

Abstract
To gain insight into the role of the cytoplasmic regions of the Kir6.2 subunit in regulating channel activity, we have expressed the sulphonylurea receptor SUR1 with Kir6.2 subunits containing systematic truncations of the N- and C-termini. Up to 30 amino acids could be truncated from the N-terminus, and up to 36 amino acids from the C-terminus without loss of functional channels in co-expression with SUR1. Furthermore, Kir6.2ΔC25 and Kir6.2ΔC36 subunits expressed functional channels in the absence of SUR1.
In co-expression with SUR1, N-terminal truncations increased Ki,atp ([ATP] causing half-maximal inhibition of channel activity) by as much as 10-fold, accompanied by an increase in the ATP-insensitive open probability, whereas the C-terminal truncations did not affect the ATP sensitivity of co-expressed channels.
A mutation in the near C-terminal region, K185Q, reduced ATP sensitivity of co-expressed channels by approximately 30-fold, and on the Kir6.2ΔN2−30 background, this mutation decreased ATP sensitivity of co-expressed channels by approximately 400-fold.
Each of these mutations also reduced the sensitivity to inhibition by ADP, AMP and adenosine tetraphosphate.
The results can be quantitatively explained by assuming that the N-terminal deletions stabilize the ATP-independent open state, whereas the Kir6.2K185Q mutation may alter the stability of ATP binding. These two effects are energetically additive, causing the large reduction of ATP sensitivity in the double mutant channels.
ATP-sensitive potassium channels (KATP) are unique among K+ channels in being rapidly and reversibly inhibited by the non-hydrolytic binding of cytoplasmic adenine nucleotides (Noma, 1983; Ashcroft, 1988; Nichols & Lederer, 1991). The realization that these channels are encoded by an ATP binding cassette (ABC) protein, the sulphonylurea receptor (SUR1 or SUR2), in addition to an inward rectifier K+ channel subunit (Kir6.1 or Kir6.2; Aguilar-Bryan et al. 1995; Inagaki et al. 1995, 1996) led to the natural presumption that inhibitory ATP binding should occur at the nucleotide binding folds (NBFs) of the SUR subunit. Mutagenic studies have now demonstrated that the NBFs of SUR1 control activation of the channel by Mg+-bound nucleotides and diazoxide, probably as a consequence of these agents mimicking, or enhancing, an effect of nucleotide hydrolysis (Nichols et al. 1996; Gribble et al. 1997; Shyng et al. 1997b). However, none of these studies has demonstrated an effect of SUR1 mutations on the intrinsic sensitivity of expressed channels to ATP inhibition. On the other hand, tandem dimeric constructs of SUR1 and Kir6.2 express channels with reduced ATP sensitivity (Clement et al. 1997; Shyng & Nichols, 1997), and, moreover, mutations of Kir6.2 can alter the inhibitory effect of ATP on channels formed by co-expression with SUR1 (Shyng et al. 1997a;Tucker et al. 1997, 1998). In the study of Shyng et al. (1997a), mutations of asparagine 160 in the second transmembrane segment alter sensitivity to polyamine-induced rectification, as well as decreasing the apparent ATP sensitivity approximately 5-fold. Tucker et al. (1997) reported that a C-terminally truncated Kir6.2 subunit (Kir6.2ΔC36) could express ATP-sensitive channels in the absence of SUR1, and reported a significant reduction of ATP sensitivity for Kir6.2ΔC36 channels with an additional mutation of lysine 185 in the truncated C-terminus. Lorenz et al. (1998) have confirmed these findings, and John et al. (1998), as well as Mikhailov et al. (1998), have most recently demonstrated that even full length Kir6.2 can form ATP-sensitive channels in the absence of SUR1. These results are fully consistent with the alternative hypothesis that ATP inhibits the channel through a direct interaction with the Kir6.2 subunit.
Shyng et al. (1997a) demonstrated a correlation between the shift in apparent Ki for ATP inhibition that results from mutations of amino acid 160 and changes in the channel open probability in the absence of ATP. This suggested that the Ki shift might result from a change in the energetics of the ATP-independent states (i.e. altering the coupling between ATP binding and channel closure), rather than from a change in ATP binding itself. In the present study we report experiments designed to systematically examine the role of the N- and C-terminal regions of the Kir6.2 subunit in regulating channel activity and ATP sensitivity, by making serial truncations at each terminus. A preliminary report of these findings has been presented to the Biophysical Society (Koster et al. 1998).
METHODS
Construction of deletion mutants
Kir6.2 constructs containing truncations in either the C- or N-terminus were prepared by deletional mutagenesis using polymerase chain reaction (PCR). The Kir6.2K185Q point mutant was prepared by overlap extension at residue 185 by sequential PCR. Resulting PCR products were subcloned into the Eco RI-Cla I sites of pCMV6b and the encoded protein expressed under control of the cytomegalovirus (CMV) promoter. SUR1 was cloned into the pECE expression vector. The nucleotide sequences of all mutant Kir6.2 constructs were verified by fluorescence-based cycle sequencing using AmpliTaq DNA polymerase, FS (Perkin-Elmer, Foster City, CA, USA) and an ABI PRISM DNA sequencer (Perkin-Elmer).
Expression of KATP channels in COSm6 cells
COSm6 cells were plated at a density of approximately 2.5 × 105 cells per well (30 mm 6-well dishes) and cultured in Dulbecco's modified Eagle's medium plus 10 mM glucose (DMEM-HG), supplemented with fetal calf serum (FCS, 10 %), penicillin (100 units ml−1) and streptomycin (100 μg ml−1). The following day, cells were transfected by incubation for 4 h at 37°C in DMEM-HG medium containing 10 % Nu-Serum I (Collaborative Biomedical Products, Bedford, MA, USA), 0.4 μg ml−1 diethylaminoethyl-dextran, 100 μM chloroquine, and 5 μg each of pCMV6b-Kir6.2, pECE-SUR1 and pECE-GFP (green fluorescent protein) cDNA. Cells were subsequently incubated for 2 min in Hepes-buffered salt solution containing DMSO (10 %), and returned to DMEM-HG plus 10 % FCS and penicillin- streptomycin.
Rubidium flux experiments
86RbCl (1 mCi ml−1) was added in fresh growth medium 48 h after transfection. Cells were incubated for 24 h before assaying Rb+ flux. For efflux measurements, cells were incubated for 30 min at 25°C in Krebs-Ringer solution with metabolic inhibitors (2.5 μg ml−1 oligomycin plus 1 mM 2-deoxy-D-glucose). At selected time points, the solution was aspirated from the cells and replaced with fresh solution. The 86Rb+ in the aspirated solution was counted in a scintillation counter.
Patch-clamp measurements
Patch-clamp experiments were made at room temperature in an oil-gate chamber which allowed the solution bathing the exposed surface of the isolated patch to be changed in less than 50 ms (Lederer & Nichols, 1989). Micropipettes were pulled from thin-walled glass (WPI Inc., New Haven, CT, USA) on a horizontal puller (Sutter Instrument, Co., Novato, CA, USA). Electrode resistance was typically 0.5-1 MΩ when filled with Kint solution (140 mM KCl, 10 mM K-Hepes, 1 mM K-EGTA, pH 7.3). Microelectrodes were sealed onto cells that fluoresced green under UV illumination, by applying light suction to the rear of the pipette. Membrane patches were voltage clamped with an Axopatch 1B patch-clamp (Axon Instruments). All currents were measured at a membrane potential of -50 mV (pipette voltage, +50 mV). Inward currents at this voltage are shown as positive-going signals. Data were normally filtered at 0.5-3 kHz, signals were digitized at 22 kHz (Neurocorder, Neurodata, NY, NY, USA) and stored on video tape. Experiments were replayed onto a chart recorder, or digitized into a microcomputer using Axotape software (Axon Instruments). The standard bath (intracellular) and pipette (extracellular) solution contained Kint, with the addition of [ATP] in the bath as indicated.
