

Abstract
The role of cystine-glutamate exchange in controlling the extracellular glutamate concentration in the central nervous system was examined by whole-cell clamping neurons in rat brain slices, and using their glutamate receptors as sensors of extracellular glutamate concentration.
Applying cystine to cerebellar slices generated a membrane current in Purkinje cells which was abolished by glutamate receptor blockers. Similar cystine-evoked currents were seen in pyramidal cells of frontal cortex slices.
Control experiments on non-N-methyl-D-aspartate (non-NMDA) receptors in enzymatically isolated Purkinje cells showed that cystine did not produce a current in slice Purkinje cells by directly activating glutamate receptors, nor by potentiating the action of background levels of glutamate on receptors. Experiments on isolated salamander Müller cells showed that cystine did not block Na+-dependent GLAST glutamate transporters (homologous to the transporters in the Bergmann glia ensheathing the Purkinje cells), nor did it block the current produced by EAAT4 and EAAC1 glutamate transporters in Purkinje cells. Thus the cystine-evoked current in Purkinje cells is not due to a rise in extracellular glutamate concentration caused by block of Na+-dependent uptake.
The dependence of cystine-evoked current on cystine concentration in slice Purkinje cells could be fitted by a Michaelis-Menten relation with a Km of 250 μM. The Km predicted from this for cystine activating glutamate efflux is less than 140 μM, because of the non-linear dependence on glutamate concentration of the Purkinje cell current. The current evoked by 1 mM cystine was little affected by removal of extracellular chloride or addition of 1 mM furosemide (frusemide), but was potentiated by 1 mM 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS).
These data suggest that external cystine generates a current in slice Purkinje cells by activating cystine-glutamate exchange in cells of the slice, releasing glutamate which activates non-NMDA receptors in the Purkinje cell membrane.
Control of the extracellular glutamate concentration, [Glu]o, in the CNS by membrane transporters is important for terminating synaptic transmission and for keeping [Glu]o below neurotoxic levels (Takahashi et al. 1997). Much work has been done on the role of the five cloned Na+-dependent glutamate transporters (Storck et al. 1992; Pines et al. 1992; Kanai & Hediger, 1992; Fairman et al. 1995; Arriza et al. 1997) in maintaining a low [Glu]o (Fig. 1A), but other transporters could contribute to extracellular glutamate homeostasis in the brain. Cystine-glutamate exchangers, in particular, can have a density comparable to that of Na+-dependent glutamate transporters (Anderson et al. 1990), and so might be capable of modulating [Glu]o significantly. These exchangers have not yet been cloned, but are likely to exist in several molecular forms since cultured neurons and glia have cystine-glutamate exchangers with different affinities and pharmacology (Murphy et al. 1990).
Figure 1. Glutamate transporters in the CNS.
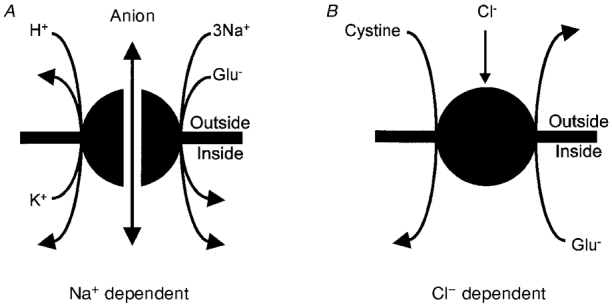
A, Na+-dependent transporters (GLAST and GLT-1 in glia, EAAC1, EAAT4 and EAAT5 in neurons) accumulate glutamate by co-transporting 3 Na+ and 1 H+, and countertransporting 1 K+ (Zerangue & Kavanaugh, 1996; Levy et al. 1998). In addition the transporters can generate current by virtue of an anion conductance in their structure which is gated by transport activity. B, Cl−-dependent transporters exchange cystine for glutamate and have not yet been cloned. They are inhibited by Cl− removal when the cystine concentration is low (Murphy et al. 1989).
The stoichiometry of cystine-glutamate exchange is uncertain, but first-order dependencies on substrate concentrations seen in radiotracing experiments (Zaczek et al. 1987; Kessler et al. 1987; Van Winkle et al. 1992) suggest that one cystine molecule (possibly as an anion: Bannai & Kitamura, 1981) may exchange with one glutamate anion (Fig. 1B). Extracellular Cl− removal reduces exchanger activity if the cystine concentration is low but not if it is high (Murphy et al. 1989), suggesting a modulation of the cystine affinity by Cl−. The concentration of cystine inside cells may be about 100-fold higher than that outside (Murphy et al. 1989), while that of glutamate is about 10 000-fold higher inside than outside (Takahashi et al. 1997), so the exchanger may normally transport glutamate out of cells and cystine in. However, with appropriate substrate gradients it can run in the reverse direction (Cho & Bannai, 1990; Kato et al. 1993) and might do so when [Glu]o rises during synaptic transmission.
Export of glutamate by cystine-glutamate exchange has been suggested as a mechanism by which microglia release glutamate and kill neurons (Piani & Fontana, 1994). Cystine-glutamate exchange is also important for providing cells with cystine, a precursor of the antioxidant glutathione (Bannai, 1984). Consequently, an elevated [Glu]o can kill neurons by preventing cystine uptake and inducing oxidative stress (Murphy et al. 1989, 1990), in addition to triggering death by the better known mechanism of excessively activating NMDA (N-methyl-D-aspartate) and non-NMDA receptors (Choi et al. 1987).
Radiotracing studies on cerebellar slices (Wyatt et al. 1996) suggest that cystine-glutamate exchange is capable of raising the extracellular glutamate concentration at a rate of 0.6 μM s−1 (see Discussion). To investigate whether cystine-glutamate exchange can alter [Glu]o enough to affect glutamate receptors, we activated the normal direction of operation of the exchanger by applying cystine in the extracellular fluid, and tried to detect any resulting change in [Glu]o by monitoring the membrane current of neurons containing glutamate receptors using whole-cell clamping.
