

Abstract
Plasma adenosine concentration increases during hypoxia to a level that excites carotid body chemoreceptors by an undetermined mechanism. We have examined this further by determining the electrophysiological responses to exogenous adenosine of sinus nerve chemoafferents in vitro and of whole-cell currents in isolated type I cells.
Steady-state, single-fibre chemoafferent discharge was increased approximately 5-fold above basal levels by 100 μM adenosine. This adenosine-stimulated discharge was reversibly and increasingly reduced by methoxyverapamil (D600, 100 μM), by application of nickel chloride (Ni2+, 2 mM) and by removal of extracellular Ca2+. These effects strongly suggest a presynaptic, excitatory action of adenosine on type I cells of the carotid body.
Adenosine decreased whole-cell outward currents at membrane potentials above -40 mV in isolated type I cells recorded during superfusion with bicarbonate-buffered saline solution at 34–36 °C. This effect was reversible and concentration dependent with a maximal effect at 10 μM.
The degree of current inhibition induced by 10 μM adenosine was voltage independent (45.39 ± 2.55% (mean ± s.e.m.) between −40 and +30 mV) and largely (∼75%), but not entirely, Ca2+ independent. 4-Aminopyridine (4-AP, 5 mM) decreased the amplitude of the control outward current by 80.60 ± 3.67% and abolished the effect of adenosine.
Adenosine was without effect upon currents near the resting membrane potential of approximately −55 mV and did not induce depolarization in current-clamp experiments.
We conclude that adenosine acts to inhibit a 4-AP-sensitive current in isolated type I cells of the rat carotid body and suggest that this mechanism contributes to the chemoexcitatory effect of adenosine in the whole carotid body.
The purine nucleoside adenosine is present in all cells as an intermediary of metabolism and is derived predominantly from the cleavage of 5′-adenosine monophosphate (AMP) by 5′-nucleotidase. Its intracellular production can be increased rapidly during hypoxia (Winn et al. 1981) leading to its increased facilitated diffusion into the extracellular space where, through activation of specific G-protein coupled membrane bound (P1) purinoceptors, it can act in an auto- or paracrine fashion or even at a more systemic level via the circulation. This link with metabolism makes adenosine an attractive candidate when investigating the cellular response to oxygen lack and activation of adenosine receptors tends towards either increasing oxygen delivery or reducing oxygen demand. A similar ‘protective’ function has been ascribed for adenosine at a systemic level, as a stimulatory action of the nucleoside upon carotid body chemoreceptor afferent discharge has been recorded in vivo (McQueen & Ribeiro, 1981) which is sufficient to increase ventilation in rats (Monteiro & Ribeiro, 1987) and humans (Watt et al. 1987). This excitatory effect is retained in vitro (Runold et al. 1990) and is thus independent of the well-established effects of adenosine upon blood flow.
Pharmacological and histochemical evidence suggests that the receptor activated at the carotid body is most likely the A2A subtype (McQueen & Ribeiro, 1986; Weaver, 1993; Sebastiao & Ribeiro, 1996) but the location of these receptors and the mechanism by which their activation initiates afferent chemoreceptor discharge is unknown. The carotid body is a highly vascular, composite receptor made up of many glomeruli containing clusters of type I cells surrounded by glial-like type II cells with an efferent and afferent innervation, in which the neural crest-derived, type I cell is now widely believed to act as the primary transducer element. Although some differences exist in the precise details, there is now a general agreement that hypoxia induces inhibition of a component of the total outward K+ current in these cells that contributes to the resting membrane potential, thus leading to membrane depolarization, voltage-gated Ca2+ entry and Ca2+-dependent neurosecretion onto (in vivo) closely apposed afferent nerve endings (Gonzalez et al. 1994; Peers & Buckler, 1995). The aim of the present study was to determine if the cellular action of adenosine mimicked that of hypoxia by firstly determining the Ca2+ dependence of its excitatory action in an in vitro whole carotid body preparation and secondly to determine the effect of adenosine upon whole-cell currents recorded in isolated type I cells.