Data analysis
Off-line analysis was performed using Axotape and Microsoft Excel programs. Stationary fluctuation analysis of macroscopic currents (Neher & Stevens, 1977; Sigworth, 1980) was performed on short (< 1 s) recordings of currents at +50 mV following a step to zero [ATP] (over which time mean channel activity rundown, or inactivation, was negligible) and in 5-10 mM [ATP]. Currents (400 pA < mean current > 7 nA, corresponding to ∼100-2000 channels) were filtered at fc (cut-off frequency) of 3 kHz and digitized at 10 kHz with 16-bit amplitude resolution. Mean patch current (I) and variance (σ2) in the absence of ATP were obtained by subtraction of mean patch current and variance of patch current in 5 or 10 mM ATP (all channels closed) from mean current and variance estimated in zero ATP. For the ATP-insensitive dual mutant channel, Kir6.2ΔN2−30/K185Q + SUR1, the baseline current was determined by irreversibly inhibiting channel activity with Kint solution supplemented with 10 mM MgCl2 and 2 mM CaCl2. Single channel current (i) was assumed to be constant at 3.75 pA, corresponding to single channel conductance of 75 pS for Kir6.2wt, Kir6.2ΔN2−30, Kir6.2K185Q and Kir6.2ΔN2−30/K185Q mutant channels (data not shown) The mean open probability (Po) was then calculated by fitting the following
| (1) |
Model simulations of steady-state [ATP], [AMP], [ADP] and [adenosine tetraphosphate] dose-response relationships were generated using Microsoft Excel. Wherever possible, data are presented as the mean ±s.e.m. (standard error of the mean). Microsoft Solver was used to fit data by a least-squares algorithm. For calculation of standard free energy change the following equation was used:
| (2) |
where R represents the gas constant (1.98 × 10−3 kcal mol−1 deg−1), T is absolute temperature (298 °K at 25°C), and K‘eq the [ATP] of half-maximal inhibition.
RESULTS
Kir6.2 N-terminal deletions decrease ATP sensitivity of SUR1 co-expressed channels
In order to determine the role of different structural regions of Kir6.2 in inhibition of the KATP channel by ATP, the Kir6.2 subunit was systematically truncated at the N- and C-termini (Fig. 1A) and the effect of these truncations on the expressed channels was examined. Up to 30 amino acids could be truncated from the N-terminus without loss of functional channels when co-expressed with SUR1. However, truncations of 40 amino acids or beyond (Kir6.2ΔN40, Kir6.2ΔN50) did not generate functional channels. Figure 1B shows representative Rb+ flux assays of KATP channel activity from various N-terminal deletion constructs, co-expressed with SUR1. In all cases, metabolic inhibition activated a flux comparable to that seen with expression of wild-type KATP currents. Only a small Rb+ efflux was observed in untransfected cells under the same experimental conditions (> 10 %). The Kir6.2ΔN2-10, Kir6.2ΔN2-20 and Kir6.2ΔN2−30 mutants were functional when co-expressed with SUR1, although currents above background were not observed with Kir6.2ΔN2-40 (data not shown). As with wild-type Kir6.2, active channels were not observed from any of the N-terminal deletions when expressed alone, either by inside-out patch-clamp (data not shown, n = 2-4 patches) or by Rb+-efflux analysis (Fig. 1B; for Kir6.2ΔN2−30). This is in contrast to C-terminal truncations (Kir6.2ΔC25 and Kir6.2ΔC36), which displayed KATP channel activity when expressed in the absence of SUR1 (data not shown) (cf. Tucker et al. 1997). These data indicate that the N-terminal 30 amino acids, unlike the C-terminus, are not involved in prohibiting functional expression of the wild-type Kir6.2 in the absence of SUR1. Interestingly, recent reports (John et al. 1998; Mikhailov et al. 1998) describe functional expression of full-length Kir6.2 in the absence of SUR1, with properties similar to Kir6.2ΔC25 channels alone. These observations may indicate that a population of KATP channels can normally form without SUR1 or simply that suppression of a normal endoplasmic reticulum retention mechanism may result from overexpression of the Kir6.2 subunit under non-physiological conditions.
Figure 1. 86Rb+ efflux from untransfected COSm6 cells and cells expressing truncated Kir6.2 subunits alone or with SUR1.
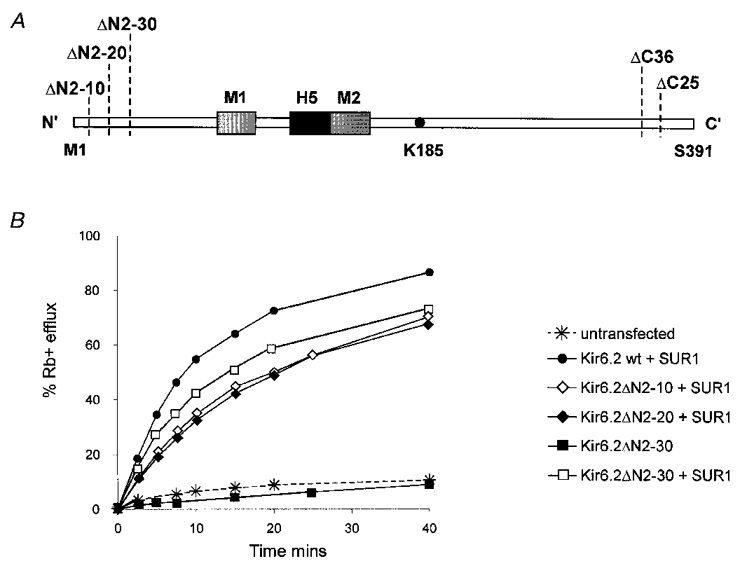
A, schematic diagram of the Kir6.2 subunit illustrating the regions in the cytoplasmic C- and N-termini that were deleted or mutated. The Kir6.2 subunit contains two transmembrane segments, M1 and M2, a pore-forming H5 segment, and cytoplasmic N- and C-termini. A lysine residue at position 185 has previously been implicated in inhibition of the KATP channel by ATP (Tucker et al. 1997). B, percentage efflux of 86Rb+ as a function of time in the presence of metabolic inhibitors (see Methods) for wild-type Kir6.2 and N-terminal deletion constructs, with or without SUR1 as indicated. All transfections were done in parallel. Graph shows results from a typical experiment (repeated 2-3 times for each construct).
The response of N-terminal deletion constructs, Kir6.2ΔN2-10 and Kir6.2ΔN2−30, to the application of cytoplasmic ATP, in the absence of Mg2+ (see Lederer & Nichols, 1989) is shown for representative inside-out patches in Fig. 2. It is clear that the sensitivity to inhibition by ATP was decreased by N-terminal deletions. Mean data from all patches (n = 5-10 for each construct) are shown as dose-response relationships in Fig. 3.
