

Abstract
The effect of insulin on K+ currents was studied with enzymatically dispersed ventricular myocytes from insulin-deficient (type I) diabetic rats. Diabetic conditions were induced by a single intravenous injection of streptozotocin (100 mg kg−1) given 8-13 days before the experiments. Measurements of plasma glucose and insulin levels confirmed the diabetic status of the animals.
A Ca2+-independent transient outward K+ current, It, and a slowly inactivating, quasi-steady-state current, Iss, which are depressed in diabetic myocytes, could be restored by exposure to 1, 10 or 100 nM insulin. This was only observed after a delay of 5-6 h, although an insulin exposure of only 1 h was sufficient to initiate its stimulatory action on It and Iss. The stimulatory effect of insulin on these K+ currents was prevented by 2 μM cycloheximide, which in itself had no direct effect on these currents.
Disruption of the actin microfilament network with 1 μM cytochalasin D (CD) also prevented the stimulatory effect of 100 nM insulin on both It and Iss. Since CD was added 1 h after insulin, inhibitory effects on insulin signalling were ruled out. Adding CD (1 μM) 5-9 h after insulin, when currents were already augmented, had no effect (up to 50 min exposure). Incubating control cells for 6-10 h with 1 μM CD had no effect on any of the currents measured.
Stabilization of the actin network by pre-exposure to 2·5 μM phalloidin restored the stimulatory effect of insulin, in the continued presence of CD, ruling out any effects of CD on components other than the cytoskeleton.
The stimulatory effect of insulin was also prevented by incubating cells with insulin in the presence of the microtubule-disrupting agent colchicine (5 μM).
These results suggest that the insulin-mediated augmentation of K+ currents in diabetic myocytes requires protein synthesis, possibly of K+ channels, as well as an intact cytoskeleton. The possibility that newly formed channels translocate to the plasma membrane in a process dependent on different elements of the cytoskeleton is discussed.
We have recently reported (Shimoni et al. 1998) that insulin reverses the depression of ventricular K+ currents which occurs in an insulin-deficient model of diabetes. The stimulating effect of insulin requires 5-6 h and is blocked by inhibition of mitogen-activated protein (MAP) kinase, a key enzyme regulating transcription (Saltiel, 1996). This suggests that new K+ channels are synthesized in myocytes from insulin-deficient diabetic rats, upon re-exposure to insulin.
A key issue in the study of cellular functions is the intracellular sorting, processing and targeting of proteins involved in a variety of cellular activities (Griffiths & Simons, 1986; Rothman, 1994; Rothman & Wieland, 1996), including ion transfer. For example, it has recently been reported (Makhina & Nichols, 1998) that regulatory subunits of the ATP-dependent K+ channel and the channel itself show independent trafficking to the plasma membrane. It has also been shown (Nagaya & Papazian, 1997) that voltage-dependent K+ channel subunits complete their assembly in the endoplasmic reticulum, prior to being transported to the plasma membrane. Furthermore, it has been suggested that alternative splicing leads to ion channel isoforms containing (different) membrane targeting signals (Ponce et al. 1997). However, the mechanisms whereby ion channels reach the plasma membrane following their synthesis and assembly are unknown.
The trafficking of newly formed proteins from the trans-Golgi network to the cell membrane has been suggested to depend on two components of the cytoskeleton, the actin microfilament system and the microtubule network (Nelson, 1992). These may serve a protein-transporting role either in parallel (Nelson, 1992) or in sequence (Fath et al. 1993). Our results demonstrating an enhancement of K+ currents in diabetic myocytes within hours of exposure to insulin (Shimoni et al. 1998) enabled us experimentally to address some of these issues.
The on-going activity of many ion channels has been shown to depend on a normal cytoskeleton, based on the sensitivity of channel activity to disruption of different elements of the cytoskeleton. Cytochalasin D, a fungal toxin which specifically inhibits actin polymerization (Cooper, 1987) and thus disrupts microfilament organization, can affect Na+ channel gating (Undrovinas et al. 1995), inward rectification of K+ channels (Mazzanti et al. 1996), and several aspects of KATP channel function (Brady et al. 1996; Terzic & Kurachi, 1996; Furukawa et al. 1996). Disruption of the microtubules with colchicine has also been found to affect the function of L-type calcium channels (Johnson & Byerly, 1993; Galli & DeFelice, 1994). This sensitivity to cytoskeletal disruption is assumed to reflect ‘architectural’ requirements of channel function, based on the known interactions between the cytoskeleton and the plasma membrane (Luna & Hitt, 1992). This interaction presumably controls the correct spatial orientation of the channels and their components. In addition, there is also evidence suggesting that the delivery, expression or clustering of ion channels in the cell membrane can be affected by changes in the cytoskeleton (reviewed in Janmey, 1998).
In the present study we set out to investigate whether the stimulatory effect of insulin on potassium currents in ventricular myocytes from diabetic rat hearts (Shimoni et al. 1998) is dependent on actin microfilaments and the microtubular system. We find that both cytochalasin D and colchicine prevent the stimulation of these potassium currents by insulin. Our results, therefore, demonstrate for the first time that an insulin-stimulated augmentation of ion currents, which presumably leads to synthesis of new channels, is dependent on the integrity of the cytoskeleton.
METHODS
All the experiments described here were done in accordance with the guidelines of the Animal Care Committee of the University of Calgary.
Experimental model
Ionic currents were measured from ventricular myocytes obtained either from control Sprague-Dawley (200-250 g) rats or from rats made diabetic by a single i.v. injection of streptozotocin (STZ, 100 mg kg−1), given 8-13 days prior to cell isolation. Measurements of plasma glucose and insulin levels confirmed the diabetic status of these animals.
Cell isolation
Animals were heparinized (2400 units kg−1i.p.), anaesthetized by methoxyflurane inhalation and then killed by cervical dislocation. The hearts were removed and the aortas cannulated on a Langendorff apparatus. The hearts were then perfused (at 37 °C; 70 cmH2O pressure) for 5 min with a solution containing (mM): 121 NaCl, 5.4 KCl, 2.8 sodium acetate, 1 MgCl2, 1 CaCl2, 5 Na2HPO4, 24 NaHCO3 and 5 glucose. This was followed by a 10 min perfusion with the same solution with CaCl2 omitted, followed by 7-8 min in the same calcium-free solution to which the following were added: 40 μM CaCl2, 10 units ml−1 collagenase (Yakult Honsha, Tokyo), 0.01 mg ml−1 protease (Sigma Type XIV) and 20 mM taurine.
