

Abstract
Arteriolar segments were isolated from pial membrane and studied within 10 h. Current-clamp and voltage-clamp measurements were made by patch-clamp recording from smooth muscle cells within arterioles. [Ca2+]i was measured from the smooth muscle cell layer by digital imaging of emission from fura-PE3 which was loaded into arterioles by pre-incubation with the acetoxymethyl ester derivative. The external diameter of arterioles was measured using a video-dimension analyser.
Endothelin-1 (ET1) was a potent constrictor of isolated arterioles and induced a sustained depolarization up to -27 mV and reduced membrane resistance (EC50 140-170 pm). At a constant holding potential of -60 mV ET-1 induced a transient followed by a sustained inward current. ET1 inhibited L-type voltage-dependent Ca2+ current.
ET1 induced a transient followed by sustained elevation of [Ca2+]i. The sustained effect was dependent on extracellular Ca2+. It occurred at a constant holding potential of -60 mV and was not inhibited by the Ca2+ antagonists nicardipine (1 μM) or D600 (10 μM). Thapsigargin (1 μM) completely depleted Ca2+ from caffeine- and ET1-sensitive sarcoplasmic reticulum but did not inhibit the ET1-induced sustained elevation of [Ca2+]i. ET1 effects on [Ca2+]i were prevented by the ETA receptor antagonist BQ123 (cyclo-D-Asp-Pro-D-Val-Leu-D-Trp).
The data suggest that ETA receptors are negatively coupled to L-type Ca2+ channels and positively coupled to receptor-operated Ca2+-permeable channels. Inhibition of L-type Ca2+ channel activity may suppress autoregulation, and Ca2+ influx through receptor-operated channels may have a major functional role in the potent long-lasting constrictor effect of endothelin-1 in the cerebral microcirculation.
The endothelin-1 peptide (ET1) is a vasoconstrictor at picomolar concentrations (Yanagisawa & Masaki, 1989; Rubanyi & Polokoff, 1994). Its effects are of substantial interest in the cerebral circulation because increased levels of ET1 occur in cerebrospinal fluid following subarachnoid haemorrhage (Seifert et al. 1995; Pluta et al. 1997), ischaemic stroke (Lampl et al. 1997) and brain injury (Armstead, 1996). ET receptor antagonists, particularly those with selectivity for the ETA class of ET receptor, reverse cerebral vasospasm in the rabbit and rat (Zuccarello et al. 1996b; Roux et al. 1997), and dilate pial arterioles and increase cerebral blood flow in the cat following focal ischaemia (Patel et al. 1996a, b). Application of ET1 to the cerebral cortex of the rat has been used to produce an animal model of stroke (Fuxe et al. 1997), and ETA receptor antagonists and endothelin-converting enzyme inhibitors are of potential therapeutic benefit for improving the outcome from stroke (Patel et al. 1996a; Caner et al. 1996). ET1 may also regulate cerebrovascular tone under physiological conditions, opposing nitric oxide-dependent vasodilatation (Thorin et al. 1998).
There appear to be multiple cellular sources of ET1 which include astrocytes, neurones, smooth muscle cells and endothelial cells (Pluta et al. 1997). Therefore, extralumenal effects of ET1 may be most relevant to the in vivo situation. Intralumenal effects of ET1 differ in character because of the barrier function of the endothelium and the presence of ET receptors on endothelial cells (Ogura et al. 1991; Patel et al. 1996b). Arterioles are defined as precapillary microvessels which have a single, circularly arranged layer of smooth muscle cells. They provide a major component of the resistance to blood flow in the brain and control the distribution of blood into the capillary beds of discrete brain regions (Mulvany & Aalkjaer, 1990). In this study we have attempted to elucidate the primary mechanisms underlying the vasoconstrictor effect of ET1 on arterioles of the mammalian cerebral cortex.
METHODS
Male Dutch dwarf rabbits (1.0-1.5 kg) were killed by an intravenous overdose of 70 mg kg−1 sodium pentobarbitone in accordance with the Code of Practice as set out by The UK Animals Scientific Procedures Act 1986. The brain was removed and placed in ice-cold Hanks’ solution bubbled with 100 % O2.
Pieces of pial membrane were removed from the cerebral cortex and incubated with 0.032 mg ml−1 protease (Type E, Sigma) and 0.2 mg ml−1 collagenase (Type 1A, Sigma) in Hanks’ solution at 37°C for 10 min. The mixture was then kept at 4°C for 15 min and subsequently agitated using a fire-polished Pasteur pipette before washing with enzyme-free Hanks’ solution and centrifugation at 1000 r.p.m. for 1 min. The supernatant was discarded and the suspension of arterioles stored on glass coverslips at 4°C in Hanks’ solution and used within 10 h. Enzymatic treatment was used to enable patch-clamp recording from smooth muscle cells embedded in arteriolar fragments (Quinn & Beech, 1998) but arterioles were prepared using the same method for all experiments whether they involved patch-clamp recording or not.
A modification of the above method has also been used for isolating a greater yield of arterioles. With this method, the enzymatic treatment involved incubation in 0.027 mg ml−1 protease and 0.17 mg ml−1 collagenase for 30 min at 37°C. Although electrophysiological measurements indicated that arterioles prepared using this 30 min method had depolarized resting membrane potentials (K. Quinn & D. J. Beech, unpublished observations), and thus were unsuitable for most of the study, it was observed that these arterioles responded routinely to caffeine. By contrast, arterioles prepared using the 10 min method responded only occasionally to caffeine. Therefore, most of the caffeine experiments described were carried out on arterioles prepared using the 30 min method. All other experiments were carried out on arterioles prepared by the 10 min method. Arterioles had an external diameter of about 30-40 μm, a thick wall of circularly arranged smooth muscle cells and evidence of longitudinally arranged endothelial cells lining the lumen (Fig. 1).
Figure 1. Patch-clamp recording and digital fluorescence imaging from the single smooth muscle cell layer of an isolated but intact precapillary arteriole.
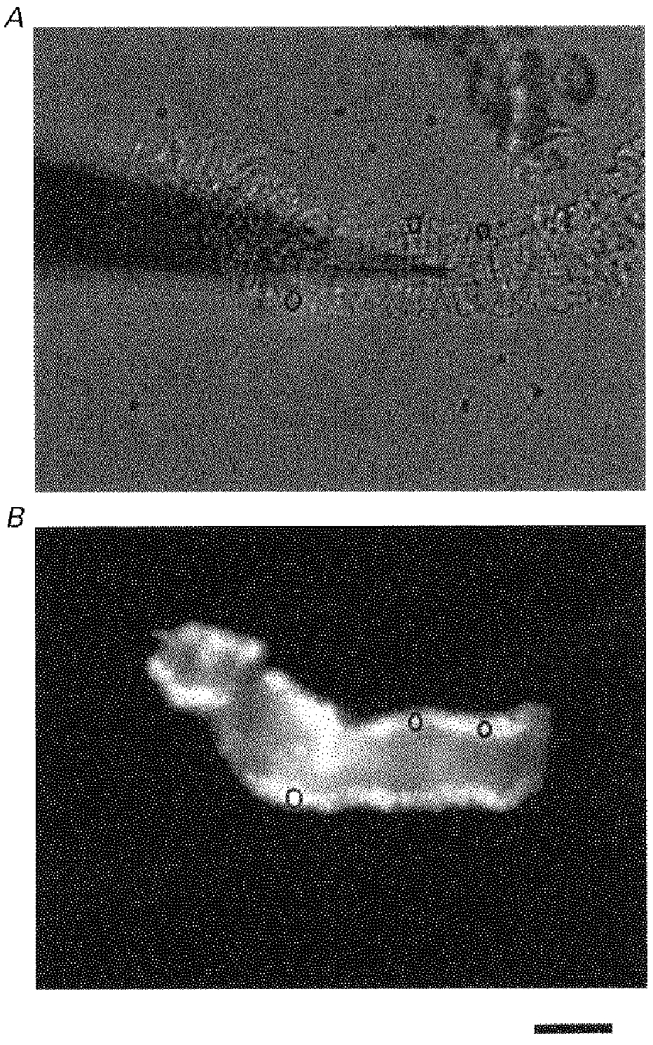
A, a brightfield image. The horizontal light grey band spanning most of the frame is an arteriole with an external diameter of about 50 μm. Cross-sectional lines are evident on the vessel and these are formed by smooth muscle cells. The less-distinct horizontal lines in the lumen may be formed by endothelial cells. This pattern is specific to arterioles and has been used to differentiate them from capillaries and venules. Three black rings are superimposed on the wall of the vessel. A gigaseal has been formed between the arteriole and a patch pipette. The patch pipette is the long black out-of-focus tapering shadow which enters the image from the left. (There is debri in the upper right-hand corner of the image.) B, fluorescence image. Emitted 510 nm light from the same arteriole as shown in A during exposure to excitation light at 380 nm. The arteriole is loaded with fura-PE3. The black circles are the same as those in A and are ROIs (see text). A section of the arteriole at the right of the frame was not loaded with fura-PE3. The horizontal scale bar is 40 μm.