Figure 2. Currents from cells co-expressing N-terminal truncated Kir6.2 subunits and SUR1.
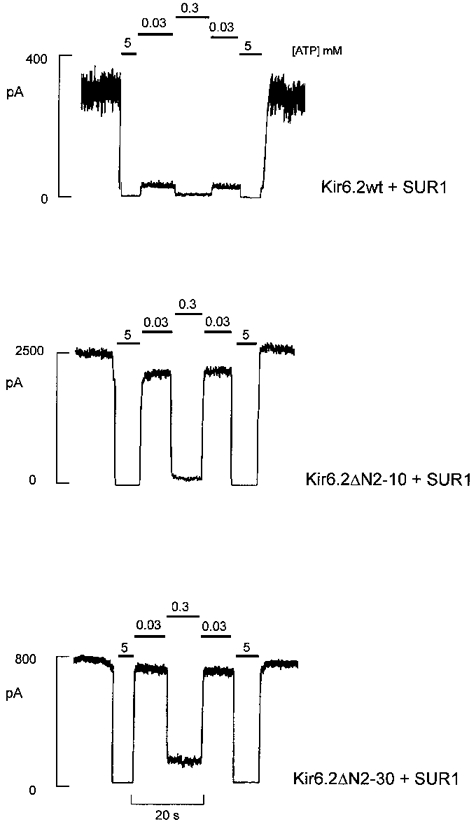
Representative currents recorded from inside-out membrane patches containing wild-type or mutant KATP channels at -50 mV in Kint solution (see Methods). Patches were exposed to differing [ATP] as indicated.
Figure 3. [ATP] dependence of N-terminal truncated KATP mutant channels.
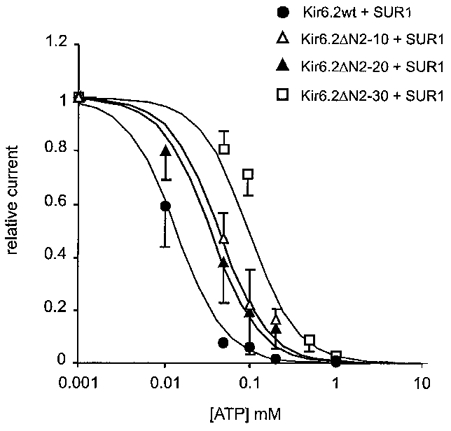
Steady-state dependence of membrane current (mean ±s.e.m. relative to current in zero ATP) on [ATP] for wild-type and mutant channels (from records such as those shown in Fig. 2). Data points represent the mean ±s.e.m. (n = 3-8 patches). The fitted lines correspond to least-squares fit of the Hill equation (relative current = 100/(1 + ([ATP]/Ki)nH), with nH fixed at 1.3, and Ki = 12 μM (wild-type), 44 μM (Kir6.2ΔN2-10), 36 μM (Kir6.2ΔN2-20 + SUR1), and 124 μM (Kir6.2ΔN2−30 + SUR1).
Previous detailed analyses of ATP sensitivity indicate that multiple ATP molecules are likely to bind sequentially to the channel (Nichols et al. 1991). In this case the Hill equation does not describe such a kinetic scheme, and is thus useful only empirically. In this paper we are primarily concerned with relative sensitivities of different constructs and nucleotides. For ATP inhibition of wild-type channels, the Hill coefficient, nH, and Ki were free to vary, and the obtained best fit (nH = 1.3) was then used to constrain subsequent fits to other constructs and nucleotides.
The Kir6.2ΔN2−30 channels were approximately 10-fold less sensitive to inhibition by ATP (Ki = 124 μM) than wild-type channels (Ki = 12 μM). Similarly, Kir6.2ΔN2-10 and Kir6.2ΔN2-20 patches displayed a 3-fold decrease in steady-state [ATP] dependence for both mutant channels (Ki = 44 and 36 μM, respectively), consistent with residues 2-10 and 20-30 being major determinants of the N-terminal regulation of ATP sensitivity.
Kir6.2 C-terminal deletions do not affect ATP sensitivity of SUR1 co-expressed channels
Systematic C-terminal deletions in Kir6.2 were generated in order to examine control of ATP sensitivity by this region of the channel. In co-expression with SUR1, deletions of up to 36 amino acids were tolerated, consistent with the previous report of Tucker et al. (1997). Figure 4B shows ATP sensitivity of currents through Kir6.2ΔC25 channels expressed alone or co-expressed with SUR1. Similar to a previous report (Tucker et al. 1997), the apparent ATP sensitivity of Kir6.2ΔC25 channels alone was decreased approximately 10-fold, as compared with the wild-type channel (Ki = 12 μM for wild-type, 125 μM for Kir6.2ΔC25 alone). However, ATP sensitivity could be restored to the wild-type level by co-expressing Kir6.2ΔC25 with SUR1 (Ki = 14 μM, Fig. 4B). The Kir6.2ΔC25 truncation was also introduced on the Kir6.2ΔN2−30 background. Figure 4A and B shows current densities and ATP sensitivity of currents through Kir6.2ΔN2−30ΔC25 channels expressed alone or co-expressed with SUR1. Consistent with a role for the N-terminus, but not the C-terminus, in controlling ATP sensitivity, the C-terminal truncation did not decrease ATP sensitivity further over the level observed for Kir6.2ΔN2−30 + SUR1 channels (Kir6.2ΔC25ΔN2-30 + SUR1, Ki = 94 μM). Moreover, when expressed in the absence of SUR1, the double deletion also did not decrease ATP sensitivity further over the level observed for Kir6.2ΔC25 alone (Kir6.2ΔC25ΔN2-30, Ki = 117 μM, Fig. 4).
Figure 4. [ATP] dependence of Kir6.2ΔN2−30ΔC25 channel expressed alone or with SUR1.
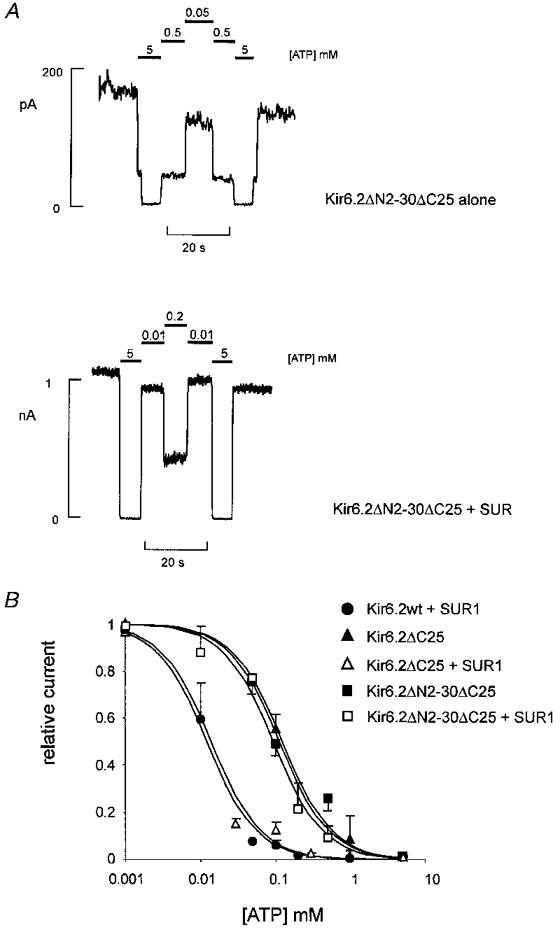
A, representative currents (from n = 3-5 patches) recorded from inside-out membrane patches containing Kir6.2ΔN2−30ΔC25 channels alone (above), or with SUR1 (below) at -50 mV in Kint solution (see Methods). Patches were exposed to differing [ATP] as shown. B, steady-state dependence of membrane current relative to current in zero ATP (mean ±s.e.m., n = 3-5 patches for each construct) on [ATP] for wild-type and Kir6.2ΔC25 mutant channels (as indicated). The data were fitted as described in Fig. 3. Ki = 12 μM (wild-type), 128 μM (Kir6.2ΔC25 alone), 14.0 μM (Kir6.2ΔC25 + SUR1), 117 μM (Kir6.2ΔN2−30ΔC25 alone) and 94 μM (Kir6.2ΔN2−30ΔC25 + SUR1).
