

Abstract
Whole-cell patch clamp recordings were used to investigate the modulation by reducing and oxidizing agents of recombinant human cardiac L-type Ca2+ channel α1C subunits stably expressed in human embryonic kidney (HEK 293) cells.
The oxidizing agents thimerosal (10 μM) and p-chloromercuribenzene sulphonic acid (PCMBS; 2 μM to 2 mM) caused irreversible inhibition of Ca2+ channel currents. The reducing agent 1,4-dithiothreitol (DTT; 2 mM) was without effect on Ca2+ channel currents, but reversed the inhibitory actions of thimerosal and PCMBS.
Ca2+ channel currents were also inhibited by pretreatment with the methanethiosulphonate compound (2-aminoethyl)methanethiosulphonate (MTSEA, 2·5 mM), but were unaffected by identical pretreatment with (2-sulphonatoethyl)methanethiosulphonate (MTSES, 10 mM). The effects of MTSEA could be fully reversed by DTT (2 mM). The degree of current inhibition caused by 200 μM PCMBS was not significantly affected by pretreatment with MTSEA, and following PCMBS treatment, MTSEA caused a similar degree of inhibition to that observed in cells that were not previously treated with PCMBS. These findings suggested that distinct thiol groups were modulated by these two agents.
Hypoxic inhibition of Ca2+ channel currents was unaffected by pretreatment of cells with MTSEA but was fully prevented by treatment with PCMBS. Our results indicate that distinct cysteine residues on the α1C subunit can undergo redox modulation and in so doing alter channel function. Some, but not all, of these residues appear to be associated with the mechanism underlying inhibition of this channel by hypoxia.
Changes in cellular metabolic status resulting from hypoxic or ischaemic conditions are well known to modulate ion channel activity in cardiac and other tissues, for example through altered channel phosphorylation state (Allen & Orchard, 1987; Trautwein & Hescheler, 1990; McDonald et al. 1994; Benndorf et al. 1997). However, in recent years several studies using a diverse range of tissues have indicated that ion channels are rapidly influenced by local oxygen levels in a membrane-limited, possibly direct manner (reviewed by Peers, 1997). Such studies were initially conducted in the carotid body arterial chemoreceptor, where oxygen-sensitive K+ channels were identified (Lopez-Barneo et al. 1988). Since then, a variety of K+ channels in these and other cells have been shown to be inhibited by hypoxia (Peers & Buckler, 1995; Haddad & Jiang, 1997; Lopez-Barneo et al. 1997; Peers, 1997).
More recently, L-type Ca2+ channels in vascular smooth muscle cells (Franco-Obregon et al. 1995; Franco-Obregon & Lopez-Barneo, 1996) and carotid body chemoreceptor cells (Montoro et al. 1996) have also been shown to be oxygen sensitive. Hypoxia inhibits these L-type Ca2+ channels in a voltage-dependent manner which appears to be associated with a slowing of channel activation kinetics. Recently, we demonstrated that hypoxia inhibits the recombinant human L-type cardiac Ca2+ channel α1C subunit stably expressed in human embryonic kidney (HEK 293) cells in a manner indistinguishable from that seen in native smooth muscle L-type Ca2+ channels (Fearon et al. 1997). Thus, the structural requirements of the channel protein for oxygen sensing reside in the α1 subunit and are shared by native smooth muscle and human cardiac variants. Furthermore, the presence of auxiliary subunits is not required to observe hypoxic inhibition.
The mechanism by which hypoxia inhibits L-type Ca2+ channels is unknown. Hypoxic inhibition of K+ channels in pulmonary smooth muscle (and other) cells has been proposed to involve redox modulation (Archer et al. 1993; Weir & Archer, 1995). In brief, this proposal suggests that in hypoxia, cellular redox couples such as glutathione (GSSG:GSH) are altered such that the reduced form of these couples (e.g. GSH) is present at higher concentrations than under normoxic conditions (see also Acker et al. 1992; Acker & Xue, 1995). These reduced forms may then interact with channel proteins to alter functioning such that channel inhibition is observed in hypoxia. In the present study, we have investigated potential redox modulation of recombinant human cardiac L-type Ca2+ channel α1C subunits, and whether such modulation is involved in hypoxic inhibition. This possibility has been suggested in part by recent studies which have indicated that both native (Lacampagne et al. 1995; Campbell et al. 1996) and recombinant (Chiamvimonvat et al. 1995; Hu et al. 1997) L-type cardiac Ca2+ channels can be modulated by oxidizing and reducing agents acting at thiol groups of cysteine residues in the channel protein.
METHODS
All experiments were carried out in HEK 293 cells stably expressing the human cardiac L-type calcium channel α1C subunit (Schultz et al. 1995). The construction of this cell line has been described previously (Fearon et al. 1997). Cells were grown in minimum essential medium with Earle's salts (Gibco, Paisley, UK), containing 9% (v/v) fetal calf serum (Globepharm, Esher, Surrey, UK), 1% (v/v) non-essential amino acids, 50 mg l−1 gentamicin, 10000 u l−1 penicillin G, 100 mg l−1 streptomycin, 0.25 mg l−1 amphotericin and 400 mg l−1 G418 (all Gibco) at 37°C in a humidified atmosphere of 5% CO2 in air. Cells were harvested from their culture flasks by trypsinization and plated out onto glass coverslips 24-48 h before use in electrophysiological studies.