The free wall of the right ventricle was then dissected and cut into smaller pieces for further incubation (at 37 °C) in calcium-free solution to which was added 0.1 mM CaCl2, 50 units ml−1 collagenase, 0.1 mg ml−1 protease, 20 mM taurine and 10 mg ml−1 albumin. Cells were collected over the next 30-60 min, and stored in calcium-free solution with no enzymes to which was added 0.1 mM CaCl2, 20 mM taurine and 5 mg ml−1 albumin.
Recording and measurements
Cells were placed in a small chamber on the stage of an inverted microscope, and perfused with a solution containing (mM): 150 NaCl, 5.4 KCl, 1.0 CaCl2, 1.0 MgCl2, 5 Hepes and 5.5 glucose, adjusted to pH 7.4 with NaOH. Currents were recorded (at 20-22 °C) using conventional whole-cell suction electrode voltage clamp methods. Electrode resistance was 2-5 MΩ. The filling solution contained (mM): 110 potassium aspartate, 30 KCl, 4 Na2ATP, 1 MgCl2, 10 EGTA, 1 CaCl2 and 5 Hepes, adjusted to pH 7.2 with KOH. This solution produces a junction potential of ≈10 mV, which was corrected for. Series resistance (Rs) was minimized by using low resistance electrodes, and by (30-50 %) compensation. Recordings with Rs of over 10 MΩ were discarded. For one series of experiments longer recording times were required. Since Rs tends to change with time, leading to changes in the magnitude of the currents being monitored, it was important to control for such drifts in Rs. This was done by interspersing test pulses periodically to monitor for changes. Small drifts in Rs could be corrected by slight adjustments of the negative pressure used in this recording method. Changes of more than 10 % led to discarding of the data.
Three potassium currents were measured in all experiments: a calcium-independent transient outward current It, which was measured in the presence of 0.3 mM CdCl2 in the perfusate and 10 mM EGTA in the pipette solution, to ensure inhibition of the calcium-dependent outward current; a slowly inactivating quasi-steady-state current Iss; and the background inward-rectifier current IK1. It was measured as the peak outward current, rather than as the difference current between peak and steady-state values. This is justified based on the following: Iss activates much more slowly than It (Apkon & Nerbonne, 1991) so that little Iss is present when peak current is measured; our earlier work (Shimoni et al. 1995) showed that the same results were obtained by measuring It as either peak current or as the difference current; in addition, It and Iss are both altered in insulin-deficient conditions and in response to insulin. These changes are not always present to the same degree, so that relating It to the steady-state level may be erroneous. Unfortunately there is no suitable pharmacological tool for inhibiting Iss, in order to separate it from It. The comparative statistics were done for current densities at +50 mV, at which no interference by the sodium current is present. IK1 was measured as the current elicited by voltage steps to -110 mV. Since this current was found to be unaffected by any of the treatments used, pulses to -110 mV could be used as indicators of drifts in Rs. An increase in Rs produced a reduction in the current elicited by these negative pulses. When this reduction was larger than 10 % (in the longer duration recordings) the cell was abandoned. In all cases current magnitudes were divided by cell capacitance, giving current densities, enabling comparisons between cells of different sizes.
Drugs and reagents
Cycloheximide, cytochalasin D, colchicine and phalloidin were purchased from Sigma. Concentrated stocks were made and aliquots were frozen. Cytochalasin D was dissolved in DMSO and diluted to a final concentration of 1 μM. This contained 0.03 % DMSO, which had no effect on its own. The other reagents were water soluble.
Statistics
One-way ANOVA (followed by a Student-Newman-Keuls multiple comparisons test) or Student's unpaired t test were used for statistical evaluation of differences between groups, with P < 0.05 considered significant. It should be noted that due to variation in the degree of insulin deficiency induced by STZ treatment, each group of treatments contained its own control (untreated) group. On each experimental day, recordings were made from several untreated cells, followed by recordings from cells subjected to a given treatment. Data obtained on different days were pooled, for a given treatment.
RESULTS
Effects of insulin
Our earlier work (Shimoni et al. 1998) showed that when myocytes from diabetic animals are exposed to 100 nM insulin for 5-9 h, the attenuation in It is fully reversed, and that of Iss partially reversed. Since higher concentrations of insulin may produce effects by stimulating insulin-like growth factor receptors in addition to insulin receptors (Steele-Perkins et al. 1988), it was important to repeat our results with a lower insulin concentration. In our first experiments (results not shown), we repeated the same protocols using 1 or 10 nM insulin. In 14 cells exposed to 1 nM insulin for 5-9 h, It density was 28.2 ± 2.4 pA pF−1 (mean ±s.e.m.), and Iss density was 8.8 ± 0.5 pA pF−1. These values are significantly (P < 0.05) larger than in untreated cells, in which mean It density was 17.6 ± 1.1 pA pF−1 (n = 30 cells), and Iss density was 6.0 ± 0.2 pA pF−1.
In addition, in the context of understanding the mechanism by which insulin increases functional ion current density at the cell membrane (see below), we investigated the required duration of exposure to insulin. We found that if cells were exposed to 10 nM insulin for 1 h, followed by removal of insulin (by centrifugation, removal of the supernatant and resuspension of the cells in insulin-free storage solution), It and Iss were still significantly (P < 0.001) augmented, when measured 6-10 h after the initial insulin exposure. Thus, at +50 mV, It density (in diabetic myocytes) was 18.4 ± 1.2 pA pF−1 (n = 26) in the absence of insulin, and 25.3 ± 1.4 pA pF−1 (n = 22) after a 1 h exposure to 10 nM insulin (recorded 6-10 h after exposure). The corresponding values for Iss were 7.5 ± 0.4 and 9.6 ± 0.7 pA pF−1. An example from two cells is shown in Fig. 1.
Figure 1. Effects of 10 nM insulin on K+ currents in ventricular myocytes from STZ-diabetic rats.