The N-terminus and lysine 185 of Kir6.2 make energetically additive contributions to ATP sensitivity
Tucker et al. (1997) demonstrated that a neutralization mutation in the near C-terminal region of Kir6.2 (K185Q) significantly reduced the ATP sensitivity of Kir6.2ΔC26 channels expressed in the absence of SUR1. It was suggested that this residue may represent a binding site for inhibitory ATP by co-ordinating and neutralizing the negatively charged phosphate group(s) of the inhibitory molecule. We introduced the K185Q mutation into the wild-type Kir6.2 and C- and N-terminal truncations and co-expressed each mutant with SUR1. As shown in Fig. 5, Kir6.2K185Q + SUR1 channels in inside-out patches are considerably less sensitive to inhibition by ATP than wild-type channels. The [ATP] dose-response relationship compiled from all patches was fitted by a Ki of 308 μM, representing an ∼30-fold decrease of ATP sensitivity compared with wild-type Kir6.2 + SUR1 channels (Fig. 6A). As with the N-terminal truncations, introduction of the C-terminal truncation on the K185Q background did not cause any further reduction of the ATP sensitivity, with Kir6.2ΔC25/K185Q + SUR1 channels exhibiting a Ki of 304 μM (Fig. 6A). Thus alteration of ATP sensitivity by both N-terminal and K185Q mutations is unaffected by introduction of these mutations onto the ΔC25 background. These results strengthen the conclusion that, while C-terminal truncation permits or enhances expression of KATP channels in the absence of SUR1, it does not alter the ATP sensitivity of expressed channels.
Figure 5. Representative current traces from cells co-expressing Kir6.2ΔN2−30, Kir6.2K185Q or Kir6.2ΔN2−30/K185Q mutant subunits and SUR1.
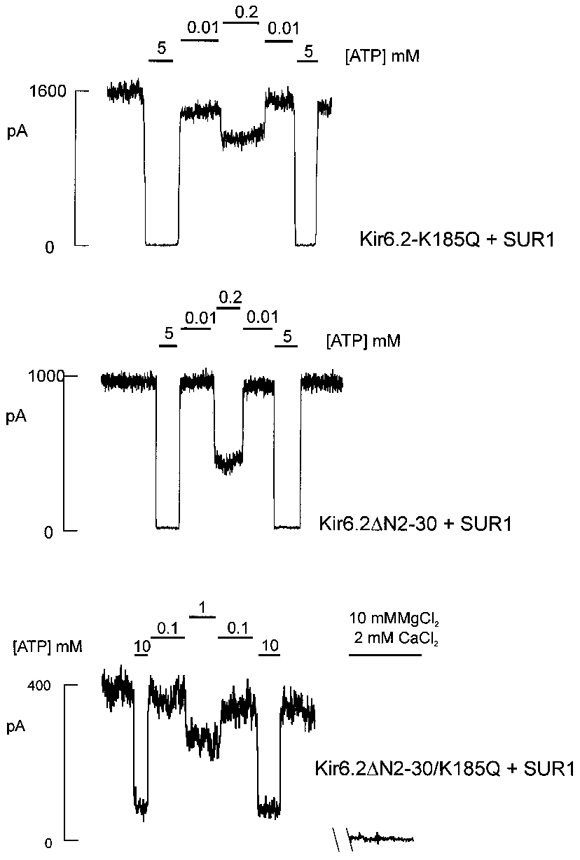
Currents recorded from inside-out membrane patches containing wild-type or mutant KATP channels at -50 mV in Kint solution (see Methods). Patches were exposed to differing [ATP] as shown. For the ATP-insensitive dual mutant channel Kir6.2ΔN2−30/K185Q + SUR1 the baseline current was determined by subsequent irreversible inhibition of channel activity with 10 mM MgCl2 and 2 mM CaCl2 (see Methods).
Figure 6. ATP dose-response relationship and mean peak open probability of N-terminal deletion and K185Q mutant channels.
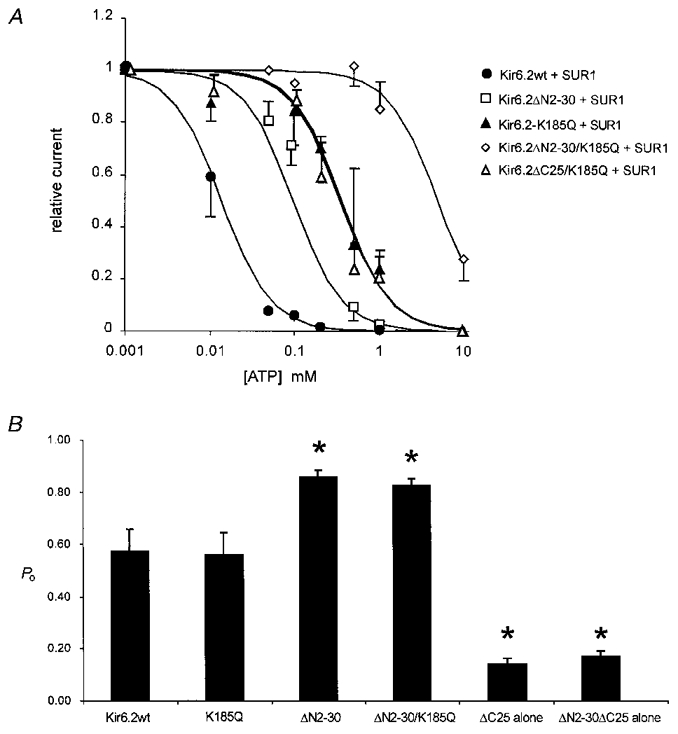
A, steady-state dependence of membrane current (relative to current in zero ATP) on [ATP] for wild-type and mutant channels (as indicated) co-expressed with SUR1. Data points represent the mean ±s.e.m. (n = 3-8 patches). The data were fitted as described in Fig. 3. Ki = 12 μM (wild-type), 124 μM (Kir6.2ΔN2−30 + SUR1), 308 μM (Kir6.2K185Q + SUR1), 304 μM (Kir6.2ΔC25/K185Q + SUR1), and 4.9 mM (Kir6.2ΔN2−30/K185Q + SUR1). B, mean peak open probability (Po) of mutant Kir6.2 subunits was measured in the absence of inhibitory ATP by steady-state noise analysis using eqn (1) (see Methods). Asterisks denote statistical difference between wild-type Po,max and mutant channel Po,max using Student's unpaired t test (*P < 0.05). Peak open probabilities (Po) were as follows (n = 4-7 patches; ±s.e.m.): Kir6.2wt = 0.57 ± 0.08; K185Q = 0.56 ± 0.08; ΔN2-30 = 0.86 ± 0.02; ΔN2-30/K185Q = 0.83 ± 0.03; ΔC25 alone = 0.14 ± 0.02; ΔN2-30/ΔC25 alone = 0.17 ± 0.02.