To measure the constrictor effect of ET1, arterioles were placed in a modified culture dish on the stage of an inverted trinocular microscope (Nikon TMS, Japan) which had an attached video camera (Sony, Japan). The external diameter of arteriolar segments was measured using a video-dimension analyser (Living Systems Instrumentation, Vermont, USA) and calibrated using a stage micrometer. The signal was captured on-line via an A-D converter (Picolog software, Pico Technology Ltd, Cambridge, UK) and stored on a computer.
To measure [Ca2+]i, arterioles were incubated with 1 μM fura-PE3 AM (Vorndran et al. 1995) for 60 min at 23°C in the standard Ca2+-containing bath solution. Arterioles were allowed to attach to a glass coverslip which was placed on the stage of an inverted epifluorescence microscope (Nikon TMD) equipped with a × 40, 1.3 NA NPlanFluor oil-immersion objective (Nikon, Japan). The source of excitation light was a xenon arc lamp (75 W) and excitation wavelengths were selected by a monochromator (Till Photonics, Planegg, Germany). Light was transmitted to the microscope via a quartz fibre-optic guide and reflected by a dichroic mirror (Omega Optical, Glen Spectra Ltd, Stanmore, UK) into the objective. Emission was collected through the objective and a 510 nm filter (40 nm band width) and digital images were sampled at 8-bit resolution by a fast scan multi format cooled CCD camera (C4880-82, Hamamatsu Photonics K. K., Hamamatsu City, Japan). Fura-PE3 was excited alternately at 355 and 380 nm and ratios of the resulting images (355/380) were produced every 5 s. Images were 320 × 244 pixels (267 × 203 μm). Regions of interest (ROIs) were used to restrict data collection to signals from smooth muscle cells and avoid endothelial cells which appeared to remain intact and line the lumen (Fig. 1). Three ROIs were selected at random for each arteriole and the ROI with the lowest ratio value was used. ROIs were moved lumenally if constriction of the arteriole occurred. All the imaging was controlled by Improvision software which included Openlab 1.7.7 (Image Processing & Vision Company Ltd, Coventry, UK) and operated on a 8600 Macintosh PowerPC.
Current- and voltage-clamp experiments were performed using amphotericin-perforated patch and conventional whole-cell recording, respectively (Quinn & Beech, 1998). Giga seals were formed directly on a smooth muscle cell embedded in an intact arteriolar segment (Fig. 1). The patch-clamp amplifier was an Axopatch 200A, and command- and data-sampling protocols were controlled by pCLAMP 6.0.4 software operating on a 486 PC, and using a Digitdata 1200 interface (Axon Instruments Inc.). Signals were filtered at 0.1 kHz and sampled at 0.5 kHz. Patch pipettes were made from borosilicate glass capillary tubing with an outside diameter of 1 mm and inside diameter of 0.58 mm (Clark Electromedical Instruments, Reading, UK). Patch pipettes had resistances, after fire-polishing and when filled with solution of: 2-3 MΩ for conventional whole-cell recording and 5-10 MΩ for amphotericin B recordings. Voltage clamp was applied to arteriolar segments of up to 300 μm in length (e.g. Figs 1 and 4). We have not tested the quality of the voltage clamp but assume the voltage was reasonably uniform along the segment because of reports that the length constant for arterioles is about 1.5 mm (Hirst & Neild, 1978). In experiments where [Ca2+]i and voltage-clamp measurements were made simultaneously the two signals are co-ordinated in time to within < 5 s. Data have been presented using Origin 4.1 software (Microcal Inc, USA).
Figure 4. Digital imaging of [Ca2+]i in an arteriole and the response to ET1.

Sequential digital images acquired every 5 s for one arteriole. The order of the images should be viewed from left to right, starting with the top left-hand corner image and finishing with the bottom right-hand corner image. Each image is the ratio of emission intensity for the 355 nm divided by the 380 nm excitation wavelengths. The ratio is shown on a colour chart (lower right-hand corner of the figure) where red indicates a high value of [Ca2+]i and green a low value. The black horizontal scale bar indicates 40 μm. A valve was switched in the perfusion system to allow inflow of ET1 (10 nM) as marked by the black dot at the bottom left-hand corner of the 8th image. ET1 probably first reached the arteriole just before the 12th image. ET1 remained in the recording chamber for the rest of the experiment.
All the experiments were performed at about 23°C, and substances were applied to arterioles by exchanging the solution in the recording chamber. Solution exchange occurred within about 30 s. All results are expressed as means ±s.e.m., and n values indicate the number of arterioles used. Groups of data were compared using Student's unpaired t test and statistical significance was concluded if P < 0.05. Mathematical functions were fitted to data points using Origin 4.1 software.
Hanks’ solution contained (mM): NaCl, 137; KCl, 5.4; CaCl2, 0.01; NaH2PO4, 0.34; K2HPO4, 0.44; D-glucose, 8; Hepes, 5. Standard bath solution contained (mM): NaCl, 130; KCl, 5; D-glucose, 8; Hepes, 10; MgCl2, 1.2; CaCl2, 1.5. Ca2+-free bath solution was prepared by substituting 0.4 mM EGTA in place of 1.5 mM CaCl2 in the standard bath solution. The 60 mM K+ bath solution was the same as the standard bath solution except with 75 mM NaCl and 60 mM KCl. The bath solution for Ca2+ current recording was the same as the standard bath solution except the 5 mM KCl was excluded and a cocktail mixture of K+ channel inhibitors was added. The cocktail included 1 mM 3,4-diaminopyridine (3,4-DAP), 1 μM glibenclamide, 100 nM apamin, 100 nM penitrem A, 4 mM tetraethylammonium chloride (TEACl) and 0.1 mM barium chloride (BaCl2). Ba2+ current was recorded in the presence of 100 μM niflumic acid and by omitting Ca2+ from the bath solution and replacing it with 1.5 mM BaCl2. In current-clamp recordings, 120 μg ml−1 amphotericin B was included in the pipette solution which contained (mM): NaCl, 5; KCl, 130; D-glucose, 8; Hepes, 10; MgCl2, 1.2; CaCl2, 1.5. Amphotericin B was prepared as a 60 mg ml−1 stock solution in 100 % DMSO. The pipette solution for conventional voltage-clamp recordings contained (mM): KCl, 130; EGTA, 0.2; MgCl2, 2; Hepes, 10; Na2ATP, 3; NaGTP, 0.5. For recordings of Ca2+ current and Ba2+ current the 130 mM KCl was replaced by 130 mM CsCl. The final pH of all the solutions was titrated to pH 7.4 using NaOH. Patch pipette solutions were filtered (pore size, 0.2 μm).
General salts were from BDH, Sigma or Aldrich. EGTA, Hepes, protease (type E), collagenase (type 1A), thapsigargin, caffeine, nicardipine, D600, glibenclamide, apamin, penitrem A, TEACl, niflumic acid and BaCl2 were from Sigma. 3,4-DAP was from Fluka Chemie AG. Fura-PE3 AM and endothelin-1 were from Calbiochem. Sarafotoxin S6C (H-Cys-Thr-Cys-Asn-Asp-Met-Thr-Asp-Glu-Glu-Cys-Leu-Asn-Phe-Cys-His-Gln-Asp-Val-Ile-Trp-OH), BQ3020 (N-Ac-Leu-Met-Asp-Lys-Glu-Ala-Val-Tyr-Phe-Ala-His-Leu-Asp-Ile-Ile-Trp-OH) and BQ123 (cyclo-d-Asp-Pro-D-Val-Leu-D-Trp) were gifts from Dr T. Warner. Nicardipine, thapsigargin, fura-PE3 AM and D600 were prepared as stock solutions in 100 % DMSO such that the final DMSO concentration was ≤ 0.1 %. Endothelin-1 was prepared as a 0.1 mM stock solution with 95 % H2O and 5 % acetic acid (bubbled with 100 % N2). Other substances were prepared directly in salt solution or in distilled water.