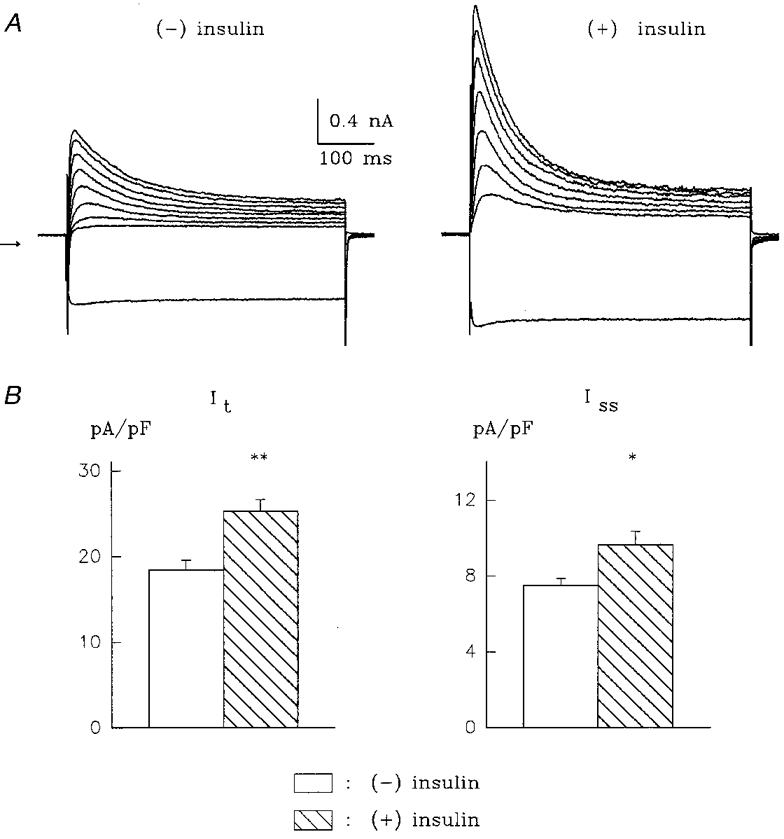
Insulin was applied for 1 h and subsequently removed by centrifugation and resuspension of myocytes in insulin-free solution. A, current traces in response to voltage steps from -80 to -110 mV (lowest trace) and from -80 to potentials ranging from -20 to +50 mV (in 10 mV steps) before (left, (-) insulin) and 6 h after (right, (+) insulin) the exposure to insulin. Zero current level is indicated by the arrow to the left. Cell capacitances were 65.8 (left) and 62.8 pF (right). B, mean +s.e.m. for It (left) and Iss (right) obtained from 26 cells in the absence of insulin and from 22 cells recorded 5-9 h after a 1 h exposure to 10 nM insulin. *P < 0.01, **P < 0.001.
This result was of importance, since it enabled us to apply several pharmacological agents (see below) 1 h after exposure to insulin, thereby eliminating concerns about possible interference of these agents with insulin binding and activation of the signalling cascade which is responsible for augmentation of It and Iss density.
Involvement of protein synthesis
Insulin is known to stimulate protein synthesis in ventricular myocytes from diabetic rats (Braun & Severson, 1992). Thus, the stimulatory effect of insulin on K+ currents (Fig. 1) may be due to the synthesis of new channels (for both currents). This notion arose from our previous findings (Shimoni et al. 1998) which showed that at least 6 h must elapse before insulin stimulates the K+ currents and that this stimulatory effect can be blocked by inhibiting the activation of MAP kinase, a key mediator of transcriptional control by insulin (Saltiel, 1996). In order to test this hypothesis more directly, we examined the effect of insulin after exposing cells to 2 μM cycloheximide, an inhibitor of protein synthesis.
Cycloheximide (2 μM) was added 1 h after the addition of 100 nM insulin, and currents were measured 5-9 h later. In 24 cells, it was found that cycloheximide completely abolished the enhancement of both It and Iss by insulin. In this group, It density at +50 mV was 17.2 ± 0.9 pA pF−1 in untreated cells (n = 42), 20.7 ± 1.1 pA pF−1 in insulin-treated cells (significantly larger, P < 0.05, n = 35) and 17.3 ± 1.2 pA pF−1 in cells exposed to insulin and cycloheximide (n = 24). The corresponding values for Iss were 6.7 ± 0.3, 8.5 ± 0.5 and 6.9 ± 0.5 pA pF−1. Exposure of cells to 2 μM cycloheximide alone did not alter any of the K+ currents measured. The mean values in the absence (n = 16) and presence (n = 14) of cycloheximide, respectively, were: It, 16.3 ± 1.0 and 15.8 ± 1.2 pA pF−1; Iss, 4.5 ± 0.2 and 4.8 ± 0.4 pA pF−1; IK1, -8.3 ± 0.6 and -9.3 ± 0.6 pA pF−1. These results, shown in Fig. 2, show that the stimulatory effect of insulin on It and Iss is dependent on protein synthesis.
Figure 2. Abolition of the stimulatory effects of insulin on K+ currents by cycloheximide.
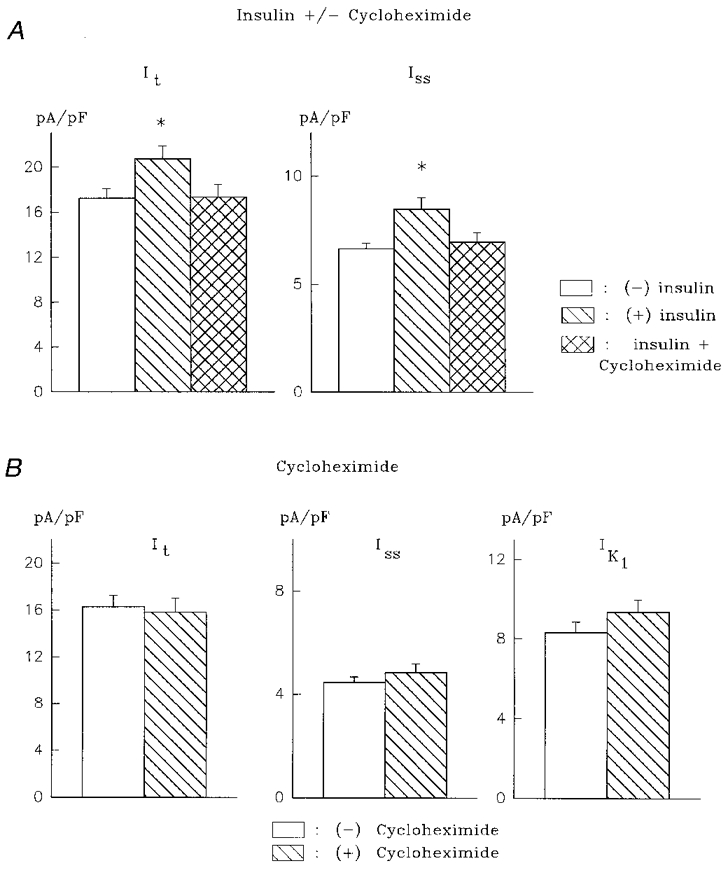
A, mean +s.e.m. of It (left) and Iss (right) in the absence of insulin (n= 42), after 5-9 h with 100 nM insulin (n= 35), and in the presence of 100 nM insulin and 2 μM cycloheximide (n= 24). Cycloheximide was added 1 h after insulin. *P < 0.05. B, cycloheximide (2 μM) alone (for 5-8 h, n= 14) has no effect on It (left), Iss (centre) and IK1 (right), in comparison to control (n= 16). Absolute values are given for IK1 here and in other figures.