As shown in Figs 5 and 6, however, combining the N-terminal deletion with the K185Q mutation has a significant, and additive, effect on apparent ATP sensitivity. The combination mutant channel Kir6.2ΔN2−30/K185Q + SUR1 is highly insensitive to inhibition by ATP with an estimated Ki of 4.9 mM (compared with a Ki of 124 and 308 μM for ΔN2-30 and K185Q mutants, respectively; Fig. 6A). This represents an ∼400-fold reduction in apparent ATP sensitivity over wild-type channels and indicates that the N-terminal deletion and the K185Q mutation are energetically additive in terms of their effect on ATP sensitivity. Each mutation alone corresponds to a reduction of 5.3 and 7.4 kJ mol−1 in apparent association energy for ATP (Kir6.2ΔN2−30 and Kir6.2K185Q, respectively), whereas the paired mutant is reduced by 13.6 kJ mol−1 compared with wild-type channels (see eqn (2) in Methods).
The open state is stabilized in the Kir6.2ΔN2−30 mutant
The data presented thus far are consistent with lysine 185 in the near C-terminus comprising part of the binding site for inhibitory ATP. However, an alternative explanation seems to underlie the decrease in sensitivity of Kir6.2ΔN2−30 channels. As previously reported, mutations of asparagine 160 in the second transmembrane segment of Kir6.2 (Shyng et al. 1997a) change the open probability of the channel in the absence of ATP, and stabilization of an open state, away from ATP-bound closed states, was postulated to account for the decreased apparent ATP sensitivity of the mutant channels. The single channel conductance of Kir6.2ΔN2−30 channels is the same as wild-type (75 pS, data not shown), but as shown in Fig. 7, the channel open probability in the absence of ATP (Po,max) is considerably higher for Kir6.2ΔN2−30 channels than for wild-type channels. Whereas, in the two records shown, wild-type channels show brief bursts of openings separated by longer closed events (τ = 72 ms, Fig. 7B), ΔN2-30 channels are well described by continuous bursting, without the longer interburst closed times. Similar results were seen in two other wild-type single channel patches, and one other ΔN2-30 channel patch. In order to obtain statistical significance, we estimated Po,max using noise analysis. In good agreement with single channel patches, Po,max≈ 0.8 (n = 5, Fig. 6B) for Kir6.2ΔN2−30 channels is considerably higher than for wild-type channels (Po,max≈ 0.5, n = 7; Fig. 6B). In contrast, the open probability of the Kir6.2K185Q mutant channel, in the absence of ATP, is not significantly different from the parental wild-type channel (Fig. 6B). Additionally, the double mutant channel, Kir6.2ΔN2−30/K185Q, exhibited an open probability similar to Kir6.2ΔN2−30 channels. As discussed in detail below, stabilization of the open state, relative to a closed state, in Kir6.2ΔN2−30 channels can account for the difference in ATP sensitivity of the deletion mutants relative to the wild-type channel as well as the energetically additive effects of the N-terminal deletion and the K185Q point mutation.
Figure 7. Kir6.2ΔN2−30 channels have a higher open probability than wild-type channels.
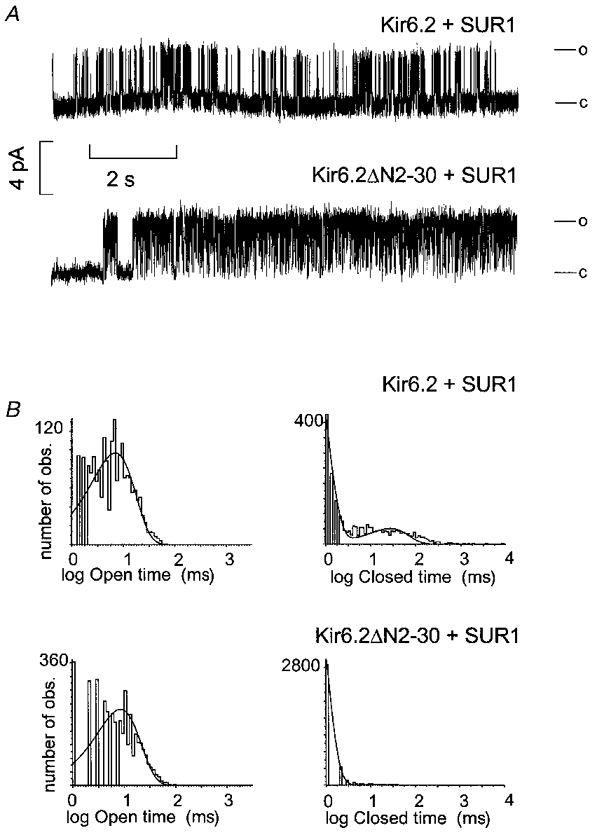
A, representative single channel currents recorded from cells expressing wild-type (Kir6.2 + SUR1) or Kir6.2ΔN2−30 + SUR1 channels, fc = 1 kHz. c and o indicate the closed and open channel levels. B, unconditional open and closed time distributions from the records shown in A, using log binning. Open times were well fitted by single exponential distributions for both wild-type (TO = 9.5 ms) and ΔN2-30 (TO = 11.9 ms). Two time constants (TC1 = 5.9 ms, relative fraction = 0.6; TC2 = 71.9 ms, relative fraction = 0.4) were necessary to describe wild-type closed distributions, but a single time constant (TC1 = 4.0 ms) was sufficient to describe ΔN2-30 channel closed time distribution.
Sensitivity to other adenosine nucleotides parallels alteration of ATP sensitivity
To define the altered nucleotide sensitivity of the mutant KATP channels further, the effect of additional adenosine nucleotides on channel activity was examined. Reducing the number of phosphate groups from three to two (ADP) or one (AMP), markedly reduces the inhibitory effect on native KATP channels (Noma, 1983; Ashcroft, 1988; Nichols & Lederer, 1991), and this is true for reconstituted and mutant KATP channels (Fig. 8). Wild-type channels are ∼10-fold less sensitive to ADP than ATP (Ki = 144 and 12 μM, respectively) and ∼140-fold less sensitive to AMP (Ki = 1.8 mM). The data are summarized in Fig. 9 for inhibition of mutant channels by ATP, ADP and adenosine tetraphosphate (AtetP). As can be seen, qualitatively similar differences are observed for the sensitivity of the mutant KATP channels, although the sensitivities to each nucleotide begin to converge for the K185Q and double-mutant channels, consistent with the K185Q mutation reducing the discrimination between adenosine nucleotides. Wild-type and mutant channels are almost insensitive to GTP (data not shown), and sensitivities to AtetP are similar to ATP sensitivities for each channel.
Figure 8. Steady-state dependence of mutant KATP channel activities on [AMP], [ADP] and [adenosine tetraphosphate].