Pieces of coverslip with attached cells were transferred to a continuously perfused (approximately 1-2 ml min−1) recording chamber (volume, 80 μl) and whole-cell patch-clamp recordings (Hamill et al. 1981) were made using patch pipettes of resistance 4-7 MΩ. Cells were perfused with a solution composed of (mmol l−1): NaCl, 95; CsCl, 5; MgCl2, 0.6; BaCl2, 20; Hepes, 5; D-glucose, 10; TEA-Cl, 20 (21-24°C, pH 7.4); and patch electrodes were filled with a solution of composition (mmol l−1): CsCl, 120; TEA-Cl, 20; MgC12, 2; EGTA, 10; Hepes, 10; ATP, 2 (pH 7.2). In experiments using PCMBS, 20 mM CaCl2 replaced BaCl2 in the extracellular bathing solution since this compound precipitates in solution containing barium ions. Cells were voltage clamped at -80 mV, and whole-cell currents were evoked by step depolarizing the membrane to various test potentials for 100 ms at a frequency of 0.1 Hz. Series resistance compensation of 70-85% was applied. Current traces were filtered at 1-2 kHz, digitized at 2-4 kHz and stored on computer for measurement of current amplitude. Leak subtraction was performed on line using a P/4 protocol for time series experiments, and by the appropriate scaling and subtraction of the average leak current evoked by small hyperpolarizing and depolarizing steps when constructing current-voltage relationships. Current amplitudes were measured over the last 10-15 ms of each step depolarization when Ba2+ was used as charge carrier, since they displayed little or no inactivation during step depolarizations. When Ca2+ was used as charge carrier, inactivation was observed and currents were measured at their peak. All voltage protocols were applied, and data acquisition and analysis performed, using an Axopatch 200A amplifier in combination with a Digidata 1200 and pCLAMP 6.0.2 software (Axon Instruments).
Bath hypoxia was achieved by bubbling the reservoir leading to the bath with nitrogen, and passing a stream of nitrogen over the surface of the bath. The level of hypoxia achieved (always stable within 30-60 s of switching solution) ranged from 15 to 25 mmHg, as measured with a commercial needle oxygen electrode (Strathkelvin Instruments, Glasgow, UK).
All drug solutions were prepared by dissolution in the extracellular perfusate. In experiments with (2-aminoethyl)methanethiosulphonate (MTSEA) and (2-sulphonatoethyl)methanethiosulphonate (MTSES) (both obtained from Toronto Research Chemicals, Totonto, Ontario, Canada) cells were incubated for 5 min at room temperature in the drug solution immediately after dissolving the drug. Cells were then transferred to the recording chamber and recordings made as described. Thimerosal and p-chloromercuribenzene sulphonic acid (PCMBS; both from Sigma) were bath applied to cells by switching to a reservoir containing the drug solution (in some cases PCMBS was applied to cells before recordings were made; see Results). Experiments with PCMBS were performed under low light intensity.
Statistical comparisons were made using Student's paired or unpaired t tests, as appropriate.
RESULTS
To investigate whether hypoxic inhibition of the recombinant human L-type Ca2+ channel α1C subunit involved redox modulation of sulfhydryl groups in cysteine residues, we first examined the ability of known reducing and oxidizing compounds to alter channel function, as determined using whole-cell patch clamp recordings. Figure 1A (representative of 13 such recordings) illustrates that the reducing agent 1,4-dithiothreitol (DTT, 2 mM) was without effect on Ca2+ channel currents recorded in HEK 293 cells. In contrast, when the oxidizing agent PCMBS (2 μM to 2 mM) was applied in the extracellular perfusate currents were inhibited (e.g. Fig. 1B). The effect of PCMBS was always irreversible (see e.g. Fig. 5C), but current amplitudes could be largely restored by bath application of 2 mM DTT (Fig. 1B), indicating that the blocking action of PCMBS arose from reaction with thiol group(s) in the channel protein. The structurally related compound thimerosal (10 μM) also had inhibitory effects (74.3 ± 4.0% inhibition, n = 16) that could be reversed with DTT (not shown).
Figure 1. Dithiothreitol reverses the inhibitory actions of PCMBS on recombinant Ca2+ currents.
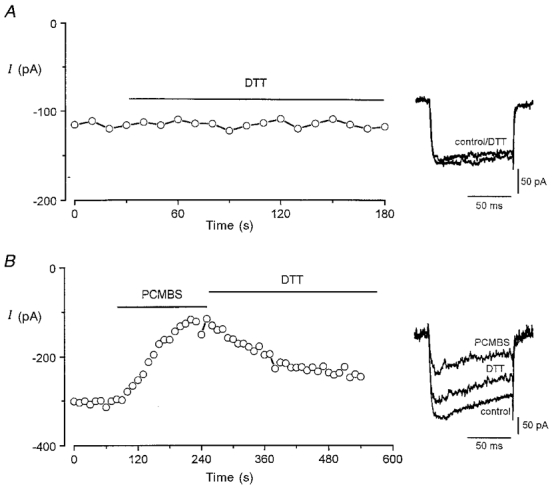
A, example time-series plot of Ca2+ channel current amplitudes; each plotted point represents the current amplitude evoked by repeated step depolarizations (100 ms, 0.2 Hz) to +10 mV from a holding potential of -80 mV. For the period indicated by the horizontal bar, the perfusate was exchanged for one containing 2 mM dithiothreitol (DTT). Inset shows example currents from this recording before and during exposure to DTT as indicated. B, as A, except that the cell was firstly perfused with 200 μM PCMBS, and then with 2 mM DTT. Inset shows example currents from this recording before and during exposure to PCMBS, then during DTT exposure as indicated.
