

Abstract
Using an in vitro model of shear stress-induced cell injury we demonstrate that application of shear to differentiated human SH-SY5Y cells leads to cell death characterized by DNA fragmentation. Controlled shear stress was applied to cells via a modified cone and plate viscometer.
We show that pulsatile shear stress leads to DNA fragmentation, as determined via flow cytometry of fluorescein-12-dUTP nick-end labelled cells, in 45 ± 4% of cells. No lactate dehydrogenase (LDH) release was observed immediately after injury; however, 24 h after injury significant LDH release was observed.
Nitric oxide production by cells subjected to pulsatile shear increased two- to threefold over that in unsheared control cells.
Inhibition of protein synthesis, nitric oxide production, Ca2+ entry into cells, and pertussis toxin-sensitive G protein activation attenuated the shear stress-induced cell injury.
Our results show for the first time that application of pulsatile shear stress to a neuron-like cell in vitro leads to nitric oxide-dependent cell death.
A number of tissues are normally under some mechanical load such as shear stress, strain, tension or compression. Lung, bone and the cardiovasculature are examples of tissues which experience mechanical loads under physiological conditions. The central nervous system, however, experiences elevated mechanical load only under pathological conditions such as traumatic head injury and glaucoma. The mechanism by which elevated mechanical load leads to neurodegeneration is poorly understood.
During head injury, high intracranial pressure associated with impact has been predicted to lead to the generation of regions of high shear stress and tissue strain which correspond to experimentally observed patterns of injury (Margulies et al. 1990; Ueno et al. 1995). Potassium efflux from cells, membrane depolarization, calcium entry into cells (Nilsson et al. 1993) and apoptosis (Garcia-Valenzuela et al. 1995) occur in high shear regions associated with experimental head injury.
Elevated intraocular pressure (IOP) typically accompanies glaucoma. Elevated IOP leads to regions of high shear stress and strain in the lamina cribrosa, the region of the eye where the optic nerve traverses the globe (Zeimer & Chen, 1987; Cahane & Bartov, 1992; Fechtner & Weinreb, 1994; Yan et al. 1994). Retinal ganglion cells, which make up the optic nerve, undergo apoptosis upon exposure to elevated IOP in rat (Brubaker, 1996) and rabbit (Quigley et al. 1995) models of glaucoma. We suppose that, as in head injury, elevated pressure leads to the generation of shear stress and strain within the retinal ganglion cell layer, and that the shear stress and strain lead to cell injury.
Shear stress induces a cascade of events in endothelial cells, vascular smooth muscle cells and osteoblasts which include frictional displacement of cell surface components, G protein activation, K+ efflux from cells, Ca2+ entry into cells, nitric oxide (NO) production, upregulated gene expression, and activation of various kinases (e.g. Berthiaume & Frangos, 1992; Alevriadou et al. 1993; Hsieh et al. 1993; Lan et al. 1994; Frangos et al. 1996; Papadaki & Eskin, 1997; Takahashi et al. 1997). Apoptosis in neurons or neuronal cell lines occurs via a similar mechanism involving membrane depolarization, opening of calcium ion channels, elevated intracellular Ca2+ levels, activation of kinases, generation of free radicals or oxidants, and G protein phosphorylation (possibly), as well as other biochemical events (Mattson et al. 1992; McConkey et al. 1996). We believe these similarities are significant and form the basis of our hypothesis of the mechanism of shear stress-induced cell injury.
We report here the development of an in vitro model of shear stress-induced cell injury. We demonstrate that pulsatile shear stress leads to DNA fragmentation in differentiated SH-SY5Y cells, that NO production increases as a result of shear application, and that inhibition of NO production, protein synthesis, pertussis toxin-sensitive G protein activation, and calcium entry into the cell decrease the fraction of cells susceptible to shear stress-induced injury.
METHODS
Materials
Eagle's Minimum Essential Media (MEM) with Earle's salts and non-essential amino acids, fetal bovine serum, penicillin, streptomycin, 2.5S Nerve Growth Factor (NGF), fungizone and trypsin-EDTA were obtained from GibcoBRL. Methanol-free formaldehyde was obtained from Poly Sciences Inc. (Warrington, PA, USA). Ethanol was purchased from Fisher Scientific (Houston, TX, USA). L-Glutamine, propidium iodide, DNase-free RNase, EDTA, EGTA, bovine serum albumin (BSA), cycloheximide, pertussis toxin, L-NAME, Triton X-100, human recombinant nerve growth factor-β (β-NGF), potassium iodide and the lactate dehydrogenase (LDH) diagnostic kit were purchased from Sigma. Dulbecco phosphate-buffered saline (PBS) was obtained from Pierce Chemical Co. (Rockford, IL, USA). The apoptosis detection kit was obtained from Promega (Apoptosis Detection System, Fluorescein; Madison, WI, USA).
Cell culture
The human neuroblastoma cell line SH-SY5Y, a kind gift from Dr Evelyn Tiffany-Castiglioni (College of Veterinary Medicine, Texas A&M University), was used for all experiments. The SH-SY5Y cell line is the neuronal subclone derived from the SK-N-SH cell line.
The cells were cultured in a humidified 5% (v/v) CO2-air environment at 37°C and grown in Eagle's MEM with 10% fetal bovine serum, 100 u ml−1 penicillin, 100 μg ml−1 streptomycin and 3 mM L-glutamine using standard techniques. Prior to shear experiments, cells were plated at a density of 400 000 cells per 60 mm tissue culture dish for all experiments. Cells were grown on tissue culture plastic without treatment with extracellular matrix proteins. Cells were differentiated in the dish for no longer than 8 days by adding 10 ng ml−1 of human β-NGF directly to the culture dish. By day 6 or 7, cells were confluent.
Shear experiment
The shear stress apparatus consisted of a GlasCol stirrer (Terre Haute, IN, USA), a controller and a cone-and-plate viscometer with a 5 deg angle machined locally (Fig. 1). The relationship between the cone angular velocity, Ω, and shear stress, τ, was determined from eqn (1) where θ1 and θ are the angle of the cone and the cell surface measured relative to the axis of rotation of the cone, respectively, μ is the viscosity of the culture medium, and φ is the angular direction in the plane of the culture dish (Bird et al. 1960):
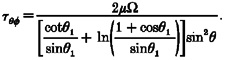 |
(1) |
Figure 1. Schematic diagram of modified cone and plate viscometer.
