.1097/00006676-199205000-00013. [DOI] [PubMed] [Google Scholar]
- McIntyre N, Holdsworth CD, Turner DS. New interpretation of oral glucose tolerance. Lancet. 1964;2:20–21. doi: 10.1016/s0140-6736(64)90011-x. [DOI] [PubMed] [Google Scholar]
- Maruyama H, Hisatomi A, Orci L, Grodsky GM, Unger RH. Insulin within islets is a physiologic glucagon release inhibitor. Journal of Clinical Investigation. 1984;74:2296–2299. doi: 10.1172/JCI111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monstein H-J, Nylander A-G, Salehi A, Chen D, Lundquist I, Håkanson R. Cholecystokinin-A and cholecystokinin-B/gastrin receptor mRNA expression in the gastrointestinal tract and pancreas of the rat and man. Scandinavian Journal of Gastroenterology. 1996;31:383–390. doi: 10.3109/00365529609006415. [DOI] [PubMed] [Google Scholar]
- Muir A. Postgastrectomy syndromes. British Journal of Surgery. 1949;37:165–178. doi: 10.1002/bjs.18003714606. [DOI] [PubMed] [Google Scholar]
- Panagiotidis G, Salehi AA, Westermark P, Lundquist I. Homologous islet amyloid polypeptide: effects on plasma levels of glucagon, insulin and glucose in the mouse. Diabetes Research and Clinical Practice. 1992;18:167–171. doi: 10.1016/0168-8227(92)90142-e. [DOI] [PubMed] [Google Scholar]
- Panagiotidis G, Stenström A, Lundquist I. Influence by β2-adrenoceptor stimulation and glucose on islet monoamine oxidase activity and insulin secretory response in the mouse. Pancreas. 1993;8:368–374. doi: 10.1097/00006676-199305000-00014. [DOI] [PubMed] [Google Scholar]
- Panagiotidis G, Stenström A, Lundquist I. In vivo action of cyclic AMP modulating secretagogues on islet monoamine oxidase activity and insulin release. Endocrine. 1994;2:571–576. [Google Scholar]
- Persson P, Håkanson R. The gastrin-gastrocalcin hypothesis. In: Håkanson R, Sundler F, editors. The Stomach as an Endocrine Organ. Amsterdam: Elsevier Science Publishers; 1991. pp. 341–350. Fernström Symposium No. 15. [Google Scholar]
- Prentki M, Corkey BE. Are the β-cells signaling molecules malonyl-CoA and cytosolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and NIDDM? Diabetes. 1996;45:273–283. doi: 10.2337/diab.45.3.273. [DOI] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM. Ca2+, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiological Reviews. 1987;67:1185–1248. doi: 10.1152/physrev.1987.67.4.1185. [DOI] [PubMed] [Google Scholar]
- Rerup C, Lundquist I. Blood glucose level in mice. I. Evaluation of a new technique of multiple serial sampling. Acta Endocrinologica (Copenhagen) 1966;52:357–367. [PubMed] [Google Scholar]
- Sako Y, Grill V. A 48-hour lipid infusion in the rat time-dependently inhibits glucose-induced insulin secretion and B-cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology. 1990;127:1580–1589. doi: 10.1210/endo-127-4-1580. [DOI] [PubMed] [Google Scholar]
- Skoglund G, Lundquist I, Ahrén B. α1- and α2-adrenoceptor activation increase plasma glucagon levels in the mouse. European Journal of Pharmacology. 1987;143:83–88. doi: 10.1016/0014-2999(87)90737-0. [DOI] [PubMed] [Google Scholar]
- Sudo T, Ishiyama K, Takemoto M, Kawamura M, Umemuar H, Shiraha S, Kuyama T, Suzuki T, Tobe T. Pancreatic endocrine function after total gastrectomy and truncal vagotomy. American Journal of Surgery. 1982;144:539–544. doi: 10.1016/0002-9610(82)90576-1. [DOI] [PubMed] [Google Scholar]
- Sundler F, Håkanson R. Peptide hormone-producing endocrine/paracrine cells in the gastro-entero-pancreatic region. In: Björklund A, Hökfelt T, Owman C, editors. The Peripheral Nervous System. Handbook of Chemical Neuroanatomy. Vol. 6. Amsterdam: Elsevier Science; 1988. pp. 219–295. [Google Scholar]
- Sundler F, Håkanson R. Gastric endocrine cell typing at the light microscopic level. In: Håkanson R, Sundler F, editors. The Stomach as an Endocrine Organ. Amsterdam: Elsevier Science Publishers; 1991. pp. 9–26. Fernström Symposium No. 15. [Google Scholar]
- Tobe T, Kouchi M, Tanimura H, Huang CH. Hyperglycemia after gastrectomy as a prediabetic state. Archives of Surgery. 1967;94:835–840. doi: 10.1001/archsurg.1967.01330120090016. [DOI] [PubMed] [Google Scholar]
- Zawalich WS, Diaz VA, Zawalich KC. Stimulatory effects of cholecystokinin on isolated perifused islets inhibited by potent and specific antagonist L 364718. Diabetes. 1988;37:1432–1437. doi: 10.2337/diab.37.10.1432. [DOI] [PubMed] [Google Scholar]
- Zhou Y-P, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. Journal of Clinical Investigation. 1994;93:870–876. doi: 10.1172/JCI117042. [DOI] [PMC free article] [PubMed] [Google Scholar]