Involvement of the cytoskeleton
We next examined whether the enhancement of It and Iss current density by insulin requires the participation of the actin microfilament network, which has been implicated in the control of ion channel function (Undrovinas et al. 1995; Brady et al. 1996; Mazzanti et al. 1996). Cytochalasin D inhibits actin polymerization and thereby shifts the dynamic balance existing between monomeric and filamentous actin towards the monomeric form (Cooper, 1987). Glucose transporters (GLUT-4) translocate to the cell membrane within minutes after insulin exposure. This translocation process depends on actin microfilaments of the cytoskeleton (Tsakiridis et al. 1994). The disruption of this intracellular actin network with cytochalasin D prevents GLUT-4 translocation in response to insulin, without affecting basal glucose transport.
Cytochalasin D elicits maximal effects on disassembly of the actin network in L6 muscle cells within 1-2 h after exposure, at concentrations of 0.1-10 μM (Tsakiridis et al. 1994), although other work suggests that cytochalasin D may act within minutes in some cells (Gottlieb et al. 1993).
In our experiments, we added 1 μM cytochalasin D 1 h after the addition of insulin, ensuring no interference with insulin binding or signal transduction. In 18 cells, the addition of cytochalasin D completely blocked the effects of insulin on both It and Iss. The current densities for It in the absence of insulin, with insulin alone, and with insulin and cytochalasin D were 18.4 ± 1.0, 25.4 ± 1.5 and 17.7 ± 1.6 pA pF−1, respectively. The corresponding values for Iss were 6.0 ± 0.2, 7.9 ± 0.4 and 5.9 ± 0.3 pA pF−1. Insulin had no effect on IK1, and the combination of insulin and cytochalasin D also did not alter IK1, suggesting that there are no non-selective effects of cytochalasin D in these myocytes (such as increased membrane leakiness). These results are shown in Fig. 3, which also shows the current- voltage relationship for It in the presence of insulin, with and without cytochalasin D.
Figure 3. Inhibition of the effects of insulin by cytochalasin D.
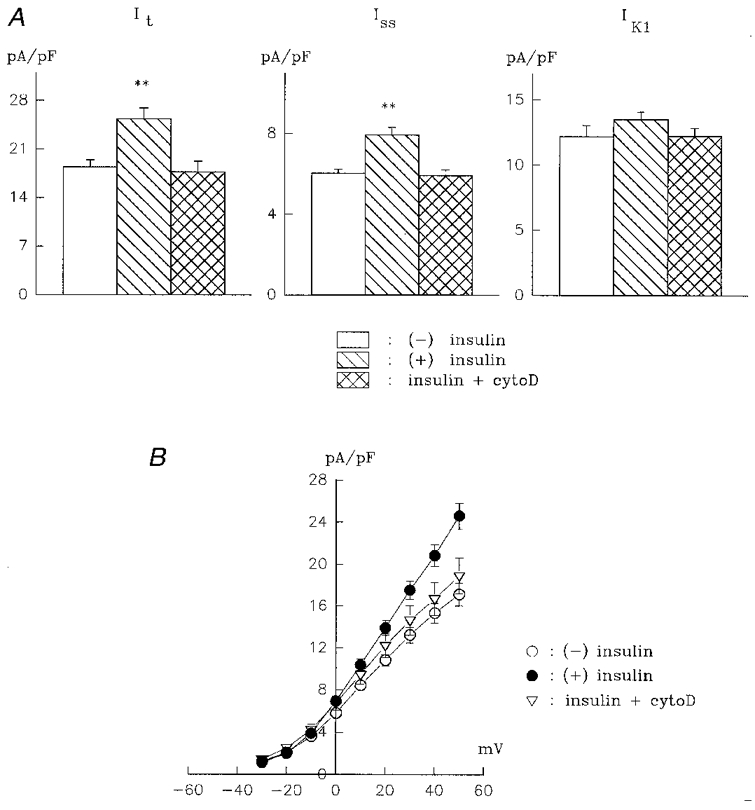
A, means +s.e.m. for It (left), Iss (centre) and IK1 (right) are shown in the absence of insulin (n= 45), after 5-9 h in 10 nM insulin (n= 16), and after 5-10 h in 10 nM insulin + 1 μM cytochalasin D (cytoD, n= 18). Cytochalasin D was added 1 h after insulin (**P < 0.001). Note that cytochalasin D abolishes the effects of insulin on It and Iss yet does not affect IK1. B, a current-voltage relationship for It (peak outward current) in the absence of insulin (n= 34), with 10 nM insulin (n= 23), and with insulin and cytochalasin D (n= 17).
Our protocol, in which cytochalasin D was added 1 h after insulin, allowed us to infer that cytochalasin D did not interfere with any of the early stages of insulin signalling. However, it is still possible that cytochalasin D could have interfered with transcription or synthesis of the K+ channels, in addition to disruption of the cytoskeleton, which may occur in some circumstances (Cooper, 1987). In order to rule out such possible additional effects of cytochalasin D, we carried out experiments in which the action of cytochalasin D is pre-empted by phalloidin, a compound that stabilizes the cytoskeleton (Cooper, 1987). It has been shown that phalloidin can prevent the action of cytochalasins on K+ channel activity in the collecting duct (Wang et al. 1994). Since phalloidin enters cells only poorly (Cooper, 1987), our experiments consisted of a prolonged (4 h) pre-exposure to phalloidin (2.5 μM), followed by 1 h exposure to insulin, and then by addition of 1 μM cytochalasin D. All recordings were done at least 5 h after exposure of cells to insulin. The results show that phalloidin prevented the inhibitory action of cytochalasin D on the stimulation of It and Iss by insulin, and restored the normal effect of insulin. Thus, mean It density (at +50 mV) in the presence of insulin, cytochalasin D and phalloidin was 22.8 ± 1.7 pA pF−1 (n = 22). The mean Iss density under these conditions was 8.3 ± 0.7 pA pF−1. These values were significantly larger than those in the untreated cells (P < 0.005), and not significantly different from cells treated with insulin alone. These results, as well as a summarizing histograms, are shown in Fig. 4.
Figure 4. Phalloidin prevents the inhibition by cytochalasin D of the stimulatory insulin effect on K+ currents.
