

Abstract
ClC proteins are a class of voltage-dependent Cl− channels with several members mutated in human diseases. The prototype ClC-0 Torpedo channel is a dimeric protein; each subunit forms a pore that can gate independently from the other one. A common slower gating mechanism acts on both pores simultaneously; slow gating activates ClC-0 at hyperpolarized voltages. The ClC-2 Cl− channel is also activated by hyperpolarization, as are some ClC-1 mutants (e.g. D136G) and wild-type (WT) ClC-1 at certain pH values.
We studied the dependence on internal Cl− ([Cl−]i) of the hyperpolarization-activated gates of several ClC channels (WT ClC-0, ClC-0 mutant P522G, ClC-1 mutant D136G and an N-terminal deletion mutant of ClC-2), by patch clamping channels expressed in Xenopus oocytes.
With all these channels, reducing [Cl−]i shifted activation to more negative voltages and reduced the maximal activation at most negative voltages.
We also investigated the external halide dependence of WT ClC-2 using two-electrode voltage-clamp recording. Reducing external Cl− ([Cl−]o) activated ClC-2 currents. Replacing [Cl−]o by the less permeant Br− reduced channel activity and accelerated deactivation.
Gating of the ClC-2 mutant K566Q in normal [Cl−]o resembled that of WT ClC-2 in low [Cl−]o, i.e. channels had a considerable open probability (Po) at resting membrane potential. Substituting external Cl− by Br− or I− led to a decrease in Po.
The [Cl−]i dependence of the hyperpolarization-activated gates of various ClC channels suggests a similar gating mechanism, and raises the possibility that the gating charge for the hyperpolarization-activated gate is provided by Cl−.
The external halide dependence of hyperpolarization-activated gating of ClC-2 suggests that it is mediated or modulated by anions as in other ClC channels. In contrast to the depolarization-activated fast gates of ClC-0 and ClC-1, the absence of Cl− favours channel opening. Lysine 556 may be important for the relevant binding site.
The ClC gene family represents a class of voltage-dependent Cl− channels, with several members involved in human hereditary diseases (for review, see Jentsch, 1996; Thakker, 1997). Mutations in the muscular channel ClC-1 lead to dominant and recessive myotonia (Koch et al. 1992; Steinmeyer et al. 1994), mutations in ClC-5 lead to Dent's disease, a disorder associated with proteinuria, hypercalciuria and kidney stones (Lloyd et al. 1996), and mutations in ClC-Kb cause a certain form of Bartter's syndrome (Simon et al. 1997), a disease associated with renal salt wasting.
The prototype ClC-0 Torpedo channel (Jentsch et al. 1990) is a dimer of two identical subunits (Ludewig et al. 1996; Middleton et al. 1996). It is a rather unusual ‘double-barrelled’ channel in which each subunit probably forms an independent pore (Ludewig et al. 1996). A common slow gating process opens and closes both pores simultaneously. This produces the typical behaviour at the single channel level in which long closed periods are interrupted by bursts with two conductance states of equal size (Miller, 1982). The gating of an individual protopore appears to be completely independent from the gating state of the neighbouring pore once the slow gate is open (Ludewig et al. 1997c).
The mechanism of the voltage dependence of ClC channels probably differs drastically from that found in typical voltage-dependent cation channels. The strong voltage-dependent activation of Na+ and K+ channels is coupled to the movement of a charged segment of the channel protein (the S4-segment, Noda et al. 1984; Stühmer et al. 1989). No similar structure is present in ClC channels (Jentsch et al. 1990). The fast gate of ClC-0, which acts on individual protopores, strongly depends on the Cl− concentration (Pusch et al. 1995; Chen & Miller, 1996; Ludewig et al. 1997a). The open probability as a function of voltage (Po(V)) and the opening rate as function of voltage (α (V)) are shifted to more positive voltages when extracellular Cl− is decreased (Pusch et al. 1995; Chen & Miller, 1996). At negative voltages, Po does not approach zero, and increasing intracellular Cl− leads to an increase of this saturating value of Po (Chen & Miller, 1996; Ludewig et al. 1997a). These observations led to the proposal that the permeating ion itself acts as the gating charge, i.e. the movement of a Cl− ion along the transmembrane electric field represents the voltage-sensitive step that is coupled to the opening of the pore (Pusch et al. 1995; Chen & Miller, 1996; Pusch, 1996). The gating of the muscular Cl− channel ClC-1 is also strongly dependent on [Cl−]o and resembles the fast gate of ClC-0 (Rychkov et al. 1996, 1998). The dependence of ClC-1 on [Cl−]i is also similar to that of the fast gate of ClC-0 at normal pH conditions (M. Pusch & T. J. Jentsch, unpublished result). Various anions interfere with the gating process of ClC-1 in a complicated manner (Rychkov et al. 1998). In contrast to ClC-0, certain impermeable anions like cyclamate and methanesulphonate may promote channel opening from the extracellular side (Rychkov et al. 1998). These anions also block the channel. Gating of ClC-1 is more complicated than that of ClC-0, and it is impossible from macroscopic measurements to separate a fast (protopore) and a slow (common pore) gating mechanism. This makes an interpretation of ion substitution experiments much more difficult.
The complex gating behaviours of ClC-0, ClC-1 and ClC-2 are summarized schematically in Fig. 1; the fast gate of ClC-0 and (under normal pH conditions) currents of ClC-1 are activated by positive voltages. In contrast, the slow gate of ClC-0 and the gate of the ubiquitous ClC-2 channel (Thiemann et al. 1992) are activated by negative voltages. An activation by hyperpolarization was also found with several mutants of ClC-0 (Ludewig et al. 1997b) and with ClC-1 (Fahlke et al. 1995). Wild-type (WT) ClC-1 also activates at negative voltages under certain pH conditions (Rychkov et al. 1996). It seems reasonable to assume that these hyperpolarization-activated gates represent a common gating mechanism. This is supported by the present experiments showing that the hyperpolarization-activated gates of ClC-0, ClC-1 and ClC-2 are all strongly stimulated by intracellular Cl−. In addition, these findings suggest that the voltage dependence of these gates may arise from a voltage-dependent movement of intracellular Cl− in the outward direction.
Figure 1. Schematic diagram summarizing the gating phenotypes of various ClC channels and mutants.
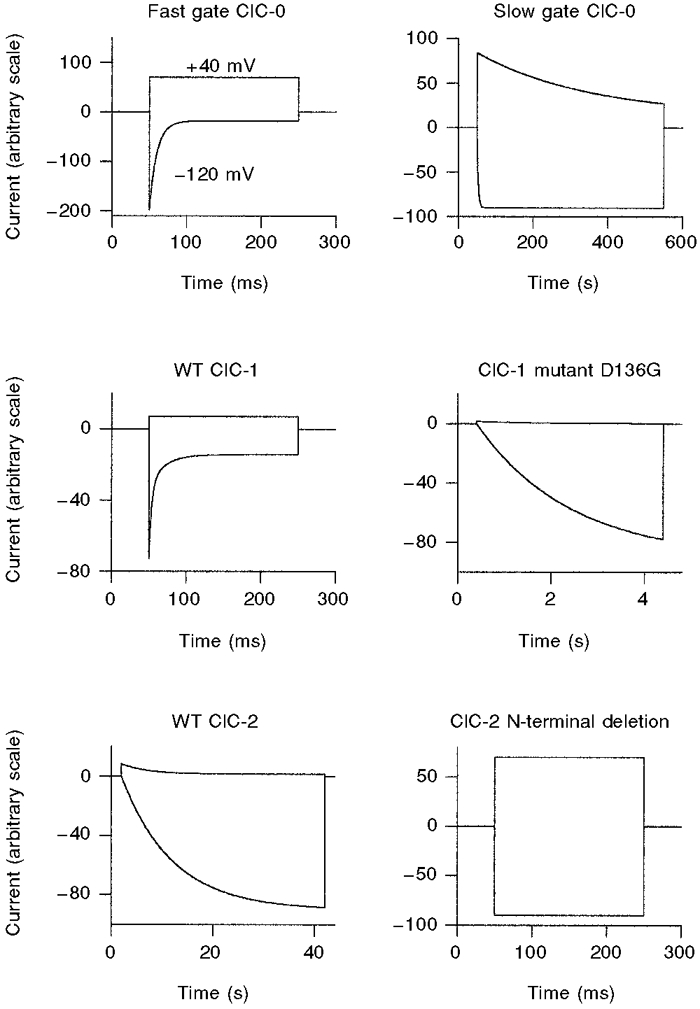
For each channel type (or gate for ClC-0), idealized voltage-clamp responses to voltage steps to -120 and +40 mV from a holding potential of -30 mV are shown. Note the various time scales. In two-electrode voltage-clamp recordings the N-terminal deletion mutant ΔNClC-2 of ClC-2 does not exhibit voltage- or time-dependent gating (as shown in the figure), in contrast to patch-clamp recordings (see Fig. 5).