METHODS
Cells
Purkinje cells in cerebellum and pyramidal cells in frontal cortex were whole-cell clamped in 200 μm thick brain slices (Penit-Soria et al. 1987; Konnerth et al. 1990; Perkel et al. 1990), cut from the brains of 12-day-old rats killed by cervical dislocation. Non-NMDA receptors in isolated Purkinje cells were studied by dissociating cerebellar slices with papain; Purkinje cells were identified by their large size soma with stumps of dendrite and axon still attached (Billups & Attwell, 1996). To study GLAST transporters in isolated retinal Müller cells from the tiger salamander (killed by decapitation followed by destruction of the brain), retinae were dissociated with papain as described by Barbour et al. (1991), and Müller cells were identified by their large size and distinctive shape.
Solutions
For experiments on rat brain slices and isolated Purkinje cells the normal external solution contained (mM): NaCl, 140; KCl, 2.5; MgCl2, 2; CaCl2, 2.5; NaH2PO4, 1; Hepes, 10; glucose, 10; bicuculline (to block GABAA receptors), 0.01; adjusted to pH 7.4 with NaOH, bubbled with O2. The pipette solution contained (mM): CsCl (to improve voltage uniformity in the Purkinje cell), 140; NaCl, 4; CaCl2, 0.5; (N-methyl-D-glucamine)2EGTA, 5; Hepes, 10; adjusted to pH 7.3 with CsOH. In experiments testing the effect of internal glutamate (see text) 10 mM sodium glutamate was present in the solution (replacing 6 mM CsCl and 4 mM NaCl). For Müller cell experiments, the external solution contained (mM): NaCl, 105; CaCl2, 3; MgCl2, 0.5; glucose, 15; Hepes, 5; BaCl2 (to block the cells’ inward rectifier K+ channels), 6; pH adjusted to 7.4 with NaOH; and the pipette solution contained (mM): KCl, 95; NaCl, 5; MgCl2, 7; CaCl2, 1; K2EGTA, 5; Na2ATP, 5; Hepes, 5; adjusted to pH 7.0 with KOH. Cystine, glutamate and other drugs were added to the external solutions used. All experiments were carried out at room temperature (23-25°C).
Electrodes
Purkinje cells and pyramidal cells were clamped with electrodes with a resistance in external solution of around 2 MΩ. In whole-cell mode the series resistance was typically 10 MΩ, and series resistance compensation was sometimes used to reduce this to around 2 MΩ. Müller cells were clamped with electrodes with resistances in external solution of 1-2 MΩ, and a series resistance in whole-cell mode of 3-5 MΩ. For the cystine- and glutamate-evoked currents reported here, series resistance voltage errors in all the cells studied were negligible. A 4 M NaCl-agar bridge was used as the earth electrode for experiments in which the extracellular chloride concentration was altered. For iontophoresis of D-aspartate onto Purkinje cells, microelectrodes with resistance around 100 MΩ filled with 100 mM Na-D-aspartate were positioned near the Purkinje cell soma, and the current through the electrode was changed from a holding current of +20 nA to an ejection current of -40 nA.
Data analysis
Effects of drugs on cystine-evoked currents were calculated by normalizing the response to cystine in the drug by the average of bracketing responses to cystine applied in control solution before and after the drug application. Cystine-evoked currents were measured at a fixed time (usually 30 or 60 s) after starting cystine application. Results are presented as means ±s.e.m.
RESULTS
Cystine evokes a current in Purkinje cells in cerebellar slices
Applying 1 mM cystine to Purkinje cells in cerebellar slices, whole-cell clamped to a negative potential, evoked an inward current (Fig. 2A). This current had a mean value of 9.1 ± 0.6 pA at -60 mV in 22 cells. It was reversibly abolished (Fig. 2A) in four cells by applying 50 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 25 μM D-(-)-2-amino-5-phosphonopentanoic acid (AP5) which are blockers, respectively, of non-NMDA and NMDA receptors (CNQX and AP5 produced no current change when applied alone). Purkinje cells of this age have been reported to have no functional NMDA receptors (Momiyama et al. 1996), suggesting that the cystine-evoked current was mediated by non-NMDA receptors. Blocking sodium-dependent and calcium-dependent action potentials with 10 μM TTX in zero calcium solution (replacing CaCl2 with MgCl2, with 2 mM EGTA added) had no effect on the cystine-evoked current (Fig. 2B; reduced by 6 ± 13 % in 4 cells), suggesting that the response was not transmitted to the Purkinje cell synaptically as a result of cystine depolarizing other cells in the slice. Polarizing the Purkinje cell to positive potentials reversed the current (Fig. 2C), which showed an I-V relation (Fig. 2D) typical of non-NMDA receptors (Takahashi et al. 1996).
Figure 2. Cystine-evoked currents in whole-cell clamped Purkinje cells.

A, in a Purkinje cell clamped to -75 mV, application of 1 mM cystine (as indicated by the horizontal bars) evokes an inward current, which is blocked when ionotropic glutamate receptors are blocked (right). B, applying TTX in zero calcium solution, to block Na+- and Ca2+-dependent action potentials, has no effect on the cystine-evoked current at -60 mV. C, the cystine-evoked current is inward at negative potentials and outward at positive potentials. D, mean I-V relation of the cystine-evoked current (ICys) in 4 cells (normalized to the value at -30 mV). Mean current at -30 mV was 7.4 ± 1.6 pA.
A simple explanation for these data would be that cystine evokes glutamate release from cells in the slice by cystine- glutamate exchange, raising [Glu]o and thus activating the Purkinje cell non-NMDA receptors. To verify this, however, we need to exclude any direct action of cystine on the cell's non-NMDA receptors, and exclude the possibility that cystine causes an increase in [Glu]o by blocking Na+-dependent glutamate transporters. The following two sections address these possibilities.
Cystine depresses non-NMDA responses to low [Glu]o and potentiates responses to high [Glu]o
Applying 1 mM cystine to Purkinje cells isolated enzymatically from cerebellar slices evoked no detectable current (6 cells), while low doses of glutamate (3 μM) evoked a readily detectable non-NMDA current (Fig. 3A) that was blocked by CNQX (data not shown). This shows, firstly, that 1 mM cystine does not directly activate non-NMDA receptors and, secondly, that the cystine is not significantly contaminated with glutamate or any other compound that might activate the channels.
Figure 3. Effects of cystine on non-NMDA receptor channels in enzymatically isolated Purkinje cells.