A preliminary report of part of this study has been published in abstract form (Vandier et al. 1998).
METHODS
Anaesthesia in adult Wistar rats (> 3 weeks old; 120-200 g) was induced with 3-4% halothane in O2 and maintained at 1.5-2.5% halothane, whilst left and right carotid bifurcations were removed as described previously (Pepper et al. 1995). The level of halothane was then increased to 4% and animals were killed by decapitation. Excised bifurcations were prepared either for single chemoafferent fibre recording or for patch-clamp recording of isolated type I cells.
Single chemoafferent fibre recording
Each carotid bifurcation was pinned on Sylgard (Dow Corning) in a small volume (∼0.2 ml) tissue bath and superfused at 3 ml min−1 with warmed (36.7-37°C), gassed (95% O2-5% CO2), bicarbonate-buffered saline solution (composition (mM): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 5 Na2SO4, 1.3 MgSO4, 24 NaHCO3, 2.4 CaCl2, 10 glucose, pH ∼7.38) whilst the excess connective tissue around the bifurcation was removed to expose the carotid body and the sinus nerve. The preparation was then partially digested in a gassed enzyme solution (0.06% collagenase (Sigma Type II), 0.02% protease (Sigma Type IX)) for 25 min at 37°C to facilitate the recording of neuronal activity.
Extracellular recordings of afferent single-fibre activity were made from the cut end of the carotid sinus nerve using glass suction electrodes and standard Neurolog modules (Digitimer Ltd, Welwyn Garden City, Hertfordshire, UK). Chemoreceptor discharge was discriminated as activity exceeding a level which was at least 50% of the amplitude of the baseline noise above the noise and which responded to a decrease in superfusate PO2 with a reversible increase in discharge frequency. Single fibres were discriminated on the basis of amplitude and shape. Action potentials were sampled digitally after conversion to TTL pulses via a window discriminator (Neurolog NL200) by a computer (Macintosh IIci with NB-MIO-16 DA and NB-MIO-8G DMA cards, National Instruments Co., Austin, TX, USA) running customized LabVIEW 2 (National Instruments Co.) software. TTL pulses were counted and binned into 10 s periods.
The PO2 and temperature of the superfusate were continually monitored in the superfusion line immediately before entering the bath by a membrane oxygen electrode (No. 733, Diamond General, Ann Arbor, MI, USA) and meter (O2/CO2 Analyser, Cameron Instrument Company, Port Aransas, TX, USA) and thermocouple (871A, Tegam Inc., Madison, OH, USA). The oxygen electrode was calibrated against, and PCO2 was determined from, superfusate samples collected just distal to the carotid body and measured on a blood gas analyser (IL 1640, Instrumentation Laboratory, Warrington, UK). Afferent spike activity and superfusate PO2 were monitored on an oscilloscope (Gould 1604) and recorded throughout each experiment on a VHS video recorder via a DC modified PCM digital unit (Applegarth Electronics).
Patch-clamp recording of isolated type I cells
Carotid bodies were removed from excised bifurcations and incubated for 10 min in magnesium-free, low Ca2+ (50 μM), bicarbonate-buffered saline solution. Collagenase (0.4 mg ml−1, Type I, Worthington) and trypsin (0.2 mg ml−1, Sigma) were then added and the solution held at 37°C for 25 min. The digested tissue was centrifuged (1000 r.p.m. for 10 min) and the enzyme solution removed and replaced by Ham's F-12 culture medium (Sigma) supplemented with 10% fetal calf serum, 100 i.u. ml−1 penicillin, 100 mg ml−1 streptomycin, 80 u l−1 insulin and 2 mg glutamine. Following trituration with fire-polished Pasteur pipettes, dispersed cells were plated onto glass coverslips and kept in a humidified incubator (5% CO2-95% air at 37°C) overnight. Cells were used within 14-24 h following isolation.