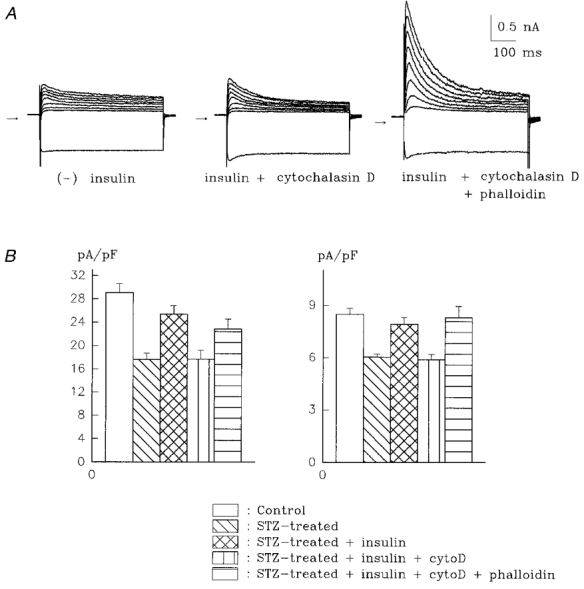
A, current traces (same voltages as in Fig. 1) from cells in the absence of insulin (left), 6 h after exposure to 10 nM insulin in the presence of 1 μM cytochalasin D (centre), and with insulin + cytochalasin D after a 4 h pre-incubation with 2.5 μM phalloidin (right). Note that cytochalasin D prevents insulin from augmenting the peak outward and steady-state currents (centre), but not if phalloidin is present (right). Cell capacitances are 51.6 (left), 57.4 (centre) and 48.8 pF (right). B, summary histogram showing current densities for It (left) and Iss (right) in control myocytes (n= 34), in untreated STZ-diabetic myocytes (n= 30), and in diabetic myocytes treated with 10 nM insulin (n= 16), with insulin + cytochalasin D (n= 18), and with the combination of insulin, cytochalasin D and phalloidin (n= 22).
This last result is significant for two reasons. First, it demonstrates that the stabilization of the actin microfilament system with phalloidin prevents subsequent disruption by cytochalasin D and normalizes the augmenting effects of insulin on the K+ currents. In addition, this result serves as a crucial control, indicating that cytochalasin D does not have non-specific actions on components other than the cytoskeleton. In other words, this result shows that cytochalasin D does not interfere with any non-microfilament-associated actions of insulin (e.g. with K+ channel assembly or processing) since the normal action of insulin on K+ currents is obtained even with cytochalasin D present, as long as the cells are pretreated with phalloidin.
In another set of experiments (results not shown), we ruled out the unlikely possibility that the prolonged exposure to phalloidin somehow upregulates both K+ currents, thereby producing the results shown in Fig. 4. In cells exposed to 2.5 μM phalloidin for 5-9 h, no changes were seen in any of the three K+ currents. It density was 16.4 ± 1.9 pA pF−1 (n = 16) in the absence, and 17.1 ± 1.8 pA pF−1 (n = 13) in the presence of phalloidin. The corresponding values for Iss were 4.9 ± 0.4 and 5.2 ± 0.3 pA pF−1; for IK1 they were -11.3 ± 0.7 and -10.2 ± 0.4 pA pF−1.
An important issue in the context of our hypothesis regarding the mechanism of current augmentation by insulin (see Discussion) relates to the timing of the effects of cytochalasin D. In an additional series of experiments we examined whether the addition of cytochalasin D 5-9 h after insulin, when It and Iss are already augmented, can reverse the effects of insulin. In 9 of 10 cells obtained from diabetic rats, addition of 1 μM cytochalasin D, between 5 and 9 h after insulin, did not diminish either current. In one cell there was a 10 % decline, due to increased series resistance. Two examples are shown in Fig. 5. Figure 5A shows current amplitudes against time, after addition of cytochalasin D, while Fig. 5B shows superimposed current traces from another cell, before and 16 min after addition of cytochalasin D. Figure 5A and B illustrates the lack of a rapid change in current amplitude after it has already been augmented by insulin (see Discussion). The lack of effect of cytochalasin D was apparent for up to the longest exposure time of 50 min (1 cell). In the other cells, no effect was seen for up to 12-18 min (bath volume exchanged within 20 s).
Figure 5. Lack of acute effect of cytochalasin D.
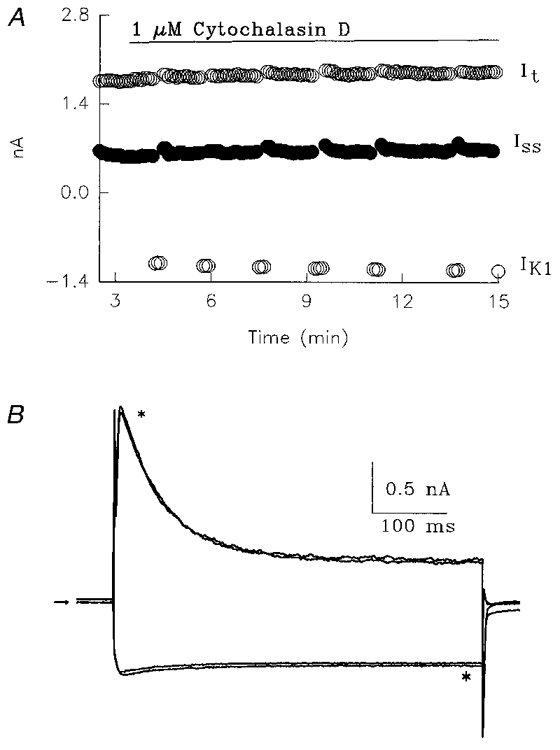
Representative results obtained from two cells from diabetic rats, exposed for > 5 h to 100 nM insulin, and then to 1 μM cytochalasin D (CD). A, this cell was preincubated with 100 nM insulin for 6 h. Current amplitudes are plotted vs. time, showing no changes in It,Iss or IK1 after adding CD to the bath. Bath volume was exchanged within 20 s. B, superimposed current traces (different cell, 8 h in insulin), in response to pulses from -80 mV to either +50 or -110 mV, before and 16 min after (*) addition of 1 μM CD. Arrow denotes zero current level.
Since longer exposure times to cytochalasin D were necessary to block the effects of insulin, we also tested whether long exposure times to cytochalasin D had any effect on the currents measured. In a subsequent set of experiments, current densities were compared in myocytes from control rats (no insulin treatment), in the absence or presence of 1 μM cytochalasin D (4-9 h). Figure 6 shows that prolonged exposure to this agent has no affect on any of the K+ currents measured.
Figure 6. Lack of effect of prolonged exposure to cytochalasin D.