RESULTS
Bath application of ET1 induced a concentration-dependent constriction of arteriolar segments (Fig. 2A). The external diameter was reduced, on average, by about 45 % and the half-effective concentration of ET1 was 140 pm (Fig. 2B).
Figure 2. ET1-induced constriction of isolated arterioles.
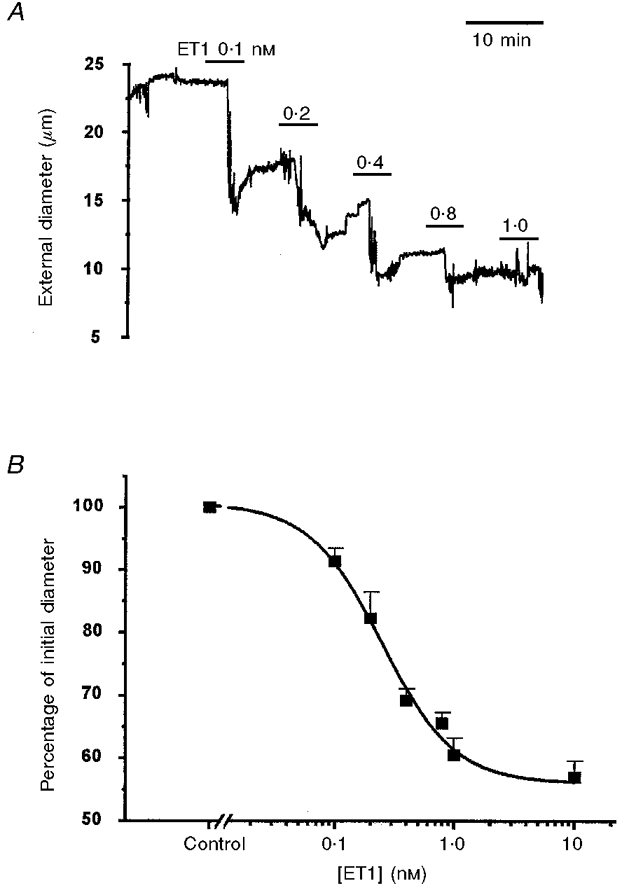
A, plot of external diameter against time for a single arteriole when the bath ET1 concentration was successively 0.1, 0.2, 0.4, 0.8 and 1 nM. B, mean ±s.e.m. (n = 6) external diameters as a percentage of the control value plotted against ET1 concentration. The smooth curve is the Hill equation with a slope of 1.4 and a mid-point of 140 pm.
The effect of ET1 on membrane potential was investigated in arteriolar segments by using patch pipettes attached directly to smooth muscle cells within the segments. This method enabled stable electrical recordings even during ET1-induced constrictions. In this series of recordings the mean resting membrane potential was -59.3 ± 3.5 mV (n = 9). ET-1 induced a concentration-dependent depolarization up to a limiting value which averaged -27.0 ± 3.6 mV (n = 9). ET1 tended to induce fluctuations in the membrane potential (Fig. 3A), and this sometimes prevented measurement of membrane resistance (Fig. 3C). The maximum effect of ET1 occurred at about 1 nM and the EC50 was 140 pm (Fig. 3A and D). Current pulses were injected during recordings and ET1 was observed to reduce the amplitude of the electrotonic potential (Fig. 3B), suggesting a reduction of membrane resistance (Fig. 3D). The EC50 for the effect of ET1 on membrane resistance was 170 pm and thus close to the EC50 for the depolarization.
Figure 3. ET1 induced depolarization and reduction in membrane resistance (Rm).
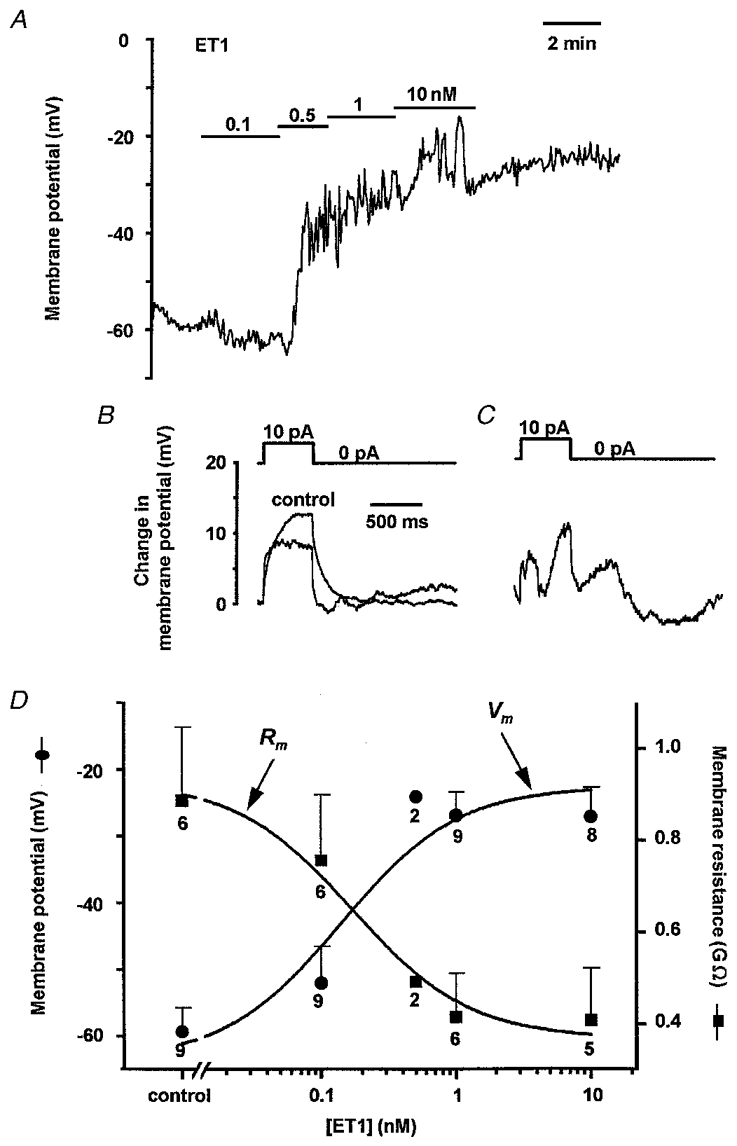
A, membrane potential plotted against time. ET1 was bath applied at 0.1, 0.5, 1 and 10 nM as indicated by the horizontal bars. Data points were sampled every 3.2 s. B, electrotonic potentials elicited by injection of 10 pA current pulses. A record is shown for control conditions and in the presence of 10 nM ET1. Each record is the mean of 3 consecutive individual records. The experiment is the same as A. C, example of another electrotonic potential from the same arteriole but where there were erratic ET1-induced variations in the membrane potential. D, concentration-dependent effect of ET1 on membrane potential (Vm; •) and membrane resistance (Rm;▪). The data points are mean ±s.e.m. values for the number of arterioles indicated by the numbers adjacent to the symbols. Error bars are not included for the n = 2 data points. The smooth curves are fitted Hill equations with slope constrained to 1. Rm values for each arteriole are the mean amplitudes of the slow component of 5 electrotonic potentials chosen at random for a given condition.
ET1-induced depolarization and reduction in membrane resistance suggested the possibility that Ca2+ influx through receptor-operated and voltage-dependent channels might play a role in the ET1 response of arterioles. This was investigated by measuring [Ca2+]i in smooth muscle cells which remained embedded in arterioles. Under basal conditions, [Ca2+]i was not uniform throughout arteriolar segments, higher Ca2+ levels appearing particularly along the edges of the arterioles or in bands across the arterioles (Fig. 4). Ca2+ levels also fluctuated in different regions of the arteriole with ‘Ca2+ hot-spots’ appearing and disappearing with time (Fig. 4). Application of 10 nM ET1 induced an increase in [Ca2+]i throughout the arteriole shown in Fig. 4. (The short delay before the effect may be due to a delay in the perfusion system). In the continued presence of ET1, [Ca2+]i declined, but not back to the basal level, remaining higher than the control values throughout the arteriole.
