

Abstract
The role of Na+–Ca2+ exchange in the regulation of the cytosolic free Ca2+ concentration ([Ca2+]i) was studied in primary cultured rat brain capillary endothelial cells. [Ca2+]i was measured by digital fluorescence imaging in cells loaded with fura-2.
ATP (100 μm) applied for a short period of time (6 s) caused a rise in [Ca2+]i from 127 ± 3 (n = 290) to 797 ± 25 nm, which then declined to the resting level, with a t½ (time required for [Ca2+]i to decline to half of peak [Ca2+]i) of 5.4 ± 0.09 s. This effect was independent of external Ca2+ and could be abolished by previously discharging the Ca2+ pool of the endoplasmic reticulum with thapsigargin (1 μm).
Application of thapsigargin (1 μm) or cyclopiazonic acid (10 μm) to inhibit the Ca2+-ATPase of the endoplasmic reticulum 6 s prior to ATP application did not influence the peak [Ca2+]i but greatly reduced the rate of decline of [Ca2+]i, with t½ values of 15 ± 1.6 and 23 ± 3 s, respectively.
In the absence of external Na+ (Na+ replaced by Li+ or N-methylglucamine) the basal [Ca2+]i was slightly elevated (152 ± 6 nm) and the restoration of [Ca2+]i after the ATP stimulation was significantly slower (t½, 7.3 ± 0.46 s in Li+ medium, 8.12 ± 0.4 s in N-methylglucamine medium).
The external Na+-dependent component of the [Ca2+]i sequestration was also demonstrated in cells stimulated by ATP subsequent to addition of cyclopiazonic acid; in a Na+-free medium [Ca2+]i remained at the peak level in 88% of the cells after stimulation with ATP.
Addition of monensin (10 μm) in the presence of external Na+ increased the resting [Ca2+]i to 222 ± 9 nm over ∼1 min and subsequent removal of extracellular sodium resulted in a further increase in [Ca2+]i to a peak of 328 ± 11 nm, which was entirely dependent on external Ca2+.
These findings indicate that a functional Na+–Ca2+ exchanger is present at the blood-brain barrier, which plays a significant role in shaping the stimulation-evoked [Ca2+]i signal and is able to work in reverse mode under pharmacological conditions.
Intracellular calcium is an important signalling mechanism in endothelial cells including the brain microvessel endothelium, leading to the generation and release of vasoactive compounds such as nitric oxide (Weih et al. 1998), prostacyclin (Wiemer et al. 1994) and endothelin (Bacic et al. 1992).
Several substances are able to activate the endothelial cells of brain microvessels that form the blood-brain barrier (BBB) leading to a rapid rise in [Ca2+]i (Abbott, 1998a). These include bradykinin (Revest et al. 1991; Wiemer et al. 1994), acetylcholine (Luiten et al. 1996), angiotensin II (Stanimirovic et al. 1996), ADP (Frelin et al. 1993), ATP and purine analogues (Vigne et al. 1994; Nobles et al. 1995; Albert et al. 1997), vasopressin (Bacic et al. 1992), histamine (Revest et al. 1991), serotonin (Olesen, 1989), bombesin (Vigne et al. 1995) and endothelin (Vigne et al. 1990; Kawai et al. 1997), as well as mechanical stimuli (Popp et al. 1992). In addition to establishing a link between stimulation and secretion, calcium signalling leads to changes in cell morphology and BBB permeability through mechanisms that may include activation of myosin light-chain kinase and other cytoskeletal components (Abbott & Revest, 1991; Fleming et al. 1995; Abbott & Romero, 1996; Bartus et al. 1996; Albert et al. 1997; Boarder & Hourani, 1998; Denker & Nigam, 1998).
The spatial and temporal characteristics of the [Ca2+]i signal are likely to play an important role in the regulation of Ca2+-dependent intracellular processes, and therefore Ca2+ transporters, which contribute to the shaping of the stimulation-evoked [Ca2+]i signal, are likely to have functional importance. The Na+-Ca2+ exchanger present in the plasma membrane has been shown to play a crucial role in regulating [Ca2+]i in several cell types including cardiac myocytes (Allen et al. 1989), smooth muscle (Batlle et al. 1991) and neurons (Reuter & Porzig, 1995; see also Blaustein, 1984). There is little information available on the Na+-Ca2+ exchange mechanism in endothelial cells. No evidence for the existence of the Na+-Ca2+ exchanger has been found in studies performed on endothelial cells cultured from bovine aorta (Schilling et al. 1988), atria (Laskey et al. 1990) or pulmonary artery (Cannell & Sage, 1989). However, it has been reported that removal of extracellular Na+ or loading the cells with Na+ resulted in an increased [Ca2+]i in cultured endothelial cells from pulmonary artery (Sage et al. 1991) and in intact endothelial cells prepared from cardiac valve (Li & Van Breemen, 1995), thus the existence of the Na+-Ca2+ exchanger has been suggested in these cells. Supporting evidence comes from an immunofluorescence study detecting cross-reaction of antibodies raised against cardiac Na+-Ca2+ exchanger in cultured endothelial cells derived from bovine aorta (Juhaszova et al. 1994).
Functional studies on cultured bovine pulmonary artery endothelial cells showed that although external Na+ had a small effect on [Ca2+]i, this appeared not to have primary importance in regulating resting [Ca2+]i (Sage et al. 1991). However, in intact endothelium from cardiac valve, robust changes in the resting [Ca2+]i were detected upon manipulating [Na+]i (Li & van Breemen, 1995). It is unclear whether this difference derives from altered characteristics of the exchanger in cultured cells as compared with that of in situ endothelial cells, or whether the distribution and the physiological role of the Na+-Ca2+ exchanger is different in endothelial cells of different origin.