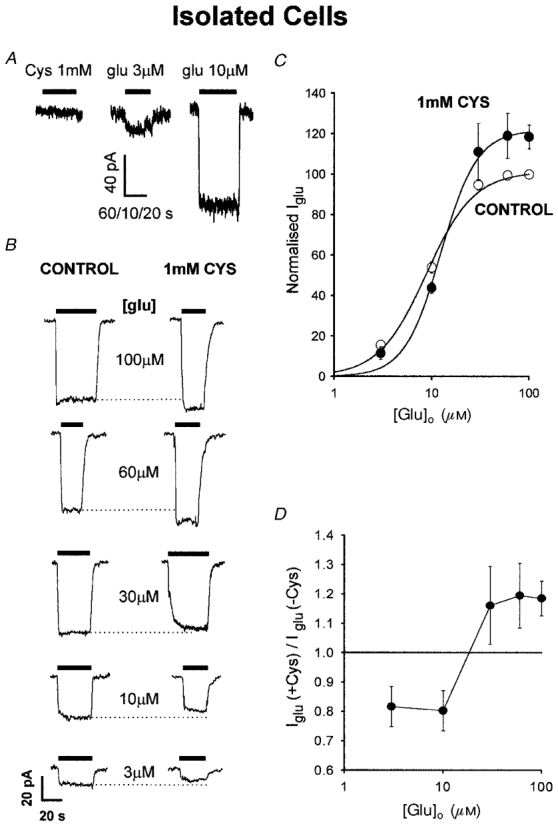
A, cystine (1 mM) evokes no current in a cell clamped to -33 mV. Responses to 3 and 10 μM glutamate are shown for comparison. The slow perfusion results in only the steady-state current being displayed. Time scale bar is 60, 10 and 20 s for the applications of cystine, 3 μM glutamate and 10 μM glutamate, respectively. B, effect of cystine on glutamate-evoked currents at -33 mV. Specimen responses to various glutamate doses are shown in the absence (left traces) and presence (right traces) of 1 mM cystine. Dotted lines extrapolate the peak response in the absence of cystine to facilitate comparison of the response in cystine. C, mean dose-response curve (± s.e.m.) for glutamate in the presence and absence of 1 mM cystine, from 5 cells studied as in B, normalized to 100 % for 100 μM glutamate in the absence of cystine. Smooth curves are best-fitted Hill equations, IGlu = Imax[Glu]n/([Glu]n+Kmn), with maximum current (Imax) = 101.3 %, half-maximal dose (Km) = 8.8 μM and Hill coefficient (n) = 1.8 in the absence of cystine, and Imax = 121.8 %, Km = 12.4 μM and n = 2.3 in the presence of cystine. D, ratio of the glutamate-evoked current in the presence of 1 mM cystine to the current in its absence (means ±s.e.m.), as a function of glutamate concentration.
Next we examined the possibility that cystine might modulate the action of glutamate on non-NMDA channels. Applying different doses of glutamate evoked an inward current that saturated at around 50 μM glutamate (Fig. 3B and C). In the presence of 1 mM cystine, the response to low doses of glutamate (< 20 μM) was reduced, while that to doses above 40 μM was potentiated (Fig. 3B-D). A possible explanation for this biphasic modulatory effect will be given in the Discussion. The value of [Glu]o in brain tissue has been reported to be in the low micromolar range (Wahl et al. 1994). Thus, applying cystine should decrease by about 20 % (Fig. 3D) any tonic current produced by this background level of glutamate acting on non-NMDA receptors. Consequently the inward current produced at negative potentials by cystine in slice Purkinje cells (Fig. 2) cannot be explained by cystine potentiating the action of the background extracellular glutamate in the slice.
Cystine has no effect on GLAST or EAAT4/EAAC1 transporters
The dendritic tree of Purkinje cells is ensheathed by Bergmann glia, which mainly express the GLAST Na+-dependent glutamate transporter (Rothstein et al. 1994). To investigate whether cystine causes an increase in [Glu]o by blocking Na+-dependent glutamate transporters, we whole-cell clamped salamander retinal Müller cells, which show a glutamate-evoked membrane current generated largely by glutamate uptake (Barbour et al. 1991). Several glutamate transporters are found in Müller cells (Eliasof et al. 1998), but the insensitivity of the glutamate-evoked current to dihydrokainate (Barbour et al. 1991; Arriza et al. 1994), and the relatively small anion conductance activated by glutamate transport (Billups et al. 1996), suggest that the glutamate-evoked current is generated largely by transporters homologous to the mammalian GLAST. Applying 1 mM cystine evoked no current change (Fig. 4A) in nine Müller cells, and did not significantly alter the current evoked by 10 μM glutamate (increased by 8.5 ± 4.5 % in 4 cells). We conclude that cystine does not cause an increase in [Glu]o by blocking GLAST transporters.
Figure 4. Cystine has no effect on GLAST and EAAT4/EAAC1 transporter currents.

A, membrane current at -63 mV of an isolated salamander retinal glial cell (which expresses mainly GLAST transporters) during the application of 10 μM glutamate (as indicated by bars) in the absence and presence of 1 mM cystine. B, membrane current at -33 mV of a Purkinje cell (in solution containing 25 μM 2,3-dihydroxy-6-nitro-7-sulphamoyl-benzene(f)quinoxaline (NBQX), 50 μM AP5 and 20 μM bicuculline) during iontophoresis of D-aspartate (D-asp, bars) which activates EAAT4 and EAAC1 transporters. The transporter currents (shown before, during and after washout (left to right) of solution containing cystine) were not affected by the presence of 1 mM cystine.
Purkinje cells themselves express both EAAT4 (Yamada et al. 1996) and EAAC1 transporters (Rothstein et al. 1994), and a block of these transporters by cystine could generate a rise in [Glu]o and current change as seen in Fig. 2A. To test whether this occurred, we monitored EAAT4/EAAC1 transporter currents in Purkinje cells as the current response to iontophoresed D-aspartate, in the presence of 25 μM NBQX, 50 μM AP5 and 20 μM bicuculline to block non-NMDA, NMDA and GABAA receptors (D-aspartate was used because it activates transporters but not non-NMDA receptors; iontophoresis was used to apply the D-aspartate to the cell body, limiting its penetration into the slice where it can release glutamate by heteroexchange on transporters and thus activate non-NMDA receptors; Takahashi et al. 1996). Applying 1 mM cystine had no effect on the transporter current (Fig. 4B; increased by 4.4 ± 5.0 % at -33 mV in 8 cells), so cystine does not cause an increase in [Glu]o by blocking EAAT4/EAAC1 transporters.