The fluctuating nature of basal [Ca2+]i and the transient followed by sustained nature of the ET1-induced elevation of [Ca2+]i are shown graphically for a single region of interest (ROI) which reflects [Ca2+]i in part of one or two arteriolar smooth muscle cells (Fig. 5A). Removal of Ca2+ from the bath solution 5 min before application of ET1 did not inhibit the transient response but completely prevented (n = 16), and in some arterioles even inverted (n = 4, data not shown), the sustained response (Fig. 5B). The peak amplitude of the transient response to ET1 (measured as an increase in the 355/380 ratio above the pre-ET1 level) was 0.089 ± 0.008 in control conditions (n = 51) and 0.11 ± 0.013 when the bath solution was made Ca2+ free 5 min before application of ET1 (n = 20). There is no significant difference between these groups. In Ca2+-containing bath solution, sustained [Ca2+]i in the presence of 10 nM ET1 was significantly higher than the pre-ET1 level (ratio of 0.93 ± 0.02 cf. 0.84 ± 0.02, n = 51). Therefore, the sustained Ca2+ response to ET1 resulted from Ca2+ influx. The Ca2+-free treatment used did not inhibit the transient response to ET1, suggesting Ca2+ was not depleted from sarcoplasmic reticulum.
Figure 5. The sustained effect of ET1 on [Ca2+]i requires Ca2+ influx.
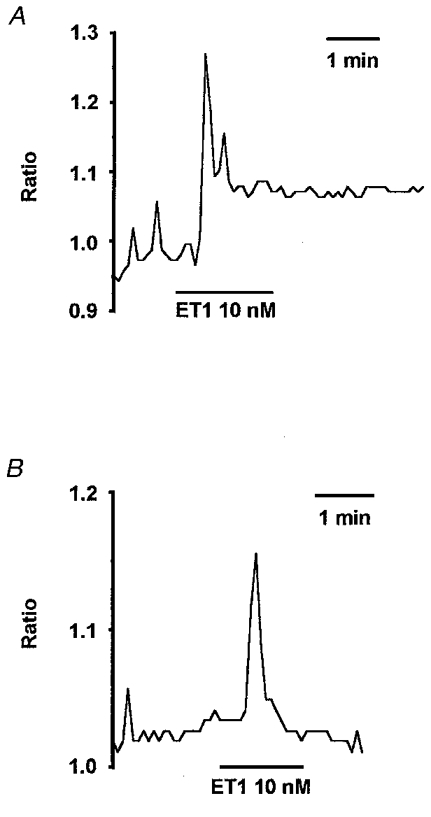
A, [Ca2+]i as indicated by the ratio of 355/380 signals for an ROI (about 5 μm in diameter) placed on the arteriole wall (see Fig. 1). The standard bath solution was used and ET1 (10 nM) was bath applied. B, the same type of experiment as A except that the standard bath solution was exchanged for a Ca2+-free bath solution, 5 min before the application of ET1.
Further evidence that the transient Ca2+ response to ET1 was due to Ca2+ release from sarcoplasmic reticulum came from the observation that it was abolished by pretreatment for 15-60 min with 1 μM thapsigargin (n = 6) or by pretreatment with 5 mM caffeine (n = 3, data not shown). Both substances deplete Ca2+ from sarcoplasmic reticulum (Treiman et al. 1998).
The effects of 10 nM ET1 on [Ca2+]i were abolished by 1 μM BQ123 (n = 9), an ETA receptor antagonist and there were no effects on [Ca2+]i of 100 nM BQ3020 (n = 6) or 100 nM sarafotoxin S6C (n = 9), two ETB receptor agonists.
The observation that the sustained elevation of [Ca2+]i, which is induced by ET1, depends entirely on Ca2+ influx from the extracellular medium could be explained by Ca2+ permeability of activated cation channels or depolarization-induced activation of L-type Ca2+ channels. To separate the two mechanisms, depolarization was prevented by experimenting in voltage-clamp mode. ET1 (10 nM) had an effect in 3 of 4 arterioles held at -60 mV and where Ca2+ measurements were made simultaneously. In the three positive cells, there was a transient inward current of -99.3 ± 37.4 pA followed by a sustained inward current of -33.6 ± 9.4 pA. The [Ca2+]i signal roughly matched the current signal, rising to a peak and declining to a sustained elevated level despite the -60 mV holding potential (Fig. 6). Therefore, ET1 could induce Ca2+ influx even when depolarization, and thus activation of L-type Ca2+ channels, was prevented.
Figure 6. Depolarization is not required for the sustained effect of ET1 on [Ca2+]i.
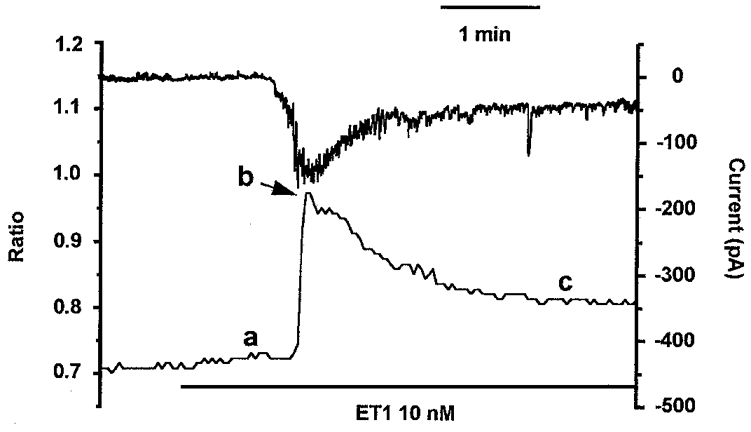
Measurement of [Ca2+]i from an ROI on the arteriole wall while maintaining a constant holding potential of -60 mV from a patch pipette attached to a smooth muscle cell adjacent to the ROI and in conventional whole-cell mode. The upper record in the plot is of holding current (right axis) and the lower record in the plot is the ratio of the 355/380 signals for the ROI (left axis). ET1 (10 nM) was bath applied as indicated by the horizontal bar. The amplitude of the ET1-induced Ca2+ signal was measured for this experiment, and all others in the study, as (b minus c) for the transient component and (c minus a) for the sustained component.
ET1 depolarized the arterioles to about -27 mV, where L-type Ca2+ channels might be expected to provide an additional Ca2+-influx pathway and contribute to the sustained ET1-induced elevation of [Ca2+]i. This hypothesis was tested by using two chemically distinct classes of Ca2+ antagonist (nicardipine and D600) at concentrations which completely block L-type Ca2+ channels. The degree of channel block was determined by observing the Ca2+ signal in response to 60 mM K+ which depolarizes the arterioles up to about -20 mV (data not shown) and causes constriction (Quinn & Beech, 1998). Application of 60 mM K+ induced a transient (355/380 ratio of 0.069 ± 0.028, n = 15) followed by a sustained (0.1 ± 0.014, n = 15) increase in [Ca2+]i above the base line. The effects were completely absent in Ca2+-free bath solution (n = 7, data not shown). Both Ca2+ antagonists completely blocked the increases in [Ca2+]i induced by 60 mM K+ but had no effect on the sustained elevation of [Ca2+]i induced by 10 nM ET1 (Fig. 7A and B).
Figure 7. Sustained effect of ET1 on [Ca2+]i which is independent of L-type Ca2+ channels.