It is not yet known whether the Na+-Ca2+ exchange mechanism exists in brain capillary endothelial cells. The brain endothelium is distinct from that of most other tissues in showing upregulation of a number of features related to its barrier-forming properties in situ. These include complex and extremely tight ‘tight junctions’ (zonulae occludentes) (Wolburg et al. 1994), and a range of specific carrier systems important in regulating transendothelial transport, including carriers mediating brain entry of glucose, amino acids and nucleosides, and efflux carriers such as P-glycoprotein protecting the brain from xenobiotics in the blood (Abbott & Romero, 1996). There is evidence that the permeability of the paracellular (tight junctional) pathway can be modulated by inflammatory mediators, and in some cases a receptor-mediated elevation of [Ca2+]i is implicated in barrier opening (Abbott & Revest 1991; Abbott & Romero, 1996; Abbott, 1998a,b).
The BBB phenotype is induced in brain endothelium in vivo by chemical factors secreted by cells of the nervous system, including the endfeet of astrocytic glia that surround the endothelium. Many of the phenotypic features of BBB endothelium are retained in primary cultured brain endothelial cells, although some are progressively lost during subsequent passage (Abbott et al. 1992; Wolburg et al. 1994).
In the present study we investigated whether a functional Na+-Ca2+ exchanger is present in primary cultured brain capillary endothelial cells. In addition to studies on the resting [Ca2+]i, the regulation of the stimulation-evoked [Ca2+]i signal was also addressed. ATP was used to stimulate cells via purinergic (P2) receptors present in the plasma membrane of rat brain capillary endothelial cells (RBCECs) (Nobles et al. 1995). We report here the existence of a Na+-Ca2+ exchange mechanism in cultured RBCECs which has only a small effect on the resting [Ca2+]i but significantly contributes to the sequestration of the stimulation-evoked [Ca2+]i rise.
METHODS
Cell culture
Procedures for obtaining the cell cultures were in accordance with Guidelines for the Use of Laboratory Animals of the Semmelweis University of Medicine, Budapest. Three- to five-month-old Lewis rats were decapitated. Primary cultures of rat brain microvascular endothelium were prepared by a modification of the method described by Abbott et al. (1992) and were characterized immunocytochemically by detection of factor VIII-related antigen. The culture medium was based on standard Dulbecco's medium composed of Dulbecco's modified Eagle's medium, 20 % plasma-derived adult bovine serum (Sigma), 75 μg ml−1 endothelial cell growth supplement, 100 i.u. ml−1 penicillin/100 μg ml−1 streptomycin, 2 mM glutamine, 80 μg ml−1 heparin grade I and supplement containing vitamin C, glutathione, insulin, transferrin and selenium. Cells were used after culturing for 7-10 days (when cells were close to confluence) on glass coverslips coated with bovine corneal endothelial extracellular matrix. This state of maturation was chosen to mimic the situation in vivo.
Preparation of extracellular matrix
This procedure was a modification of the method described by Gospodarowicz (1984). Briefly, fresh bovine eyes kept in ice were obtained from an abattoir, primary cultures of corneal endothelium were prepared, passaged 2-3 times and finally cultured on untreated glass coverslips. After reaching confluence, cells were lysed to expose the extracellular matrix by treating the preparations with 20 mM NH4OH in distilled water. Matrix-coated coverslips were kept under sterile conditions in isotonic salt solution for up to 3 months at 4°C (Dömötör et al. 1998).
Measurement of [Ca2+]i
To monitor changes in [Ca2+]i, cells on coverslips were incubated for 1 h at 37°C with 6 μM fura-2 AM in Dulbecco's modified Eagle's medium. Cells were transferred into a perfusion chamber fitted to the stage of an inverted Nikon Diaphot 200 microscope and superfused with normal Hepes buffer at 37°C for 10 min prior to experiments. All drugs and chemicals were introduced to the close proximity of cells at 37°C under computer control via a glass multitube system (tip diameter, 0.5 mm; perfusion rate constant at 6 μl s−1). [Ca2+]i was measured by digital image fluorescence microscopy (objective, Fluor 40/1.3 oil; Nikon). Excitation wavelengths were 340 and 380 nm, generated by a polychromator illumination system with a resolution of 12 nm (Visitron Sv. GmbH, Pucheim, Germany). Fluorescence emission was monitored at 500 nm. Digital imaging and data analysis were carried out using a 512 × 512 frame transfer CCD camera (Princeton Instruments) and Metafluor software (Universal Imaging Corp., West Chester, PA, USA). A ratio image was aquired every 2 s. Fluorescence under each experimental condition was monitored in cells from two to three coverslips (10-15 cells in a frame). In order to assess whether 2 s sampling gave sufficient resolution, some experiments were repeated with 1 s fluorescence sampling (see legend to Fig. 2).
Figure 2. [Ca2+]i in response to ATP in a normal and in a Na+-free, Li+-containing solution.

[Ca2+]i in response to ATP (100 μM, 6 s) in normal solution (trace a, n = 290) and in a Na+-free, Li+-containing solution (trace b, n = 54). Perfusion with normal medium was switched to Na+-free medium 20 s prior to stimulation with ATP. Inset: columns represent the means of individually determined t½ values (+s.e.m.) measured in normal control medium (C) and in medium where Na+ was replaced by either Li+ or N-methylglucamine (NMGA) (n = 50) as indicated. Repetition of the protocol in Li+ medium with a 1 s sampling rate gave the same pattern of results (t½(Li) = 7.6 s, n = 45); for convenience further studies were all done using 2 s sampling. *P < 0.05, **P < 0.0001 (significantly different from control).
Ratios were converted to free [Ca2+]i using the equation: [Ca2+]i = Kd[(R - Rmin)/(Rmax - R)](Sf2/Sb2) (Grynkiewicz et al. 1985), in which R is the 340 nm/380 nm fluorescence ratio. The maximum (Rmax) and minimum ratio (Rmin) and the ratio of fluorescence for Ca2+-bound/Ca2+-free dye measured at 380 nm (Sf2/Sb2) were determined using an in vitro calibration method and were corrected for viscosity (Poenie, 1994). Experimental results are expressed as [Ca2+]i.