Dose-response curve for the action of cystine in slices
The data above support the idea that the current evoked by cystine in Purkinje cells in cerebellar slices is produced by cystine-glutamate exchange releasing glutamate which activates non-NMDA receptors in the Purkinje cell membrane. To investigate the level of cystine needed to release glutamate, we measured the Purkinje cell response to different cystine doses. A detectable inward current was produced by 100 μM cystine (Fig. 5A). The dose-response curve obtained from four Purkinje cells could be fitted by a Michaelis-Menten curve with a Km of around 250 μM (Fig. 5B, dashed curve).
Figure 5. Dose-response data for the cystine-evoked current at -63 mV in Purkinje cells.
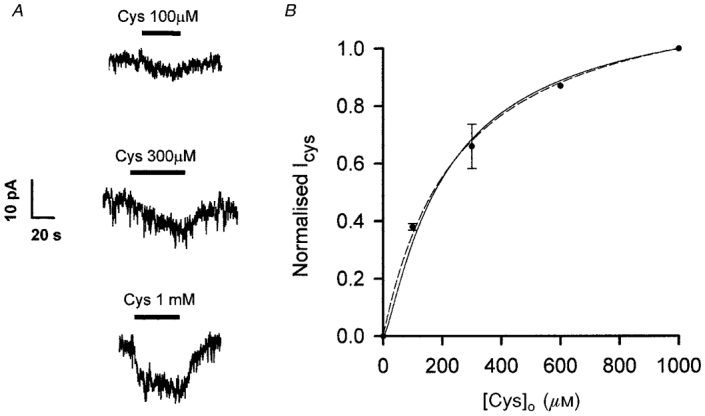
A, specimen data from 1 cell. B, mean dose-response curve from 4 cells (measured 30 s after start of the cystine application) normalized to the response to 1 mM cystine. Data are fitted with a Michaelis-Menten relation, ICys = Imax[Cys]/([Cys]+Km), with Imax = 1.25 and Km = 252 μM (dashed curve), and with a Michaelis-Menten relation raised to the power 1.5 (to compensate for the non-linear dependence on [Glu] of non-NMDA receptor responses) ICys =Imax([Cys]/([Cys]+Km))1.5, with Imax = 1.14 and Km = 140 μM (continuous curve).
On theoretical grounds this Km value is expected to be higher than that for the cystine dependence of glutamate efflux on the cystine-glutamate exchanger for two reasons. First, the rise of [Glu]o produced by cystine-glutamate exchange is not proportional to the glutamate efflux rate because of removal of glutamate by Na+-dependent glutamate transporters which saturate at high [Glu]o. Second, the non-NMDA receptors used to monitor the rise of [Glu]o generate a current which is a non-linear function of [Glu]o. The expected alteration of the dose-response curve for cystine by these effects can be calculated with simple assumptions as follows. Glutamate efflux on the exchanger represented in Fig. 1B is assumed to have a Michaelis-Menten dependence on external cystine concentration [Cys]o:
| (1) |
where VCys and KCys are the maximum rate and EC50 for cystine, respectively, of the cystine-glutamate exchanger. This efflux must equal the rate of glutamate removal by Na+-dependent uptake and by diffusion to the bulk solution. For simplicity we assume that diffusion is small compared with Na+-dependent uptake (the rise in [Glu]o that generates the current detected probably occurs in small extracellular spaces in poor diffusive contact with the bulk solution). Thus:
| (2) |
where VGlu and KGlu are the maximum rate and EC50 for glutamate, respectively, of Na+-dependent glutamate transport. Equating glutamate efflux and removal from eqns (1) and (2) we find that:
| (3) |
where [Glu]o,max = KGluVCys/(VGlu - VCys) and the value of [Cys]o which produces a half-maximal rise in [Glu]o is given by:
| (4) |
and so is increased from KCys by a factor which is larger when the maximum rate of cystine-glutamate exchange is closer to the maximum rate of Na+-dependent uptake. For the small rises in [Glu]o which presumably produce the small currents we detect when cystine is applied, non-NMDA receptors generate steady-state currents which (in hippocampal pyramidal cells) are approximately proportional to [Glu]o1.5 (Patneau & Mayer, 1990; the data in Fig. 3C are best fitted by Hill equations with Hill coefficients of 1.8 (control data) and 2.3 (in cystine), but there are not enough data at low [Glu]o values to define accurately the power of the dependence on [Glu]o). Thus, the current generated by cystine, ICys, is predicted to be proportional to eqn (3) raised to the power 1.5:
| (5) |
where Imax is the maximum current evoked by a high cystine concentration. Fitting this equation to the data in Fig. 5B (continuous line) gives a value for of 140 μM. Equation (4) predicts that the EC50 for cystine activating glutamate release is given by:
| (6) |
Accurate values for VCys/VGlu, the ratio of the Vmax values for cystine-glutamate exchange and Na+-dependent glutamate transport are not available for cerebellum, but using VCys = 450 μmol l−1 h−1 (for rat cerebellar slices, Wyatt et al. 1996) and VGlu= 2109 μmol l−1 h−1 (for cerebellar synaptosomes from 15-day-old rats, Sandoval et al. 1984), calculated assuming 95 mg protein (g wet tissue)−1), we estimate that KCys = 110 μM. For comparison, the Km value measured for uptake of radioactive cystine into rat cerebellar slices is 77 μM (Wyatt et al. 1996).
Adding 10 mM glutamate to the whole-cell pipette solution did not increase the current evoked by 1 mM cystine (mean value at -30 mV was 6.6 ± 0.3 pA in 5 cells, compared with 7.1 ± 1.3 pA in 5 cells studied with the internal solution lacking glutamate). This implies either that the cystine-glutamate exchange raising the extracellular glutamate concentration in these experiments is mainly in cells other than Purkinje cells (e.g. it might be in surrounding glia), or else that the Purkinje cell does express cystine- glutamate exchange but that dialysis of the Purkinje cell soma with glutamate-free solution does not effectively remove internal glutamate from the dendrites of the cell (which constitute the majority of its membrane area).