Figure 5. Hypoxic inhibition of Ca2+ channel currents is unaffected by MTSEA but abolished by PCMBS.
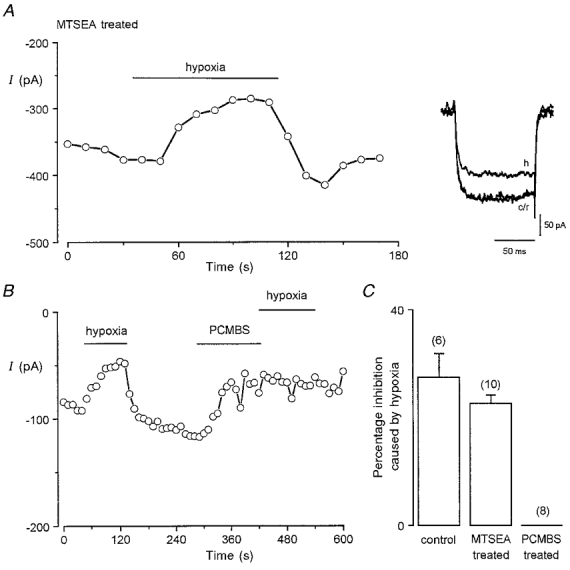
A, example time series plot of Ca2+ channel current amplitudes evoked in a cell by repeated step depolarizations to +10 mV from a holding potential of -80 mV (100 ms, 0.2 Hz). The cell had been previously treated with 2.5 mM MTSEA for 5 min. Period of exposure of cell to hypoxic solution (PO2 ca. 20 mmHg) indicated by horizontal bar. Inset shows example currents from the same cell before (c), during (h) and after (r) exposure to hypoxic solution. B, as A, except cells were not pretreated with any agent. Instead, the cell was firstly exposed to hypoxia, then PCMBS (200 μM) was bath applied for period indicated by horizontal bar, and then the cells were exposed again to hypoxia. C, mean (with vertical s.e.m. bar) percentage inhibition caused by hypoxia in untreated cells (control), in cells pretreated with 2.5 mM MTSEA and in cells pretreated with 200 μM PCMBS. Inhibition calculated from time series experiments such as in B. Number of cells tested indicated above each bar.
Current-voltage relationships were constructed in 11 cells before and after application of 200 μM PCMBS (Fig. 2A). It is noteworthy that when using 20 mM Ca2+ as charge carrier for these studies, peak inward currents were observed at the very positive test potential of +40 mV. This is likely to be due to the strong surface charge screening effect of Ca2+ (Hille et al. 1975) since when the same concentration of Ba2+ was used, current-voltage relationships typically peaked at around 0 or +10 mV (see e.g. Fig. 3). Despite this, it was apparent that PCMBS inhibited currents at all activating test potentials studied (Fig. 2A). A concentration-response relationship was also determined for PCMBS (Fig. 2B), and the inhibitory action of PCMBS was not significantly increased by raising the concentration from 20 μM to 2 mM. Thus, the effects of PCMBS approached saturation and this agent was not able to fully block Ca2+ currents.
Figure 2. PCMBS inhibition of recombinant human cardiac Ca2+ channel α1C subunits.
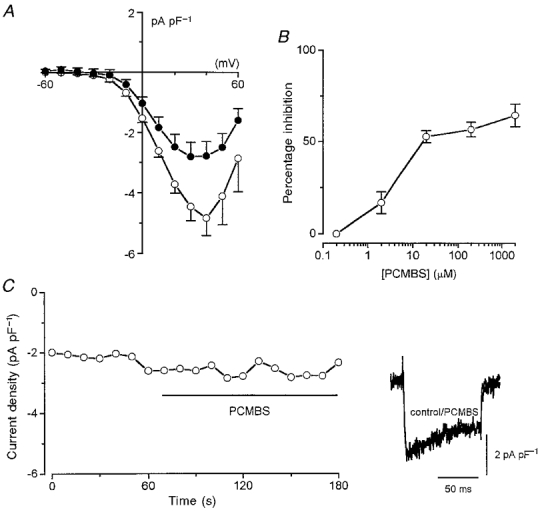
A, mean (with vertical s.e.m. bars) Ca2+ current density versus voltage before (control, ○, n = 11 cells) and after (PCMBS, •, n = 11 cells) bath application of 200 μM PCMBS. B, percentage inhibition of Ca2+ current caused by bath application of PCMBS at a range of concentrations. Each point is the measured percentage inhibition with vertical s.e.m. bars taken from between 6 and 9 cells. Inhibition was calculated from measurements of currents evoked by step depolarizations from -80 to +10 mV. C, time series plot of Ca2+ current amplitudes evoked by successive step depolarizations in a representative cell which had been pretreated with 200 μM PCMBS. Period of bath application of 200 μM PCMBS indicated by horizontal bar. Inset shows example currents before and during bath application of PCMBS, as indicated.