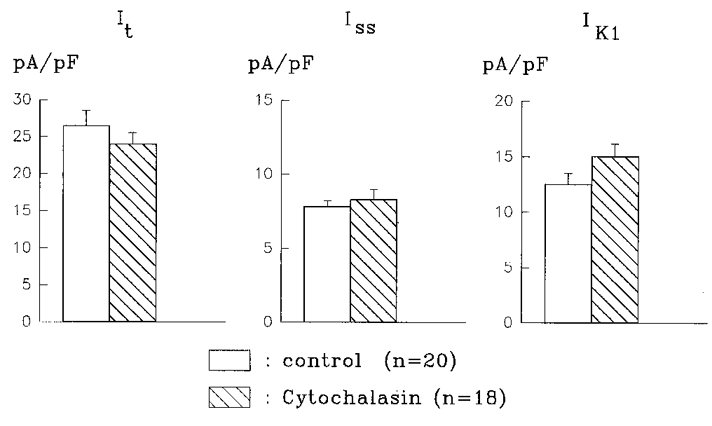
Histograms show the mean current densities obtained from control myocytes or following 4-9 h in 1 μM CD. Densities for It (left), Iss (middle) and IK1 (right) are not significantly (P > 0.05) altered by CD.
In the final set of experiments we investigated whether the integrity of the microtubular system is also required for the enhancement of It and Iss by insulin. Cells from diabetic rats were incubated either with 100 nM insulin alone (for at least 5 h), or with insulin and colchicine (5 μM). Colchicine was added 1 h after insulin, and recordings made between 5 and 10 h after addition of insulin. The treatment with colchicine was also found to abolish the enhancement of both It and Iss by insulin, as shown in Fig. 7. No deleterious effects of colchicine were observed, since the cells maintained a normal resting potential, and IK1 was unaffected. At -110 mV, the mean density of IK1 was 12.3 ± 0.7 (n = 28), 11.9 ± 0.6 (n = 29) and 11.0 ± 0.8 pA pF−1 (n = 21) in untreated, insulin-treated, and insulin + colchicine-treated cells, respectively.
Figure 7. Block of insulin effects by colchicine.
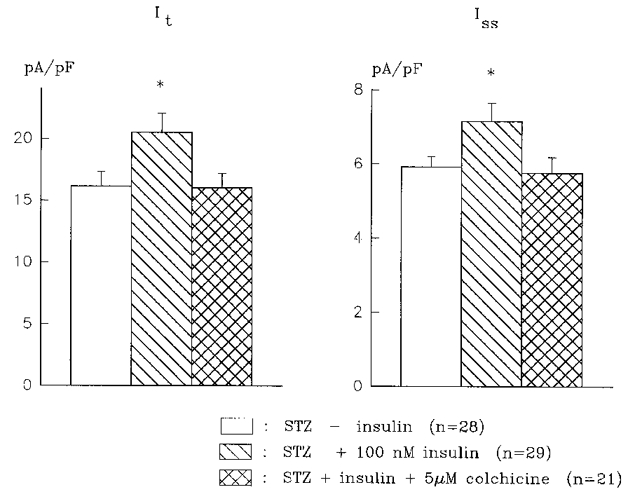
Summary histograms showing that the augmentation of both It (left) and Iss (right) by 100 nM insulin is abolished by 5 μM colchicine. Cells were incubated for at least 5½ h with insulin. Colchicine was added 1 h after insulin. The density of both currents in the presence of insulin is significantly (* P < 0.05) larger than the density in the absence of insulin, or the density in insulin + colchicine.
DISCUSSION
Summary of findings
Our present results further characterize our previous findings by showing that depressed K+ currents in ventricular myocytes from insulin-deficient rats can be restored by 1-10 nM insulin (in contrast to 100 nM used previously). First, it is shown (Fig. 1) that although these effects can only be measured with a delay of at least 5-6 h, an insulin exposure time of only 1 h is sufficient to initiate the stimulatory effect on K+ currents. This effect is sensitive to cycloheximide (Fig. 2), implying the involvement of protein synthesis, presumably of new K+ channels. Our most significant result is that the stimulatory effect of insulin on It and Iss is sensitive to cytochalasin D (Fig. 3) and colchicine (Fig. 7), agents that disrupt the actin microfilament and microtubule networks, respectively. The inhibitory effect of cytochalasin D can be blocked by an actin microfilament stabilizer, phalloidin, so that the normal stimulatory effect of insulin can be restored (Fig. 4). Cytochalasin D has no effect once the currents have already been augmented by insulin, nor does it alter currents in control myocytes, even with prolonged (4-9 h) exposure.
Implications and significance
Our results suggest that K+ currents are attenuated in the insulin-deficient diabetic state due to a reduction in the number of active channels. Addition of insulin stimulates the synthesis of new channels, which are presumably translocated to the cell membrane. This process depends on both the microtubule and the actin microfilament systems, based on the sensitivity of the insulin effects to cytochalasin D and colchicine.
However, this interpretation must be made with caution. Cytochalasin D disruption of the actin cytoskeleton has been reported to interfere with the initial events in insulin signalling, including activation of MAP kinase (Tsakiridis et al. 1997). Since in our experiments insulin addition to the cardiomyocytes preceded cytochalasin D by 60 min, it would appear that the subsequent cytochalasin D-sensitive steps involve more distal events, possibly the movement of newly synthetized K+ channels to the plasma membrane. Other possible effects of cytochalasin D, unrelated to cytoskeletal disruption, were ruled out by the finding (Fig. 4) that insulin exerted its normal stimulating action in the presence of cytochalasin D when the cytoskeleton was stabilized with phalloidin. Nevertheless, it is still possible that cytochalasin D inhibits (channel) protein synthesis, if this process is itself dependent on an intact cytoskeleton. Cytochalasin D has been shown by Ornelles et al. (1986) to release mRNA from actin microfilaments, but these results were obtained at a concentration (10 μM) considerably higher than used here (their KD values were 14 and 100 μM, whereas in the present experiments only 1 μM cytochalasin was used).
Earlier work on ion channels had shown that a polarized distribution of ion channels in Xenopus oocytes can also be disrupted by anticytoskeletal agents (Peter et al. 1991), although it was unclear from that work if the distribution of mRNA or of processed protein was affected by the disruption of the cytoskeleton. Earlier work showed that trafficking of transporting proteins depends on the integrity of the cytoskeleton (Bradbury & Bridges, 1994; Janmey, 1998). Our results show that this is also the case for voltage-dependent ion channels.