METHODS
Molecular biology and oocyte injection
The D136G mutation of ClC-1 (Fahlke et al. 1995) was introduced by recombinant PCR and verified by sequencing. Mutation P522G of ClC-0 (Ludewig et al. 1997a), mutation ΔNClC-2 (deletion of amino acids 16-61) (Gründer et al. 1992) and mutation K566Q of ClC-2 (Jordt & Jentsch, 1997) have been described previously. All constructs were cloned into a vector containing Xenopusβ-globin untranslated sequences (Lorenz et al. 1996). They were linearized with MluI or SnaBI and capped cRNA was transcribed in vitro using SP6 RNA polymerase and the mMessage mMachine kit (Ambion, Austin, TX, USA). cRNA was diluted to about 30-100 mg l−1 before injection. Ovaries were obtained from frogs that had been anaesthetized with tricaine (0.17%) for 15 min. After surgery, frogs were allowed to recover from anaesthesia and suitable aftercare was given. Stage V and VI Xenopus oocytes were then manually defolliculated and injected with 50 nl of cRNA solution. Oocytes were kept in Barth's solution (88 mM NaCl, 2.4 mM NaHCO3, 1.0 mM KCl, 0.41 mM CaCl2, 0.33 mM Ca(NO3)2, 0.82 mM MgSO4 and 10 mM Hepes; pH 7.6) at 14-19°C.
For patch clamping, defolliculated oocytes were collagenase treated (1 g l−1 Sigma type II for 10-15 min) before injection, and the vitelline membrane was removed manually after incubation of oocytes in hypertonic medium directly before the experiment (Methfessel et al. 1986). Patch pipettes were pulled from borosilicate glass and had resistances of 1.5-4 MΩ. Patches usually contained many channels (> 50) such that ‘quasi-macroscopic’ currents could be recorded.
Electrophysiology
Patch-clamp experiments were performed at room temperature 1-5 days after injection using the inside-out or outside-out configuration with an Axopatch-200 amplifier and pCLAMP 5.5 software. In all figures, zero current is indicated by dashed lines. Standard solutions for inside-out patch recordings were as follows. Bath (=‘intracellular’) solution contained 100 mM NMDG-Cl, 2 mM MgCl2, 5 mM Na-Hepes and 5 mM EGTA at pH 7.4, or was a solution in which NMDG-Cl was partially replaced by NMDG-glutamate. In some experiments, [Cl−]i was raised to 160 mM by addition of NMDG-Cl, thus raising also the ionic strength and osmolarity. Currents were only slightly changed by this procedure in a manner consistent with the overall effects of changing [Cl−]i, suggesting that raising osmolarity or ionic strength per se had no effect on channel gating.
Extracellular (pipette) solution contained 100 mM NMDG-Cl, 5 mM MgCl2 and 5 mM Na-Hepes at pH 7.4, or was a solution in which NMDG-Cl was partially replaced by NMDG-glutamate. Solution changes on the same patch were accomplished by a local perfusion system. Data were low pass filtered at half of the sample frequency and analysed using home-written software (written in Visual C++, Microsoft) and the SigmaPlot program (Jandel Scientific, San Rafael, CA, USA). ClC-2 currents were measured using standard two-electrode voltage clamp with a TurboTec (npi Electronics, Tamm, Germany) high-voltage amplifier and pCLAMP 5.5 software. Voltage-clamp protocols are described in the Figure legends. The holding potential was generally 0 mV; for the experiments with ΔNClC-2 it was 60 mV. Apparent open probability (Po) was obtained from experiments as shown in Fig. 2. After stepping the voltage to various test values, channel activation was monitored at a constant ‘tail’ voltage (usually +60 mV). Extrapolated peak currents at this voltage were fitted using a Boltzmann distribution of the form I (V) =Io+ (Imax - Io)/(1 + exp (zF (V½ - V)/RT)), where Imax is the (extrapolated) current at maximal stimulation, z is the apparent gating charge, V½ is the voltage of half-maximal activation, and Io is a constant offset. From these macroscopic current measurements only a ‘relative’Po can be estimated by normalizing with Imax: Po(V) =I(V)/Imax.
Figure 2. Effect of [Cl−]i on the slow gating of WT ClC-0.
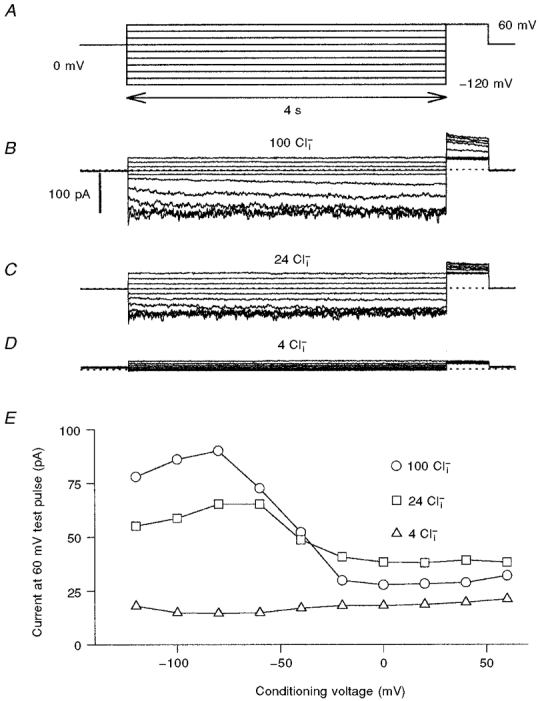
Steady-state slow gate open probability as a function of voltage was assessed with the pulse protocol shown in A. Progressive hyperpolarization cumulatively activates the slow gate. The degree of slow gate activation was then assessed at the fixed tail pulse of +60 mV. At this voltage, the fast gate opens maximally within less than 10 ms, even at low [Cl−]i. In B, C and D, recordings from the same inside-out patch that was superfused are shown, with the indicated Cl− concentrations. In E, the (averaged) current at the +60 mV tail pulse is plotted as a function of the conditioning voltage. Similar results were obtained in six independent experiments.
RESULTS
[Cl−]i dependence of slow gating of ClC-0
The slow gate of ClC-0 determines the probability of bursts during which the two protopores are visible. Po,slow increases at negative voltages. Unfortunately, quantitative measurements of the slow gating are very difficult at the single channel level due to the extremely slow kinetics at voltages more positive than -50 mV (Pusch et al. 1997). Macroscopically the slow gate can be analysed using pulse protocols as described in Pusch et al. (1997). Po,slow(V) can be fairly well described by a Boltzmann distribution. However, it seems that the slow gate does not close completely at positive voltages (Pusch et al. 1997). A dependence of slow gating of ClC-0 on [Cl−]o was demonstrated by Chen & Miller (1996), who found that decreasing [Cl−]o leads to a drastic shortening of burst duration.