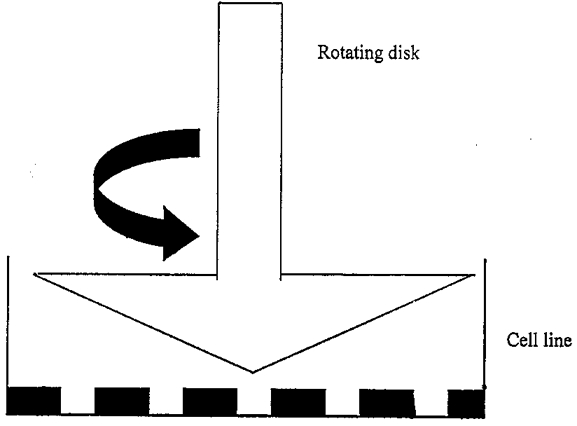
Cells are grown in the tissue culture dish, which is placed beneath the cone. The cone forms a 5 deg angle with the culture dish. Tissue culture medium fills the space between the cone and the culture dish. The angular velocity of the cone is controlled to generate a uniform shear field.
A monolayer of human neuroblastoma cells cultured in a 60 mm polystyrene culture dish was placed under the rotating cone with the tip of the cone in contact with the centre of the dish. The rotating cone creates a shear field, and the force experienced by the cells is inversely proportional to the distance from the centre of the cone and proportional to the distance from the bottom of the dish to the bottom of the cone. To create a uniform shear stress field across the bottom of the culture dish, the cone was made with an angle so that for cells in the centre of the dish there was a smaller distance between the bottom of the cone and the dish, while for cells far from the centre of the dish there was a larger distance between the bottom of the cone and the dish. There was some disturbance to the cells located at the centre of the dish where the cone was in contact with the dish. However, the surface area affected was no more than 2% of the total area. No significant cell detachment was observed otherwise.
The medium in each plate was changed prior to shearing. The control plate was treated exactly the same as the sheared plate except for the application of shear. Except when otherwise noted, all cells were subjected to pulsatile shear for 90 min. To generate pulsatile shear, the rotating cone was started and stopped automatically such that the cone was rotating for 13 min then stopped for 2 min repeatedly for a period of 90 min. During the 13 min ‘on’ period, the cone was rotated at a constant angular velocity. For a shear stress of 10 dyn cm−2, the angular velocity of the cone was controlled at 90 revolutions min−1. Experiments were carried out in a 37°C incubator. During the interval between shear stress and assays, cells were placed in the humidified CO2 incubator.
Viability
The LDH released from cells was measured using a prepackaged assay (Sigma) according to the manufacturer's directions. Except for the control cells, all cells were exposed to pulsatile shear for 90 min at 5, 10 or 15 dyn cm−2. The LDH release measurement was taken at 0, 5 and 24 h after shear application.
To determine the maximum possible LDH release, culture medium was removed from cells and either replaced with water to lyse cells via osmotic swelling, or with PBS containing 1% Triton X-100. LDH release was then measured as directed by the manufacturer. There was no difference in the maximum amount of LDH release determined using water or Triton X-100 to lyse cells.
DNA fragmentation
DNA fragmentation was measured using a TdT-mediated dUTP nick-end labelling (TUNEL) assay kit from Promega (Fitchburg, WI, USA). The nicked 3′ OH termini were labelled with fluorescein-12-dUTP and whole DNA 3′ ends were labelled with propidium iodide (PI) according to the procedure outlined for cell suspension with flow cytometry given by the manufacturer. For each measurement, 2-3 million cells were used. To prepare cells for the TUNEL assay, the culture medium was removed from the culture dish and trypsin-EDTA was added to detach the cell monolayer gently from the dish. The cell suspension was then centrifuged and washed with PBS. Afterwards, cells were fixed with methanol-free formaldehyde for 20 min. Another wash with PBS was performed after fixing the cells. Finally, the cells were permeabilized by adding ethanol. The cells were then stored in a -20°C freezer for at least 4 h before carrying out the rest of the procedure outlined by the manufacturer.
Results were analysed on a FACS Caliber flow cytometer (Becton Dickinson) using a 15 mW argon laser at a 488 nm excitation wavelength. Emission filters of 530 nm bandpass for green fluorescence and 585 nm for red fluorescence were used. Electronic compensation was used to prevent bleed-through fluorescence. Forward and side light scatter, green (fluorescein-12-dUTP) fluorescence (FL1) and red (PI) fluorescence (FL2) were collected and analysed on 15 000 cells sample−1 with Cellquest software (Becton Dickinson). The fraction of cells with fragmented DNA was determined by counting cells with green (fluorescein) fluorescence whose intensity was above a threshold determined from control cells. The two parameter histogram of FL2 area versus FL2 width was used to gate ‘on’ single cells.
Nitric oxide
NO formed by cells is rapidly converted to nitrite in cell culture medium. Nitrite was measured chemiluminescently by injection of 100 μl of sample into a Sievers Nitric Oxide Analyser (NOA) 270B (Boulder, CO, USA). In an acidic environment, nitrite was reduced by potassium iodide to NO and then transferred to a reaction cell to react with ozone. The light produced by this reaction was detected via a photomultiplier tube. The signal from the photomultiplier tube was calibrated against nitrite standards.
Except for the control cells, all cells were exposed to pulsatile shear for 90 min at 5, 10 or 15 dyn cm−2. NO measurements were taken at 0, 5 and 24 h after shear application.
Inhibition of shear-induced cell injury
Cycloheximide (10 μg ml−1) and pertussis toxin (10 ng ml−1) were incubated with differentiated cells for 4 h prior to application of shear. L-NAME (1 mM) and EGTA in Ca2+-free medium (1 mM) were incubated with differentiated cells for 90 min prior to application of shear. The culture medium was changed prior to incubation with the inhibitors. Cells were then subjected to pulsatile shear at 10 dyn cm−2 for 90 min. Immediately after shear cells were assayed for DNA fragmentation. Control cells were treated identically except for the application of shear.
Data analysis
All data presented are expressed as the mean ± the standard error of the mean (s.e.m.) of n independent determinations. Every independent determination was a unique culture dish exposed to shear and/or pharmacological agent for the specified amount of time. The value of n for each data set is reported in the figure legends. To determine whether a given data set was significantly different from a control data set, Student's t test with unequal variance was used. Unless otherwise noted a criterion of P < 0.05 was employed to determine whether the two sets of data were different. To compare multiple data sets relative to each other, Duncan's test for multiple comparisons was performed using SAS software (Cary, NC, USA).
RESULTS
We examined the relationship between magnitude and duration of shear stress on cell injury. As shown in Fig. 2, a significant percentage of cells exposed to a shear stress of 10 dyn cm−2 for as short a period as 10 min had fragmented DNA (13 ± 0.2%) compared with unsheared controls (1.9 ± 0.4%). A short duration of shear at a high shear level resulted in greater cell injury than that induced by a longer duration of shear at a moderate shear level (P < 0.01). When shear was applied for 10 min at 100 dyn cm−2, 75 ± 6% of cells had fragmented DNA, while 45 ± 4% of cells sheared for 90 min at 10 dyn cm−2 had fragmented DNA. For all subsequent experiments we chose to use pulsatile shear at 5-15 dyn cm−2, 90 min duration. Experiments could be better controlled at the moderate shear levels while still producing significant cellular injury. Very high shear levels (100 dyn cm−2) were avoided as significant cell detachment occurred which would have confounded our results.