Figure 3. Inhibition of recombinant human cardiac Ca2+ channel α1C subunits by methanethiosulphonate compounds.
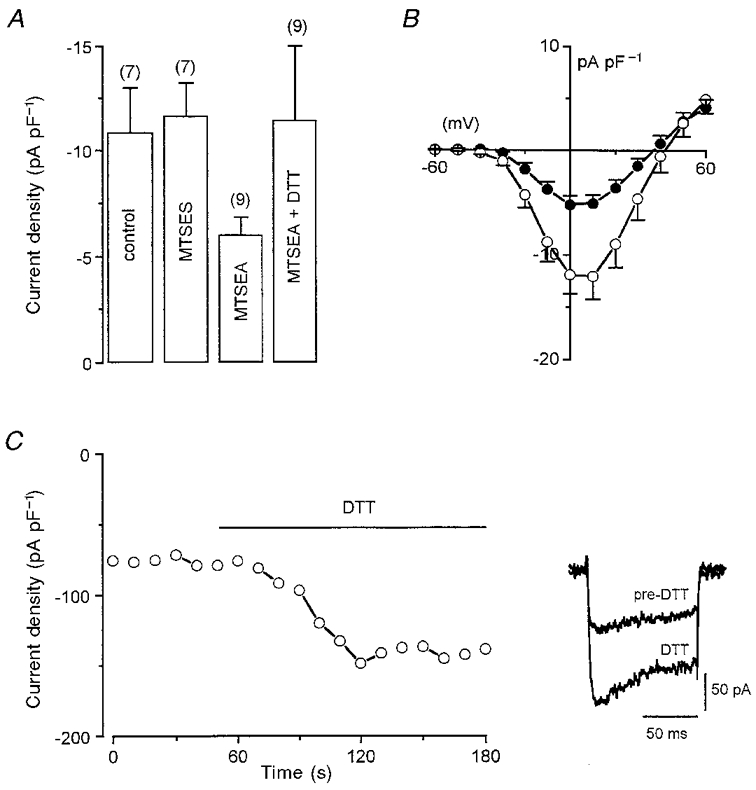
A, mean (with vertical s.e.m. bars) Ca2+ channel current density (recorded using currents evoked by step depolarizations to +10 mV from a holding potential of -80 mV, and Ba2+ as charge carrier) in untreated cells, cells pretreated with 10 mM MTSES, cells pretreated with 2.5 mM MTSEA, and cells pretreated with MTSEA and subsequently exposed to 2 mM DTT, as indicated. Number of cells tested in each case is indicated above each bar. B, mean (with vertical s.e.m. bars) Ca2+ channel current density versus voltage plots in untreated cells (○, n = 8) and cells pretreated with 2.5 mM MTSEA (•, n = 14). C, time series plot of Ca2+ channel current amplitudes evoked by successive step depolarizations in a representative cell which had been pretreated with 2.5 mM MTSEA. Period of bath application of 2 mM DTT indicated by horizontal bar. Inset shows example currents before and during bath application of DTT.
These experiments were all conducted by bath applying PCMBS whilst the cell was repeatedly step depolarized. The inhibitory action could therefore arise because PCMBS reacts with a cysteine residue which is exposed during depolarization when the channel is in its open conformation. To investigate this possibility further, we pretreated cells with PCMBS (200 μM) for 1 min without applying depolarizing steps and then recorded currents in the absence of extracellular PCMBS, using the repeated step depolarization protocol (to +10 mV). Pretreatment with PCMBS significantly (P < 0.001, unpaired t test) reduced current density in these experiments from a control value of -5.28 ± 0.57 pA pF−1 (n = 22 cells which were not pretreated, measured at a test potential of +10 mV) to -2.93 ± 0.48 pA pF−1 (n = 12 pretreated cells). This current density value following PCMBS pretreatment was indistinguishable from that observed following bath application of PCMBS to nine cells during repeated step depolarizations to +10 mV (-2.78 ± 0.39 pA pF−1). Furthermore, bath application of 200 μM PCMBS caused no further inhibition of current in cells pretreated with this agent (Fig. 2C, representative of 6 cells tested). These findings suggest that channel activation is not required for PCMBS to access cysteine residues and thereby cause inhibition.