Figure 2A illustrates the pulse protocol used to measure the steady-state activation of the slow gate. Relatively long conditioning pulses to various potentials (starting from positive values) lead to a progressive opening of the slow gate. The relative degree of slow gate activation was then monitored using a fixed pulse to +60 mV. At +60 mV the fast gate opens maximally within less than a millisecond. Thus the current at +60 mV is proportional to the number of channels with an open slow gate at the end of the conditioning pulse. A family of current traces recorded with this protocol in symmetrical 100 mM Cl− is shown in Fig. 2B, and the voltage dependence is plotted in Fig. 2E (○). The typical activation at negative voltage is apparent. The slight decrease of the activation at the most negative voltages (-100 and -120 mV) was observed consistently, but was quite variable in quantitative terms. This phenomenon has already been noted by Ludewig et al. (1997a) and Fong et al. (1998) using two-electrode voltage-clamp recordings.
The dependence of slow gating on [Cl−]i was studied by superfusing inside-out patches with different solutions. Apart from possible effects on slow gating, changing [Cl−]i obviously also changes the open-channel current and fast gating open probabilities. These effects are, however, minimized using the protocol of Fig. 2A because the single channel current at +60 mV is mostly dependent on [Cl−]o (which was fixed in these experiments) and the fast gate opens maximally at +60 mV, even at low [Cl−]i (Ludewig et al. 1997a). Current traces for 24 mM [Cl−]i and 4 mM [Cl−]i from the same patch are shown in Fig. 2C and D and the resulting activation curves are shown in Fig. 2E. The open probability was drastically reduced to the extent that at 4 mM [Cl−]i, channels could not be activated at all, even at -120 mV.
Once the slow gate has been activated, one has to wait several minutes before applying a second pulse protocol (Pusch et al. 1997). This makes measurements on WT ClC-0 rather difficult. It can be seen, for example in Fig. 2E, that the currents for 24 mM [Cl−]i are larger at positive voltages than those at 100 mM. This was possibly caused by an insufficient waiting period between the two recordings (∼2 min). We therefore performed similar measurements with the ClC-0 mutant P522G, which has drastically accelerated slow gating kinetics (Ludewig et al. 1997a), facilitating our measurements. The dependence of slow gating on [Cl−]i was very similar to that of WT, as shown in Fig. 3. In most patches a certain degree of ‘run-down’ was present. This can be seen from the incomplete recovery of the currents after washing with 160 mM [Cl−]i (Fig. 3F). The decrease of Po,slow at low [Cl−]i was, nevertheless, evident. As for WT ClC-0, the slight decrease of the activation at the most negative voltages prevented a fit of a Boltzmann distribution over the complete voltage range. Fitting Boltzmann distributions only up to the maximum of the activation to the data for 160 mM [Cl−]i, 100 mM [Cl−]i and 24 mM [Cl−]i indicated that the voltage of half-maximal activation, V½, was shifted to more negative values at low [Cl−]i (from -52 ± 8 mV (mean ±s.d., n = 7) at 100 mM to -82 ± 11 mV (n = 6) at 24 mM). Reducing [Cl−]i thus seems to have two effects: overall reduction of Po,slow and shifting of the activation curve to more negative voltages. A quantitative interpretation of this apparent shift is, however, complicated by the decrease of the activation at the most negative voltages that could also be affected by [Cl−]i and thereby unspecifically change the parameters of the Boltzmann fit (Fong et al. 1998).
Figure 3. Effect of [Cl−]i on the slow gating of the ClC-0 mutant P522G.
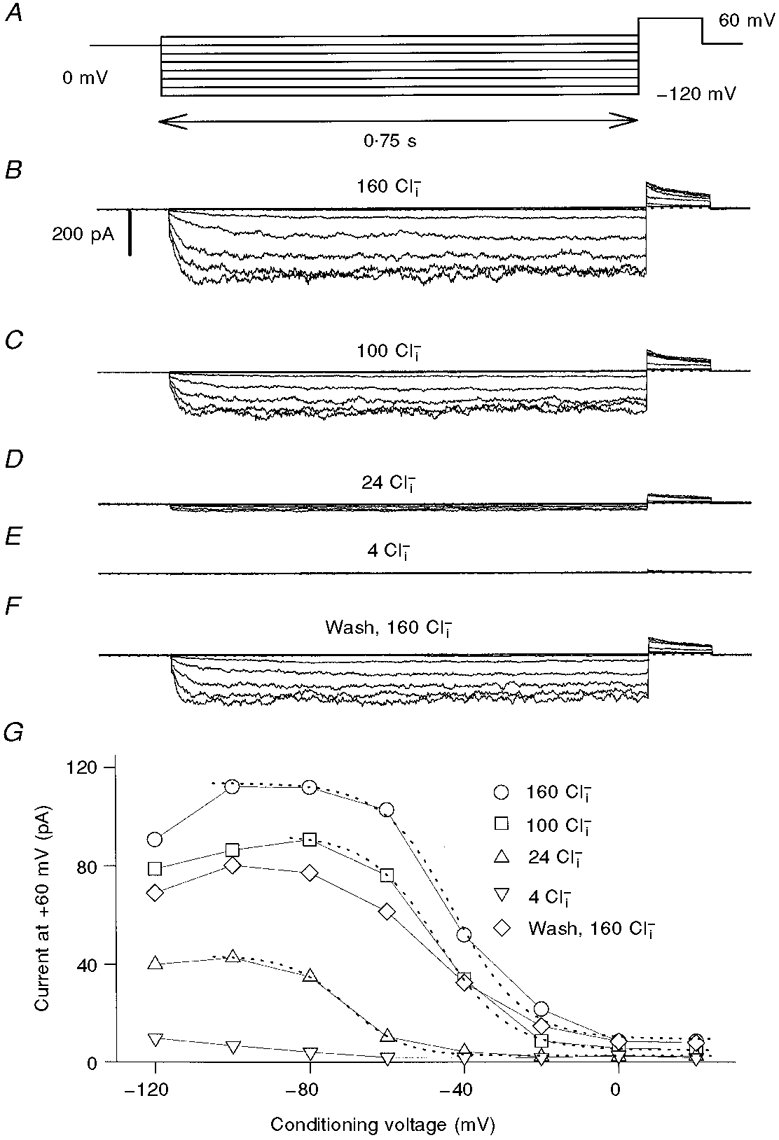
The pulse-protocol (A) is similar to that used for WT ClC-0 (Fig. 2), although with shorter pulse durations. B-F, current traces obtained from one patch perfused with the indicated solutions in chronological order. G, the (averaged) current at the +60 mV tail pulse is plotted as a function of the conditioning voltage. Note the incomplete recovery after the final wash. The dotted lines represent Boltzmann fits with the parameters obtained from several experiments given in the text. Similar results were obtained in seven independent experiments.
[Cl−]i dependence of the ClC-1 mutant D136G
Gating of ClC-1 is similar to the fast gating of ClC-0 in that it also activates with depolarization (Steinmeyer et al. 1991; Pusch et al. 1994; Rychkov et al. 1996). Under normal conditions, ClC-1 lacks a slow hyperpolarization-activated gate. However, a slow hyperpolarization-activated gate becomes visible at low external pH and positive holding potentials (Rychkov et al. 1996). Also, several mutants of ClC-1 have a drastically altered gating that is dominated by a slow activation at negative voltages (Fahlke et al. 1995, 1997b). One of these mutants, D136G, causes recessive myotonia (Fahlke et al. 1995). We investigated the [Cl−]i dependence of this mutant using similar pulse protocols as for ClC-0.