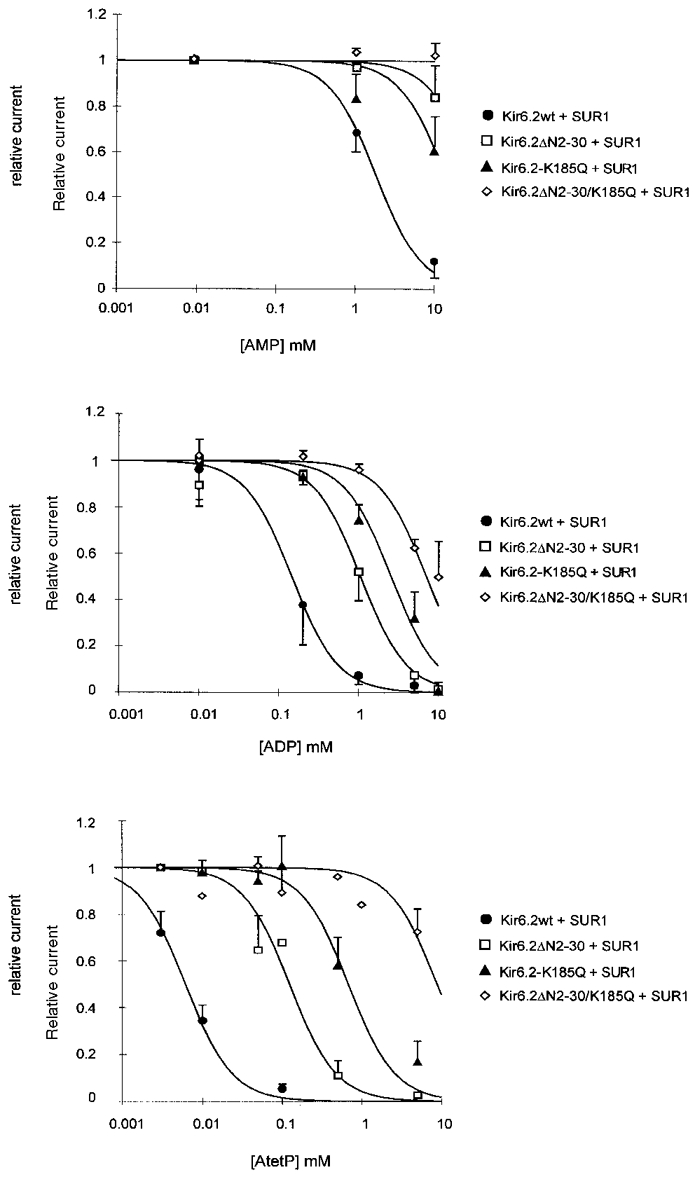
Dependence of membrane current (relative to current in zero adenosine nucleotide) on [adenosine nucleotides] for wild-type and mutant channels (as indicated). Data points represent the mean ±s.e.m. (n = 3-10 patches). The data were fitted as described in Fig. 3. Ki for AMP = 1.8 mM (wild-type), 31.9 mM (Kir6.2ΔN2−30 + SUR1), and 13.6 mM (Kir6.2K185Q + SUR1). Not determined for Kir6.2ΔN2−30/K185Q + SUR1. Ki for ADP = 144 μM (wild-type), 1.1 mM (Kir6.2ΔN2−30 + SUR1), 2.5 mM (Kir6.2K185Q + SUR1), and 7.1 mM (Kir6.2ΔN2−30/K185Q + SUR1). Ki for adenosine tetraphosphate = 6 μM (wild-type), 124 μM (Kir6.2ΔN2−30 + SUR1), 680 μM (Kir6.2K185Q + SUR1), and 8.8 mM (Kir6.2ΔN2−30/K185Q + SUR1).
Figure 9. Relative sensitivity of mutant KATP channels to inhibition by adenosine nucleotides.
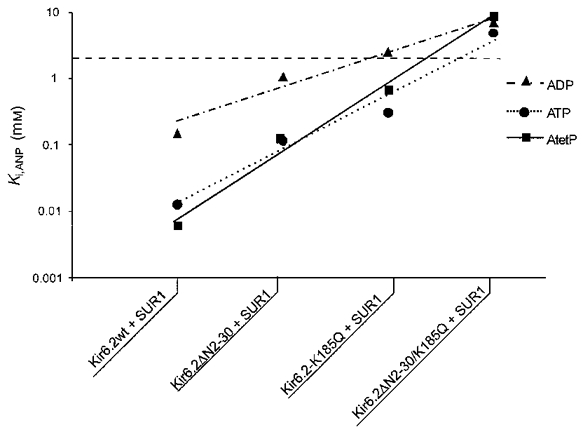
Calculated Ki for ATP, ADP and AtetP from Fig. 8 for mutant KATP channels. The dashed line above 3 mM indicates unreliability of estimated parameters. The lines connect values predicted by adjustments to the model of Fig. 10 (see text for details).
DISCUSSION
Control of nucleotide sensitivity by the Kir6.2 subunit
The pore-forming Kir6.2 subunit of the KATP channel consists of two transmembrane segments, M1 and M2, with an extracellular P-loop, and cytoplasmic N- and C-termini. While previous studies have implicated the pore-lining M2 segment in controlling the gating and rectification properties of the channel (Shyng et al. 1997a), little is known of the role of the cytoplasmic domains, particularly the N-terminus, in channel activity. The present study demonstrates that Kir6.2 subunits with deletions of up to 30 to 40 amino acids from either the cytoplasmic N- or C-termini can be co-expressed with SUR1 to generate functional ATP-sensitive K+ channels. Moreover, deletions of 25 or 36 amino acids from the C-terminus (cf. Tucker et al. 1997), but not the N-terminus, lead to the generation of significant ATP-sensitive K+ channel current in the absence of SUR1. These data confirm a role of the C-terminus, and not the N-terminus, in preventing expression of channel activity from Kir6.2 subunits in the absence of co-expressed SUR1. Deletions of the N-terminus result in up to a 10-fold reduction in ATP sensitivity, the critical amino acids being in residues 2-10 and 20-30, associated with a significant increase in open probability in the absence of ATP. In contrast, even though deletions of the C-terminus permit expression of functional channels without SUR1, the ATP sensitivity of co-expressed channels is indistinguishable from the sensitivity of wild-type channels, suggesting that the C-terminal 36 amino acids are not involved in controlling intrinsic ATP sensitivity. Consistent with a previous report (Tucker et al. 1997), lysine 185 in the proximal C-terminus beyond the second transmembrane region (M2), when mutated to a glutamine, causes an ∼30-fold reduction in ATP sensitivity, though, unlike the Kir6.2ΔN2−30 channels, this mutation is not associated with a change in open probability (see below).
The cloning of the SUR1 subunit (Aguilar-Bryan et al. 1995), and demonstration that it represents a requisite component of the functional KATP channel complex (Inagaki et al. 1995) suggested the obvious possibility that ATP inhibition, the defining property of these channels, results from an interaction of Mg+-free ATP with the nucleotide binding folds (NBF) of the SUR1 subunit. Consistent with this possibility was the further demonstration that co-expression of Kir6.2 with SUR2A generated channels with a slightly lower Ki for ATP than co-expression with SUR1 (Inagaki et al. 1996). However, several studies have now demonstrated that mutations of the nucleotide binding folds, while changing or abolishing channel activation by Mg-ADP, MgATP and potassium channel openers (KCO), do not alter the intrinsic sensitivity to inhibition by ATP (Nichols et al. 1996; Gribble et al. 1997, 1998; Shyng et al. 1997a). In contrast, mutations of both pore-forming, and cytoplasmic residues of the Kir6.2 subunit have been shown to significantly alter the intrinsic ATP sensitivity of co-expressed channels (Shyng et al. 1997b; Tucker et al. 1997, 1998).