Figure 2. Effect of duration and intensity of shear on the percentage of cells with fragmented DNA as determined using the TUNEL assay.
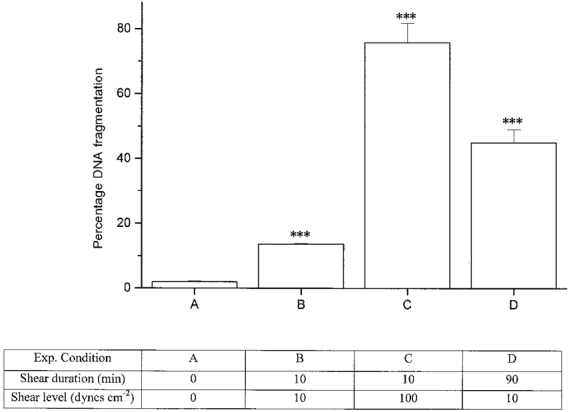
Column A represents unsheared control cells. Steady shear was applied for 10 min to B and C. Pulsatile shear was applied for 90 min to D. B and D were exposed to shear of 10 dyn cm−2 while C was exposed to shear of 100 dyn cm−2. DNA fragmentation was assayed immediately after shear application. n = 9 for A, 7 for B, and 3 for C and D. *** Significant result relative to unsheared control cells (A) (P < 0.005).
The percentage of TUNEL positive cells (with fragmented DNA) as a function of time after application of pulsatile shear stress is shown in Fig. 3. Cells subjected to pulsatile shear stress had a marked increase in the percentage of cells with fragmented DNA as compared with unsheared cells. Immediately after shear application, there was a significant difference in the number of TUNEL positive cells sheared at 5 dyn cm−2 and cells sheared at 10 or 15 dyn cm−2 as determined by Duncan's test for multiple comparisons (P < 0.01). Application of shear stress at 10 and 15 dyn cm−2 resulted in similar percentages of TUNEL positive cells. In all cases, the percentage of cells with fragmented DNA was greatest immediately after shear application or remained unchanged at the times measured.
Figure 3. Percentage of TUNEL positive cells (with fragmented DNA) as a function of time and shear level.
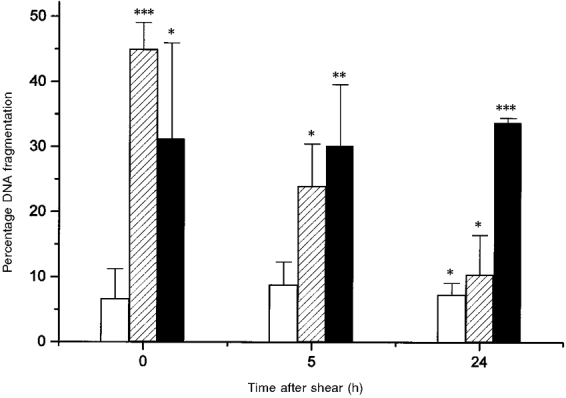
Time was measured starting immediately after shear application was terminated. Cells were exposed to pulsatile shear for 90 min at 5 (□), 10 ( ) and 15 dyn cm−2 (▪). For all data at 5 dyn cm−2, n = 3; for all others, n = 3-7. Asterisks indicate the significance of the result relative to unsheared control cells: * P < 0.05, ** P < 0.01 and *** P < 0.005.
) and 15 dyn cm−2 (▪). For all data at 5 dyn cm−2, n = 3; for all others, n = 3-7. Asterisks indicate the significance of the result relative to unsheared control cells: * P < 0.05, ** P < 0.01 and *** P < 0.005.
Representative histograms from flow cytometry of cells exposed to pulsatile or no shear and labelled using the TUNEL assay are shown in Fig. 4. The FL2 area was assumed to be proportional to the length of DNA in a cell. Counts in the region labelled R5 would indicate cells with the shortest DNA. Cells in region R5 also stained most intensely with fluorescein-12-dUTP and made up the TUNEL positive (cells with fragmented DNA) fraction of cells. Regions labelled R2, R3 and R4 correspond to cells in the resting (G1) phase, DNA synthesis (S) phase, and mitosis (G2/M) phase of the cell cycle, respectively, (Darzynkiewicz et al. 1992). Fifteen to forty per cent of sheared SH-SY5Y cells had fragmented DNA, compared with 2% of control cells at times measured from 0 to 24 h after shear application. As seen in Fig. 4, unsheared cells were in all phases of the cell cycle. Immediately after shear application, there was a decrease in the number of cells in the G1 phase, although cells were lost from all phases of the cell cycle. This qualitative trend was observed in all shear experiments.
Figure 4. Representative flow cytometry data.
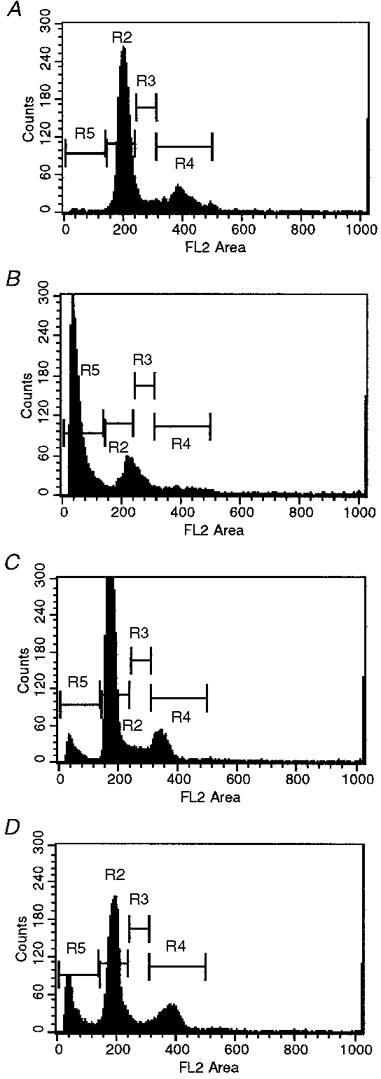
The cells were unsheared (A), or sheared at 10 dyn cm−2 for 90 min and stained using the TUNEL assay 0 (B), 5 (C) and 24 h (D) after shear application. The frequency of cells is plotted against the length of DNA (FL2 area) in a given cell; 15 000 cells were counted per time point. The regions labelled R2, R3 and R4 indicate cells in G1, S and G2/M phase, respectively. Region R5 is the population of fragmented DNA.
As shown in Fig. 5A, SH-SY5Y cells subjected to shear stress did not release a significant amount of LDH immediately after shear application relative to unsheared (and unlysed) control cells. This trend was observed for all magnitudes of shear measured. Only at 24 h after shear application was significant LDH release observed compared with unsheared controls (Fig. 5B). The maximum amount of LDH released when cells were lysed via osmotic swelling was 3.02 ± 0.11 times that released from unsheared (and unlysed) control cells. Assuming the LDH released via osmotic swelling represents the maximum possible LDH release, then application of shear led to cell lysis in 27 ± 10% of cells 24 h after shear application.