Figure 4 shows results from an inside-out patch containing D136G mutant channels in 160, 100, 24 and 4 mM [Cl−]i. Similar to the slow gate of ClC-0, the open probability of the D136G was drastically reduced by lowering [Cl−]i. Because gating did not reach steady state even at -120 mV, no quantitative analysis of the voltage dependence was performed. In contrast to the strong effect on mutant D136G, WT ClC-1 was only relatively little affected by changing [Cl−]i similar to the fast gate of ClC-0, with increasing [Cl−]i leading to an increase of the saturating value of Po at negative voltages (M. Pusch & T. J. Jentsch, unpublished result; see also Introduction).
Figure 4. Effect of [Cl−]i on the gating of the ClC-1 mutant D136G.
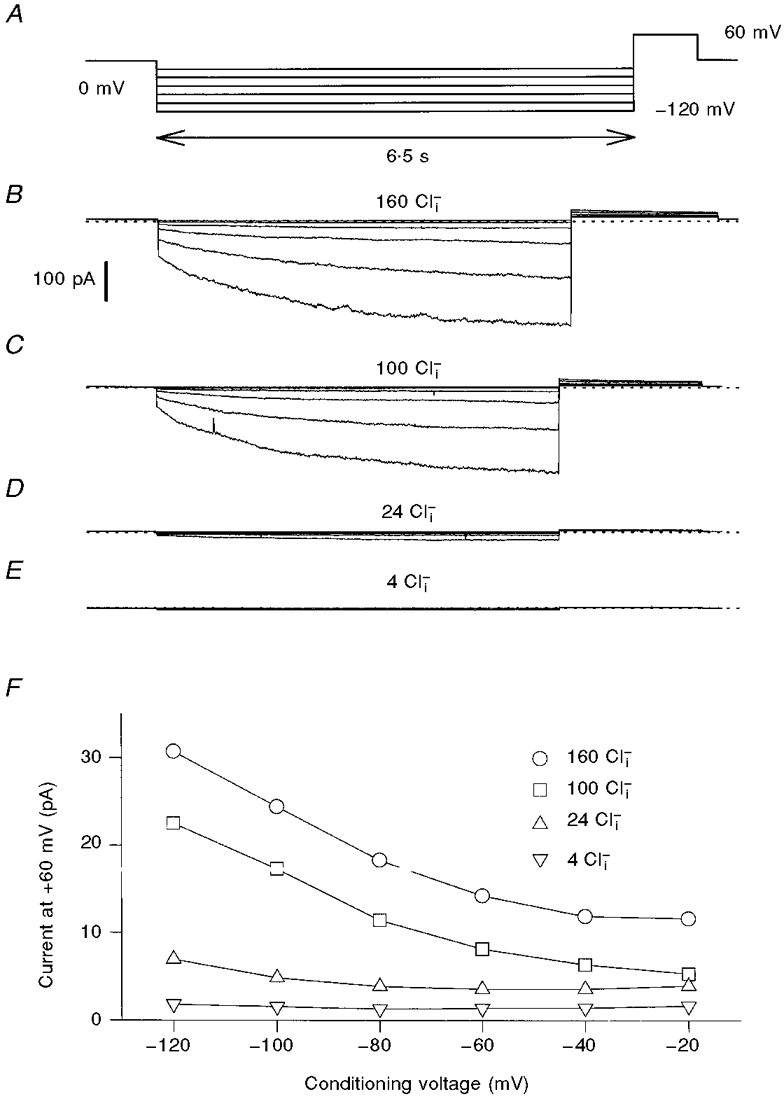
Activation of D136G was studied using a similar pulse protocol as for ClC-0 (A). Currents are from the same patch. Similar results were obtained in four independent experiments.
[Cl−]i dependence of ClC-2 containing an N-terminal deletion
ClC-2 expressed in Xenopus oocytes is activated by strong hyperpolarization, by incubation of oocytes in a hypotonic medium, and by acidic extracellular pH (Gründer et al. 1992; Thiemann et al. 1992; Jordt & Jentsch, 1997). We also tried to study the dependence of the hyperpolarization-activated gate of ClC-2 on intracellular Cl− in the inside-out configuration. However, we encountered several experimental limitations. After excision of patches from the oocyte membrane, the channels displayed an unstable gating behaviour. In some cases, ClC-2 channels activated spontaneously, and in other cases we observed channel rundown after a few minutes of exposure to the test medium (data not shown). These problems precluded a systematic analysis of gating. In contrast to ClC-0 and ClC-1, ClC-2 is activated by oocyte swelling. The effects on gating upon patch excision might therefore be explained by a wash-out of an intracellular factor essential for gating stability or by a slow disintegration of submembraneous cytoskeletal components essential for mediating cell volume changes to the channel protein.
The N-terminus of ClC-2 contains a region that is essential for both activation by hyperpolarization and activation by hypotonic extracellular medium. Its deletion led to ‘constitutively’ activated channels that could not be activated further by hyperpolarization or cell swelling when analysed in two-electrode voltage-clamp experiments (Gründer et al. 1992). We investigated one of the N-terminal deletion mutants (amino acids 16-61 deleted, hereafter called ΔNClC-2) using the patch-clamp technique. This mutant could be stably recorded in inside-out patches with only little ‘run-down’ or spontaneous activation occurring during periods of more than 30 min. Interestingly, and in contrast to the two-electrode voltage-clamp recordings where almost no gating is visible (Gründer et al. 1992; Jordt & Jentsch, 1997), the channels were activated by hyperpolarization under these conditions. Channels were closed by applying a positive holding potential of +60 mV, and channel opening was achieved by stepping the voltage to more negative values (Fig. 5A). The kinetics of this activation was, however, much faster than the slow activation of WT ClC-2. Cl− accumulation/depletion effects may underlie these differences, problems that may be less severe under patch-clamp conditions. Thus the amino-terminal deletion did not completely destroy ClC-2 gating and a relatively fast (compared with WT ClC-2) hyperpolarization-activated gate was maintained.
Figure 5. Effect of [Cl−]i on the gating of N-terminally deleted ClC-2.
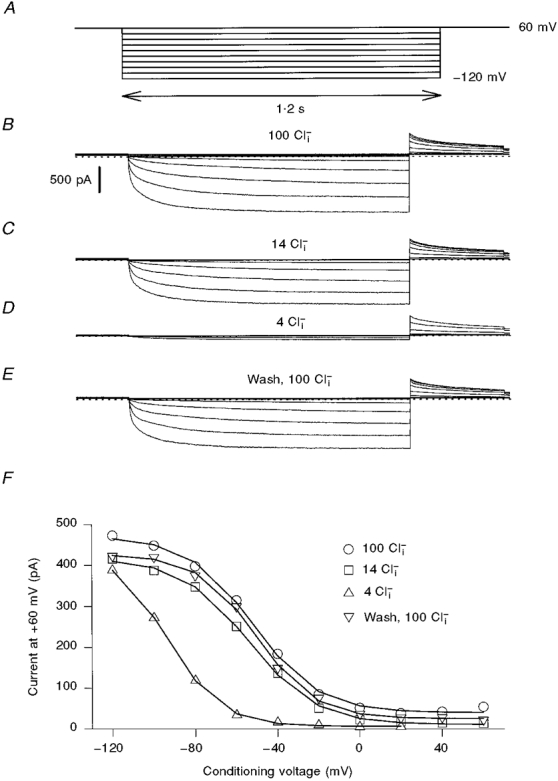
Activation of ΔNClC-2 was studied using a similar pulse protocol as for ClC-0 (A). Currents are from the same patch. Similar results were obtained in five independent experiments.