Solutions and drugs
Normal Hepes buffer contained (mM): NaCl, 135; KCl, 5.4; CaCl2, 1.8; MgCl2, 1.2; glucose, 10; and Hepes, 20; pH adjusted to 7.4. For experiments in nominally calcium-free extracellular solution, CaCl2 was omitted. For a Na+-free medium Na+ was replaced by an equimolar amount of Li+ or N-methyl-D-glucamine (NMGA). Chemicals were obtained from Sigma.
Data analysis and statistics
Data analysis was performed with Sigmaplot 4.0 software. t½ (the time required for [Ca2+]i to decline to half of the peak [Ca2+]i) values were calculated from the individual [Ca2+]i traces, then averaged.
Results are given as means ±s.e.m. Statistical significance was evaluated by Student's t test and differences with P < 0.05 were taken as significant.
RESULTS
Agonist-stimulated elevation in [Ca2+]i
It is well established that both primary and immortalized rat brain capillary endothelial cells express at least two P2 receptor subtypes where extracellular ATP can act to cause a rapid increase in cytosolic [Ca2+] (Vigne et al. 1994; Nobles et al. 1995; Albert et al. 1997). In our primary endothelial cell preparation, ATP (100 μM) applied for 70 s caused a characteristic elevation in [Ca2+]i from the basal 136 ± 4.8 nM (n = 40) to a peak of 664 ± 27 nM, which was followed by a biphasic decay with a sustained plateau phase (Fig. 1A). When cells were perfused with a nominally Ca2+-free medium, the plateau phase of the ATP-evoked [Ca2+]i signal was abolished but the peak value (646 ± 33.5 nM, n = 28) was not significantly different from that obtained in the presence of Ca2+ (Fig. 1B). This is consistent with previous reports that the plateau phase of the [Ca2+]i transient results from Ca2+ entry across the plasma membrane (Hallam & Pearson, 1986; Colden-Stanfield et al. 1989). The complex pattern of the response was simplified by applying ATP (100 μM) for a short period of time (6 s) which resulted in [Ca2+]i transients having the same peak values and general characteristics in the presence and absence of extracellular Ca2+ (691 ± 9 nM, n = 289; 642 ± 18 nM, n = 85, respectively) (Fig. 1C and D). This showed that influx of extracellular calcium was not involved in the [Ca2+]i signal when ATP was applied as a brief pulse.
Figure 1. [Ca2+]i transients in response to 100 μM ATP in rat brain capillary endothelial cells.
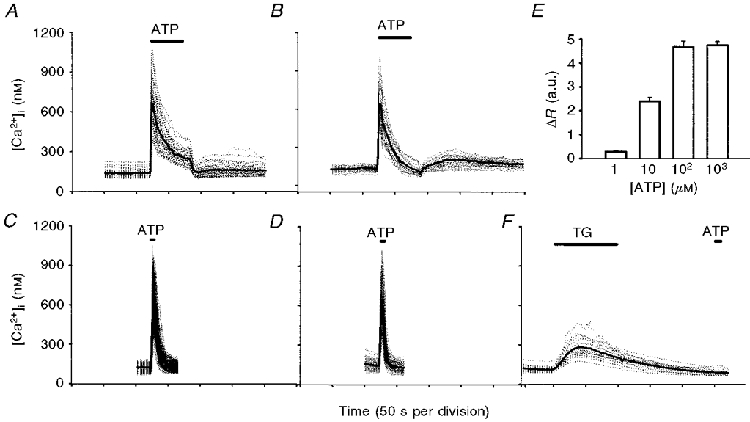
In A and B, cells were exposed to ATP for 70 s in a normal Hepes buffer (A;n = 40) or in nominally calcium-free Hepes buffer (B; n = 28). In C and D, ATP was applied for 6 s in Ca2+-containing (C;n = 289) or Ca2+-free (D;n = 85) Hepes buffer. In F, thapsigargin (1 μM) was added prior to ATP as indicated. Dotted traces represent recordings from individual cells, thick continuous traces represent mean values. In E, columns represent mean +s.e.m. of peak [Ca2+]i shown as a change in the 340 nm/380 nm fluorescence ratio (ΔR), induced by ATP (n = 20). a.u., arbitrary units.
In order to confirm that under these conditions Ca2+ was mobilized from the endoplasmic reticulum (ER), thapsigargin (TG, 1 μM), an inhibitor of the ER Ca2+-ATPase, was added prior to the ATP stimulus. The small [Ca2+]i transient caused by TG indicated depletion of the ER calcium pools, and subsequent application of ATP failed to elevate [Ca2+]i despite the presence of extracellular Ca2+ (Fig. 1F). Similar results were obtained using cyclopiazonic acid (CPA, 10 μM) which inhibits both isoforms of the ER Ca2+-ATPase (Papp et al. 1993) (data not shown). These results indicate that the [Ca2+]i transient caused by a short ATP pulse is a result of mobilization of Ca2+ from the TG- and CPA-sensitive calcium stores of the ER. The effect of ATP on [Ca2+]i was dependent on the concentration used, with a maximal response at 100 μM ATP (Fig. 1E).
In the experiments presented below, ATP was applied at 100 μM as a short pulse (6 s) to stimulate RBCECs and generate a Ca2+ transient attributable entirely to release of Ca2+ from the ER. It is important to note that in order to obtain reproducible and comparable [Ca2+]i responses, it was necessary to use cell preparations at the same stage of maturation (7-10 days after seeding).