Figure 5. LDH release as a function of shear stress (A) and as a function of time (B).
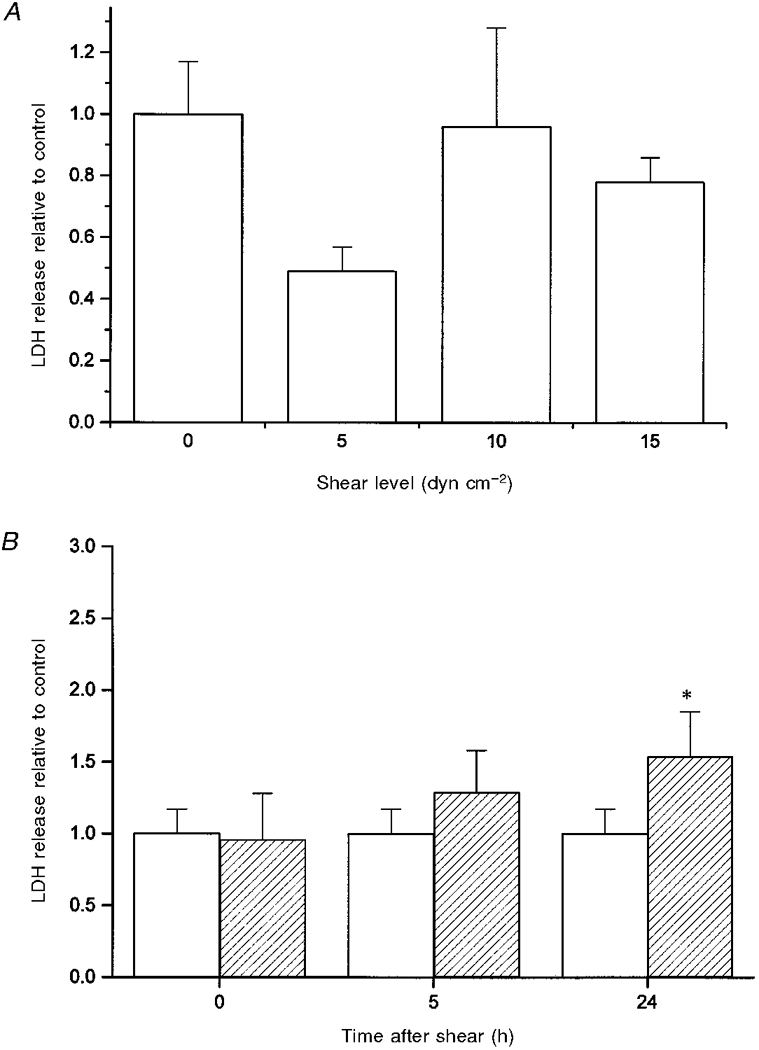
A, cells were unsheared (0 dyn cm−2, n = 10) or sheared at 37 °C for 90 min at 5 (n = 3), 10 (n = 7) or 15 dyn cm−2 (n = 4). LDH release was measured immediately after shear was terminated. For all data, the ratio of LDH release from the sheared cells relative to the control cells is given. B, cells were exposed to no shear (□) or shear for 90 min at 10 dyn cm−2 ( ). For unsheared controls, n = 10. For sheared samples at 0, 5 and 24 h after shear exposure, n = 7, 3 and 4, respectively. * Significant result relative to the unsheared control at 24 h (P < 0.05).
). For unsheared controls, n = 10. For sheared samples at 0, 5 and 24 h after shear exposure, n = 7, 3 and 4, respectively. * Significant result relative to the unsheared control at 24 h (P < 0.05).
As an indicator of the total amount of NO produced by cells, we measured the amount of nitrite in the culture medium of cells at 0, 5 and 24 h after shearing was terminated. As shown in Fig. 6A, immediately after shearing was terminated the amount of nitrite measured in the culture medium was elevated compared with that for unsheared cells although the increase was only significant when shear was applied at 10 and 15 dyn cm−2 (P < 0.03). The amount of nitrite in the culture medium at 10 dyn cm−2 was not different from that at 15 dyn cm−2 (P > 0.3) but was significantly different from that at 5 dyn cm−2 (P < 0.05). The total amount of nitrite measured in the culture medium of the unsheared cells did not significantly increase within 24 h. The amount of nitrite in the culture medium of sheared cells remained elevated for the next 24 h without further increase (P > 0.1) (Fig. 6B).
Figure 6. Effect of shear stress on NO level in culture medium as a function of shear level (A) and time (B).
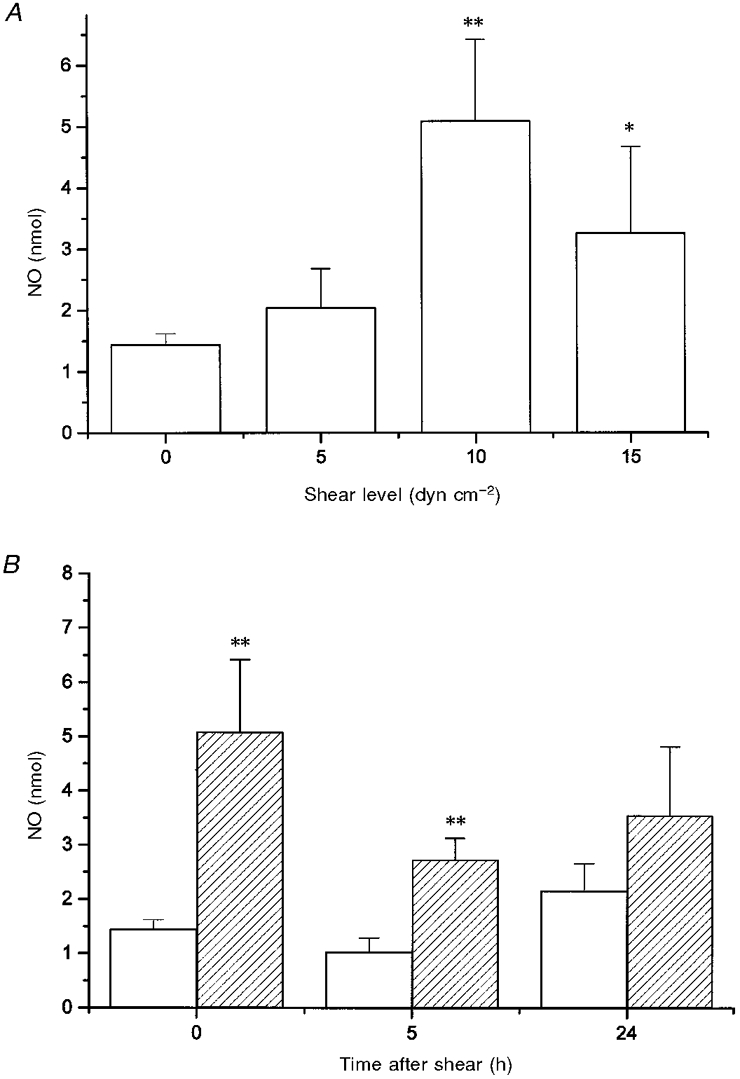
A, shear was applied at 37 °C for 90 min at 5, 10 and 15 dyn cm−2. The quantity of NO accumulated in the culture medium was determined from the amount of nitrite measured immediately after shear application was terminated. For all data, n = 7-12. Asterisks indicate the significance of the result compared with unsheared controls: * P < 0.05 and ** P < 0.01. B, representative plot of the effect of shear stress on NO level in the culture medium as a function of time after application of shear. Cells were exposed to no shear (□) or shear for 90 min at 10 dyn cm−2 ( ). At times 0, 5 and 24 h, n = 12, 3 and 4, respectively. ** Significant result relative to unsheared controls at the same time (P < 0.01).