We have also investigated the effects on whole cell Ca2+ channel currents of two methanethiosulphonate (MTS) compounds, the negatively charged MTSES and the positively charged MTSEA. These agents are highly reactive and extremely unstable in solution, causing a large acidic shift in solution pH. For this reason, we did not attempt to bath-apply these compounds, but instead pre-exposed cells to them for a brief period (5 min) immediately after dissolving them. We found that pretreatment of cells with MTSES (10 mM) had no effect on Ca2+ channel current density (Fig. 3A). By contrast, pretreatment with MTSEA (2.5 mM) caused a significant decrease (P < 0.001) in Ca2+ current density but, as for PCMBS, currents could not be fully blocked (Fig. 3A). A comparison of current-voltage relationships in control and MTSEA-treated cells indicated that this agent, like PCMBS (Fig. 2A), caused incomplete inhibition of currents throughout the range of activating test potentials studied (Fig. 3B). Also like PCMBS (Fig. 1), the inhibitory action of MTSEA occurred via an interaction with thiol groups on cysteine residues, since current densities could be enhanced in MTSEA-treated cells with the reducing agent DTT (2 mM; Fig. 3A and C). Indeed, current density in cells pretreated with MTSEA and then exposed to DTT in the perfusate were not significantly different from those found in untreated cells (Fig. 3A).
Previous studies (Akabas et al. 1992; Holmgren et al. 1996) have indicated that maximal effects of MTSEA on ion channel function should be observed under our experimental conditions (exposure of cells to 2.5 mM MTSEA for 5 min). We were unable to test this for ourselves since longer exposures, or exposures to higher concentrations of MTSEA, invariably caused cell detachment from coverslips and a general degradation in their appearance. However, the observations that neither MTSEA nor PCMBS could fully inhibit Ca2+ channel currents prompted us to investigate whether they might be acting at the same or different thiol groups on the channel protein. To address this, we first pretreated cells with 2.5 mM MTSEA, then bath applied PCMBS (200 μM). As illustrated in Fig. 4, PCMBS was able to inhibit currents in MTSEA-treated cells (by 48.2 ± 5.7%, n = 7). This degree of inhibition was not significantly different from that seen in cells which had not previously been exposed to MTSEA (P = 0.19, Student's unpaired t test). In a further series of experiments, we measured current densities in cells either untreated, or pretreated with MTSEA (2.5 mM) or PCMBS (200 μM; a maximally effective concentration) or pretreated with 2.5 mM MTSEA following pretreatment with 200 μM PCMBS. These studies were all conducted using Ca2+ as charge carrier, and results are summarized in Fig. 4B. The most important observation was that pretreatment with PCMBS did not abolish the inhibitory action of MTSEA. Indeed, the effect of MTSEA to reduce current density (by approximately 32%; P < 0.02, unpaired t test) was similar following PCMBS treatment (38%; P < 0.04, unpaired t test, Fig. 4B). These findings further support the idea that these two agents act to inhibit Ca2+ currents by acting at distinct sites on the α1C protein.
Figure 4. Pretreatment with MTSEA does not prevent Ca2+ current inhibition by PCMBS, and vice versa.

A, time series plot of Ca2+ current amplitudes evoked in a representative HEK 293 cell by repeated step depolarizations (100 ms, 0.2 Hz) to +10 mV from a holding potential of -80 mV. The cell had previously been exposed to 2.5 mM MTSEA. Period of bath application of 200 μM PCMBS is indicated by horizontal bar. Superimposed traces show example currents before and during bath application of PCMBS. B, bar graph plotting mean (±s.e.m.) current density obtained in cells by step depolarizations from -80 mV to +10 mV using 20 mM Ca2+ as charge carrier. Values were obtained from the number of cells indicated below each bar under control conditions, following pretreatment with either PCMBS (200 μM) or MTSEA (2.5 mM), or following pretreatment with PCMBS and then MTSEA at the same concentrations, as indicated.
As described in the Introduction, it has been suggested that hypoxic inhibition of ion channels might involve redox modulation of cysteine residues within the channel protein. To study this possibility, we have investigated whether hypoxia could modulate Ca2+ channel currents following pretreatment of cells with either MTSEA or PCMBS. As illustrated in Fig. 5A, pretreatment of cells with MTSEA did not prevent currents being inhibited by hypoxia: in 10 MTSEA-treated cells tested, lowering of perfusate PO2 levels to 15-25 mmHg caused a reversible inhibition of currents by 27.4 ± 4.5%, a value not significantly different from the degree of inhibition previously reported (Fearon et al. 1997) at this PO2 level when Ba2+ was used as charge carrier (P = 0.23, unpaired t test; see also Fig. 5C). In contrast to this finding, exposure of cells (n = 8) to PCMBS completely abolished hypoxic inhibition of currents (Fig. 5B and C). Thus, redox modulation of channels with PCMBS, but not with MTSEA, prevented channel inhibition by hypoxia.
DISCUSSION
Modulation by reducing and oxidizing agents of both native and recombinant L-type Ca2+ channels has previously been investigated by other groups, and differing effects are apparent. Using myocytes isolated from ferret ventriculum, Campbell et al. (1996) recently demonstrated that DTT caused inhibition of L-type Ca2+ currents, an effect which could be reversed by the oxidizing agent 5,5′-dithio-bis[2-nitrobenzoic acid] (DTNB). Indeed, DTNB applied alone was able to enhance Ca2+ currents. Their observation that DTT could inhibit currents contrasts with our finding that DTT was without effect on currents that were not previously pretreated with PCMBS or MTSEA (Fig. 1A). However, our results are in excellent agreement with those of Lacampagne et al. (1995), who reported that DTT was without effect on L-type Ca2+ currents recorded in guinea-pig ventricular myocytes. Furthermore, these workers found that Ca2+ currents could be inhibited by p-hydroxy-mercuric-phenylsulphonic acid (PHMPS, structurally extremely similar to PCMBS and thimerosal used in the present study) and also showed that this inhibition by PHMPS could be reversed by DTT. Thus our observations using recombinant human L-type cardiac Ca2+ channel α1C subunits are in accordance with results obtained from guinea-pig but not ferret cardiac myocytes.