ATP-evoked [Ca2+]i signals in the absence of external Na+
In order to examine whether extracellular Na+ made any contribution to the sequestration of the [Ca2+]i transient generated by ATP, cells were stimulated in a Na+-free medium where Na+ was replaced by Li+ or NMGA. Removal of Na+ caused a small but significant rise in resting [Ca2+]i (from 127 ± 3 to 152 ± 6 nM; Fig. 2), in agreement with findings in an intact cardiac endothelial monolayer (Li & van Breemen, 1995) but in contrast with those on cultured pulmonary artery endothelial cells (Sage et al. 1991). Cells perfused with normal, Na+-containing medium responded to ATP with an initial rapid increase in [Ca2+]i from 127 ± 3 nM to a peak of 797 ± 25 nM (n = 290), followed by a return to the basal value with a t½ of 5.4 ± 0.09 s (Fig. 2). Replacement of external Na+ by Li+ had no influence on the amplitude of the ATP-evoked [Ca2+]i transient (890 ± 71 nM), but slightly increased the resting [Ca2+]i (152 ± 6 nM) and reduced the rate of decline to the basal level as indicated by a significantly greater t½ (7.3 ± 0.46 s) (Fig. 2, inset). An increased t½ was also observed when NMGA was used to substitute for Na+ (t½ = 8.12 ± 0.4 s) (Fig. 2, inset), but in this case the amplitude of the ATP-evoked calcium rise was somewhat smaller than those observed in the control or Li+-based medium (541 ± 13 nM).
The contribution of an external Na+-dependent Ca2+ sequestration process to the shaping of the stimulation-evoked [Ca2+]i signal was confirmed using either TG or CPA to inhibit the ER Ca2+-ATPase (Inesi & Sagara, 1994). TG (1 μM) or CPA (10 μM) was given 6 s prior to an ATP pulse, which enabled these agents to inhibit the Ca2+-ATPase without significantly altering the ER Ca2+ pool. Stimulation with ATP gave rise to a [Ca2+]i signal with a similar peak amplitude to the control (788 ± 38 nM, n = 42), both in the presence of CPA (790 ± 20 nM, n = 49) and TG (723 ± 23 nM, n = 50), respectively. However, the sequestration of [Ca2+]i transients was greatly reduced as indicated by increased t½ values (t½(CPA) = 23 ± 3 s, n = 49; t½(TG) = 15.1 ± 1.6 s, n = 50) (Fig. 3). In 22 % of cells [Ca2+]i failed to decline to 50 % of the peak within 100 s when CPA was applied. The slight flattening of the recovery curves in TG (and CPA) medium around 30-40 s after application may reflect the ability of these agents to cause slight and slowly rising elevation of [Ca2+]i (Fig. 1F), but the influence on the recovery curves is small. These results show that uptake of Ca2+ by the ER is a major mechanism for regulation of the stimulation-evoked [Ca2+]i signal, but when the ER Ca2+-ATPase is inhibited, [Ca2+]i can still be reduced, although at a much slower rate.
Figure 3. [Ca2+]i in response to ATP in normal solution and in the presence of thapsigargin or cyclopiazonic acid.

[Ca2+]i in response to ATP (100 μM, 6 s) in normal solution (trace a;n = 42) and in the presence of thapsigargin (TG, 1 μM; trace b, n = 50) or cyclopiazonic acid (CPA, 10 μM; trace c, n = 49). Inhibitors were added to cells 6 s prior to the ATP stimulus and were present for 1 min. The inset shows the respective t½ values for the recovery phase as mean +s.e.m. for control and for experiments with TG or CPA as indicated. *P < 0.0001 (significantly different from control).
In the following experiments we examined whether the slow [Ca2+]i decline in the absence of Ca2+ reuptake by the ER was dependent on external Na+. For these experiments, CPA was applied in a Na+-free (Li+) medium 6 s prior to ATP, which did not influence the amplitude of the ATP-evoked [Ca2+]i transient (771 ± 31 nM, n = 49) as compared with the control (788 ± 38 nM, n = 42) or with the response when CPA was added in the normal medium (790 ± 20 nM, n = 49). [Ca2+]i following the ATP stimulus remained at a sustained elevated plateau level in 43 out of 49 cells treated with CPA in a Na+-free medium (Fig. 4). These results revealed an external Na+-dependent component of the sequestration of [Ca2+]i which appears to be responsible for the slow reduction of [Ca2+]i following an ATP stimulus when uptake of Ca2+ by the ER is inhibited.
Figure 4. [Ca2+]i in response to ATP in normal solution, 6 s after the application of CPA in normal medium or when CPA was added in Na+-free solution.

A, [Ca2+]i (nM ±s.e.m.) in response to ATP (100 μM, 6 s) in normal solution (trace a;n = 42), 6 s after the application of cyclopiazonic acid (CPA, 10 μM) in normal medium (trace b; n = 49) or when CPA was added in Na+-free solution (Na+ replaced by Li+) (trace c;n = 49). Na+-free solution was introduced 20 s prior to stimulation with ATP. B, individual recordings in cells stimulated with ATP (100 μM, 6 s) after application of CPA in Na+-free solution (also used for trace c in A). See Fig. 1 for explanation of continuous and dotted lines.
Effect of a reduced Na+ electrochemical gradient on [Ca2+]i
Since transport of ions by the Na+-Ca2+ exchanger is dependent on the electrochemical gradients of Na+ and Ca2+ across the cell membrane, an increase in [Na+]i is expected to increase [Ca2+]i in cells whose plasma membrane contains a functional Na+-Ca2+ exchange mechanism. Loading RBCECs with Na+ in Na+-containing medium can be accomplished by the cation ionophore monensin (10 μM), which in our cells (Fig. 5) increased the basal [Ca2+]i from 120 ± 5 to 222 ± 9 nM (n = 22), consistent with a Na+-Ca2+ exchanger working in reverse mode (Na+ out, Ca2+ in). Upon subsequent switch to a sodium-free perfusion medium, an additional increase in [Ca2+]i to 328 ± 11 nM was observed, which then decreased slowly (Fig. 5, trace a). In this case the large transient outward Na+ gradient created by Na+ removal is likely to serve as a driving force for Ca2+ entry. In the absence of extracellular Ca2+ these manipulations of the Na+ gradient failed to cause any change in [Ca2+]i, indicating that the [Ca2+]i rise shown in Fig. 5 (trace a) is indeed the result of Ca2+ entry from the extracellular medium.