). At times 0, 5 and 24 h, n = 12, 3 and 4, respectively. ** Significant result relative to unsheared controls at the same time (P < 0.01).
As shown in Fig. 7, addition of cycloheximide, pertussis toxin, L-NAME and EGTA in Ca2+-free medium, which are inhibitors of protein synthesis, Go/Gi protein activation, nitric oxide production and Ca2+ entry into the cell, respectively, attenuated shear-induced cell injury. The measured percentage DNA fragmentation relative to unsheared cells was significantly different from that measured for untreated sheared cells (P < 0.06 for EGTA and P < 0.005 for all others). In addition to protecting cells from shear-induced injury, pertussis toxin also reduced shear-induced nitrite accumulation in the culture medium from 5.1 ± 1.3 nmol for cells sheared in the absence of pertussis toxin to 0.33 ± 0.11 nmol for cells sheared in the presence of pertussis toxin.
Figure 7. Percentage of cells with DNA fragmentation as a function of inhibitors of cell injury.
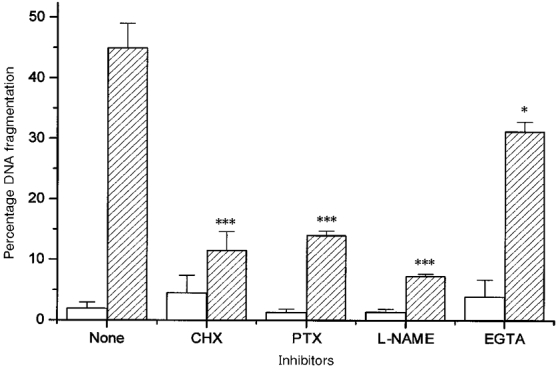
Cells were incubated with the inhibitors for 1.5-4 h and then sheared for 90 min at 10 dyn cm−2 and physiological temperature. Cells were assayed for DNA fragmentation immediately after shear application. Cells were unsheared (□) or sheared (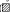 ) and exposed to no inhibitor (None), cycloheximide (CHX), pertussis toxin (PTX), L-NAME or EGTA in Ca2+-free medium (EGTA). The unsheared controls were not significantly different from each other (P > 0.3). For unsheared samples treated with no inhibitor, cycloheximide, pertussis toxin, L-NAME and EGTA, n = 9, 4, 3, 5 and 3, respectively. For sheared samples using the same inhibitors, n = 7, 8, 3, 4 and 3, respectively. Asterisks indicate the significance of the result relative to the unsheared controls treated with the same inhibitor: * P < 0.06 and *** P < 0.005.
) and exposed to no inhibitor (None), cycloheximide (CHX), pertussis toxin (PTX), L-NAME or EGTA in Ca2+-free medium (EGTA). The unsheared controls were not significantly different from each other (P > 0.3). For unsheared samples treated with no inhibitor, cycloheximide, pertussis toxin, L-NAME and EGTA, n = 9, 4, 3, 5 and 3, respectively. For sheared samples using the same inhibitors, n = 7, 8, 3, 4 and 3, respectively. Asterisks indicate the significance of the result relative to the unsheared controls treated with the same inhibitor: * P < 0.06 and *** P < 0.005.
DISCUSSION
Shear stress was chosen as the mechanical force to evaluate because of its known relevance in head injury (e.g. White & Krause, 1993; LaPlaca et al. 1997). A number of investigators have demonstrated, using a variety of modelling techniques, that high shear levels are generated during traumatic head injury (Margulies et al. 1990; Chu et al. 1994; Ueno et al. 1995). Levels of shear predicted during head injury are in the order of 4 × 105 dyn cm−2.
Shear stress is also a relevant mechanical force experienced during glaucoma. It has been shown that elevated pressure within the eye during glaucoma leads to elevated stress and strain in the sclera and lamina cribrosa (Cahane & Bartov, 1992). The levels of stress within the sclera during glaucoma are predicted to be at least an order of magnitude lower than those predicted during traumatic head injury.
In both traumatic head injury and glaucoma, the levels of stress predicted in the tissue are several orders of magnitude higher than the levels chosen for use in this study. We assumed that within a tissue the extracellular matrix carries the majority of the mechanical load while the cells themselves experience much lower stresses. Based on our experience, no isolated cells would survive a shear stress of 105 dyn cm−2 for even the briefest application; thus we chose a much lower level of stress in order to examine the mechanism of shear stress-induced injury.
In both head injury and glaucoma, regions of high shear stress correspond to regions of high strain. Strain is the dimensionless stretching of tissue expressed as the ratio of the deformed length to the undeformed length, and is related to the applied stress and the viscoelastic properties of the cell or tissue. Although we have not specifically measured strain in our in vitro model, based on work by others using a similar device, we would expect about a 20% cell strain in response to the shear stress we applied (LaPlaca et al. 1997). A 20% strain is similar to the degree of deformation of the optic nerve head upon application of elevated pressure (Yan et al. 1994). A 10% strain is predicted to be the critical strain above which injury occurs during diffuse axonal injury (Margulies & Thibault, 1992). We believe that both shear stress and strain may contribute to the cell injury observed in vivo during glaucoma and head injury.
Differentiated SH-SY5Y cells were chosen as a model cell because they express G proteins, have neuron-like ion channels such as L-type Ca2+ channels, and, when differentiated, have long processes similar to those observed in neurons (e.g. Hong, 1997; Prince & Oreland, 1997; Nikonorov et al. 1998). The length of the processes reaches a maximum after 7-8 days of differentiation (Hong, 1997). It has been noted that upon differentiation, SH-SY5Y cells continue to divide (Ridge et al. 1996). Despite this unneuron-like behaviour, SH-SY5Y cells have been used extensively to model neuronal function and differentiation, neuronal Ca2+ signalling, neurodegeneration and neurotoxicity (e.g. Miller, 1996; Prince & Oreland, 1997; Hong, 1997; Mackrill, 1997; Ehrich & Correll, 1998). In this study, initial experiments were performed on undifferentiated cells; however, no shear stress-induced loss of viability or increase in NO production was observed (data not shown). We conclude that some property or properties of differentiated SH-SY5Y cells is/are critical in the determination of cell sensitivity to shear stress-induced cell injury.