Figure 5 shows the [Cl−]i sensitivity of ΔNClC-2 gating. It was only slightly affected when [Cl−]i was reduced to 14 mM. At 4 mM, there was a strong decrease of Po at most voltages. At -120 mV, channels could be activated as much as in higher [Cl−]i, indicating that the main effect of a reduction of [Cl−]i was to shift the Po(V) curve to more negative voltages (V½= -31 ± 11 mV (mean ±s.d., n = 5) at 100 mM and V½= -70 ± 15 mV (n = 4) at 4 mM [Cl−]i).
Dependence of ClC-2 on extracellular halides
To investigate the anion dependence of WT ClC-2 gating we varied the extracellular anion composition in two-electrode voltage-clamp recordings. [Cl−]o was reduced to 1 mM by replacement with the presumably inert anion glutamate. Surprisingly, in contrast to the effects of [Cl−]o reduction on other ClC channels, ClC-2 was apparently activated by low [Cl−]o (Fig. 6A). After reducing [Cl−]o to 1 mM, a higher proportion of channels was already open at the resting potential and the current at the end of the hyperpolarizing pulses was larger in 5 and 1 mM [Cl−]o compared with 104 mM (Fig. 6B). This indicates that binding of extracellular Cl− favours a closed state.
Figure 6. Modulation of WT ClC-2 gating by extracellular halides.
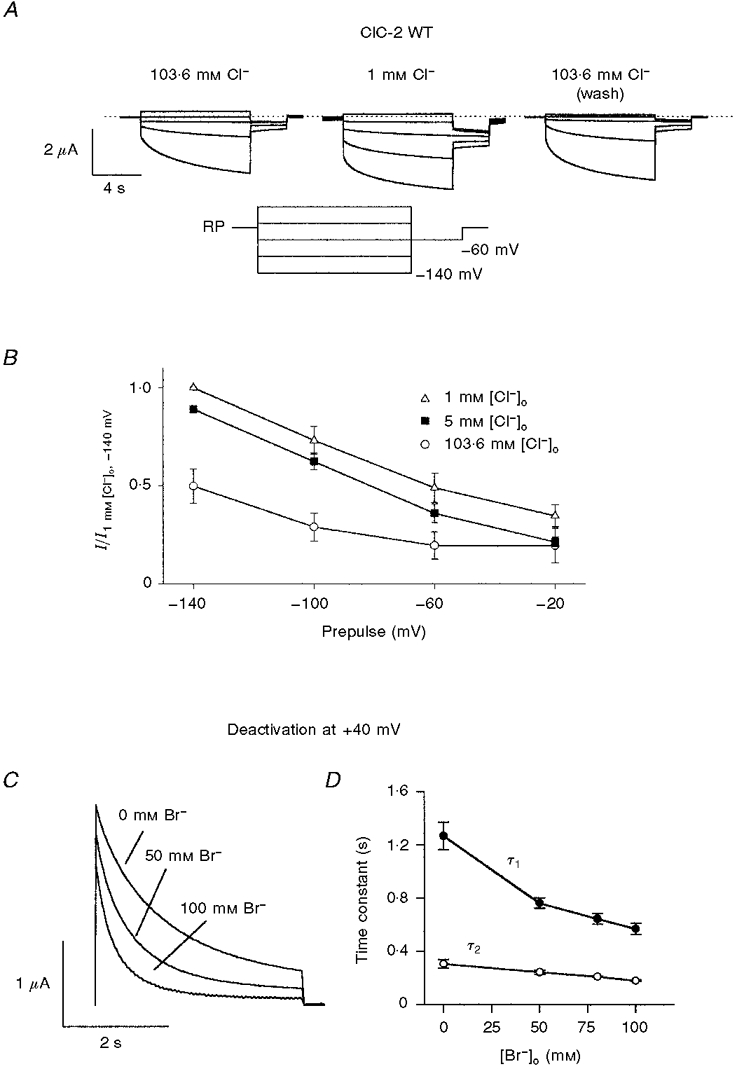
A, dependence of ClC-2 on extracellular Cl−: voltage-clamp traces from a typical oocyte expressing rat ClC-2 after 1 min perfusion with ND96 (left), ND96 with 103.6 mM Cl− substituted with glutamate (middle), and again in ND96 (right). Voltage was clamped for 9 s each from the resting membrane potential to values between +20 mV and -140 mV in steps of -40 mV, followed by a 3 s test pulse to -60 mV (see inset). B, activation of rat ClC-2 by a reduction in [Cl−]o: tail currents at -60 mV were measured after activation for 9 s at the given prepulses in solutions with [Cl−]o substituted by increasing amounts of glutamate (○, 103.6 mM (ND96); ▪, 5 mM; ▵, 1 mM). Averaged results from experiments as shown in A from five oocytes are shown. Currents were normalized to the tail current after a prepulse of -140 mV and 1 mM [Cl−]o. Data were corrected for liquid junction potentials. C, dependence of deactivation kinetics on extracellular halides: superimposed deactivation currents at +40 mV after 9 s activation at -140 mV dependent on the substitution of extracellular Cl− with bromide. The extracellular solution contained 100 mM NaX− (X = Cl−+ Br−), 1 mM MgCl2, 1 mM CaCl2 and 5 mM Na-Hepes; pH 7.4. The total halide concentration is 104 mM. Deactivation accelerates with Cl− substituted with increasing concentrations of bromide. D, deactivation time constants at +40 mV in relation to extracellular Cl− substituted with bromide. Currents recorded from four oocytes as in panel C were fitted to the bi-exponential equation 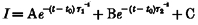 . Both time constants decrease with increasing concentrations of bromide.
. Both time constants decrease with increasing concentrations of bromide.
Replacing [Cl−]o partially or fully by bromide led to a drastic acceleration of channel deactivation at +40 mV after an activating pulse to -120 mV (Fig. 6C). The deactivation time course could be fitted by a double exponential function, indicating that the gating of ClC-2 involved more than two states. Both time constants decreased in higher [Br−]o (Fig. 6D). This suggests that extracellular bromide stabilizes the above-mentioned closed state even more strongly than Cl−, probably because bromide binds to the channel with a higher affinity.
Similar studies were performed with the K566Q mutant of ClC-2. The highly conserved lysine at the intracellular end of the D12 transmembrane domain is an important determinant of pore properties (rectification) and voltage-dependent gating in ClC-0, -1 and -2 (Pusch et al. 1995; Middleton et al. 1996; Rychkov et al. 1996; Jordt & Jentsch, 1997). Similar to the equivalent mutations in ClC-0 and -1, neutralization of K566 (K566Q) in ClC-2 shifts the voltage dependence of gating towards positive voltages (+80 mV), leaving a large fraction of channels opened at the resting membrane potential. This phenotype thus resembles WT ClC-2 in low [Cl−]o (Fig. 7A; Jordt & Jentsch, 1997). Reducing [Cl−]o to 1 mM opened the mutant even more strongly (Fig. 7A and B), even though the effect was less pronounced than for WT ClC-2. Again similar to WT, replacing extracellular Cl− by bromide led to a closure (Fig. 7C) and faster deactivation at +40 mV (Fig. 7E and F) of K566Q. Iodide, which blocks outward currents (Thiemann et al. 1992), also promoted channel closure at the resting potential (Fig. 7C). In high extracellular bromide, the currents carried by the mutant were just like WT currents in high extracellular Cl−.
Figure 7. Modulation of ClC-2 mutant K566Q by extracellular halides.