We have demonstrated previously that mutations of residue 160, within the pore-forming M2 segment, alter the ATP sensitivity of expressed mutant channels, by altering the relative stability of the open channel (Shyng et al. 1997b). According to the proposed model, Kir6.2N160X mutant channels with increased peak open probabilities would spend less time in distant ATP-bound closed states, resulting in an apparent decrease in ATP affinity. A similar mechanism seems to be responsible for the decrease in the Ki for ATP that results from N-terminal truncation. Figure 10A shows a simplified version of a kinetic model initially proposed by Nichols et al. (1991) and modified to explain the N160 mutational effects (for details see Shyng et al. 1997b). This model satisfactorily accounts for the following, generally observed, kinetic features of KATP channels: (1) increase of inter-burst closed times with increasing [ATP]; (2) lack of dependence of intra-burst closed and open times on [ATP]; and (3) steady-state dependence of channel on [ATP] that is steeper than a Hill coefficient of 1. The model does not explicitly provide an explanation for the stimulatory actions of diazoxide or Mg-ADP, acting through interactions with SUR1, and at this juncture there is no unifying kinetic model to explain all such data. For Kir6.2ΔN2−30 + SUR1 mutant channels, both the decreased steady-state [ATP] dependence and increased peak open probability can be explained by a shift in the coupling transition (KCO) between the ATP-unbound closed state (C) and the open state (O) (Fig. 10C). In this case the ATP binding constant (KA) remains unaltered. In contrast, for Kir6.2K185Q + SUR1 channels a shift in the ATP-binding constant alone (KA) can explain the decrease in apparent ATP affinity, with unmodified peak open probability. Finally, the striking effect of the double mutant Kir6.2ΔN2−30/K185Q on ATP sensitivity of co-expressed channels, in which the Ki for ATP is reduced 400-fold, is then explained by the additive effect of the two mutations, acting at different steps in the linearly coupled kinetic process (KCO and KA). As shown in Fig. 10B, the proposed model accurately simulates the kinetic behaviour of both the wild-type and mutant KATP channels with respect to observed peak open probability and ATP sensitivity.
Figure 10. Model simulation of the observed kinetic behaviour of Kir6.2wt, Kir6.2ΔN2−30, Kir6.2K185Q, and Kir6.2ΔN2−30/K185Q mutant channels.
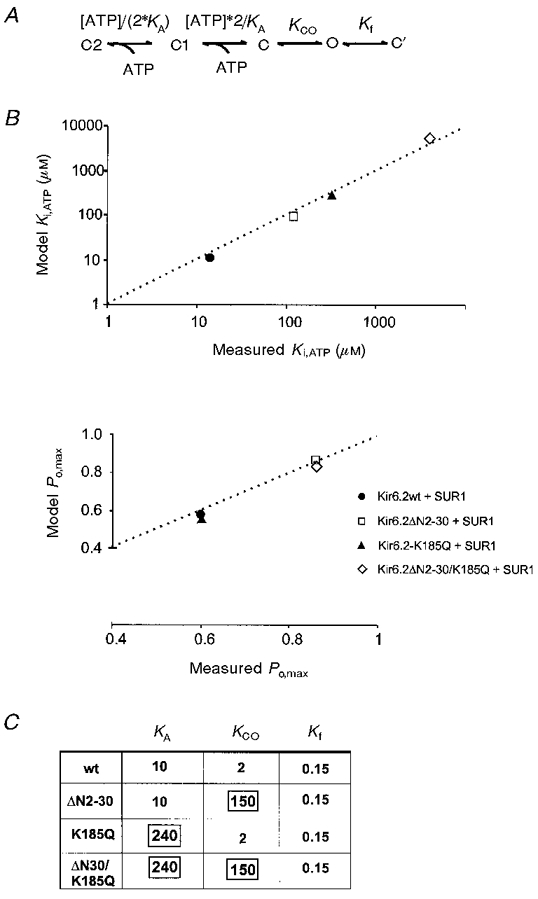
A, model scheme for simulation of KATP channel activity (see Shyng et al. 1997a). Only two sequential ATP binding steps are necessary to approximate the steepness of steady-state dependence of channel activity on [ATP]. C’ refers to the rapid closed state observed within a burst. Equilibrium constants for each transition are indicated. B, model predicted Ki[ATP] and Po,max plotted against measured Ki[ATP] and Po,max, respectively (see Fig. 6A and B. C), table showing the model parameters for equilibrium constants (KCO and Kf) and the ATP binding constant (KA). Only the boxed constants were altered to produce the necessary simulations of mutant channel activity.
One goal of these studies is to understand the physical basis of gating, and the question arises as to how the N-terminus, which is not directly connected to the pore of the channel, is able to control open probability. Analysis of control of channel activity by the SUR1 subunit (Shyng et al. 1997b), led us to hypothesize that SUR1 exerts a hypersensitizing effect on the ATP inhibition of Kir6.2 channel activity that reduces the effective Ki from around 100 μM (as seen with Kir6.2ΔC25 expressed without SUR1) to around 10 μM. This 10-fold increase of ATP sensitivity could result from a shift in the coupling between ATP binding and the open state, in which the interaction of SUR1 destabilizes the open state relative to the ATP accessible closed state (i.e. a reduction in KCO in the model of Fig. 10).
In light of the realization that the expression of Kir6.2 in the absence of SUR1 results in channels which actually have a considerably lower Po,max (Trapp et al. 1998; John et al. 1998), we are forced to suggest that expression of Kir6 in the absence of SUR has an additional effect that results in the lowering of Po to much lower values (∼0.1), by an effect besides that of a simple shift in the equilibrium between the open state and ATP accessible states, i.e. there is an additional lowering of the open-state stability that may be unrelated to transitions to ATP-accessible closed states. Given the clearly intimate physical interaction between SUR and Kir6 subunits (Clement et al. 1997), such additional kinetic effects may not be surprising.
That the N-terminus of Kir6.2 might be involved in transducing this effect is suggested by the subsequent observation that tethering the N-terminus of Kir6.2 to the C-terminus of SUR1 may also serve to uncouple the hypersensitizing action, the ATP sensitivity of channels formed from SUR1-Kir6.2 dimers being reduced 2- to 8-fold (Shyng & Nichols, 1997; Clement et al. 1997; Inagaki et al. 1997). Thus we might envisage that the normal ‘role’ of the N-terminus is to destabilize the open state under the influence of SUR1, and that this action is blocked by physical tethering of the N-terminus to SUR1, and is therefore absent both after deletion of the Kir6.2 N-terminus, or upon expression of Kir6.2 without SUR1. This would explain the lack of additivity of the effect of N- and C-terminal deletions expressed without SUR1 (Figs 4 and 6B).