Effects of agents thought to block cystine-glutamate exchange
Under some circumstances, activity of the cystine- glutamate exchanger is reduced by removal of external chloride (Kessler et al. 1987; Zaczek et al. 1987; Murphy et al. 1989). We found, however, no significant effect of replacing Cl− by methanesulphonate ions on the current evoked in three Purkinje cells by 1 mM cystine (current decreased by 12 ± 19 %; Fig. 6A).
Figure 6. Effects of anion substitution and blockers on the current evoked by 1 mM cystine in Purkinje cells clamped to -63 mV.
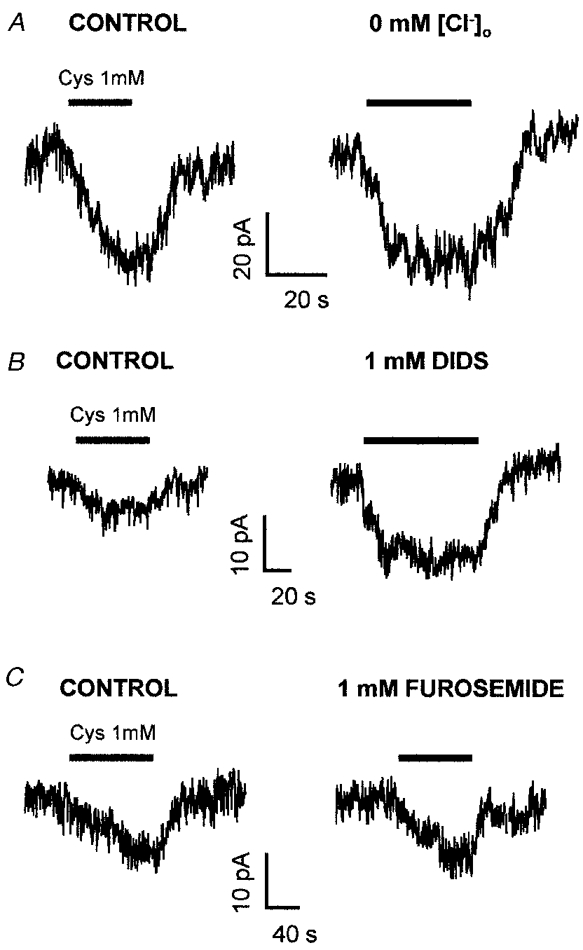
A, replacing external Cl− (left trace) with methanesulphonate (right trace) had no effect on the cystine-evoked current. B, the cystine-evoked current (left trace) was potentiated by the anion transporter blocker DIDS (1 mM, right trace). C, the cystine-evoked current (left trace) was little affected by the anion transporter blocker furosemide (1 mM, right trace).
DIDS and furosemide have been reported to block glutamate binding to, or homoexchange on, cystine- glutamate exchange (Recasens et al. 1987; Koyama et al. 1995), with IC50 values for inhibition of binding of 15 μM and 200 μM, respectively. We found, however, that 1 mM DIDS actually potentiated the Purkinje cell response to 1 mM cystine (the current was increased by 103 ± 35 % in 6 cells; Fig. 6B), while 1 mM furosemide had little effect (current increased by 2.1 ± 0.6 % in 2 cells; Fig. 6C).
Possible explanations for the lack of a blocking effect of these manipulations are considered in the Discussion. We also tried to block the Purkinje cell response to cystine with the cystine-glutamate exchange blocker L-α-aminoadipate (1 mM) (Koyama et al. 1995), but this agent induced a large inward current in the Purkinje cell probably by blocking, or evoking heteroexchange of glutamate on, the EAAT4 glutamate transporter (Fairman et al. 1995) and raising the local glutamate concentration.
Effects of cystine in the frontal cortex
In the molecular layer of the cerebellum (i.e. around the Purkinje cell dendritic trees) the density of cystine- glutamate exchangers is approximately 10 % of that of Na+-dependent glutamate transporters (Anderson et al. 1990). In frontal cortex, however, the density of Na+-dependent transporters is 3.2-fold smaller than in the cerebellar molecular layer, while that of cystine-glutamate exchangers is 2.4-fold higher, so the ratio of cystine-glutamate exchangers to Na+-dependent transporters is 7.5-fold higher in cortex than in cerebellum (Anderson et al. 1990).
Cystine evoked a current in pyramidal cells of frontal cortex slices which, as in Purkinje cells, was blocked by CNQX and AP5 (Fig. 7A). As a crude comparison of the relative importance of cystine-glutamate exchange and Na+-dependent glutamate transporters in the cerebellum and the cortex, we compared the currents produced in Purkinje cells and in cortical pyramidal cells when we applied cystine and L-trans-pyrrolidine-2,4-dicarboxylic acid (PDC). PDC is a glutamate analogue that is transported slowly on Na+-dependent glutamate transporters (Sarantis et al. 1993). Largely by heteroexchange with intracellular glutamate on these transporters (Volterra et al. 1996), it raises the extracellular glutamate concentration and thus produces a current in neurons (Sarantis et al. 1993). In seven cerebellar Purkinje cells 1 mM cystine produced a current change that was 5.5 ± 1.7 % of that evoked by 300 μM PDC (Fig. 7B). In five cortical pyramidal cells, the cystine-evoked current was a slightly larger fraction (8.5 ± 3.0 %) of that evoked by PDC (Fig. 7C), but this difference was not statistically significant (P = 0.37). Although this might be surprising, given the greater density of cystine-glutamate exchangers relative to Na+-dependent transporters in cortex, the ratio of responses to cystine and PDC is also affected by factors other than the relative densities of the two transporter types. First, the different Na+-dependent transporters present in cerebellum (largely GLAST, EAAT4 and EAAC1) and in cortex (largely GLT-1 and EAAC1) might show different amounts of heteroexchange of PDC with glutamate. Second, the non-linear [Glu]o dependence of membrane current (and hence the relative responses to different [Glu]o rises) may differ in pyramidal and Purkinje cells, due to different non-NMDA receptor properties or due to the contribution of NMDA receptors to the response in the cortex (there are no functional NMDA receptors on Purkinje cells at this age: Momiyama et al. 1996).
Figure 7. Comparison of the currents produced by glutamate release by cystine (on the cystine-glutamate exchanger) and PDC (which undergoes heteroexchange with glutamate on Na+-dependent glutamate transporters) in the cerebellum and frontal cortex.