Figure 5. The effect of monensin and subsequent removal of external Na+ on [Ca2+]i in Ca2+-containing or nominally Ca2+-free medium.

The effect of monensin (10 μM) and subsequent removal of external Na+ (replaced by Li+) on [Ca2+]i (means ±s.e.m.) in Ca2+-containing (trace a;n = 22) or nominally Ca2+-free medium (trace b;n = 15). The traces were obtained from two different cell cultures from the same preparation batch.
Incubation of cells with the Na+,K+-ATPase inhibitor ouabain (500 μM) for 1 h was without effect on [Ca2+]i, and ouabain plus monensin raised [Ca2+]i to the same extent as monensin alone (data not shown). However, after preincubating the cells with ouabain (500 μM) for 50 min or treating cells with monensin (10 μM) for 1 min and subsequently with sodium-free medium (1 min), ATP application resulted in [Ca2+]i transients with significantly increased t½ values (9.9 ± 0.3 s after ouabain; 18.34 ± 1.5 s after monensin + 0 Na+). The amplitude of the [Ca2+]i signal in response to ATP was also reduced under these conditions (418 ± 14 nM, n = 30).
DISCUSSION
The present study performed on primary cultured brain microvascular endothelium is the first to present evidence for the existence of a Na+-Ca2+ exchange mechanism at the blood-brain barrier, and shows that the exchanger contributes both to the control of resting Ca2+ and to the regulation of [Ca2+]i in agonist-stimulated cells.
There is controversy in the literature as to the presence and function of the Na+-Ca2+ exchanger in endothelial cells. In most studies, removal of extracellular Na+, which is expected to reveal the function of a Na+-Ca2+ exchanger by driving Ca2+ into the cells, failed to increase [Ca2+]i in cultured endothelial cells (Schilling et al. 1988; Laskey et al. 1990). Not only was the resting [Ca2+]i unaltered in these cells but the transient [Ca2+]i signal in response to stimulation by bradykinin was also unaffected by a Na+-free medium (Schilling et al. 1988; Laskey et al. 1990). These experiments reinforce the view that the Na+-Ca2+ exchanger does not significantly contribute to the regulation of [Ca2+]i in endothelial cells (Cannell & Sage, 1989; Laskey et al. 1990). Circumstantial evidence for a possible physiological role of Na+-Ca2+ exchange in endothelial cells was presented by Winquist et al. (1985), who showed that the acetylcholine-evoked relaxation of aortic rings was abolished by dichlorobenzamil, a compound used to inhibit the Na+-Ca2+ exchanger. Clear evidence for a Na+-dependent Ca2+ entry in cultured endothelial cells was provided by Sage et al. (1991) who demonstrated that removal of external Na+ subsequent to loading the cells with sodium promoted Ca2+ entry, but this mechanism did not appear to be of fundamental importance in maintaining resting [Ca2+]i. By contrast, the large increase in [Ca2+]i upon reducing the transmembrane Na+ gradient in intact cardiac valvular endothelium indicated that in these cells Na+-Ca2+ exchange is likely to have functional significance (Li & van Breemen, 1995). As an explanation for this discrepancy, it was suggested that important membrane functions might be altered in cultured cells whereas cells in intact endothelium not exposed to enzymatic or mechanical treatments would be expected to give more physiologically relevant results (Li & van Breemen, 1995).
Our experiments performed on primary cultures of brain endothelial cells aimed to establish whether a Na+-dependent mechanism plays a role in the recovery of [Ca2+]i from an elevated level produced by stimulation with ATP. We demonstrated that the t½ of the decline of [Ca2+]i to the basal level after stimulation with ATP was increased by 35-50 % when external Na+ was replaced by Li+ or NMGA, respectively (Fig. 2). This indicated that an external Na+-dependent Ca2+ extrusion made a contribution to the sequestration of the stimulation-evoked [Ca2+]i rise. However, removal of the extracellular Na+ may also reduce pHi due to the lack of H+ extrusion via Na+-H+ exchange, which may interfere with processes regulating [Ca2+]i. This is unlikely in our case since Li+ is able to substitute for Na+ on the exchanger and drive the efflux of H+ (Aronson, 1985). The effect of extracellular Li+ on [Ca2+]i regulation also needs to be considered. Li+ can inhibit cytoplasmic inositol phosphate monoesterase (Berridge et al. 1982) resulting in an increased IP3 accumulation, and consequently an enhanced agonist-evoked [Ca2+]i response. This possibility was avoided using NMGA to replace Na+, and yet the rate of decline in [Ca2+]i under these conditions was similar to that observed in a Li+-substituted medium. This suggests that the altered ATP-induced [Ca2+]i signal in a Na+-free medium resulted directly from the lack of Na+, and is consistent with the suggestion that Ca2+ extrusion via the Na+-Ca2+ exchanger working in the forward mode contributes to the sequestration of the stimulation-evoked [Ca2+]i rise.
The major mechanism by which the ATP-induced peak [Ca2+]i is reduced appears to be Ca2+ uptake by the ER, as indicated by a marked reduction in the rate of [Ca2+]i decline in the presence of inhibitors of the ER Ca2+-ATPase (Fig. 3). This pharmacological manipulation also provided further evidence for the role of the Na+-Ca2+ exchanger in extruding Ca2+. When the ER Ca2+-ATPase is not functional, the sequestration of [Ca2+]i after stimulation with ATP is greatly impaired but still significant (Fig. 3). The remaining [Ca2+]i sequestration appears to be entirely dependent on external Na+ in the majority of cells; in the simultaneous presence of CPA and absence of extracellular Na+, cells responded to ATP with a [Ca2+]i rise but were unable to reduce [Ca2+]i, which then remained at an elevated level (Fig. 4). The recovery seen in a minority of cells (7 out of 49) under these conditions may indicate a small contribution of other mechanisms, e.g. plasmalemmal Ca2+-ATPase.