The present results differ from previous reports which showed that ion channel function can be affected by disruption in the microfilament or microtubule systems (Galli & Defelice, 1994; Undrovinas et al. 1995; Furukawa et al. 1996). Previous work dealt with cytoskeletal disruption affecting channels already in place. Our results suggest that such disruption prevents the targeting of new channels to the cell membrane following their formation. When cytochalasin D is added acutely to cells from diabetic animals, after at least 5 h in insulin, when It and Iss are already augmented, there is no effect on current amplitudes (Fig. 5). This suggests that once the new channels are inserted into the cell membrane, they are no longer sensitive to cytoskeletal disruption, during a short-term exposure to cytochalasin D (12-50 min). This strongly supports our hypothesis that it is the translocation of insulin-induced, newly synthesized channels that is disrupted by cytochalasin D and perhaps by colchicine. The lack of effect of prolonged exposure to cytochalasin D (Fig. 6) shows that the on-going activity of these channels does not depend on the integrity of the cytoskeleton. This further suggests that the turnover rate of these channels is slow, and unaffected by such disruption. It is possible that much longer periods of disruption would impair the renewal of channels in the membrane, and contribute to a faster run-down.
Our results have important implications for the normal and pathological control of the processing of ion channels and their targeting to the cell membrane. Actin microfilament disruption has also been found to affect channel activity in hippocampal neurons (Rosenmund & Westbrook, 1993), suggesting that our results may apply to other excitable tissues. It is now recognized that abnormalities in cytoskeletal proteins play a role in cardiomyopathies (Towbin, 1998). Our results suggest that insulin-deficient diabetes may induce functional abnormalities in cytoskeletal elements due to reduced levels of insulin, as has been suggested to occur in skeletal muscle (Chen-Zion et al. 1994). Tsakiridis et al. (1994) showed that the cytoskeletal microfilament system was essential for the rapid translocation of pre-formed glucose transporters to the cell membrane, in response to insulin. Following exposure to insulin, they observed a re-arrangement of the actin network. Future studies will have to address the issue of whether a deficiency in insulin affects the structure of the actin network in cardiomyocytes as well.
Limitations of this study
The major limitation of this study is that we are unable as yet to provide more direct evidence either for the increase in synthesis of K+ channels by insulin, or for the translocation of newly synthesized K+ channels to the cell membrane. Although we demonstrate that protein synthesis inhibition blocks the enhancing effect of insulin on It and Iss, and suggest that new K+ channels are formed, other interpretations are possible. For example, it is possible that in type I diabetes there is a deficiency not in K+ channels per se, but in an accessory protein required for the activation of channels, or for transporting latent channels from an inactive pool to the membrane. In this scenario, addition of insulin would augment the synthesis of this accessory protein, so that more active channels are present in the membrane. This transfer of latent channels to the membrane may be the process requiring the cytoskeleton. Since the channel isoforms underlying It in rat ventricle have been identified as Kv4.2 and Kv4.3 (Dixon et al. 1996; Fiset et al. 1997), it may be possible in the near future to use isoform-specific antibodies to examine specific K+ channel content by immunoblotting. This may indicate whether new channels are synthesized in response to insulin. Tagging these channel isoforms with fluorescent probes may enable a visualization of their proposed translocation to the cell membrane.
Another limitation is the lack of direct visualization of actin microfilaments and associated proteins. This will be attempted in future studies, although separating the actin microfilament network elements from contractile elements will be extremely difficult in cardiac myocytes (Rothen-Rutishauser et al. 1998). Staining of these elements is usually reported for non-contractile cells or muscle cells maintained in culture (Tsakiridis et al. 1994; Fernandez-Borja et al. 1995), so that tagging of other cytoskeleton-associated proteins may be required.
Finally, our results depend on the selectivity of the pharmacological tools at our disposal. Nevertheless, the combination of the effects of cytochalasin D in the absence and presence of phalloidin (Figs 3 and 4) make a compelling case for the involvement of the cytoskeleton in the insulin-dependent augmentation of the K+ currents. The results suggest that cytoskeletal elements are involved in the translocation of newly formed K+ channels to the cell membrane.
Acknowledgments
Some of this work was done in the laboratory of Dr W. Giles, whose support we gratefully acknowledge. Grant support from the Canadian Diabetes Association in honour of Mary Selina Jamieson (to Y. S.) and from the Medical Research Council of Canada (to D. S.) is also gratefully acknowledged.
References
- Apkon M, Nerbonne JM. Characterization of two distinct depolarization-activated K+ currents in isolated adult rat ventricular myocytes. Journal of General Physiology. 1991;97:973–1011. doi: 10.1085/jgp.97.5.973. 10.1085/jgp.97.5.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury NA, Bridges RJ. Role of membrane trafficking in plasma membrane solute transport. American Journal of Physiology. 1994;267:C1–24. doi: 10.1152/ajpcell.1994.267.1.C1. [DOI] [PubMed] [Google Scholar]
- Brady PA, Alekseev AE, Aleksanderova LA, Gomez LA, Terzic A. A disrupter of actin microfilaments impairs sulfonylurea-inhibitory gating of cardiac KATP channels. American Journal of Physiology. 1996;271:H2710–2716. doi: 10.1152/ajpheart.1996.271.6.H2710. [DOI] [PubMed] [Google Scholar]
- Braun JEA, Severson DL. Lipoprotein lipase release from cardiac myocytes is increased by decavanadate but not insulin. American Journal of Physiology. 1992;262:E663–670. doi: 10.1152/ajpendo.1992.262.5.E663. [DOI] [PubMed] [Google Scholar]
- Chen-Zion M, Livnat T, Beitner R. Effects of long-term streptozotocin diabetes on cytoskeletal and cytosolic phosphofructokinase and the levels of glucose 1,6-biphosphate and fructose 2,6-biphosphate in different rat muscles. Biochemical Medicine and Metabolic Biology. 1994;53:137–144. doi: 10.1006/bmmb.1994.1069. [DOI] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. Journal of Cell Biology. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JE, Shi W, Wang HS, Mcdonald C, Yu H, Wymore RS, Cohen IS, Mckinnon D. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circulation Research. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- Fath KR, Mamajiwalla SN, Burgess DR. The cytoskeleton in development of epithelial cell polarity. Journal of Cell Science. 1993;17(suppl.):65–73. doi: 10.1242/jcs.1993.supplement_17.10. [DOI] [PubMed] [Google Scholar]