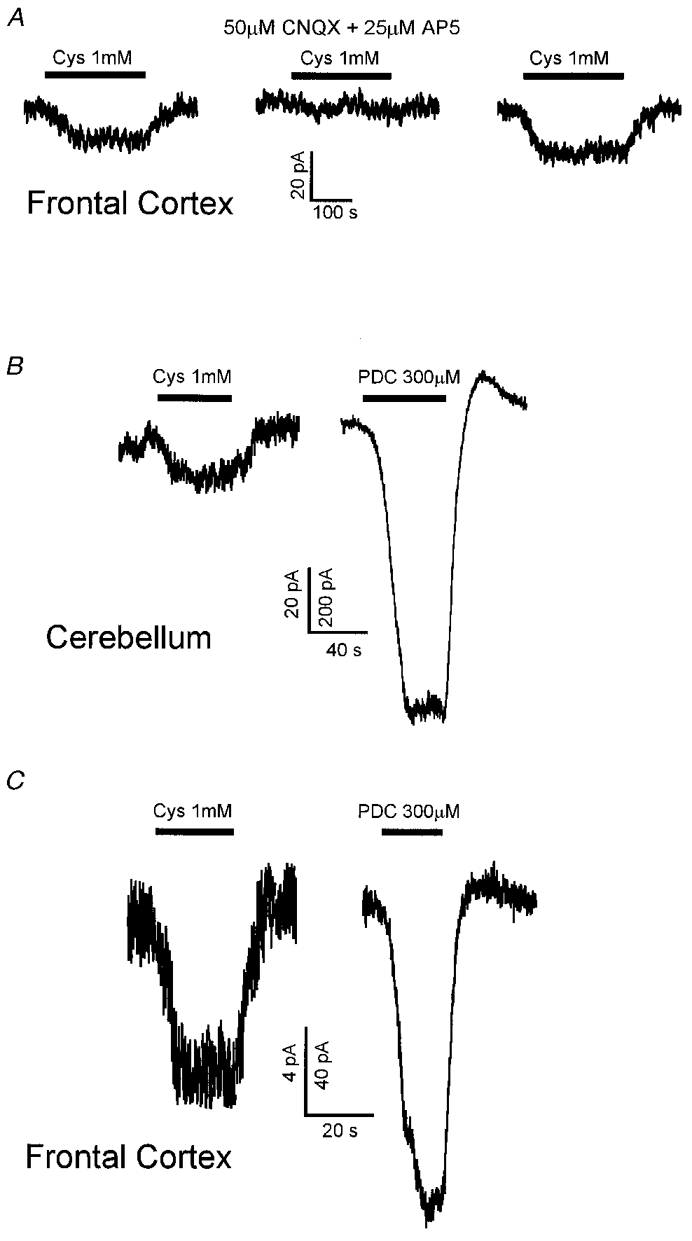
A, current evoked by 1 mM cystine in a frontal cortex pyramidal cell clamped to -33 mV in control solution, in the presence of 50 μM CNQX and 25 μM AP5, and with those blockers washed out again. B, currents evoked by 1 mM cystine and 300 μM PDC in a Purkinje cell clamped to -73 mV. C, currents evoked by 1 mM cystine and 300 μM PDC in a frontal cortex pyramidal cell clamped to -33 mV. For both B and C the current scale for the cystine response is 10-fold smaller than for the PDC response.
DISCUSSION
Cystine-glutamate exchange in cerebellar slices
Most work on transporters controlling the glutamate concentration in the extracellular space has focused on the Na+-dependent transporters. However, the presence of cystine-glutamate exchangers at a density which is 10-75 % (in different brain locations) of that of Na+-dependent transporters (Anderson et al. 1990) suggests that these may also have a significant effect on the extracellular glutamate concentration. Radiotracing studies on cerebellar slices have shown a maximum uptake rate for cystine of roughly 450 μmol l−1 h−1 (Wyatt et al. 1996). Assuming the stoichiometry of Fig. 1, so that glutamate leaves cells at the same rate, this will increase the glutamate concentration in the extracellular space (with volume fraction 0.2) at a rate of 450/(3600 × 0.2) μM s−1 = 0.6 μM s−1. Depending on the extent to which the released glutamate is taken up by Na+-dependent transporters, this efflux rate may be large enough to activate or desensitize NMDA and non-NMDA receptors. To test this we carried out this first electrophysiological study of the role of cystine-glutamate exchange in the CNS.
We found that applying 1 mM cystine to cerebellar slices generated an inward current in Purkinje cells which was blocked by glutamate receptor blockers. Control experiments showed that the current was not due to a direct action of cystine on the non-NMDA receptors in Purkinje cells (see below), nor due to a block of Na+-dependent glutamate transporters. We attribute this current to activation of cystine-glutamate exchange, releasing glutamate which activates the Purkinje cell non-NMDA receptors.
Which cells release glutamate?
Sagara et al. (1993) have suggested that cystine is supplied to neurons via glial cells, with glia taking up cystine, then converting it to and exporting cysteine, which neurons take up and convert back to cystine to make into glutathione. On this model, the source of the glutamate which generates an inward current in Purkinje cells might be cystine-glutamate exchangers in Bergmann glia. By contrast, Murphy et al. (1990) claim that neurons express a high affinity cystine- glutamate exchange which might also contribute to the cystine-evoked rise of [Glu]o which we observed (although adding glutamate to the solution in the pipette used to whole-cell clamp the Purkinje cell did not increase the cystine-evoked current: see p. 790).
Effects of anion replacement and blockers
The cystine-glutamate exchanger is often defined by its Cl− dependence: removing external Cl− has been shown to block transport of glutamate and cystine by the exchanger (Kessler et al. 1987; Murphy et al. 1989; Koyama et al. 1995). We found no significant effect of Cl− removal on the inward current evoked in Purkinje cells by 1 mM cystine (Fig. 6A), which might seem at odds with earlier work. However, Murphy et al. (1989) also found no significant effect of Cl− removal on the uptake of 1 mM cystine by cystine-glutamate exchange in a neuronal cell line, although the uptake of 10 μM cystine was reduced by 43 %, suggesting that Cl− removal inhibits the cystine-glutamate exchanger by lowering the affinity for cystine. We conclude that when the cystine-glutamate exchanger is saturated with cystine it is not Cl− dependent, consistent with our data. We could not examine the Cl− dependence of the current evoked in Purkinje cells by doses of cystine as low as 10 μM, because the current would be undetectably small.