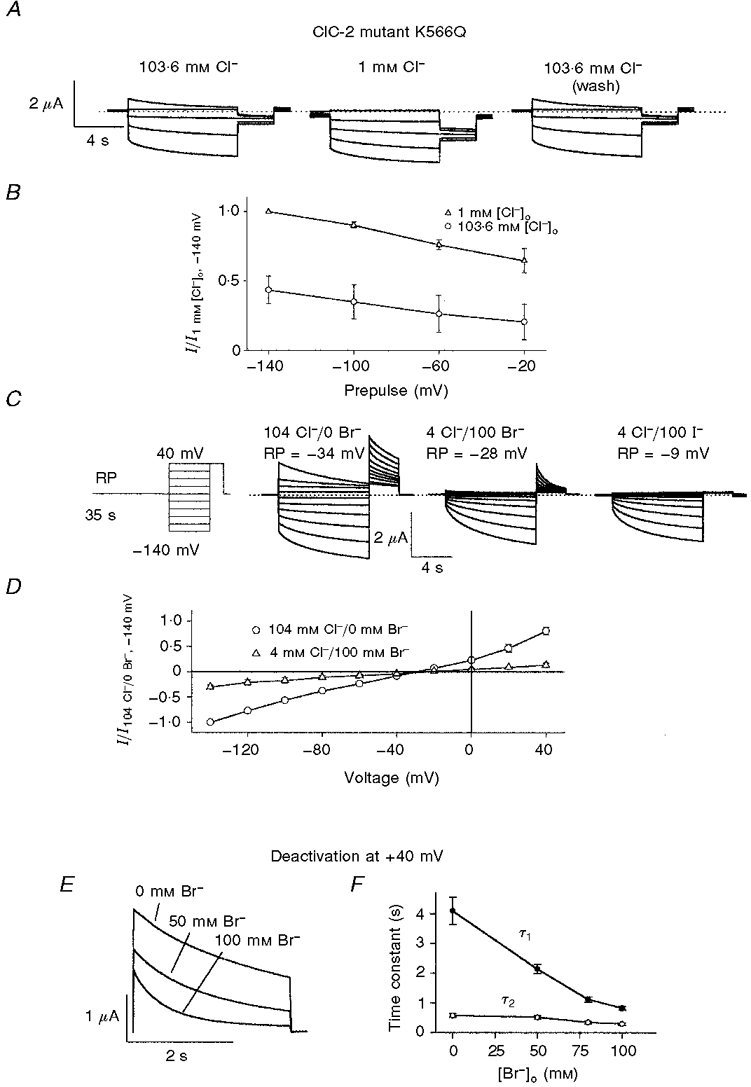
A, dependence of K566Q on extracellular Cl−: voltage-clamp traces from a typical oocyte expressing mutant K566Q measured as in Fig. 6A. B, averaged tail currents of K566Q mutant at different values of [Cl−]o. Experiments were done as in Fig. 6A and averaged results from four oocytes are shown. Data have been corrected for liquid junction potentials. Activation by hyperpolarization appears to be shifted to more positive potentials at low Cl− concentrations. C, shift in voltage dependence of the K566Q mutant by extracellular halides. Voltage protocol (left): each test pulse (from +40 to -140 mV in steps of -20 mV for 9 s) was preceded by a long 35 s period at the resting membrane potential and concluded by a +40 mV pulse for 3 s. Voltage-clamp traces from a typical oocyte after 1 min perfusion with extracellular solution containing Cl− (left), Cl− substituted by bromide (middle) and Cl− substituted by iodide (right) are shown. Solutions are as in Fig. 6C. Whereas in Cl− a large fraction of channels are already open at the resting membrane potential, substitution by bromide and iodide shifts the voltage dependence of activation to ClC-2 wild-type levels. D, comparison of instantaneous currents after the prepulse at the resting membrane potential in 104 mM Cl− and 4 mM Cl−/100 mM bromide-containing solution measured as in C. Currents are normalized to values at -140 mV in 104 mM Cl− (n = 3). E and F, dependence of deactivation kinetics of the K566Q mutant on extracellular halides. Currents and time constants are determined as in Fig. 6C and D.
DISCUSSION
Although they belong to the same gene family and are about 50% identical at the amino acid level, the voltage dependencies of ClC-0, ClC-1 and ClC-2 Cl− channels differ significantly. Thus the Torpedo channel ClC-0 has two readily distinguishable gates: a slow gate activates the channel upon hyperpolarization, and a fast gate opens the channel with depolarization. ClC-1, the major skeletal muscle Cl− channel, is normally activated by depolarization, and the nearly ubiquitously expressed ClC-2 Cl− channel is activated by hyperpolarization.
However, these different gating behaviours may obscure underlying common mechanisms. In contrast to ClC-0, activation of ClC-1 by depolarization cannot be described by a single rate constant, suggesting the presence of several different gating mechanisms as well (Fahlke et al. 1995; Rychkov et al. 1996). Interestingly, ClC-1 can show an activation by hyperpolarization at certain pH conditions (Rychkov et al. 1996). In addition, a ClC-1 point mutation (D136G) imparts an activation by hyperpolarization. This suggests that ClC-1 and ClC-0 share an intrinsic mechanism that is able to open the channel upon hyperpolarization.
In the present work, we have shown that the activation by hyperpolarization depends on the intracellular Cl− concentration in all three channels. Even though in some channels this becomes apparent only after mutagenesis, it suggests a common gating mechanism.
Unfortunately, the low single-channel conductance of ClC-1 and ClC-2 has precluded an analysis of gating at the single-channel level. This has only been possible for the Torpedo channel ClC-0, whose conductance is about 10 pS. This revealed that the channel has two pores (it is a ‘double-barrelled’ channel). The fast gate operates on the single ‘proto’ pores, while the slow hyperpolarization-activated gate closes both pores simultaneously. By analogy, other ClC channels like ClC-1, which was suggested to be a dimer as well (Fahlke et al. 1997a), are probably also ‘double-barrelled’ channels. The [Cl−]i dependence of the various hyperpolarization-activated gates suggests that they function similarly to the slow gate of ClC-0 and act simultaneously on the two pores of double-barrelled channels, as has also been suggested by Foskett (1998). However, this remains to be shown directly. In contrast to this hypothesis, a one-pore structure of ClC-1 has been suggested recently by Fahlke et al. (1998). These authors used various channel modifying reagents in homo- and heteromeric ClC-1 mutant constructs and concluded that their results were incompatible with a channel structure with two physically distinct conduction pathways. We think, however, that the results of Fahlke et al. (1998) could be equally well explained by an effect of the reagents on channel gating or by the modification of a (hypothetical) entrance vestibule that is common to both pores, interpretations that are fully compatible with a double-barrelled structure. Single-channel measurements are probably needed to further clarify the putative double-barrelled structure of ClC-1.
The fast depolarization-activated gate that acts on single ClC-0 protopores strongly depends on [Cl−]o. The permeating anion may act as the gating charge responsible for the voltage dependence of this gate (Pusch et al. 1995; Chen & Miller, 1996). In this model, binding of extracellular Cl− to the channel and its subsequent inward movement is coupled to the opening of the pore. Since the anion travels along the electric field, gating becomes voltage and concentration dependent. One could speculate that a similar mechanism accounts for the voltage dependence of the hyperpolarization-activated gates, only that Cl− is now moving from the intracellular side into the pore. The larger voltage dependence could be related to a movement of two Cl− ions, one for each pore, consistent with the fact that the slow gate (of ClC-0) acts on both protopores simultaneously. Such a mechanism could offer explanations for several phenomena, e.g. the irreversibility of ClC-0 gating that has been detected by Richard & Miller (1990). The complex behaviour of ClC-1 in anion substitution experiments (Rychkov et al. 1998) has to be interpreted eventually in the framework of a double-barrelled model (see also Foskett (1998) for a discussion of this problem).