Using α1 subunits of L-type Ca2+ channels from rabbit lung (vascular smooth muscle) stably transfected in Chinese hamster ovary (CHO) cells, Chiamvimonvat et al. (1995) found that two oxidizing agents inhibited whole-cell Ca2+ currents. These workers found similar inhibitory actions of thimerosal, which is a poorly membrane-permeable oxidizing agent, and 2,2′-dithiodipyridine (DTDP), a hydrophobic, membrane-permeable oxidizing agent. Furthermore, these effects could be reversed by DTT, indicating that inhibition of currents was dependent on oxidation of an extracellularly accessible cysteine residue. In a subsequent study (Hu et al. 1997) this same group found that rabbit cardiac L-type α1C subunits, co-expressed with either the rabbit skeletal muscle β subunit (β1a) or the rat cardiac β subunit (β2a) were modestly inhibited by MTSEA and virtually unaffected by MTSES (the two methanethiosulphonate compounds used in the present study). These findings are in reasonable agreement with the present study, although the magnitude of current inhibition is variable. Thus, despite variable factors such as differences in recombinant expression systems, splice variations between the different α1 subunits studied, species variation and the fact that some experiments were conducted using co-expression of α1 subunits with auxiliary subunits, redox modulation of the major, pore-forming subunit of the L-type Ca2+ channel is well established in recombinant studies.
Given that some of the agents used in the present study are membrane permeable (DTT, MTSEA), the possibility that Ca2+ current inhibition is secondary to other, intracellular actions of these agents should be considered. A previous report has indicated that the oxidizing agent thimerosal can cause release of Ca2+ from intracellular stores in a pituitary cell line (Karhapaa et al. 1996; NB these cells required permeabilization to allow access of thimerosal), a finding which raises the possibility that current inhibition could be due to Ca2+-induced current inactivation arising from Ca2+ mobilization from internal stores. We consider that this possibility is unlikely, since our previous studies have shown that, although another Ca2+ mobilizing agent (isobutylmethylxanthine) can cause inhibition of the α1C subunit stably expressed in HEK 293 cells (Fearon et al. 1998), this is entirely independent of any action on intracellular Ca2+ stores. Furthermore, the redox modulating agents used here had no appreciable effect on Ca2+ current inactivation rates, which would be expected to become accelerated if Ca2+-induced inactivation was evoked. Methanethiosulphonate compounds such as MTSEA and MTSES used here are assumed to act directly on cysteine residues of the protein of interest (i.e. the α1C subunit in the present study); numerous reports in the literature make the same assumption, and in many cases this is strongly supported by the fact that cysteine-substituted mutant proteins respond in dramatically different ways to these agents, indicating a direct effect on recombinant membrane proteins (see e.g. Akabas et al. 1992; Xu & Akabas, 1996).
The present study is, to our knowledge, the first to compare directly the effects of two structurally distinct agents which react with cysteine residues. Our finding that PCMBS could inhibit Ca2+ currents in cells pretreated with MTSEA argues strongly that these two agents react with different cysteine residues. This conclusion rests on the assumption that MTSEA was maximal in its effect under our conditions of application (preincubation for 5 min at a concentration of 2.5 mM). We were unable to increase either exposure time or MTSEA concentration without causing cell damage, but it is reasonable to assume that this treatment would cause maximal effects, given that previous studies have indicated that at this concentration MTSEA exerts maximal effects with exposure periods of only a few seconds (Hu et al. 1997). Our conclusion that these agents act at different residues is also supported by previous studies on cardiac Na+ channels which were found to be unaffected by thimerosal or DTDP (Chiamvimonvat et al. 1995), but were strongly inhibited by MTSEA (Hu et al. 1997). It is most likely that these agents discriminate between different cysteine residues because of the nature of the local environment surrounding the cysteine residues. Both the PCMBS-sensitive and the MTSEA-sensitive residues appear to be accessible to these reagents whist the channel is in the closed state, since both agents were effective in causing channel inhibition when applied to cells prior to channel activation using depolarizing voltage steps. Indeed, exposure to PCMBS-pretreated cells of PCMBS via the perfusate caused no further inhibition (Fig. 2C). Available evidence indicates that PCMBS is only poorly membrane permeable and so our findings indicate it acts at an extracellularly accessible cysteine residue. By contrast, MTSEA has been shown to be membrane permeable (Holmgren et al. 1996) and so we cannot discount an effect of this agent on an intracellularly located cysteine residue.