Pulsatile application of shear stress was used in all experiments. Based on preliminary results, pulsatile shear was at least twice as effective as steady shear at causing cell injury (data not shown). In studies reported by other investigators, pulsatile shear has been shown to have a more dramatic effect on cell physiology than steady shear. G protein activation is specifically triggered by changes in shear (Frangos et al. 1996). Oscillations in Ca2+ concentration within endothelial cells are more pronounced in pulsatile flow (Helmlinger et al. 1996). Nitric oxide production exhibits a biphasic response to shear, with the more dramatic increase in production associated with a change in shear (Kuchan et al. 1994; Frangos et al. 1996). We made no attempt to optimize the frequency of stress application, but simply demonstrated the usefulness of pulsatile shear application in producing cell injury. Pulsatility of mechanical load occurs in vivo at frequencies associated with the cardiac cycle and frequencies associated with diurnal variations in IOP and blood pressure.
As shown in Fig. 2, we demonstrated that application of moderate shear stress for 90 min as well as a high shear stress for 10 min resulted in a significant amount of DNA fragmentation. We believe the results from our experimental system could be extrapolated in either direction to provide insight into the mechanism of shear stress- or strain-induced injury in an acute disorder such as traumatic head injury and a chronic disorder such as glaucoma.
Figure 3 shows that DNA fragmentation was greatest immediately after shear application. As time progressed, the fraction of TUNEL positive cells decreased slightly, possibly because the cells that survive divide and multiply, thus increasing the total number of cells with time. A second possible explanation is that apoptotic cells decrease in size with time, thus increasing the likelihood that the cells would be lost during the centrifugation/wash steps of the TUNEL assay.
The flow cytometry data, depicted in Fig. 4, show that the unsheared cells were in all phases of the cell cycle. Immediately after shear application a reduction in the number of cells in all phases of the cell cycle was observed.
As shown in Fig. 5A and B, application of shear stress to SH-SY5Y cells did not lead to an immediate release of LDH, while a modest release was observed 24 h after shear stress application. Shear stress applied to the NT2-N neuronal cell line at high loading rates (exceeding 200 dyn cm−2 s−1) led to an immediate LDH release (LaPlaca et al. 1997). Sustained LDH release was observed only for high loading rates, while for generation of shear occurring over greater than 2 s, no sustained LDH release was observed (LaPlaca et al. 1997). In the experiments reported here, the loading rate was not controlled but was expected to be well below the level required to cause sustained LDH release.
The lack of extensive release of LDH at physiological temperature immediately after shear stress (Fig. 5A) suggests that necrosis had not occurred at that time since membrane integrity was maintained. At the same time, almost half of the cells had fragmented DNA. The DNA fragmentation observed may be indicative of apoptosis. The percentage of cells with DNA fragmentation observed immediately after injury was significantly greater than the percentage of cells we estimated may have lysed based on LDH data at 24 h after injury. Cell injury or death may occur via multiple mechanisms in our in vitro model. Cell death via apoptosis is relevant in neurodegeneration in vivo. During experimental glaucoma, it has been observed that retinal ganglion cells in the glaucomatous eye die via apoptosis (Garcia-Valenzuela et al. 1995). Moreover, autopsy of 37 cases of fatal head injury showed selective neuronal apoptosis in the hippocampal region of the brain (Kotapka et al. 1993).
An increase in nitrite in the cell culture medium was observed in response to shear in SH-SY5Y cells, as shown in Fig. 6A and B. The elevated nitrite levels in the medium are probably the consequence of increased NO production in response to shear. The same result has been seen in endothelial cells (Frangos et al. 1996). Nitrite levels in the culture medium continued to remain elevated without further increase for 24 h following application of shear indicating that NO production did not remain elevated after termination of shear stress application.
As shown in Fig. 7, cycloheximide, pertussis toxin, L-NAME and EGTA in Ca2+-free medium were all successful at preventing shear-induced cell injury. The results show that inhibition of protein synthesis, G protein activation, NO production and Ca2+ entry into the cells were able to lower the extent of shear-induced cell injury (P < 0.001 for all except EGTA (P < 0.06)). The effectiveness of EGTA at preventing shear-induced cell injury was significantly less than that of cycloheximide, pertussis toxin and L-NAME as determined by Duncan's test for multiple comparisons (P < 0.001).
There is growing evidence that exposure to oxidative stress and/or free radicals such as nitric oxide, hydrogen peroxide and peroxynitrite can induce apoptosis in neurons (e.g. Satoh et al. 1996; Desole et al. 1997; Kruman et al. 1997). It has been demonstrated that nanomolar concentrations of NO, the same concentrations produced by SH-SY5Y cells upon application of shear, induce apoptosis in neuronal cell lines (Desole et al. 1997). Incorporating a nitric oxide synthase inhibitor, L-NAME, resulted in a reduction in the amount of DNA fragmentation indicating that indeed NO production plays an important role in the mechanism of shear-induced apoptosis.
Pertussis toxin, a specific inhibitor of GTP hydrolysis and Go/Gi activation, was also protective against shear-induced cell injury. Shear induces G protein activation in endothelial cells (Berthiaume & Frangos, 1992), osteoblasts (Reich et al. 1997) and SH-SY5Y cells (M. E. Edwards, personal communication). G proteins are known to affect apoptosis bidirectionally in some neuronal cell populations (Yan et al. 1995). As presented here, in SH-SY5Y cells inhibition of G protein activation blocks shear stress-induced NO production. Pertussis toxin may attenuate shear stress-induced cell injury via its effect on NO production.
Inhibition of new protein synthesis with cycloheximide completely blocked shear-induced loss of viability. c-fos expression is known to be induced by shear in some cell types (Hsieh et al. 1993). Presumably, other as yet unidentified proteins are newly expressed in response to shear. Expression of c-fos and heat shock proteins or mRNA for heat shock proteins is associated with experimental head injury (Lowenstein et al. 1994; Mikawa et al. 1995; Linsberg et al. 1996). Within SH-SY5Y cells, new protein expression appears to be necessary for cell injury.
The presence of EGTA in the extracellular space prevents Ca2+ entry into the cell, which, based on our results, is sufficient to reduce shear-induced cell death. A variety of evidence suggests that Ca2+ entry into cells is an early step in the cellular response to shear (Papadaki & Eskin, 1997). In addition, shear-induced changes in intracellular Ca2+ have been measured directly in NT2-N cells, where a greater than fivefold increase in intracellular Ca2+ concentration was observed for low shear loading rates during a 4 s interval (LaPlaca et al. 1997). Even greater increases in intracellular Ca2+ levels were observed for higher shear loading rates. Increased intracellular Ca2+ concentration was also observed upon application of strain, again at a high rate of deformation, in differentiated NG105-15 cells (Cargill & Thibault, 1996). Loss of Ca2+ homeostasis has been implicated in neuronal apoptosis (Mattson et al. 1992). Ca2+ entry into cells, possibly through an excitotoxic mechanism, has been implicated in cell death associated with head injury (Nilsson et al. 1996). Thus, our findings are consistent with the current understanding of the role of Ca2+ in shear and neuronal cell death.