DIDS and furosemide have been shown to block glutamate binding to and homoexchange on the cystine-glutamate exchanger (Recasens et al. 1987; Koyama et al. 1995), but apparently their effect on cystine transport has not been investigated. We found no effect of furosemide on the cystine-evoked current in Purkinje cells, while DIDS actually potentiated the current. Conceivably these drugs need to interfere with glutamate binding to block the activity of the exchanger, and in our experiments they do not block because we are running the exchanger in the direction where cystine enters and glutamate binds at the inner membrane surface. The potentiation of the current produced by DIDS could be explained by less removal by Na+-dependent glutamate transporters of the glutamate which cystine releases, since DIDS is known to inhibit these transporters in Bergmann glia (Ruiz & Ortega, 1995).
Effects of cystine on Purkinje cell non-NMDA channels
Cystine inhibited the response of non-NMDA channels in isolated Purkinje cells to low doses of glutamate (< 20 μM) by about 20 % (Fig. 3), so the inward currents evoked by glutamate release in slices (where the resting [Glu]o is likely to be in the low micromolar range) would be 20 % larger in the absence of this inhibition. By contrast, cystine increased the response to glutamate concentrations above 40 μM (Fig. 3). A similar biphasic dose-response curve has been seen for the action of DNQX on non-NMDA receptors (Fig. 6 of Geoffroy et al. 1991): this antagonist competes with glutamate for binding at low glutamate doses, but at high glutamate doses its dominant effect is to remove desensitization. Thus, we attribute the effect of cystine we observe to cystine being a weak antagonist at non-NMDA receptors.
Physiological significance of glutamate release by cystine-glutamate exchange
The EC50 of 250 μM that we found for cystine evoking an inward current in Purkinje cells implies a Km for cystine activating glutamate efflux of less than 140 μM (we estimate 110 μM), because of the non-linear dependence of non-NMDA receptor current on [Glu]o and the non-linear dependence of [Glu]o on glutamate release rate due to uptake by Na+-dependent transporters (see Results). Wyatt et al. (1996) found a Km of 77 μM for uptake of radioactive cystine into cerebellar slices. The normal cystine concentration in the extracellular space of the brain is uncertain, but it presumably lies closer to the value in the cerebrospinal fluid (CSF) than to the blood plasma value (0.2 and 80 μM, respectively; Murphy et al. 1989). Trafficking of cystine between neurons and glia (see above) may result in the brain [Cys]o being substantially higher than that in the CSF, but it is unlikely to be a significant fraction of the EC50 of 250 μM, suggesting that export of glutamate into the extracellular space by cystine-glutamate exchange will normally have a negligible effect on the membrane current of Purkinje cells.
Acknowledgments
This study was supported by The Wellcome Trust and an MRC studentship to O. W. We thank Brian Billups for help with isolated Purkinje cell experiments, Lynn Bindman and Shanida Morris for advice on making cortical slices, and Alasdair Gibb and Angus Silver for comments on the manuscript.
References
- Anderson KJ, Monaghan DT, Bridges RJ, Tavoularis AL, Cotman CW. Autoradiographic characterization of putative excitatory amino acid transport sites. Neuroscience. 1990;38:311–322. doi: 10.1016/0306-4522(90)90030-8. 10.1016/0306-4522(90)90030-8. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Eliasof S, Kavanaugh MP, Amara S. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proceedings of the National Academy of Sciences of the USA. 1997;94:4155–4160. doi: 10.1073/pnas.94.8.4155. 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparison of three glutamate transporter subtypes cloned from human motor cortex. Journal of Neuroscience. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubry A, Batini C, Billard JM, Kado RT, Morain P. Tetrodotoxin induced calcium spikes: in vitro and in vivo studies of normal and deafferented Purkinje cells. Experimental Brain Research. 1991;84:297–302. doi: 10.1007/BF00231449. [DOI] [PubMed] [Google Scholar]
- Bannai S. Transport of cystine and cysteine in mammalian cells. Biochimica et Biophysica Acta. 1984;779:289–306. doi: 10.1016/0304-4157(84)90014-5. [DOI] [PubMed] [Google Scholar]
- Bannai S, Kitamura E. Role of proton dissociation in the transport of cystine and glutamate in human diploid fibroblasts in culture. Journal of Biological Chemistry. 1981;256:5770–5772. [PubMed] [Google Scholar]
- Barbour B, Brew H, Attwell D. Electrogenic uptake of glutamate and aspartate into glial cells isolated from the salamander retina. The Journal of Physiology. 1991;436:169–193. doi: 10.1113/jphysiol.1991.sp018545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billups B, Attwell D. Modulation of non-vesicular glutamate release by pH. Nature. 1996;379:171–174. doi: 10.1038/379171a0. [DOI] [PubMed] [Google Scholar]
- Billups B, Rossi D, Attwell D. Anion conductance behavior of the glutamate uptake carrier in salamander retinal glial cells. Journal of Neuroscience. 1996;16:6722–6731. doi: 10.1523/JNEUROSCI.16-21-06722.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Bannai S. Uptake of glutamate and cystine in C-6 glioma cells and in cultured astrocytes. Journal of Neurochemistry. 1990;55:2091–2097. doi: 10.1111/j.1471-4159.1990.tb05800.x. [DOI] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. Journal of Neuroscience. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasof S, Arriza JL, Leighton BH, Kavanaugh MP, Amara SG. Excitatory amino acid receptors of the salamander retina: identification, localisation and function. Journal of Neuroscience. 1998;18:698–712. doi: 10.1523/JNEUROSCI.18-02-00698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- Geoffroy M, Lambolez B, Audinat E, Hamon B, Crepel F, Rossier J, Kado RT. Reduction of desensitization of a glutamate ionotropic receptor by antagonists. Molecular Pharmacology. 1991;39:587–591. [PubMed] [Google Scholar]