Results presented in Fig. 5 provide further evidence for the suggestion that PCMBS and MTSEA react with different cysteine residues. Thus, hypoxic inhibition of Ca2+ channel currents was observed in MTSEA-treated cells, and the degree of hypoxic inhibition was comparable to our previously reported observations in untreated cells (Fearon et al. 1997). This indicates that the cysteine residues that react with MTSEA are in no way involved in hypoxic inhibition of Ca2+ channels, but are in some way involved in normal channel function, since MTSEA significantly reduced current amplitudes. By contrast, in PCMBS-treated cells hypoxic inhibition was abolished. It should be noted that in order to avoid precipitation, experiments involving PCMBS were conducted using Ca2+ rather than Ba2+ as charge carrier and our previous study demonstrated that Ca2+ (as opposed to Ba2+) currents are more sensitive to inhibition by hypoxia (Fearon et al. 1997), so our present finding that PCMBS abolished this effect is not due to the fact that different charge carriers were used. We therefore conclude that cysteine residues sensitive to PCMBS treatment (but not those sensitive to MTSEA treatment) are of importance in both normal channel functioning and the response of the channel to acute hypoxia.
The mechanism by which acute hypoxia inhibits Ca2+ channels or indeed other ion channel types remains to be determined. At present, it is not known whether oxygen sensing is a property of ion channels themselves, or a closely associated but distinct oxygen-sensing element endogenously expressed by the host cell (Lopez-Barneo, 1994). Available evidence argues in favour of the latter possibility: Patel et al. (1997) recently identified the oxygen-sensitive K+ channel in pulmonary vascular myocytes as Kv2.1 (found in association with the electrically silent Kv9.3). When expressed in a recombinant system (COS cells), these channels were only found to be inhibited by hypoxia in a subpopulation of cells, whilst all other biophysical and pharmacological properties were consistent in each transfected cell. The conclusion drawn by these authors was that COS cells do not always express an endogenous ‘oxygen sensor’, and hence the recombinant K+ channels do not always appear oxygen sensitive. If this is the case, our results would indicate that HEK 293 cells consistently express endogenous ‘oxygen sensors’, since we have always found hypoxia to inhibit the recombinant α1C subunit (Fearon et al. 1997). Furthermore, the present study would also suggest that the cysteine residues susceptible to redox modulation by PCMBS are important not only in normal channel functioning, but also in the functional coupling of the L-type Ca2+ channel α1C subunit to a putative oxygen sensor.
Acknowledgments
We are grateful to A. Schwartz for helpful discussions during these studies. This work was supported by the British Heart Foundation and the NIH (NIH HL22619-19). I. M. F. holds a British Heart Foundation PhD Studentship and A. J. B. is a British Heart Foundation Senior Lecturer.
References
- Acker H, Bolling B, Delpiano MA, Dufau E, Gorlach A, Holtermann G. The meaning of H2O2 generation in carotid body cells for PO2 chemoreception. Journal of the Autonomic Nervous System. 1992;41:41–52. doi: 10.1016/0165-1838(92)90125-z. 10.1016/0165-1838(92)90125-Z. [DOI] [PubMed] [Google Scholar]
- Acker H, Xue D. Mechanisms of O2 sensing in the carotid body in comparison with other O2-sensing cells. News in Physiological Sciences. 1995;10:211–216. [Google Scholar]
- Akabas MH, Stauffer DA, Xu M, Karlin A. Acetylcholine-receptor channel structure probed in cysteine-substitution mutants. Science. 1992;258:307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- Allen DG, Orchard CH. Myocardial contractile function during ischemia and hypoxia. Circulation Research. 1987;60:153–168. doi: 10.1161/01.res.60.2.153. [DOI] [PubMed] [Google Scholar]
- Archer SL, Huang J, Henry T, Peterson D, Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circulation Research. 1993;73:1100–1112. doi: 10.1161/01.res.73.6.1100. [DOI] [PubMed] [Google Scholar]
- Benndorf K, Thierfelder S, Doepner B, Gebhardt C, Hirche H. Role of cardiac K-ATP channels during anoxia and ischemia. News in Physiological Sciences. 1997;12:78–83. [Google Scholar]
- Campbell DL, Stamler JS, Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes – dual mechanism regulation by nitric-oxide and s-nitrosothiols. Journal of General Physiology. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamvimonvat N, O'rourke B, Kamp TJ, Kallen RG, Hofmann F, Flockerzi V, Marban E. Functional consequences of sulfhydryl modification in the pore-forming subunits of cardiovascular Ca2+ and Na+ channels. Circulation Research. 1995;76:325–334. doi: 10.1161/01.res.76.3.325. [DOI] [PubMed] [Google Scholar]
- Fearon IM, Palmer ACV, Balmforth AJ, Ball SG, Mikala G, Peers C. Inhibition of recombinant human cardiac L-type Ca2+ channel α1C subunits by 3-isobutyl-1-methylxanthine. European Journal of Pharmacology. 1998;342:353–358. doi: 10.1016/s0014-2999(97)01497-0. 10.1016/S0014-2999(97)01497-0. [DOI] [PubMed] [Google Scholar]