It is surprising that the ClC-2 mutant ΔNClC-2 gates differently when measured with two-electrode voltage-clamp (Gründer et al. 1992; Jordt & Jentsch, 1997) or patch-clamp techniques (Fig. 5). If Cl− accumulation/depletion effects play a role, the patch-clamp method is probably more suitable as these effects will be minimized. Alternatively, disruption of the cytoskeleton with patch excision or the loss of cytosolic factors must be taken into account. Although ΔNClC-2 is still activated by hyperpolarization of patches, its gating is much faster than that of WT ClC-2 measured in Xenopus oocytes (Thiemann et al. 1992). It could be that ClC-2 possesses two different hyperpolarization-activated gates, a very slow one that is also activated by hypotonicity and which is destroyed by the N-terminal deletions and mutations in the D7/D8 linker (Jordt & Jentsch, 1997), and another fast one that becomes visible only when the slow gate is abolished. Alternatively, N-terminal deletions and mutations in D7/D8 could drastically accelerate the hyperpolarization-activated gate.
Another novel finding is that ClC-2 gating also depends on extracellular halides. This is superficially similar to the external anion dependence of ClC-0 (Pusch et al. 1995; Chen & Miller, 1996) and ClC-1 (Rychkov et al. 1996, 1998), but - in contrast to those channels - extracellular Cl− closes ClC-2. An interpretation, however, is complicated, because the relationship of this gating process with either the fast gate or slow gate of ClC-0 is unclear. The direction of the voltage dependence and the slow kinetics would favour the identification of the very slow activation of WT ClC-2 with the slow gate of ClC-0. Several arguments, however, speak against this. First, the slow gate of ClC-0 does not depend on osmolarity. Second, the relatively fast hyperpolarization-activated gate of the deletion mutant ΔNClC-2 is more reminiscent of the slow gate of ClC-0, but is clearly different from WT ClC-2 gating. Third, the slow gate of ClC-0 is inhibited by low [Cl−]o. The very slow hyperpolarization-activated gate of ClC-2 may represent a distinct gating mechanism that is absent from ClC-0 and ClC-1.
Quite surprisingly, it appears that the open probability of WT ClC-2 is reduced if Cl− is bound to the channel and that the relevant binding site is accessible from the extracellular solution. Replacing extracellular Cl− by bromide promotes channel closure. Bromide may bind with higher affinity to the channel than Cl−, such that a replacement of Cl− by bromide is equivalent to an increase in the Cl− concentration. Also, ClC-1 has a relatively high affinity for bromide, as is evident from a strong block by external bromide (Steinmeyer et al. 1994; Rychkov et al. 1998). We have not analysed quantitatively the voltage dependence of channel opening. Qualitatively, it seems that higher anion concentrations are associated with a ‘shift’ of the open probability-voltage curve to more negative voltages. The effects of the mutant K566Q on channel activation and halide dependence can be interpreted by a reduction of the Cl−/bromide affinity of the binding site that is involved in the halide sensitivity. Interestingly, homologous mutations in ClC-0 (K519E) and ClC-1 (K585E) apparently also affect an anion binding site that is, however, related to the anion dependence of open pore properties and the fast single-protopore gate of these channels (Pusch et al. 1995; Middleton et al. 1996; Rychkov et al. 1996; M. Pusch & T. J. Jentsch, unpublished results). These results suggest a certain interaction of the fast single-protopore gate and the slow hyperpolarization activation and osmosensitivity of ClC-2. More detailed studies of ClC-2, including measurements of ClC-2 expressed in smaller cells, are definitely necessary for a more profound understanding of its complex gating mechanisms.
In several cell types, native hyperpolarization-activated Cl− currents that resemble ClC-2 strongly depend on [Cl−]i (Chesnoy-Marchais, 1983; Dinudom et al. 1993; Staley, 1994; Fritsch & Edelman, 1996; Ferroni et al. 1997). Our findings support the hypothesis that these currents are indeed mediated by ClC-2.
What could be the functional role of a stimulation by intracellular Cl−? In some epithelia, cotransporters at the apical membrane use the sodium gradient to raise intracellular Cl− above the equilibrium potential. Cl− then leaves the cell via basolateral Cl− channels, resulting in a net transepithelial transport. If basolateral Cl− channels are activated by high intracellular Cl−, this would turn on the conductance when it is needed. Indeed, the basolateral Cl− channel in the thick ascending loop of Henle, a nephron segment with a high rate of NaCl reabsorption, was shown to be activated by intracellular Cl− (Reeves et al. 1995). In lung epithelium and pancreatic acinar cells, ClC-2 channels are distributed along apical membranes and could provide a Cl− efflux pathway stimulated by high intracellular Cl− concentration (Murray et al. 1995; Carew & Thorn, 1996). Furthermore, an activation of Cl− channels by intracellular Cl− may play an important role in neuronal excitability. It was suggested that ClC-2 may serve as a regulator of intracellular Cl− concentration, allowing a passive efflux of Cl− when it exceeds its electrochemical equilibrium. This would prevent GABAergic synapses from being excitatory (Staley et al. 1996; Clark et al. 1998; Foskett, 1998). Thus an activation of Cl− channels by intracellular Cl− may be of considerable physiological importance.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie (to T. J. J.) and by Telethon Italy (to M. P.).
References
- Carew MA, Thorn P. Identification of ClC-2-like chloride currents in pig pancreatic acinar cells. Pflügers Archiv. 1996;433:84–90. doi: 10.1007/s004240050252. [DOI] [PubMed] [Google Scholar]
- Chen T-Y, Miller C. Non-equilibrium gating and voltage dependence of ClC-0 Cl− channel. Journal of General Physiology. 1996;108:237–250. doi: 10.1085/jgp.108.4.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnoy-Marchais D. Characterization of a chloride conductance activated by hyperpolarization in Aplysia neurones. The Journal of Physiology. 1983;342:277–308. doi: 10.1113/jphysiol.1983.sp014851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S, Jordt SE, Jentsch TJ, Mathie A. Characterization of the hyperpolarization-activated chloride current in dissociated rat sympathetic neurons. The Journal of Physiology. 1998;506:665–678. doi: 10.1111/j.1469-7793.1998.665bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinudom A, Young JA, Cook DI. Na+ and Cl− conductances are controlled by cytosolic Cl− concentration in the intralobular duct cells of mouse mandibular glands. Journal of Membrane Biology. 1993;135:289–295. doi: 10.1007/BF00211100. [DOI] [PubMed] [Google Scholar]
- Fahlke C, Knittle T, Gurnett CA, Campbell KP, George AL., Jr Subunit stoichiometry of human muscle chloride channels. Journal of General Physiology. 1997a;109:93–104. doi: 10.1085/jgp.109.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke C, Rhodes TH, Desai RR, George AL., Jr Pore stoichiometry of a voltage-gated chloride channel. Nature. 1998;394:687–690. doi: 10.1038/29319. [DOI] [PubMed] [Google Scholar]
- Fahlke C, Rüdel R, Mitrovic N, Zhou M, George AL. An aspartic residue important for voltage-dependent gating of human muscle chloride channels. Neuron. 1995;15:463–472. doi: 10.1016/0896-6273(95)90050-0. [DOI] [PubMed] [Google Scholar]
- Fahlke C, Yu HT, Beck CL, Rhodes TH, George AL., Jr Pore-forming segments in voltage-gated chloride channels. Nature. 1997b;390:529–532. doi: 10.1038/37391. [DOI] [PubMed] [Google Scholar]