- Kanai Y, Hediger MA. Primary structure and functional characterization of a high affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- Kato S, Ishita S, Sugawara K, Mawatari M. Cystine/glutamate antiporter expression in retinal Müller cells: implications for DL-alpha-aminoadipate toxicity. Neuroscience. 1993;57:473–482. doi: 10.1016/0306-4522(93)90080-y. 10.1016/0306-4522(93)90080-Y. [DOI] [PubMed] [Google Scholar]
- Kessler M, Baudry M, Lynch G. Use of cystine to distinguish glutamate binding from glutamate sequestration. Neuroscience Letters. 1987;81:221–226. doi: 10.1016/0304-3940(87)91002-0. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proceedings of the National Academy of Sciences of the USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y, Ishibashi T, Baba A. Increase in chloride-dependent L-glutamate transport activity in synaptic membrane after in vitro ischemic treatment. Journal of Neurochemistry. 1995;65:1798–1804. doi: 10.1046/j.1471-4159.1995.65041798.x. [DOI] [PubMed] [Google Scholar]
- Levy LM, Warr O, Attwell D. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for endogenous Na+-dependent glutamate uptake. Journal of Neuroscience. 1998;18:9620–9628. doi: 10.1523/JNEUROSCI.18-23-09620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momiyama A, Feldmeyer D, Cull-Candy SG. Identification of a native low-conductance NMDA channel with reduced sensitivity to Mg2+ in rat central neurones. The Journal of Physiology. 1996;494:479–492. doi: 10.1113/jphysiol.1996.sp021507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB Journal. 1990;4:1624–1633. [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at NMDA and quisqualate receptors. Journal of Neuroscience. 1990;10:2385–2393. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penit-Soria J, Audinat E, Crepel F. Excitation of rat prefrontal neurons by dopamine: an in vitro electrophysiological study. Brain Research. 1987;425:263–274. doi: 10.1016/0006-8993(87)90509-9. 10.1016/0006-8993(87)90509-9. [DOI] [PubMed] [Google Scholar]
- Perkel DJ, Hestrin S, Sah P, Nicoll RA. Excitatory synaptic transmission in Purkinje cells. Proceedings of the Royal Society. 1990;B 241:116–121. doi: 10.1098/rspb.1990.0074. [DOI] [PubMed] [Google Scholar]
- Piani D, Fontana A. Involvement of the cystine transport system x(c)/- in the macrophage induced glutamate-dependent toxicity to neurons. Journal of Immunology. 1994;152:3578–3585. [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. Cloning and expression of a rat brain L-glutamate transporter. Nature. 1992;360:464–467. doi: 10.1038/360464a0. 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- Recasens M, Pin JP, Bockaert J. Chloride transport blockers inhibit the chloride-dependent glutamate binding to rat brain membranes. Neuroscience Letters. 1987;74:211–216. doi: 10.1016/0304-3940(87)90151-0. 10.1016/0304-3940(87)90151-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Dykes-Hoberg M, Lin L, Wu D, Nash N, Kumcl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Ruiz M, Ortega A. Characterization of an Na+-dependent glutamate/aspartate transporter from cultured Bergmann glia. NeuroReport. 1995;6:2041–2044. doi: 10.1097/00001756-199510010-00021. [DOI] [PubMed] [Google Scholar]
- Sagara JI, Miura K, Bannai S. Maintenance of neuronal glutathione by glial cells. Journal of Neurochemistry. 1993;61:1672–1676. doi: 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- Sandoval ME, Torner CA, Medrano L. High affinity uptake and Ca2+-dependent release of glutamic acid in the developing cerebellum. Neuroscience. 1984;11:867–875. doi: 10.1016/0306-4522(84)90197-0. 10.1016/0306-4522(84)90197-0. [DOI] [PubMed] [Google Scholar]
- Sarantis M, Ballerini L, Miller B, Silver RA, Edwards M, Attwell D. Glutamate uptake from the synaptic cleft does not shape the decay of the non-NMDA component of the synaptic current. Neuron. 1993;11:541–549. doi: 10.1016/0896-6273(93)90158-n. 10.1016/0896-6273(93)90158-N. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proceedings of the National Academy of Sciences of the USA. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Billups B, Rossi D, Sarantis M, Hamann M, Attwell D. The role of glutamate transporters in glutamate homeostasis in the brain. Journal of Experimental Biology. 1997;200:401–409. doi: 10.1242/jeb.200.2.401. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Sarantis M, Attwell D. Postsynaptic glutamate uptake in rat cerebellar Purkinje cells. The Journal of Physiology. 1996;497:523–530. doi: 10.1113/jphysiol.1996.sp021785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Winkle L J, Mann D F, Wasserlauf H G, Patel M. Mediated Na+-independent transport of L-glutamate and L-cystine in 1- and 2-cell mouse conceptuses. Biochimica et Biophysica Acta. 1992;1107:299–304. doi: 10.1016/0005-2736(92)90416-j. [DOI] [PubMed] [Google Scholar]
- Volterra A, Bezzi P, Rizzini BL, Trotti D, Ullensvang K, Danbolt NC, Racagni G. The competitive transport inhibitor L-trans-pyrrolidine-2, 4-dicarboxylate triggers excitotoxicity in rat cortical neuron-astrocyte co-cultures via glutamate release rather than uptake inhibition. European Journal of Neuroscience. 1996;8:2019–2028. doi: 10.1111/j.1460-9568.1996.tb01345.x. [DOI] [PubMed] [Google Scholar]
- Wahl F, Obrenovitch TP, Hardy AM, Plotkine M, Boulu R, Symon L. Extracellular glutamate during focal cerebral ischaemia in rats: time course and calcium-dependency. Journal of Neurochemistry. 1994;63:1003–1011. doi: 10.1046/j.1471-4159.1994.63031003.x. [DOI] [PubMed] [Google Scholar]
- Wyatt I, Gyte A, Simpson MG, Widdowson PS, Lock EA. The role of glutathione in L-2-chloropropionic acid induced cerebellar granule cell necrosis in the rat. Archives of Toxicology. 1996;70:724–735. doi: 10.1007/s002040050333. 10.1007/s002040050333. [DOI] [PubMed] [Google Scholar]
- Yamada K, Watanabe M, Shibata T, Tanaka K, Wada K, Inoue Y. EAAT4 is a post-synaptic glutamate transporter at Purkinje cell synapses. NeuroReport. 1996;7:2013–2017. doi: 10.1097/00001756-199608120-00032. [DOI] [PubMed] [Google Scholar]
- Zaczek R, Balm M, Arlis S, Drucker H, Coyle JT. Quisqualate-sensitive, chloride-dependent transport of glutamate into rat brain synaptosomes. Journal of Neuroscience Research. 1987;18:425–431. doi: 10.1002/jnr.490180307. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]