We have demonstrated that differentiated SH-SY5Y cells, a neuron-like cell line, respond to shear stress via DNA fragmentation. Some of the key steps in the pathway of cell injury include NO production, G protein activation, new protein synthesis and entry of calcium into the cell. These results may have implications for the treatment of neurodegenerative disorders where elevated mechanical forces play a role.
Acknowledgments
Financial support was provided by Texas A&M University. Technical assistance from Michael Edwards and Jane Miller is gratefully acknowledged.
References
- Alevriadou BR, Eskin SG, McIntire LV, Schilling WP. Effect of shear stress on Rb2+ efflux from calf pulmonary artery endothelial cells. Annals of Biomedical Engineering. 1993;21:1–7. doi: 10.1007/BF02368159. [DOI] [PubMed] [Google Scholar]
- Berthiaume F, Frangos JA. Flow-induced prostacyclin production is mediated by a pertussis toxin-sensitive G protein. FEBS Letters. 1992;308:277–279. doi: 10.1016/0014-5793(92)81292-t. [DOI] [PubMed] [Google Scholar]
- Bird RB, Steward W, Lightfoot E. Transport Phenomena. New York: John Wiley & Sons; 1960. [Google Scholar]
- Brubaker RF. Delayed functional loss in glaucoma. LII Edward Jackson Memorial Lecture. American Journal of Ophthalmology. 1996;12:473–483. doi: 10.1016/s0002-9394(14)75421-2. [DOI] [PubMed] [Google Scholar]
- Cahane M, Bartov E. Axial length and scleral thickness effect on susceptibility to glaucomatous damage: a theoretical model implementing Laplace's Law. Ophthalmic Research. 1992;24:280–284. doi: 10.1159/000267179. [DOI] [PubMed] [Google Scholar]
- Cargill RS, II, Thibault L. Acute alterations in [Ca2+]i in NG108–15 cells subjected to high strain rate deformation and chemical hypoxia: an in vitro model for neural trauma. Journal of Neurotrauma. 1996;13:395–407. doi: 10.1089/neu.1996.13.395. [DOI] [PubMed] [Google Scholar]
- Chu C, Lin M, Huang H, Lee M. Finite element analysis of cerebral contusion. Journal of Biomechanics. 1994;27:187–194. doi: 10.1016/0021-9290(94)90208-9. [DOI] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- Desole MS, Sciola L, Delogu MR, Sircana S, Migheli R, Miele E. Role of oxidative stress in the manganese and 1-methyl-4-(2′-ethylphenyl)-1,2,3,6-tetrahydropyridine-induced apoptosis in PC12 cells. Neurochemistry International. 1997;31:169–176. doi: 10.1016/s0197-0186(96)00146-5. 10.1016/S0197-0186(96)00146-5. [DOI] [PubMed] [Google Scholar]
- Ehrich M, Correll L. Inhibition of carboxylesterases in SH-SY5Y human and NB41A3 mouse neuroblastoma cells by organophosphorus esters. Journal of Toxicology and Environmental Health. 1998;53:385–399. doi: 10.1080/009841098159240. [DOI] [PubMed] [Google Scholar]
- Fechtner R, Weinreb R. Mechanisms of optic nerve damage in primary open angle glaucoma. Surveys in Ophthalmology. 1994;39:23–42. doi: 10.1016/s0039-6257(05)80042-6. [DOI] [PubMed] [Google Scholar]
- Frangos JA, Huang TY, Clark CB. Steady shear and step changes in shear stimulate endothelium via independent mechanisms - superposition of transient and sustained nitric oxide production. Biochemical and Biophysical Research Communications. 1996;224:660–665. doi: 10.1006/bbrc.1996.1081. 10.1006/bbrc.1996.1081. [DOI] [PubMed] [Google Scholar]
- Garcia-Valenzuela E, Shareef S, Walsh J, Sharma SC. Programmed cell death of retinal ganglion cells during experimental glaucoma. Experimental Cell Research. 1995;61:33–44. doi: 10.1016/s0014-4835(95)80056-5. [DOI] [PubMed] [Google Scholar]
- Helmlinger G, Berk B, Nerem R. Pulsatile and steady flow induced calcium oscillations in single cultured endothelial cells. Journal of Vascular Research. 1996;33:360–369. doi: 10.1159/000159164. [DOI] [PubMed] [Google Scholar]
- Hong MS. Bioremediation and Neurotoxicological Characterization of Organophosphorus Compounds. Texas: A&M University; 1997. PhD dissertation. [Google Scholar]
- Hsieh HJ, Li NQ, Frangos JA. Pulsatile and steady flow induces c-fos expression in human endothelial cells. Journal of Cellular Physiology. 1993;154:143–151. doi: 10.1002/jcp.1041540118. [DOI] [PubMed] [Google Scholar]
- Kotapka MJ, Graham DI, Adams JH, Doyle D, Gennarelli TA. Hippocampal damage in fatal pediatric head injury. Neuropathology and Applied Neurobiology. 1993;19:128–133. doi: 10.1111/j.1365-2990.1993.tb00417.x. [DOI] [PubMed] [Google Scholar]
- Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. Journal of Neuroscience. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchan M, Jo H, Frangos J. Role of G proteins in shear stress mediated nitric oxide production by endothelial cells. American Journal of Physiology. 1994;267:C753–758. doi: 10.1152/ajpcell.1994.267.3.C753. [DOI] [PubMed] [Google Scholar]
- Lan Q, Mercurius KO, Davies PF. Stimulation of transcription factors NFκB and AP1 in endothelial cells subjected to shear stress. Biochemical and Biophysical Research Communications. 1994;201:950–956. doi: 10.1006/bbrc.1994.1794. 10.1006/bbrc.1994.1794. [DOI] [PubMed] [Google Scholar]
- LaPlaca M, Lee VY, Thibault LE. An in vitro model of traumatic neural injury: loading rate-dependent changes in acute cytosolic calcium and lactate dehydrogenase release. Journal of Neurotrauma. 1997;14:355–368. doi: 10.1089/neu.1997.14.355. [DOI] [PubMed] [Google Scholar]
- Linsberg P, Frerichs K, Siren A, Hallenbeck J, Nowak TS., Jr Heat shock protein and c-fos expression in focal microvascular brain damage. Journal of Cerebral Blood Flow and Metabolism. 1996;16:82–91. doi: 10.1097/00004647-199601000-00010. [DOI] [PubMed] [Google Scholar]
- Lowenstein D, Gwinn R, Seren M, Simon R, McIntosh T. Increased expression of mRNA encoding calbindin-D28K, the glucose regulated proteins, or the 72 kDa heat shock protein in three models of acute CNS injury. Brain Research. Molecular Brain Research. 1994;22:299–308. doi: 10.1016/0169-328x(94)90058-2. [DOI] [PubMed] [Google Scholar]