- Ferroni S, Marchini C, Nobile M, Rapisarda C. Characterization of an inwardly rectifying chloride conductance expressed by cultured rat cortical astrocytes. Glia. 1997;21:217–227. doi: 10.1002/(sici)1098-1136(199710)21:2<217::aid-glia5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Fong PY, Rehfeldt A, Jentsch TJ. Determinants of slow gating in ClC-0, the voltage-gated chloride channel of Torpedo marmorata. American Journal of Physiology. 1998;43:C966–973. doi: 10.1152/ajpcell.1998.274.4.C966. [DOI] [PubMed] [Google Scholar]
- Foskett JK. ClC and CFTR chloride channel gating. Annual Review of Physiology. 1998;60:689–717. doi: 10.1146/annurev.physiol.60.1.689. [DOI] [PubMed] [Google Scholar]
- Fritsch J, Edelman A. Modulation of the hyperpolarization-activated Cl− current in human intestinal T-84 epithelial cells by phosphorylation. The Journal of Physiology. 1996;490:115–128. doi: 10.1113/jphysiol.1996.sp021130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründer S, Thiemann A, Pusch M, Jentsch TJ. Regions involved in the opening of ClC-2 chloride channel by voltage and cell volume. Nature. 1992;360:759–763. doi: 10.1038/360759a0. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Chloride channels: a molecular perspective. Current Opinion in Neurobiology. 1996;6:303–310. doi: 10.1016/s0959-4388(96)80112-7. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Steinmeyer K, Schwarz G. Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus oocytes. Nature. 1990;348:510–514. doi: 10.1038/348510a0. [DOI] [PubMed] [Google Scholar]
- Jordt S-E, Jentsch TJ. Molecular dissection of gating in the ClC-2 chloride channel. EMBO Journal. 1997;16:1582–1592. doi: 10.1093/emboj/16.7.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MC, Steinmeyer K, Lorenz C, Ricker K, Wolf F, Otto M, Zoll B, Lehmann-Horn F, Grzeschik K-H, Jentsch TJ. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257:797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- Lloyd SE, Pearce SHS, Fisher SE, Steinmeyer K, Schwappach B, Scheinmann SJ, Harding B, Bolino A, Devoto M, Goodyer P, Rigden SPA, Wrong O, Jentsch TJ, Craig IW, Thakker RV. Mutations in the chloride channel ClC-5 are associated with X-linked hypercalciuric nephrolithiasis. Nature. 1996;379:445–449. doi: 10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- Lorenz C, Pusch M, Jentsch TJ. Heteromeric ClC channels with novel properties. Proceedings of the National Academy of Sciences of the USA. 1996;93:13362–13366. doi: 10.1073/pnas.93.23.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig U, Jentsch TJ, Pusch M. Analysis of a protein region involved in permeation and gating of the voltage-gated chloride channel ClC-0. The Journal of Physiology. 1997a;498:691–702. doi: 10.1113/jphysiol.1997.sp021893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig U, Jentsch TJ, Pusch M. Inward rectification in ClC-0 chloride channels caused by mutations in several protein regions. Journal of General Physiology. 1997b;110:165–171. doi: 10.1085/jgp.110.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig U, Pusch M, Jentsch TJ. Two physically distinct pores in the dimeric ClC-0 channel. Nature. 1996;383:340–343. doi: 10.1038/383340a0. [DOI] [PubMed] [Google Scholar]
- Ludewig U, Pusch M, Jentsch TJ. Independent gating of single pores in ClC-0 chloride channels. Biophysical Journal. 1997c;73:789–797. doi: 10.1016/S0006-3495(97)78111-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methfessel C, Witzemann V, Takahashi T, Mishina M, Numa S, Sakmann B. Patch clamp measurements on Xenopus laevis oocytes: currents through endogenous channels and implanted acetylcholine receptor and sodium channels. Pflügers Archiv. 1986;407:577–588. doi: 10.1007/BF00582635. [DOI] [PubMed] [Google Scholar]
- Middleton RE, Pheasant DJ, Miller C. Homodimeric structure of a ClC-type channel. Nature. 1996;383:337–340. doi: 10.1038/383337a0. [DOI] [PubMed] [Google Scholar]
- Miller C. Open-state substructure of single chloride channels from Torpedo electroplax. Philosophical Transactions of the Royal Society B. 1982;299:401–411. doi: 10.1098/rstb.1982.0140. [DOI] [PubMed] [Google Scholar]
- Murray CB, Morales MM, Flotte TR, McGrath-Morrow SA, Guggino WB, Zeitlin PL. ClC-2: A developmentally dependent chloride channel expressed in the fetal lung and downregulated after birth. American Journal of Respiratory Cellular and Molecular Biology. 1995;12:597–604. doi: 10.1165/ajrcmb.12.6.7766424. [DOI] [PubMed] [Google Scholar]
- Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Miniamino N, Kangawa K, Matsuo H, Raftery MA, Hirose T, Inayama S, Hayashida H, Miyata T, Numa S. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- Pusch M. Knocking on channel's door: The permeating anion acts as the gating charge in ClC-0. Journal of General Physiology. 1996;108:233–236. doi: 10.1085/jgp.108.4.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Ludewig U, Jentsch TJ. Temperature dependence of fast and slow gating relaxations of ClC-0 chloride channels. Journal of General Physiology. 1997;109:105–116. doi: 10.1085/jgp.109.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Ludewig U, Rehfeldt A, Jentsch TJ. Gating of the voltage-dependent chloride channel ClC-0 by the permeant anion. Nature. 1995;373:527–531. doi: 10.1038/373527a0. [DOI] [PubMed] [Google Scholar]
- Pusch M, Steinmeyer K, Jentsch TJ. Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophysical Journal. 1994;66:149–152. doi: 10.1016/S0006-3495(94)80753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves WB, Winters CJ, Filipov DM, Andreoli TE. Cl− channels in basolateral renal medullary vesicles IX. Channels from mouse MTAL cell patches and medullary vesicles. American Journal of Physiology. 1995;269:F621–627. doi: 10.1152/ajprenal.1995.269.5.F621. [DOI] [PubMed] [Google Scholar]
- Richard EA, Miller C. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 1990;247:1208–1210. doi: 10.1126/science.2156338. [DOI] [PubMed] [Google Scholar]
- Rychkov GY, Pusch M, Astill DJ, St, Roberts ML, Jentsch TJ, Bretag AH. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. The Journal of Physiology. 1996;497:423–435. doi: 10.1113/jphysiol.1996.sp021778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov GY, Pusch M, Roberts ML, Jentsch TJ, Bretag AH. Permeation and block of the skeletal muscle chloride channel, ClC-1, by foreign anions. Journal of General Physiology. 1998;111:653–665. doi: 10.1085/jgp.111.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP. Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nature Genetics. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- Staley K. The role of an inwardly rectifiying chloride conductance in postsynaptic inhibition. Journal of Neurophysiology. 1994;72:273–84. doi: 10.1152/jn.1994.72.1.273. [DOI] [PubMed] [Google Scholar]
- Staley K, Smith R, Schaack J, Wilcox C, Jentsch TJ. Alteration of GABA (a) receptor function following gene-transfer of the ClC-2 chloride channel. Neuron. 1996;17:543–551. doi: 10.1016/s0896-6273(00)80186-5. [DOI] [PubMed] [Google Scholar]
- Steinmeyer K, Ortland C, Jentsch TJ. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature. 1991;354:301–304. doi: 10.1038/354301a0. [DOI] [PubMed] [Google Scholar]
- Steinmeyer K, Pusch M, Koch MC, Jentsch TJ. Multimeric structure of ClC-1 chloride channel revealed by mutations in dominant myotonia congenita (Thomsen) EMBO Journal. 1994;13:737–743. doi: 10.1002/j.1460-2075.1994.tb06315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;239:598–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Thakker RV. Chloride channels cough up. Nature Genetics. 1997;17:125–127. doi: 10.1038/ng1097-125. [DOI] [PubMed] [Google Scholar]
- Thiemann A, Gründer S, Pusch M, Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356:57–60. doi: 10.1038/356057a0. [DOI] [PubMed] [Google Scholar]