- Fearon IM, Palmer ACV, Balmforth AJ, Ball SG, Mikala G, Schwartz A, Peers C. Hypoxia inhibits the recombinant α1C subunit of the human cardiac L-type Ca2+ channel. The Journal of Physiology. 1997;500:551–556. doi: 10.1113/jphysiol.1997.sp022041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Obregon A, Lopez-Barneo J. Low PO2 inhibits calcium channel activity in arterial smooth muscle cells. American Journal of Physiology. 1996;271:H2290–2299. doi: 10.1152/ajpheart.1996.271.6.H2290. [DOI] [PubMed] [Google Scholar]
- Franco-Obregon A, Urena J, Lopez-Barneo J. Oxygen-sensitive calcium channels in vascular smooth muscle and their possible role in hypoxic arterial relaxation. Proceedings of the National Academy of Sciences of the USA. 1995;92:4715–4719. doi: 10.1073/pnas.92.10.4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad GG, Jiang C. O2-sensing mechanisms in excitable cells: role of plasma membrane K+ channels. Annual Review of Physiology. 1997;59:23–42. doi: 10.1146/annurev.physiol.59.1.23. 10.1146/annurev.physiol.59.1.23. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hille B, Woodhull AM, Shapiro BI. Negative surface charge near sodium channels of nerve: divalent ions, monovalent ions and pH. Philosophical Transactions of The Royal Society B. 1975;270:301–318. doi: 10.1098/rstb.1975.0011. [DOI] [PubMed] [Google Scholar]
- Holmgren M, Liu Y, Xu Y, Yellen G. On the use of thiol-modifying agents to determine channel topology. Neuropharmacology. 1996;35:797–804. doi: 10.1016/0028-3908(96)00129-3. [DOI] [PubMed] [Google Scholar]
- Hu H, Chiamvimonvat N, Yamagishi T, Marban E. Direct inhibition of expressed cardiac L-type Ca2+ channels by s-nitrosothiol nitric oxide donors. Circulation Research. 1997;81:742–752. doi: 10.1161/01.res.81.5.742. [DOI] [PubMed] [Google Scholar]
- Karhapaa L, Titievsky A, Kaila K, Tornquist K. Redox modulation of calcium entry and release of intracellular calcium by thimerosal in GH4C1 pituitary cells. Cell Calcium. 1996;20:447–457. doi: 10.1016/s0143-4160(96)90086-x. [DOI] [PubMed] [Google Scholar]
- Lacampagne A, Duittoz A, Bolanos P, Peineau N, Argibay J A. Effect of sulfhydryl oxidation on ionic and gating currents associated with L-type calcium channels in isolated guinea-pig ventricular myocytes. Cardiovascular Research. 1995;30:799–806. [PubMed] [Google Scholar]
- Lopez-Barneo J. Oxygen-sensitive ion channels – how ubiquitous are they? Trends in Neurosciences. 1994;17:133–135. doi: 10.1016/0166-2236(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Lopez-Lopez JR, Urena J, Gonzalez C. Chemotransduction in the carotid-body – K+ current modulated by PO2 in type-I chemoreceptor cells. Science. 1988;241:580–582. doi: 10.1126/science.2456613. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Ortega-Saenz P, Molina A, Franco-Obregon A, Urena J, Castellano A. Oxygen sensing by ion channels. Kidney International. 1997;51:454–461. doi: 10.1038/ki.1997.61. [DOI] [PubMed] [Google Scholar]
- Mcdonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Montoro RJ, Urena J, Fernandez-Chacon R, De Toledo GA, Lopez-Barneo J. Oxygen sensing by ion channels and chemotransduction in single glomus cells. Journal of General Physiology. 1996;107:133–143. doi: 10.1085/jgp.107.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AJ, Lazdunski M, Honore E. Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO Journal. 1997;16:6615–6625. doi: 10.1093/emboj/16.22.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C. Oxygen-sensitive ion channels. Trends in Pharmacological Sciences. 1997;18:405–408. doi: 10.1016/s0165-6147(97)01120-6. [DOI] [PubMed] [Google Scholar]
- Peers C, Buckler KJ. Transduction of chemostimuli by the type-I carotid-body cell. Journal of Membrane Biology. 1995;144:1–9. doi: 10.1007/BF00238411. [DOI] [PubMed] [Google Scholar]
- Schultz D, Mikala G, Yatani A, Engle DB, Iles DE, Segers B, Sinke RJ, Weghuis DO, Klockner U, Wakamori M, Wang JJ, Melvin D, Varadi G, Schwartz A. Cloning, chromosomal localization, and functional expression of the α1 subunit of the L-type voltage-dependent calcium channel from normal human heart. Proceedings of the National Academy of Sciences of the USA. 1995;92:4715–4719. doi: 10.1073/pnas.90.13.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein W, Hescheler J. Regulation of cardiac L-type calcium current by phosphorylation and G proteins. Annual Review of Physiology. 1990;52:257–274. doi: 10.1146/annurev.ph.52.030190.001353. [DOI] [PubMed] [Google Scholar]
- Weir EK, Archer SL. The mechanism of acute hypoxic pulmonary vasoconstriction – the tale of 2 channels. FASEB Journal. 1995;9:183–189. doi: 10.1096/fasebj.9.2.7781921. [DOI] [PubMed] [Google Scholar]
- Xu M, Akabas MH. Identification of channel-lining residues in the M2 membrane-spanning segment of the GABAA receptor α1 subunit. Journal of General Physiology. 1996;107:195–205. doi: 10.1085/jgp.107.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]