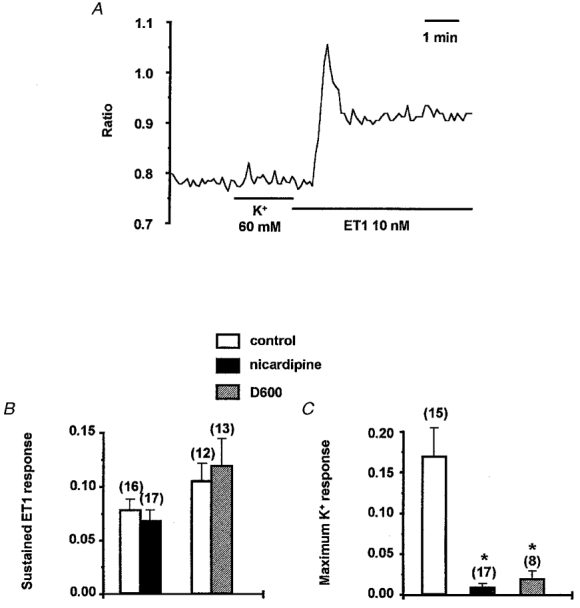
A, [Ca2+]i as indicated by the ratio of 355/380 signals for an ROI (about 5 μm in diameter) placed on the arteriole wall (see Fig. 1). The arteriole was incubated in 10 μM D600. K+ (60 mM) and ET1 (10 nM) were bath applied as indicated by the horizontal bars. B, mean ±s.e.m. sustained response to 10 nM ET1 given as a change in 355/380 ratio (c minus a; as Fig. 6). C, mean ±s.e.m. response to 60 mM K+ given as the increase in the 355/380 ratio above control value. Data are shown for in the absence of a Ca2+ antagonist (□), and in the presence of 1 μM nicardipine (▪) or 10 μM D600 ( ). The numbers of arterioles contributing to the mean values are given in parentheses. Ca2+ antagonists were applied for 15 to 60 min before application of ET1 or 60 mM K+. Experiments with ET1 made an alternate comparison between control conditions and in the presence of one Ca2+ antagonist and thus data columns for each Ca2+ antagonist have their own control for comparison. Significant inhibition of the response is indicated by *.
). The numbers of arterioles contributing to the mean values are given in parentheses. Ca2+ antagonists were applied for 15 to 60 min before application of ET1 or 60 mM K+. Experiments with ET1 made an alternate comparison between control conditions and in the presence of one Ca2+ antagonist and thus data columns for each Ca2+ antagonist have their own control for comparison. Significant inhibition of the response is indicated by *.
It was also noted that the transient increase in [Ca2+]i which was induced by ET1 was inhibited by 1 μM nicardipine (P = 0.008, n = 17) (data not shown) but not by 10 μM D600 (P = 0.49, n = 13).
The implication from the above experiments is that ET1 did not activate L-type Ca2+ channels even though it induced sufficient depolarization to activate them. The possibility that ETA receptors are negatively coupled to L-type Ca2+ channels was investigated more directly by making Ba2+-current recordings in voltage-clamp mode. K+ channel currents were attenuated by a cocktail of K+ channel inhibitors: 3,4-DAP to block Kv channels, glibenclamide to block ATP-sensitive K+ channels, apamin to block SKCa channels, penitrem A and TEA+ to block BKCa channels, and Ba2+ to block inward rectifier K+ channels. Ca2+-activated Cl− current was inhibited by 100 μM niflumic acid. Inward current which occurred during the depolarizing ramp protocol was inhibited by D600 (Fig. 8) and thus seemed to be carried by L-type Ca2+ channels. ET1 (1 nM) almost abolished the voltage-dependent inward current over a period of about 10 min (Fig. 9A-E). In this experiment, inward current induced by ET1 (Fig. 6) was small or absent and so it did not complicate the study of voltage-dependent Ca2+ channel current (Fig. 9B cf. 9C). In Ca2+-containing solution, and in the absence of Ba2+ and niflumic acid, ET1 had a similar inhibitory effect on the inward current (Fig. 9F). No effect was observed during a 10 min application of the ETB agonist BQ3020 (100 nM; n = 3, data not shown).
Figure 8. The voltage-dependent inward current was inhibited by D600.
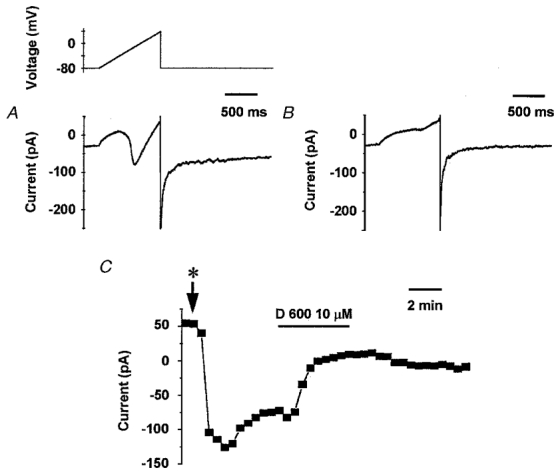
In Ba2+-containing solution including 100 μM niflumic acid and K+ channel blockers, a 1 s voltage ramp was applied every 30 s from the -80 mV holding potential to +40 mV. The current change during the ramp is plotted against time after 5 min (A) of control perfusion and 4 min after application of 10 μM D600 (B). The absolute amplitude of the peak inward current during each ramp was measured isochronically and plotted against time in C. Initiation of bath perfusion with the Ba2+ solution containing 100 μM of niflumic acid and the cocktail of K+ channel blockers is indicated by *. D600 (10 μM) was bath applied as indicated by the horizontal bar.
Figure 9. Inhibition of Ca2+ channel current by ET1.
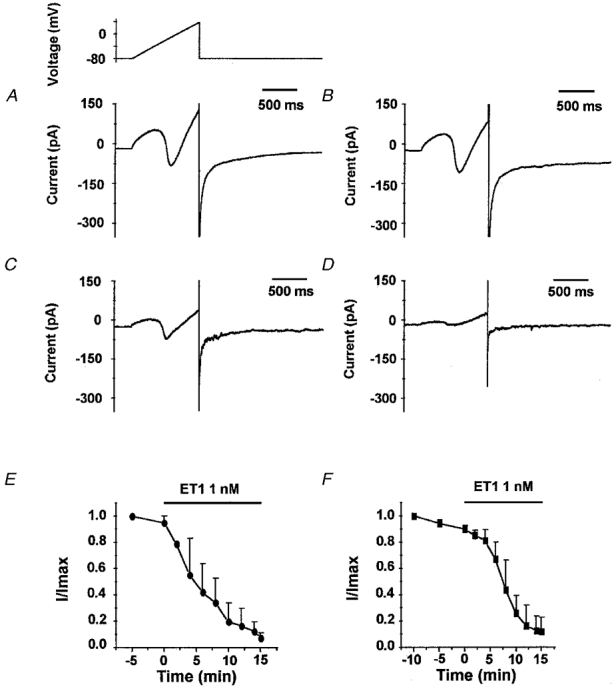
In Ba2+-containing solution and in the presence of K+ channel blockers and niflumic acid (100 μM), ET1 (1 nM) inhibited the inward current induced by a 1 s voltage ramp protocol from -80 mV to +40 mV (holding potential -80 mV) every 30 s (A-D). The current elicited during the voltage ramp protocol is shown for 4 min (A) and 7 min (B) after control perfusion, and 8 min (C) and 11 min (D) after application of ET1 (1 nM). E and F, mean data for the effect of ET1. Leakage current was estimated from the linear portion of the current at voltages between -80 mV and -45 mV. ICa and IBa were measured as the difference between the peak inward current and the extrapolated value of leak current at the same voltage. Each current amplitude (I) was then divided by the amplitude of the current recorded in the absence of ET1 (Imax). E, I/Imax (means ±s.e.m., n = 3) for the experimental conditions described above (A-D). F, also I/Imax (means ±s.e.m., n = 3) but for the effect of ET1 (1 nM) on ICa recorded in Ca2+ solution without niflumic acid but with K+ channels blockers. The voltage protocol was a 1 s voltage ramp applied every 30 s from -100 mV to + 40 mV (holding potential -60 mV).