- McConkey DJ, Zhivotovsky B, Orrenius S. Apoptosis - molecular mechanisms and biomedical implications. Molecular Aspects of Medicine. 1996;17:1–110. doi: 10.1016/0098-2997(95)00006-2. 10.1016/0098-2997(95)00006-2. [DOI] [PubMed] [Google Scholar]
- Mackrill JJ, Challiss RAJ, O'Connel DA, Lai FA, Nahorski SR. Differential expression and regulation of ryanodine receptor and myo-inositol 1,4,5-triphosphate receptor Ca2+ release channels in mammalian tissues and cell lines. Biochemical Journal. 1997;327:251–258. doi: 10.1042/bj3270251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies S, Thibault L. A proposed tolerance criterion for diffuse axonal injury in man. Journal of Biomechanics. 1992;25:917–923. doi: 10.1016/0021-9290(92)90231-o. 10.1016/0021-9290(92)90231-O. [DOI] [PubMed] [Google Scholar]
- Margulies S, Thibault L, Gennarelli T. Physical model simulations of brain injury in the primate. Journal of Biomechanics. 1990;23:823–836. doi: 10.1016/0021-9290(90)90029-3. 10.1016/0021-9290(90)90029-3. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Rydel RE, Lieberburg I, Smith-Swintosky VL. Altered calcium signaling and neuronal injury: stroke and Alzheimer's disease as examples. Annals of the New York Academy of Sciences. 1992;679:1–21. doi: 10.1111/j.1749-6632.1993.tb18285.x. [DOI] [PubMed] [Google Scholar]
- Mikawa S, Sharp F, Kamii H, Kinouchi H, Epstein C, Chan PK. Expression of c-fos and hsp70 mRNA after traumatic brain injury in transgenic mice overexpressing CuZn superoxide dismutase. Brain Research. Molecular Brain Research. 1995;33:288–294. doi: 10.1016/0169-328x(95)00146-j. [DOI] [PubMed] [Google Scholar]
- Miller SW, Trimmer PA, Parker WD, Davis RE. Creation and characterization of mitochondrial DNA-depleted cell lines with ‘neuronal-like’ properties. Journal of Neurochemistry. 1996;67:1897–1907. doi: 10.1046/j.1471-4159.1996.67051897.x. [DOI] [PubMed] [Google Scholar]
- Nikonorov IM, Blanck TJ, Recio-Pinto E. The effect of halothane on single human neuronal L-type calcium channels. Anesthesia and Analgesia. 1998;86:885–895. doi: 10.1097/00000539-199804000-00038. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Hillered L, Olsson Y, Sheardown MJ, Hansen AJ. Regional changes in interstitial K+ and Ca2+ levels following cortical compression contusion trauma in rats. Journal of Cerebral Blood Flow and Metabolism. 1993;13:183–192. doi: 10.1038/jcbfm.1993.22. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Laursen H, Hillered L, Hansen AJ. Calcium movements in traumatic brain injury: the role of glutamate-receptor operated ion channels. Journal of Cerebral Blood Flow and Metabolism. 1996;16:262–270. doi: 10.1097/00004647-199603000-00011. [DOI] [PubMed] [Google Scholar]
- Papadaki M, Eskin SG. Effects of fluid shear stress on gene regulation of vascular cells. Biotechnology Progress. 1997;13:209–221. doi: 10.1021/bp970029f. 10.1021/bp970029f. [DOI] [PubMed] [Google Scholar]
- Prince JA, Oreland L. Staurosporine differentiated human SH-SY5Y neuroblastoma cultures exhibit transient apoptosis and trophic factor independence. Brain Research Bulletin. 1997;43:515–523. doi: 10.1016/s0361-9230(97)00328-6. 10.1016/S0361-9230(97)00328-6. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, Zack DJ. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Investigative Ophthalmology and Visual Science. 1995;36:774–786. [PubMed] [Google Scholar]
- Reich K, McAllister T, Guidi S, Frangos J. Activation of G proteins mediates flow induced prostaglandin E2 production in osteoblasts. Endocrinology. 1997;138:1014–1018. doi: 10.1210/endo.138.3.4999. 10.1210/en.138.3.1014. [DOI] [PubMed] [Google Scholar]
- Ridge J, Terle DA, Dragunsky E, Levenbook I. Effects of gamma-IFN and NGF on subpopulations in a human neuroblastoma cell line; flow cytometric and morphological analysis. In Vitro Cellular and Developmental Biology Animal. 1996;32:238–248. doi: 10.1007/BF02722952. [DOI] [PubMed] [Google Scholar]
- Satoh T, Sakai N, Enokido Y, Uchiyama Y, Hatanaka H. Free radical independent protection by nerve growth factor and bcl-2 of PC12 cells from hydrogen-peroxide-triggered apoptosis. Journal of Biochemistry. 1996;120:540–546. doi: 10.1093/oxfordjournals.jbchem.a021447. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Ishida T, Traub O, Corson MA, Berk BC. Mechanotransduction in endothelial cells: temporal signaling events in response to shear stress. Journal of Vascular Research. 1997;34:212–219. doi: 10.1159/000159225. [DOI] [PubMed] [Google Scholar]
- Ueno K, Melvin JW, Li L, Lighthall JW. Development of tissue level brain injury criteria by finite element analysis. Journal of Neurotrauma. 1995;12:695–706. doi: 10.1089/neu.1995.12.695. [DOI] [PubMed] [Google Scholar]
- White B, Krause G. Brain injury and repair mechanisms: the potential for pharmacologic therapy in closed head trauma. Annals of Emergency Medicine. 1993;22:970–979. doi: 10.1016/s0196-0644(05)82737-4. [DOI] [PubMed] [Google Scholar]
- Yan D, Coloma F, Metheetrairut A, Trope G, Heathcoat J, Ethier C. Deformation of the lamina cribrosa by elevated intraocular pressure. British Journal of Ophthalmology. 1994;78:643–648. doi: 10.1136/bjo.78.8.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan GM, Lin SZ, Irwin RP, Paul SM. Activation of G proteins bidirectionally affects apoptosis of cultured cerebellar granule neurons. Journal of Neurochemistry. 1995;65:2425–2431. doi: 10.1046/j.1471-4159.1995.65062425.x. [DOI] [PubMed] [Google Scholar]
- Zeimer R, Chen K. Comparison of a noninvasive measurement of optic nervehead mechanical compliance with an invasive method. Investigative Ophthalmology and Visual Science. 1987;28:1735–1739. [PubMed] [Google Scholar]