- Fernandez-Borja M, Bellido D, Makiya R, David G, Olivecrona G, Reina M, Vilaro S. Actin cytoskeleton of fibroblasts organizes surface proteoglycans that bind basic fibroblast growth factor and lipoprotein lipase. Cell Motility and the Cytoskeleton. 1995;30:89–107. doi: 10.1002/cm.970300202. [DOI] [PubMed] [Google Scholar]
- Fiset C, Clark RB, Shimoni Y, Giles WR. Shal-type channels contribute to the Ca2+-independent transient outward K+ current in rat ventricle. The Journal of Physiology. 1997;500:51–64. doi: 10.1113/jphysiol.1997.sp021998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Yamane T, Terai T, Katayama Y, Hiraoka M. Functional linkage of the cardiac ATP-sensitive K+ channel to the actin cytoskeleton. Pflügers Archiv. 1996;431:504–512. doi: 10.1007/BF02191896. [DOI] [PubMed] [Google Scholar]
- Galli A, Defelice LJ. Inactivation of L-type Ca channels in embryonic chick ventricle cells: dependence on the cytoskeletal agents colchicine and taxol. Biophysical Journal. 1994;67:2296–2304. doi: 10.1016/S0006-3495(94)80715-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb TA, Ivanov IE, Adesnik M, Sabatini DD. Actin microfilaments play a critical role in endocytosis in the apical but not the basolateral surface of polarized epithelial cells. Journal of Cell Biology. 1993;120:695–710. doi: 10.1083/jcb.120.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, Simons K. The trans Golgi network: sorting at the exit site of the Golgi complex. Science. 1986;234:438–442. doi: 10.1126/science.2945253. [DOI] [PubMed] [Google Scholar]
- Janmey PA. The cytoskeleton and cell signaling: component localization and mechanical coupling. Physiological Reviews. 1998;78:763–781. doi: 10.1152/physrev.1998.78.3.763. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Byerly L. A cytoskeletal mechanism for Ca2+ channel metabolic dependence and inactivation by intracellular Ca2+ Neuron. 1993;10:797–804. doi: 10.1016/0896-6273(93)90196-x. [DOI] [PubMed] [Google Scholar]
- Luna EJ, Hitt AL. Cytoskeleton-plasma membrane interactions. Science. 1992;258:955–963. doi: 10.1126/science.1439807. [DOI] [PubMed] [Google Scholar]
- Makhina EN, Nichols CG. Independent trafficking of KATP channel subunits to the plasma membrane. Journal of Biological Chemistry. 1998;273:3369–3374. doi: 10.1074/jbc.273.6.3369. [DOI] [PubMed] [Google Scholar]
- Mazzanti M, Assandri R, Ferroni A, Difrancesco D. Cytoskeletal control of rectification and expression of four substates in cardiac rectifier K+ channels. FASEB Journal. 1996;10:357–361. doi: 10.1096/fasebj.10.2.8641571. [DOI] [PubMed] [Google Scholar]
- Nagaya N, Papazian DM. Potassium channel α and β subunits assemble in the endoplasmic reticulum. Journal of Biological Chemistry. 1997;272:3022–3027. doi: 10.1074/jbc.272.5.3022. [DOI] [PubMed] [Google Scholar]
- Nelson WJ. Regulation of cell surface polarity from bacteria to mammals. Science. 1992;258:948–954. doi: 10.1126/science.1439806. [DOI] [PubMed] [Google Scholar]
- Ornelles DA, Fey EG, Penman S. Cytochalasin releases mRNA from the cytoskeletal framework and inhibits protein syntheis. Molecular and Cellular Biology. 1986;6:1650–1662. doi: 10.1128/mcb.6.5.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter AB, Schittny JC, Niggli V, Reuter H, Sigel E. The polarized distribution of poly(A+)-mRNA-induced functional ion channels in Xenopus oocyte plasma membranes is prevented by anticytoskeletal drugs. Journal of Cell Biology. 1991;114:455–464. doi: 10.1083/jcb.114.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce A, Vega-Saenz de Miera E, Kentros C, Moreno H, Thornhill B, Rudy B. K+ channel subunit isoforms with divergent carboxy-terminal sequences carry distinct membrane targeting signals. Journal of Membrane Biology. 1997;159:149–159. doi: 10.1007/s002329900278. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- Rothen-Rutishauser BM, Ehler E, Perriard E, Messerli JM, Perriard JC. Different behaviour of the non-sarcomeric cytoskeleton in neonatal and adult rat cardiomyocytes. Journal of Molecular and Cellular Cardiology. 1998;30:19–31. doi: 10.1006/jmcc.1997.0596. [DOI] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- Saltiel AR. Diverse signalling pathways in the cellular actions of insulin. American Journal of Physiology. 1996;270:E375–385. doi: 10.1152/ajpendo.1996.270.3.E375. [DOI] [PubMed] [Google Scholar]
- Shimoni Y, Ewart HS, Severson D. Type I and II models of diabetes produce different modifications of K+ currents in rat heart: role of insulin. The Journal of Physiology. 1998;507:485–496. doi: 10.1111/j.1469-7793.1998.485bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoni Y, Severson D, Giles W. Thyroid status and diabetes modulate regional differences in potassium currents in rat ventricle. The Journal of Physiology. 1995;488:673–688. doi: 10.1113/jphysiol.1995.sp020999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele-Perkins G, Turner J, Edman JC, Hari J, Pierce SB, Stover C, Rutter WJ, Roth RA. Expression and characterization of a functional human insulin-like growth factor I receptor. Journal of Biological Chemistry. 1988;288:11486–11492. [PubMed] [Google Scholar]
- Terzic A, Kurachi Y. Actin microfilament disrupters enhance KATP channel opening in patches from guinea-pig cardiomyocytes. The Journal of Physiology. 1996;492:395–404. doi: 10.1113/jphysiol.1996.sp021316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin JA. The role of cytoskeletal proteins in cardiomyopathies. Current Opinion in Cell Biology. 1998;10:131–139. doi: 10.1016/s0955-0674(98)80096-3. [DOI] [PubMed] [Google Scholar]
- Tsakiridis T, Vranic M, Klip A. Disassembly of the actin network inhibits insulin-dependent stimulation of glucose transport and prevents recruitment of glucose transporters to the plasma membrane. Journal of Biological Chemistry. 1994;269:29934–29942. [PubMed] [Google Scholar]
- Tsakiridis T, Wang Q, Taha C, Grinstein S, Downey G, Klip A. Cytoskeletal Regulation of Membrane Function. Rockefeller University Press; 1997. Involvement of the actin network in insulin signalling; pp. 257–271. [PubMed] [Google Scholar]
- Undrovinas AJ, Shander GS, Makielski JC. Cytoskeleton modulates gating of voltage-dependent sodium channel in heart. American Journal of Physiology. 1995;269:H203–214. doi: 10.1152/ajpheart.1995.269.1.H203. [DOI] [PubMed] [Google Scholar]
- Wang WH, Cassola A, Giebisch G. Involvement of cytoskeleton in modulation of apical K+ channel activity in rat collecting duct. American Journal of Physiology. 1994;267:F592–598. doi: 10.1152/ajprenal.1994.267.4.F592. [DOI] [PubMed] [Google Scholar]