The activation mechanism for ETA receptors coupling to Ca2+-permeable channels could be linked to Ca2+ depletion from sarcoplasmic reticulum and be a general feature of any substance which releases Ca2+ from stores (i.e. it could reflect capacitative Ca2+ entry). To investigate if capacitative Ca2+ entry exists in these arterioles we depleted Ca2+ from sarcoplasmic reticulum using thapsigargin and tested the effect of changing from the Ca2+-free bath solution to the standard bath solution, which contained 1.5 mM Ca2+. In control conditions, adding 1.5 mM Ca2+ caused a small, and perhaps unconvincing, elevation of [Ca2+]i (Fig. 10A). After pretreatment with 1 μM thapsigargin for 1 h, adding 1.5 mM Ca2+ always caused an obvious increase in [Ca2+]i (Fig. 10B). The increase in the 355/380 ratio above baseline upon addition of 1.5 mM Ca2+ was 0.037 ± 0.012 (n = 7) in control conditions and significantly larger at 0.092 ± 0.018 (n = 7) in the presence of thapsigargin. Further evidence for the presence of capacitative Ca2+ entry came from the effect of caffeine which induced a transient and a sustained elevation of [Ca2+]i (Fig. 10C). The sustained effect of caffeine appeared to be smaller but was not statistically different (P = 0.059) in Ca2+-free solution: the 355/380 ratio above baseline was 0.0052 ± 0.0052 (n = 6), compared with 0.063 ± 0.021 (n = 10) in standard bath solution. Thus, the data seem consistent with the existence of capacitative Ca2+ entry in these arterioles.
Figure 10. Sustained effect of ET1 on [Ca2+]i and its relation to depletion of Ca2+ from sarcoplasmic reticulum.
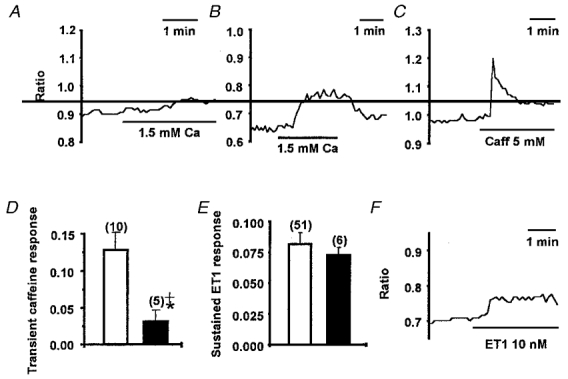
Effect on [Ca2+]i of changing from the Ca2+-free bath solution to the standard bath solution (containing 1.5 mM Ca2+) under control conditions (A) and after pretreatment with 1 μM thapsigargin for 1 h (B). C, effect of bath applied 5 mM caffeine on [Ca2+]i in standard bath solution. D, means ±s.e.m. increase in ratio (increase in [Ca2+]i) at the peak of the response to 5 mM caffeine in control conditions (□) and after pretreatment with 1 μM thapsigargin (TG) for 10-60 min (▪). Significant inhibition of the caffeine response is indicated by *, and no significant difference between zero and the caffeine response in the presence of thapsigargin is indicated by +. E, means ±s.e.m. increase in ratio for the sustained [Ca2+]i response to 10 nM ET1 in control conditions (□) and after pretreatment with 1 μM thapsigargin for 10-60 min (▪). F, example of a single recording showing the effect of 10 nM ET1 on [Ca2+]i after incubation with 1 μM thapsigargin for 1 h. In all cases, [Ca2+]i is indicated by the ratio of 355/380 signals for an ROI (about 5 μm in diameter) placed on the arteriole wall (see Fig. 1).
To investigate if the sustained ET1 response involved capacitative Ca2+ entry, we attempted to deplete Ca2+ completely from sarcoplasmic reticulum, thus maximally inducing capacitative Ca2+ entry, prior to application of ET1. Arterioles were pre-incubated in thapsigargin, and caffeine was used to test if the sarcoplasmic reticulum was empty. Thapsigargin strongly inhibited the response to 5 mM caffeine and the residual effect of caffeine was not significantly different from zero (Fig. 10D). In control conditions, 10 mM caffeine had no effect after a prior application of 5 mM caffeine (n = 3, data not shown), showing that caffeine was maximally effective at a concentration of 5 mM. Pretreatment with 1 μM thapsigargin had no significant effect on the sustained response (Fig. 10E and F). Thus, ET1 appeared to activate a Ca2+-influx pathway which was separate from capacitative Ca2+ entry.
DISCUSSION
The experiments demonstrate that ion channels and Ca2+-handling mechanisms can be studied in smooth muscle cells of functional 30 μm diameter precapillary arterioles without the need for isolation of cells from the intact tissue. The focus of this study was on the mechanism of action of ET1. ET1 constricted arteriolar segments and induced depolarization and a decrease in membrane resistance all with similar EC50 values of about 150 pm. Although the depolarization was sufficient to activate L-type voltage-dependent Ca2+ channels these channels played no role in carrying the sustained Ca2+ influx which was characteristic of a maximally effective concentration of ET1. The ET receptors were negatively coupled to L-type Ca2+ channels. The ET1-induced Ca2+ influx was instead mediated by separate Ca2+-permeable channels, the activation of which also caused depolarization. These channels may be classified as receptor-operated Ca2+ channels which are positively coupled to the ET receptor. They were not activated simply because ET1 caused Ca2+ release from sarcoplasmic reticulum.
The constrictor potency of ET1 on isolated arterioles was similar to that reported previously, for example for guinea-pig ileum submucosal arterioles (Hill et al. 1997). The receptor pharmacology of the ET1 effects suggests involvement only of the ETA subtype of ET receptor in pial arterioles. There is a similar dominant role of ETA receptors in mediating vasoconstriction, and expression of ETA receptors in vascular smooth muscle cells, in human and rabbit cerebral arteries (Yu et al. 1995; Petersson et al. 1996; reviewed by Rubanyi & Polokoff, 1994). There are reports of a dilatory function of ETB receptor activation (Patel et al. 1996b), but an ETB receptor effect was not evident in our recordings from cerebral arterioles, perhaps because ETB agonists were applied only under basal conditions or while ETA receptors were also activated by ET1.
The ET1-induced initial transient rise in [Ca2+]i was not the focus of this study but comparisons with other studies on peripheral arteries suggest the mechanism involves inositol 1,4, 5-trisphosphate (InsP3)-mediated Ca2+ release from sarcoplasmic reticulum. ET1 induced a rapid transient formation of inositol phosphates in A7r5 cells (smooth muscle cells from embryonic rat aorta), rabbit aorta and other vascular preparations (Van Renterghem et al. 1988; Rubanyi & Polokoff, 1994), and activation of Ca2+-dependent Cl− current by ET1 in mesenteric artery myocytes was blocked by intracellular heparin, which inhibits activation of the InsP3 receptor (Klöckner & Isenberg, 1991). The observation that nicardipine, but not D600, inhibited the transient effect of ET1 on [Ca2+]i in arterioles may be explained by a lack of selectivity of nicardipine for the L-type Ca2+ channel at a concentration of 1 μM. Xuan et al. (1989) observed that 1 μM but not 0.1 μM nicardipine inhibited inositol phosphate formation in A10 cells and suggested that Ca2+ influx through L-type Ca2+ channels was in some way involved in the maintenance of the inositol phosphate response. However, it is difficult to reconcile this idea with resistance of the transient response in arterioles to D600 unless the voltage-dependence of the Ca2+ channel block, which is different for nicardipine compared with D600, is important.
At a holding potential of -60 mV, ET1 induced a transient and then a sustained inward current, and in current clamp the depolarization induced by ET1 was associated with increased conductance. Therefore, ET1 activated plasma membrane ion channels which passed current with a reversal potential positive of -60 mV. The fact that the transient inward current had approximately the same time course as the transient increase in [Ca2+]i suggests involvement of a Ca2+-activated channel. The channel may be similar to the Ca2+-dependent cation channels or Ca2+-dependent Cl− channels observed in other blood vessels (Loirand et al. 1991; Klöckner & Isenberg, 1991). The primary argument that the channels which carried the sustained ET1-induced current (and maintained the depolarization) were different from those carrying the transient current is that there was no sustained elevation of [Ca2+]i which was independent of Ca2+ influx and thus no Ca2+ to activate the Ca2+-dependent cation or Cl− channels. That is, there must have been a sustained influx of Ca2+ through a pathway which was not activated by Ca2+ release from sarcoplasmic reticulum. Previous evidence that ET1 activates a receptor-operated Ca2+ channel in blood vessels comes from studies of rat and rabbit aorta (Chen & Wagoner, 1991; Enoki et al. 1995). It is unclear whether or not the resulting Ca2+ influx is sufficient to activate Ca2+-dependent channels so that these channels also carry part of the sustained inward current.