The exchanger in RBCECs was also able to function in a reverse mode when Ca2+ influx was driven by a reversed Na+ electrochemical gradient. For this demonstration, [Na+]i was increased by monensin, which resulted in an increase in [Ca2+]i. The further increase in [Ca2+]i observed upon removal of external Na+ in the presence of monensin (Fig. 5) could be best interpreted as the effect of an even larger outward Na+ electrochemical gradient forcing the operation of the Na+-Ca2+ exchanger in reverse mode.
It is difficult to explain satisfactorily why exposure to ouabain, assumed to raise [Na+]i due to inhibition of plasmalemmal Na+,K+-ATPase, failed to elevate [Ca2+]i as also reported by Sage et al. (1991). It is possible that the extent of increase in [Na+]i in the presence of ouabain is insufficient to reverse the Na+ electrochemical gradient. Previous reports from our group showed that inhibition of the Na+,K+-ATPase in isolated nerve terminals by ouabain (Deri & Adam-Vizi, 1993) or by an insufficient supply of ATP (Tretter et al. 1998) resulted in an ∼30 mM increase in [Na+]i which proved to be insufficient to cause Ca2+ influx (Adam-Vizi & Ligeti, 1986) in spite of the presence of a functional Na+-Ca2+ exchanger in the plasma membrane (Gill et al. 1981). In cultured endothelial cells having a smaller surface-to-volume ratio than nerve terminals, the rise in [Na+]i may be even smaller. Given the observed increase in [Ca2+]i in response to ouabain in intact endothelium, it cannot be excluded that differences exist in the regulation of [Ca2+]i between cultured and in situ endothelial cells, as suggested by Li & van Breemen (1995). Nevertheless, whereas the assumed increase in [Na+]i due to ouabain proved to be insufficient to drive Ca2+ entry in RBCECs, it was able to increase the t½ of [Ca2+]i decline after stimulation with ATP, suggesting it was adequate to reduce Ca2+ extrusion when [Ca2+]i was increased.
In conclusion, our results show that a Na+-dependent mechanism plays a significant role in the recovery phase of the ATP-evoked [Ca2+]i signal in cultured brain microvascular endothelial cells. We suggest that the physiological role of the Na+-Ca2+ exchanger at the blood-brain barrier is to extrude Ca2+, and thus to contribute to the regulation of [Ca2+]i in resting and agonist-stimulated cells. Under appropriate pharmacological conditions (Na+ removal or substantial increase in [Na+]i) the exchanger can also operate in the reverse mode, driving influx of Ca2+ into the cells.
Acknowledgments
We thank Katalin Takacs for her excellent technical assistance. This work was supported by grants from OTKA, ETT, MTA, MKM to V. A.-V., and from the British Council to N. J. A. and V. A-V. N. J. A. was supported by the Medical Research Council, the Wellcome Trust and the Royal Society.
References
- Abbott NJ. Role of intracellular calcium in regulation of blood-brain barrier permeability. In: Pardridge WM, editor. An Introduction to the Blood- Brain Barrier: Methodology and Biology. Cambridge University Press; 1998a. pp. 345–353. [Google Scholar]
- Abbott NJ. Receptor-mediated modulation of blood-brain barrier permeability. Cell and Molecular Neurobiology. 1998b. in the Press. [DOI] [PubMed]
- Abbott NJ, Hughes CCW, Revest PA, Greenwood J. Development and characterisation of a rat brain capillary endothelial culture: towards an in vitro blood-brain barrier. Journal of Cell Science. 1992;103:23–38. doi: 10.1242/jcs.103.1.23. [DOI] [PubMed] [Google Scholar]
- Abbott NJ, Revest PA. Control of brain endothelial permeability. Cerebrovascular Brain Metabolism Reviews. 1991;3:39–72. [PubMed] [Google Scholar]
- Abbott NJ, Romero IA. Transporting therapeutics across the blood-brain barrier. Molecular Medicine Today. 1996;2:106–113. doi: 10.1016/1357-4310(96)88720-x. 10.1016/1357-4310(96)88720-X. [DOI] [PubMed] [Google Scholar]
- Adam-Vizi V, Ligeti E. Calcium uptake of synaptosomes as a function of membrane potential under different depolarizing conditions. The Journal of Physiology. 1986;372:363–377. doi: 10.1113/jphysiol.1986.sp016013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert JL, Boyle JP, Roberts JA, Challis RAJ, Gubby SE, Boarder MR. Regulation of brain capillary endothelial cells by P2Y receptors coupled to Ca2+, phospholipase C and mitogen-activated protein kinase. British Journal of Pharmacology. 1997;122:935–941. doi: 10.1038/sj.bjp.0701453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TJA, Noble D, Reuter H. Sodium-Calcium Exchange. Oxford University Press; 1989. [Google Scholar]
- Aronson PS. Kinetic properties of the plasma membrane sodium-hydrogen exchanger. Annual Review of Physiology. 1985;47:545–560. doi: 10.1146/annurev.ph.47.030185.002553. [DOI] [PubMed] [Google Scholar]
- Bacic F, Uematsu S, McCarron RM, Spatz M. Secretion of immunoreactive endothelin-1 by capillary and microvascular endothelium of human brain. Neurochemical Research. 1992;17:699–702. doi: 10.1007/BF00968008. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Elliott P, Hayward N, Dean R, McEwen EL, Fisher SK. Permeability of the blood-brain barrier by the bradykinin agonist, RMP-7; evidence for a sensitive, auto-regulated, receptor-mediated system. Immunopharmacology. 1996;33:270–278. doi: 10.1016/0162-3109(96)00070-7. [DOI] [PubMed] [Google Scholar]