Reduced sensitivity to other adenosine nucleotides
As shown in Fig. 8, qualitative discrimination between adenosine nucleotides is maintained for both N-terminal truncations and for the K185Q mutation. Given the inability to examine the effects of nucleotide concentrations greater than about 10 mM (due to effects on free [K+]), estimated Ki becomes unreliable above ∼3 mM. If conclusions above are justified ((1) that N-terminal deletions affect open probability; (2) that N-terminal deletions and K185Q mutant effects are independent and additive) then appropriate decreases of the binding affinity of the wild-type channel for ADP (by a factor of 1/12), and AMP (by a factor of 1/122 = 1/144) should also predict the affinity of ΔN2-30 channels for these additional nucleotides. Figure 9 shows that appropriate changes of KA (the nucleotide binding constant) lead to reasonable predictions of the observed sensitivity (Ki) of wild-type and mutant channels to different nucleotides. Consistent with the K185Q mutation acting by reducing the intrinsic binding affinity for nucleotide phosphates, similar, although quantitatively smaller, reductions of binding affinity for ADP (by a factor of 1/5), and for AMP (by a factor of 1/52, i.e. 1/25) reasonably predict not only the sensitivity of K185Q mutant channels to these nucleotides, but also the double mutant channels (Fig. 9).
The data are consistent with K185 forming part of the inhibitory ATP binding site, but this remains a point of speculation. The Kir6.2 subunit does not contain a consensus ATP-binding motif (Inagaki et al. 1995), and there is thus far no evidence for ATP binding to Kir6.2. In agreement with a preliminary study by Babenko & Bryan, (1998), we observe strong labelling of SUR1, but not Kir6.2, with a photo-affinity derivative of ATP, 8-azido-ATP (J. C. Koster & C. G. Nichols, unpublished data).
In conclusion, the present results demonstrate that truncations of the N- and C-termini of the Kir6.2 subunits can generate KATP channels with novel properties. Deletions of the C-terminus, while not affecting the intrinsic channel ATP sensitivity, allow the generation of functional channels in the absence of SUR1, confirming the results of Tucker et al. (1997). Deletions of the N-terminus, though not generating functional channels without SUR1, nevertheless stabilize the open channel, leading to a reduction of apparent ATP sensitivity in the presence of SUR1. This leads us to suggest that the N-terminus may function to transduce a destabilizing effect of SUR1 on the open state. A mutation in the near C-terminal region (K185Q) reduced the ATP sensitivity of SUR1-co-expressed channels without changing peak open probability, consistent with a reduction of ATP binding affinity. Finally, a dual mutant channel containing both the N-terminal truncation and lysine 185 mutation has very low ATP sensitivity, resulting from the energetically additive effects of open-state stabilization and reduction of apparent ATP affinity.
Acknowledgments
We are grateful to Dr S. Seino (Department of Molecular Medicine, Chiba University Graduate School of Medicine, Japan) for providing us with the Kir6.2 clone, and to Jeremiah Shepard for technical assistance. This work was supported by a fellowship from the NIH (NIH, Washington University DRTC Training grant, to J. C. K.), a Career Development Award from the American Diabetes Association (S.-L. S.), a grant HL45742 from the NIH (C. G. N.), and an Established Investigatorship from the American Heart Association (C. G. N.). We are grateful to the Washington University Diabetes Research and Training Centre for continued molecular biology support.
References
- Aguilar Bryan L, Nichols CG, Wechsler SW, Clement JP, IV, Boyd AE, III, Gonzalez G, Herrera Sosa H, Nguy K, Bryan J, Nelson DA. The β cell high affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM. Adenosine 5′-triphosphate-sensitive potassium channels. Annual Review of Neuroscience. 1988;11:97–118. doi: 10.1146/annurev.ne.11.030188.000525. 10.1146/annurev.ne.11.030188.000525. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Bryan J. What have we learned from C-terminus truncated Kir6.2 channels? Biophysical Journal. 1998;74:A114. [Google Scholar]
- Clement JP, IV, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of KATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO Journal. 1997;16:1145–1152. doi: 10.1093/emboj/16.6.1145. 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the beta cell K-ATP channel by interaction with its SUR1 subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzales G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. 10.1016/S0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic β-cell ATP-sensitive K+ channel. FEBS Letters. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. 10.1016/S0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- John SA, Monck JR, Weiss JN, Ribalet B. The sulfonylurea receptor SUR1 regulates ATP-sensitive mouse Kir6.2 K+ channels linked to green fluorescent protein in human embryonic kidney cells (HEK 29 3) The Journal of Physiology. 1998;510:333–345. doi: 10.1111/j.1469-7793.1998.333bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster JC, Shyng S-L, Sha Q, Nichols CG. Involvement of the N-terminus of Kir6.2 in regulating ATP-sensitivity of KATP channels. Biophysical Journal. 1998;74:A230. [Google Scholar]
- Lederer WJ, Nichols CG. Nucleotide modulation of the activity of rat heart KATP channels in isolated membrane patches. The Journal of Physiology. 1989;419:193–211. doi: 10.1113/jphysiol.1989.sp017869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz E, Alekseev AE, Krapivinsky GB, Carrasco A, Clapham DE, Terzic A. Evidence for direct physical association between a K+ channel (kir6.2) and an ATP-binding cassette protein (SUR1) which affects cellular distribution and kinetic behavior of an ATP-sensitive K+ channel. Molecular and Cellular Biology. 1998;18:1652–1659. doi: 10.1128/mcb.18.3.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov MV, Proks P, Ashcroft FM, Ashcroft SJ. Expression of functionally active ATP-sensitive K-channels in insect cells using baculovirus. FEBS Letters. 1998;429:390–394. doi: 10.1016/s0014-5793(98)00640-1. 10.1016/S0014-5793(98)00640-1. [DOI] [PubMed] [Google Scholar]
- Neher E, Stevens C. Conductance fluctuations and ionic pores in membranes. Annual Review of Biophysics and Bioengineering. 1977;6:345–381. doi: 10.1146/annurev.bb.06.060177.002021. 10.1146/annurev.bb.06.060177.002021. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lederer WJ. ATP-sensitive potassium channels in the cardiovascular system. American Journal of Physiology. 1991;261:H1675–1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lederer WJ, Cannell MB. The ATP-dependence of KATP channel kinetics in isolated membrane patches from rat ventricle. Biophysical Journal. 1991;60:1164–1177. doi: 10.1016/S0006-3495(91)82152-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG, Shyng S-L, Nestorowicz A, Glaser B, Clement JP, IV, Gonzalez G, Aguilar-Bryan L, Permutt AM, Bryan JP. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- Shyng S-L, Ferrigni T, Nichols CG. Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. Journal of General Physiology. 1997a;110:141–153. doi: 10.1085/jgp.110.2.141. 10.1085/jgp.110.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S-L, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP: distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. Journal of General Physiology. 1997b;110:643–654. doi: 10.1085/jgp.110.6.643. 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S-L, Nichols CG. Octameric stoichiometry of the KATP channel. Journal of General Physiology. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth RJ. The conductance of sodium channels under conditions of reduced current at the node of Ranvier. The Journal of Physiology. 1980;307:131–142. doi: 10.1113/jphysiol.1980.sp013427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Proks P, Tucker SJ, Ashcroft FM. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. Journal of General Physiology. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, Reimann F, Ashcroft FM. Molecular determinants of K-ATP channel inhibition by ATP. EMBO Journal. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]