Although our experiments with thapsigargin and caffeine suggest that capacitative Ca2+ entry exists in pial arterioles, there are reasons to be cautious about making a definite conclusion. There appeared to be a small basal flux of Ca2+ into the arterioles. Thapsigargin may have amplified the signal from this basal flux because it inhibited sarcoplasmic reticulum Ca2+-ATPase and thus removed a superficial buffer barrier created by sarcoplasmic reticulum in close proximity to the plasma membrane (van Breemen et al. 1995). The alternative explanation is that it is the thapsigargin-induced depletion of Ca2+ from sarcoplasmic reticulum which, in some way, triggered the opening of plasma membrane Ca2+-permeable channels (i.e. capacitative Ca2+ entry). The sustained elevation of [Ca2+]i which was induced by caffeine supports the capacitative Ca2+ entry hypothesis.
ET1-induced Ca2+ influx was separate from any capacitative Ca2+ entry mechanism because its amplitude remained the same even when sarcoplasmic reticulum had previously been depleted of Ca2+ and was so barren of Ca2+ that neither ET1 nor caffeine could elicit Ca2+ release. A further implication of these data is that ET1 did not induce capacitative Ca2+ entry even though it released Ca2+ from sarcoplasmic reticulum. Perhaps if capacitative Ca2+ entry does exist in pial arterioles it is only activated by strong depletion of Ca2+ from sarcoplasmic reticulum and this level of depletion did not occur with ET1.
ET1 was initially thought to produce contraction of vascular smooth muscle by acting like an endogenous BAY K 8644, stimulating L-type Ca2+ channels (Goto et al. 1989). It is now apparent that ET receptors couple to multiple mechanisms (Rubanyi & Polokoff, 1994) but further reports have supported the conclusion that ET1 can stimulate L-type Ca2+ channels in, for example, guinea-pig portal vein (Inoue et al. 1990). ET1 has also been observed to induce transient, but not sustained, inhibition of L-type Ca2+ channel current in, for example, coronary and renal artery smooth muscle cells (Klöckner & Isenberg, 1991; Gordienko et al. 1994). This transient effect is most likely to occur because of Ca2+-dependent inactivation of the Ca2+ channels during the transient ET1-induced elevation of [Ca2+]i. Our conclusion that ET1 induces long-lasting inhibition, or even abolition, of L-type Ca2+ channel activity in pial arterioles is supported by two primary lines of evidence: (1) Ca2+ antagonists completely blocked Ca2+ influx through L-type Ca2+ channels but had no effect on the ET1-induced sustained elevation of [Ca2+]i which was accompanied by depolarization; (2) voltage-clamp recordings from arteriolar segments showed sustained ET1-induced inhibition of niflumic acid-resistant inward current which could also be blocked by D600, an L-type calcium channel blocker. It remains to be tested whether very low concentrations of ET1 might cause only partial inhibition of voltage-dependent Ca2+ channels and yet still cause depolarization. The difference between our observation and those of previous studies could be explained by species or vessel differences, or because we used an intact multicellular arteriole preparation rather than isolated single arterial smooth muscle cells exposed to conventional whole-cell recording. The suggestion is not, however, new that spasmogenic agonists at inositol phosphate-coupled receptors cause inhibition of L-type voltage-dependent Ca2+ channels; histamine, for example, contracts the guinea-pig ileum and yet has strong inhibitory effects on the L-type Ca2+ channel (Beech, 1993).
The knowledge that ET1 can inhibit L-type Ca2+ channel activity may help to explain why there is a confusing overall picture from Ca2+-antagonist studies aimed at determining the role of L-type Ca2+ channels in ET1-induced contraction; either no effect or a partial inhibitory effect of Ca2+-antagonists is reported (Edwards & Trizna, 1990; Ogura et al. 1991). Perhaps, whether there is an effect of Ca2+ antagonists depends on the relative balance between the amount of ET1-induced depolarization and ET1-induced inhibition of L-type Ca2+ channels. The consequence of complete L-type Ca2+ channel inhibition by ET1 will be suppression of depolarization - and presumably pressurization - induced constriction and, therefore, inhibition of autoregulation. This will be most severe in small arteries and arterioles of the cerebral circulation which are considered to have a high dependence on L-type Ca2+ channels. Such an effect may contribute to the observed loss of autoregulation during cerebral ischaemia (Zhu & Auer, 1995).
The function of the positive coupling of ETA receptors to receptor-operated Ca2+ channels may be to provide Ca2+ influx for the refilling of sarcoplasmic reticulum, to provide Ca2+ directly for activation of the contractile apparatus or to effect long-term cellular change. Although ET1-induced opening of receptor-operated channels was associated with depolarization, the observation that ET1 simultaneously inhibited L-type voltage-dependent Ca2+ channels seems to indicate that the depolarization could simply be a consequence of activating a cation channel and have no functional consequence. ET1-induced activation of a receptor-operated Ca2+ channel may be of major importance for the in vivo effects of ET1 and the role of ET1 in vasospasm because it has been observed that ET1-induced vasoconstriction and vasospasm are strongly inhibited by Ca2+-free solution but not by the Ca2+ antagonist verapamil (Zuccarello et al. 1996a).
This report shows that detailed studies of the mechanisms underlying vasoconstrictor action are possible in functional precapillary pial arterioles. The data support conclusions from studies of the aorta that receptor-operated Ca2+-permeable channels are a major feature of the sustained action of ET1. We also present evidence that opening of the Ca2+-permeable channels is not a response to Ca2+ depletion from sarcoplasmic reticulum. In fact we found no evidence that ET1 induces capacitative Ca2+ entry. Furthermore, we observed an inhibitory effect of ET1 on voltage-dependent Ca2+ channels which may go some way to explaining why Ca2+ antagonists are relatively ineffective against ET1-induced vasospasm and could explain the loss of autoregulation which occurs in ischaemic brain regions. The observed dominant role of receptor-operated Ca2+-permeable channels in the action of ET1 on cerebral microvessels encourages the idea that blockers of these channels would have a beneficial effect following cerebrovascular accidents such as subarachnoid haemorrhage and focal cerebral ischaemia where ET1 appears to have a deleterious effect via ETA receptors.
Acknowledgments
This research is funded by The Wellcome Trust (grant reference no. 050035). We thank E. V. Reynolds for vessel diameter measurements.