- Batlle DC, Godinich M, LaPointe MS, Munoz E, Carone F, Mehring N. Extracellular Na+ dependency of free cytosolic Ca2+ regulation in aortic vascular smooth muscle cells. American Journal of Physiology. 1991;261:C845–856. doi: 10.1152/ajpcell.1991.261.5.C845. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Downes CP, Hanley MR. Lithium amplifies agonist-dependent phosphatidylinositol responses in brain and salivary glands. Biochemical Journal. 1982;206:587–595. doi: 10.1042/bj2060587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP. The energetics and kinetics of sodium- calcium exchange in barnacle muscles, squid axons and mammalian heart: the role of ATP. In: Blaustein MP, Lieberman M, editors. Electrogenic Transport: Fundamental Principles and Physiological Implications. New York: Raven Press Publishers; 1984. pp. 129–147. [PubMed] [Google Scholar]
- Boarder MR, Hourani SMO. The regulation of vascular function by P2 receptors: multiple sites and multiple receptors. Trends in Pharmacological Sciences. 1998;19:99–107. doi: 10.1016/s0165-6147(98)01170-5. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Sage SO. Bradykinin-evoked changes in cytosolic calcium and membrane currents in bovine pulmonary artery endothelial cells. The Journal of Physiology. 1989;419:555–568. doi: 10.1113/jphysiol.1989.sp017886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colden-Stanfield M, Schilling WP, Ritchie AK, Eskin SG, Navarro LT. Bradykinin-induced increases in cytosolic calcium and ionic currents in cultured bovine aortic endothelial cells. Circulation Research. 1989;61:632–640. doi: 10.1161/01.res.61.5.632. [DOI] [PubMed] [Google Scholar]
- Denker BM, Nigam SK. Molecular structure and assembly of the tight junction. American Journal of Physiology. 1998;274:F1–9. doi: 10.1152/ajprenal.1998.274.1.F1. [DOI] [PubMed] [Google Scholar]
- Deri Z, Adam-Vizi V. Detection of intracellular free Na+ concentration of synaptosomes by a fluorescent indicator, sodium-binding benzofuran isophthalate: the effect of veratridine, ouabain and alpha-latrotoxin. Journal of Neurochemistry. 1993;61:818–825. doi: 10.1111/j.1471-4159.1993.tb03592.x. [DOI] [PubMed] [Google Scholar]
- Dömötör E, Sipos I, Kittel Á, Abbott NJ, Adam-Vizi V. Improved growth of cultured brain microvascular endothelial cells on glass coated with a biological matrix. Neurochemistry International. 1998;33:473–478. doi: 10.1016/s0197-0186(98)00057-6. 10.1016/S0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Busse R. Calcium signaling in endothelial cells involves activation of tyrosine kinases and leads to activation of mitogen-activated protein kinase. Circulation Research. 1995;75:522–529. doi: 10.1161/01.res.76.4.522. [DOI] [PubMed] [Google Scholar]
- Frelin C, Breittmayer JP, Vigne P. ADP induces inositol phosphate-independent intracellular Ca2+ mobilization in brain capillary endothelial cells. Journal of Biological Chemistry. 1993;268:8787–8792. [PubMed] [Google Scholar]
- Gill DL, Grollman EF, Kohn LD. Calcium transport mechanisms in membrane vesicles from guinea pig brain synaptosomes. Journal of Biological Chemistry. 1981;256:184–192. [PubMed] [Google Scholar]
- Gospodarowicz D. Preparation of extracellular matrices produced by cultured bovine corneal endothelial cells and PF-HR-9 endodermal cells: their use in cell culture. In: Barnes DA, editor. Methods for Preparation of Media Supplements and Substrata for Serum-free Animal Cell Culture. Alan R. Liss; 1984. pp. 275–293. [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of calcium indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hallam TJ, Pearson JD. Exogenous ATP raises cytoplasmic free calcium in fura-2-loaded piglet aortic endothelial cells. FEBS Letters. 1986;207:95–99. doi: 10.1016/0014-5793(86)80019-9. 10.1016/0014-5793(86)80019-9. [DOI] [PubMed] [Google Scholar]
- Inesi G, Sagara Y. Specific inhibitors of intracellular Ca2+ transport ATPases. Journal of Membrane Biology. 1994;141:1–6. doi: 10.1007/BF00232868. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Ambesi A, Lindenmayer GE, Bloch RJ, Blaustein MP. Na+-Ca2+ exchanger in arteries; identification by immunoblotting and immunofluorescence microscopy. American Journal of Physiology. 1994;266:C234–242. doi: 10.1152/ajpcell.1994.266.1.C234. [DOI] [PubMed] [Google Scholar]
- Kawai N, Yamamoto T, Yamamoto H, McCarron RM, Spatz M. Functional characterization of endothelin receptors on cultured brain capillary endothelial cells of the rat. Neurochemistry International. 1997;31:597–605. doi: 10.1016/s0197-0186(97)00018-1. 10.1016/S0197-0186(97)00018-1. [DOI] [PubMed] [Google Scholar]
- Laskey RE, Adams DJ, Johns A, Rubanyi GM, Van Breemen C. Membrane potential and Na+-K+ pump activity modulate resting and bradykinin-stimulated changes in cytosolic free calcium in cultured endothelial cells from bovine atria. Journal of Biological Chemistry. 1990;265:2613–2619. [PubMed] [Google Scholar]
- Li L, Van Breemen C. Na+-Ca2+ exchange in intact endothelium of rabbit cardiac valve. Circulation Research. 1995;76:396–404. doi: 10.1161/01.res.76.3.396. [DOI] [PubMed] [Google Scholar]
- Luiten PG, De Jong GI, Van der Zee EA, Van Dijken H. Ultrastructural localization of cholinergic muscarinic receptors in rat brain cortical capillaries. Brain Research. 1996;720:225–229. doi: 10.1016/0006-8993(96)00195-3. 10.1016/0006-8993(96)00195-3. [DOI] [PubMed] [Google Scholar]