References
- Armstead WM. Role of endothelin in pial artery vasoconstriction and altered responses to vasopressin after brain injury. Journal of Neurosurgery. 1996;85:901–907. doi: 10.3171/jns.1996.85.5.0901. [DOI] [PubMed] [Google Scholar]
- Beech DJ. Inhibitory effects of histamine and bradykinin on calcium current in smooth muscle cells isolated from guinea-pig ileum. The Journal of Physiology. 1993;463:565–583. doi: 10.1113/jphysiol.1993.sp019611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caner HH, Kwan AL, Arthur A, Jeng AY, Lappe RW, Kassell NF, Lee KS. Systemic administration of an inhibitor of endothelin-converting enzyme for attenuation of cerebral vasospasm following experimental subarachnoid hemorrhage. Journal of Neurosurgery. 1996;85:917–922. doi: 10.3171/jns.1996.85.5.0917. [DOI] [PubMed] [Google Scholar]
- Chen C, Wagoner PK. Endothelin induces a nonselective cation current in vascular smooth muscle cells. Circulation Research. 1991;69:447–454. doi: 10.1161/01.res.69.2.447. [DOI] [PubMed] [Google Scholar]
- Edwards R, Trizna W. Response of isolated intracerebral arterioles to endothelins. Pharmacology. 1990;41:149–152. doi: 10.1159/000138711. [DOI] [PubMed] [Google Scholar]
- Enoki T, Miwa S, Sakamoto A, Minowa T, Komuro T, Kobayashi S, Ninomiya H, Masaki T. Long-lasting activation of cation current by low concentration of endothelin-1 in mouse fibroblasts and smooth muscle cells of rabbit aorta. British Journal of Pharmacology. 1995;115:479–485. doi: 10.1111/j.1476-5381.1995.tb16358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxe K, Bjelke B, Andbjer B, Grahn H, Rimondini R, Agnati LF. Endothelin-1 induced lesions of the frontoparietal cortex of the rat. A possible model of focal cortical ischemia. NeuroReport. 1997;8:2623–2629. doi: 10.1097/00001756-199707280-00040. [DOI] [PubMed] [Google Scholar]
- Gordienko DV, Clausen C, Goligorsky MS. Ionic currents and endothelin signaling in smooth muscle cells from rat renal resistance arteries. American Journal of Physiology. 1994;266:F325–341. doi: 10.1152/ajprenal.1994.266.2.F325. [DOI] [PubMed] [Google Scholar]
- Goto K, Kasuya Y, Matsuki N, Takuwa Y, Kurihara H, Ishikawa T, Kimura S, Yanagisawa M, Masaki T. Endothelin activates the dihydropyridine-sensitive, voltage-dependent Ca2+ channel in vascular smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1989;86:3915–3918. doi: 10.1073/pnas.86.10.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CE, Kirton A, Wu DD, Vanner SJ. Role of maxi-K+ channels in endothelin-induced vasoconstriction of mesenteric and submucosal arterioles. American Journal of Physiology. 1997;36:G1087–1093. doi: 10.1152/ajpgi.1997.273.5.G1087. [DOI] [PubMed] [Google Scholar]
- Hirst GDS, Neild TO. An analysis of excitatory junctional potentials recorded from arterioles. The Journal of Physiology. 1978;280:87–104. doi: 10.1113/jphysiol.1978.sp012374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Oike M, Nakao K, Kitamura K, Kuriyama H. Endothelin augments unitary calcium channel currents on the smooth muscle cell membrane of guinea-pig portal vein. The Journal of Physiology. 1990;423:171–191. doi: 10.1113/jphysiol.1990.sp018017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöckner U, Isenberg G. Endothelin depolarizes myocytes from porcine coronary and human mesenteric arteries through a Ca-activated chloride current. Pflügers Archiv. 1991;418:168–175. doi: 10.1007/BF00370467. [DOI] [PubMed] [Google Scholar]
- Lampl Y, Fleminger G, Gilad R, Galron R, Sarova-Pinhas I, Sokolovsky M. Endothelin in cerebrospinal fluid and plasma of patients in the early stage of ischemic stroke. Stroke. 1997;28:1951–1955. doi: 10.1161/01.str.28.10.1951. [DOI] [PubMed] [Google Scholar]
- Loirand G, Pacaud P, Baron A, Mironneau C, Mironneau J. Large conductance calcium-activated non-selective cation channel in smooth muscle cells isolated from rat portal vein. The Journal of Physiology. 1991;437:461–475. doi: 10.1113/jphysiol.1991.sp018606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Aalkjaer C. Structure and function of small arteries. Physiological Reviews. 1990;70:921–961. doi: 10.1152/physrev.1990.70.4.921. [DOI] [PubMed] [Google Scholar]
- Ogura K, Takayasu M, Dacey RG. Differential effects of intra- and extraluminal endothelin on cerebral arterioles. American Journal of Physiology. 1991;261:H531–537. doi: 10.1152/ajpheart.1991.261.2.H531. [DOI] [PubMed] [Google Scholar]
- Patel TR, Galbraith S, Mcauley MA, Mcculloch J. Endothelin-mediated vascular tone following focal cerebral-ischemia in the cat. Journal of Cerebral Blood Flow and Metabolism. 1996a;16:679–687. doi: 10.1097/00004647-199607000-00019. [DOI] [PubMed] [Google Scholar]
- Patel TR, Mcauley MA, Mcculloch J. Endothelin receptor mediated constriction and dilatation in feline cerebral resistance arterioles in vivo. European Journal of Pharmacology. 1996b;307:41–48. doi: 10.1016/0014-2999(96)00188-4. [DOI] [PubMed] [Google Scholar]
- Petersson J, Hanson GC, Lindberg BF, Hogestatt ED. Contractile effect of big endothelin-1 and its conversion to endothelin-1 in rabbit cerebral-arteries. Naunyn-Schmiedeberg's Archives of Pharmacology. 1996;354:656–661. doi: 10.1007/BF00170842. [DOI] [PubMed] [Google Scholar]
- Pluta RM, Boock RJ, Afshar JK, Clouse K, Bacic M, Ehrenreich H, Oldfield EH. Source and cause of endothelin-1 release into cerebrospinal fluid after subarachnoid hemorrhage. Journal of Neurosurgery. 1997;87:287–293. doi: 10.3171/jns.1997.87.2.0287. [DOI] [PubMed] [Google Scholar]
- Quinn K, Beech D. A method for direct patch-clamp recording from smooth muscle cells embedded in functional brain microvessels. Pflügers Archiv. 1998;435:564–569. doi: 10.1007/s004240050553. [DOI] [PubMed] [Google Scholar]
- Roux S, Breu V, Giller T, Neidhart W, Ramuz H, Coassolo P, Clozel JP, Clozel M. Ro 61–1790, a new hydrosoluble endothelin antagonist: general pharmacology and effects on experimental cerebral vasospasm. Journal of Pharmacology and Experimental Therapeutics. 1997;283:1110–1118. [PubMed] [Google Scholar]
- Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacological Reviews. 1994;46:325–415. [PubMed] [Google Scholar]
- Seifert V, Löffler B, Zimmerman M, Roux S, Stolke D. Endothelin concentrations in patients with aneurysmal subarachnoid hemorrhage. Journal of Neurosurgery. 1995;82:55–62. doi: 10.3171/jns.1995.82.1.0055. [DOI] [PubMed] [Google Scholar]
- Thorin E, Nguyen T, Bouthillier A. Control of vascular tone by endogenous endothelin-1 in human pial arteries. Stroke. 1998;29:175–180. doi: 10.1161/01.str.29.1.175. [DOI] [PubMed] [Google Scholar]
- Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends in Pharmacological Sciences. 1998;19:131–135. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- Van Breemen C, Chen Q, Laher I. Superficial buffer barrier function of smooth-muscle sarcoplasmic reticulum. Trends in Pharmacological Sciences. 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- Van Renterghem C, Vigne P, Barhanin J, Schmid-Alliana A, Frelin C, Lazdunski M. Molecular mechanism of action of the vasoconstrictor peptide endothelin. Biochemical and Biophysical Research Communications. 1988;157:977–985. doi: 10.1016/s0006-291x(88)80970-7. [DOI] [PubMed] [Google Scholar]
- Vorndran C, Minta A, Poenie M. New fluorescent calcium indicators designed for cytosolic retention or measuring calcium near membranes. Biophysical Journal. 1995;69:2112–2124. doi: 10.1016/S0006-3495(95)80082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Y, Whorton AR, Watkins WD. Inhibition by nicardipine on endothelin-mediated inositol phosphate formation and Ca2+ mobilization in smooth muscle cell. Biochemical and Biophysical Research Communications. 1989;160:758–764. doi: 10.1016/0006-291x(89)92498-4. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Masaki T. Endothelin, a novel endothelium-derived peptide. Biochemical Pharmacology. 1989;38:1877–1883. doi: 10.1016/0006-2952(89)90484-x. [DOI] [PubMed] [Google Scholar]
- Yu JCM, Pickard JD, Davenport AP. Endothelin ET(A) receptor expression in human cerebrovascular smooth-muscle cells. British Journal of Pharmacology. 1995;116:2441–2446. doi: 10.1111/j.1476-5381.1995.tb15093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CZ, Auer RN. Graded hypotension and MCA occlusion duration: effect in transient focal ischemia. Journal of Cerebral Blood Flow and Metabolism. 1995;15:980–988. doi: 10.1038/jcbfm.1995.124. [DOI] [PubMed] [Google Scholar]
- Zuccarello M, Boccaletti R, Tosun M, Rapoport RM. Role of extracellular Ca2+ in subarachnoid hemorrhage-induced spasm of the rabbit basilar artery. Stroke. 1996a;27:1896–1902. doi: 10.1161/01.str.27.10.1896. [DOI] [PubMed] [Google Scholar]
- Zuccarello M, Soattin GB, Lewis AI, Breu V, Hallak H, Rapoport RM. Prevention of subarachnoid hemorrhage-induced cerebral vasospasm by oral administration of endothelin receptor antagonists. Journal of Neurosurgery. 1996b;84:503–507. doi: 10.3171/jns.1996.84.3.0503. [DOI] [PubMed] [Google Scholar]