- Nobles M, Revest PA, Couraud PO, Abbott NJ. Characteristics of nucleotide receptors that cause elevation of cytoplasmic calcium in immortalized rat brain endothelial cells (RBE4) and in primary cultures. British Journal of Pharmacology. 1995;115:1245–1252. doi: 10.1111/j.1476-5381.1995.tb15032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen SP. An electrophysiological study of microvascular permeability and its modulation by chemical mediators. Acta Physiologica Scandinavica. 1989;(suppl. 579):1–28. [PubMed] [Google Scholar]
- Papp B, Paszti K, Kovacs T, Sarkadi B, Gardos G, Enouf J, Enyedi A. Characterization of the inositol trisphosphate-sensitive and insensitive calcium stores by selective inhibition of the endoplasmic reticulum-type calcium pump isoforms in isolated platelet membrane vesicles. Cell Calcium. 1993;14:531–538. doi: 10.1016/0143-4160(93)90074-g. 10.1016/0143-4160(93)90074-G. [DOI] [PubMed] [Google Scholar]
- Poenie M. Alteration of intracellular fura-2 fluorescence by viscosity: a simple correction. Cell Calcium. 1994;11:85–91. doi: 10.1016/0143-4160(90)90062-y. 10.1016/0143-4160(90)90062-Y. [DOI] [PubMed] [Google Scholar]
- Popp R, Hoyer J, Meyer J, Galla HJ, Gögelein H. Stretch-activated non-selective cation channels in the antiluminal membrane of porcine cerebral capillaries. The Journal of Physiology. 1992;454:435–449. doi: 10.1113/jphysiol.1992.sp019272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter H, Porzig H. Localization and functional significance of the Na+/Ca2+ exchanger in presynaptic boutons of hippocampal cells in culture. Neuron. 1995;15:1077–1084. doi: 10.1016/0896-6273(95)90096-9. 10.1016/0896-6273(95)90096-9. [DOI] [PubMed] [Google Scholar]
- Revest PA, Abbott NJ, Gillespie JI. Receptor-mediated changes in intracellular [Ca2+]i in cultured rat brain capillary endothelial cells. Brain Research. 1991;549:159–161. doi: 10.1016/0006-8993(91)90614-2. 10.1016/0006-8993(91)90614-2. [DOI] [PubMed] [Google Scholar]
- Sage SO, van Breemen C, Cannel MB. Sodium-calcium exchange in cultured bovine pulmonary artery endothelial cells. The Journal of Physiology. 1991;440:569–580. doi: 10.1113/jphysiol.1991.sp018725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling WP, Ritchie AK, Navarro LT, Eskin SG. Bradykinin-stimulated calcium influx in cultured bovine aortic endothelial cells. American Journal of Physiology. 1988;255:H219–227. doi: 10.1152/ajpheart.1988.255.2.H219. [DOI] [PubMed] [Google Scholar]
- Stanimirovic D, Morley P, Ball R, Hamel E, Mealing G, Durkin JP. Angiotensin II-induced fluid phase endocytosis in human cerebromicrovascular endothelial cells is regulated by the inositol-phosphate signaling pathway. Journal of Cell Physiology. 1996;169:455–467. doi: 10.1002/(SICI)1097-4652(199612)169:3<455::AID-JCP6>3.0.CO;2-N. 10.1002/(SICI)1097-4652(199612)169:3<455::AID-JCP6>3.3.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Tretter L, Chinopoulos C, Adam-Vizi V. Plasma membrane depolarization and disturbed Na+ homeostasis induced by the protonophore FCCP in isolated nerve terminals. Molecular Pharmacology. 1998;53:734–741. doi: 10.1124/mol.53.4.734. [DOI] [PubMed] [Google Scholar]
- Vigne P, Feolde E, Breittmayer JP, Frelin C. Characterization of the effects of 2-methylthio-ATP and 2-chloro-ATP on brain capillary endothelial cells: similarities to ADP and differences from ATP. British Journal of Pharmacology. 1994;112:775–780. doi: 10.1111/j.1476-5381.1994.tb13146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigne P, Feolde E, Van Renterghem C, Breittmayer JP, Frelin C. Properties and functions of a neuromedin-B-preferring bombesin receptor in brain microvascular endothelial cells. European Journal of Biochemistry. 1995;233:414–418. doi: 10.1111/j.1432-1033.1995.414_2.x. [DOI] [PubMed] [Google Scholar]
- Vigne P, Marsault R, Breittmayer JF, Frelin C. Endothelin stimulates phosphatidylinositol hydrolysis and DNA synthesis in brain capillary endothelial cells. Biochemical Journal. 1990;266:415–420. doi: 10.1042/bj2660415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weih MK, Weikert S, Freyer D, Dirnagl U. Chemiluminescence detection of nitric oxide production from rat cerebral cortical endothelial cells in culture. Brain Research Protocol. 1998;2:175–182. doi: 10.1016/s1385-299x(97)00037-8. 10.1016/S1385-299X(97)00037-8. [DOI] [PubMed] [Google Scholar]
- Wiemer G, Popp R, Scholkens BA, Gogelein H. Enhancement of cytosolic calcium, prostacyclin and nitric oxide by bradykinin and the ACE inhibitor ramiprilat in porcine brain capillary endothelial cells. Brain Research. 1994;638:261–266. doi: 10.1016/0006-8993(94)90658-0. 10.1016/0006-8993(94)90658-0. [DOI] [PubMed] [Google Scholar]
- Winquist RJ, Bunting PB, Schofield TL. Blockade of endothelium-dependent relaxation by the amiloride analog dichlorobenzamyl: possible role of Na+/Ca2+ exchange in the release of endothelium-derived relaxant factor. Journal of Pharmacology and Experimental Therapeutics. 1985;235:644–650. [PubMed] [Google Scholar]
- Wolburg H, Neuhaus J, Kniesel U, Krauss B, Schmid EM, Ocalan M, Farrel C, Risau W. Modulation of tight junction structure in blood-brain barrier endothelial cells. Effects of tissue culture, second messengers and cocultured astrocytes. Journal of Cell Science. 1994;107:1347–1357. doi: 10.1242/jcs.107.5.1347. [DOI] [PubMed] [Google Scholar]