Solutions and drugs
The bicarbonate-buffered, extracellular saline solution contained (mM): 117 NaCl, 4.5 KCl, 23 NaHCO3, 1 MgCl2, 2.5 CaCl2 and 11 glucose and was equilibrated with 5% CO2 and 95% air (pH ∼7.4). For Ca2+-free solutions, CaCl2 was removed and 1 mM EGTA and 3.5 mM MgCl2 were added. Adenosine (Sigma) and 4-AP (Sigma) were added directly in saline solution. The pipette filling intracellular solution used for whole-cell recordings contained (mM): 6 NaCl, 107 KCl, 2 Na2ATP, 11 Hepes, 5 EGTA, 2 MgCl2, 2 CaCl2 (pH 7.2 adjusted with KOH). pCa ≡ 7 was calculated using CAMG software (W. H. Martin, Yale University, CT, USA).
Extracellular solutions were superfused at ∼2 ml min−1 in a 200 μl bath chamber. Given a dead space of 600 μl and assuming that a total change of bath solution occurs with 800 μl of perfused solution (i.e. 4 × bath chamber volume), bath concentrations would begin to change at ∼18 s and new solution equilibria would be reached ∼42 s after switching to the new solution. All experiments were performed at 34-36°C and at a PO2∼150 mmHg (confirmed on IL 1640 blood gas analyser).
Electrophysiology
Isolated type I cells were identified as spherical in shape and approximately 10 μm in diameter and in preliminary studies (data not shown) by amine fluorescence (de la Torre & Surgeon, 1976). The whole-cell patch-clamp configuration was used to record currents. Electrodes of 5-7 MΩ were made using Clarke GC150F-10 borosilicate glass capillaries (Clarke Electromedical, Reading, UK) and were fire-polished before use. The junction potentials between the electrode and the bath were cancelled using the voltage pipette offset control of the amplifier. Electrode capacitances were electronically compensated (∼4 pF). Cell capacitance was calculated by integration of the current obtained during pulses between -60 and -70 mV, divided by the pulse amplitude. To take account of cell size, current amplitudes were normalized by dividing by cell capacitance. Mean cell capacitance was calculated as 2.74 ± 0.09 pF (n = 29).
Data acquisition and analysis
Experiments were conducted using an Axopatch-1D (Axon Instruments Inc.) amplifier and currents were filtered with a Bessel low-pass filter. Records were simultaneously displayed on an oscilloscope (Gould 1602), digitized with Digidata-1200 A/D converter (Axon Instruments Inc.) and stored on disk in an PC computer using pCLAMP version 6.02 software (Axon Instruments Inc.). The membrane potentials of type I cells were held at -70 mV and the current-voltage (I-V) relation between -90 and 30 mV was obtained using ramp protocols (average of 5 ramps at 0.24 mV ms−1 every 10 s) and 10 mV increment pulse protocols (average of the last 20 ms of 300 ms duration pulses every 6 s). Currents were sampled at 5 kHz and filtered at 1 kHz. The membrane potential was recorded using the amplifier in current-clamp mode (I = 0) and stored on standard VHS videocassette via a DC modified PCM digital unit (Applegarth Electronics). Analyses of the data were performed using CLAMPFIT and FETCHAN (pCLAMP software) and ORIGIN 4.1 software (Microcal Inc., Northampton, MA, USA).
All data are expressed as means ±s.e.m. and tested for significant differences (P < 0.05) with regression analysis or one factor ANOVA with post hoc tests as indicated, using Statview II (Abacus Concepts, Berkeley, CA, USA) software.
RESULTS
Ca2+ dependence of adenosine in the in vitro carotid body
Chemoreceptor discharge was recorded from 24 single-fibre chemoafferents from 14 carotid body preparations. In all cases, spontaneous basal discharge (0.14 ± 0.02 impulses s−1) was obtained during hyperoxic (PO2 > 400 mmHg) normocapnia (PCO2 = 34.8 ± 0.4 mmHg) and was increased significantly by reducing superfusate PO2. No protocol lasted longer than 60 min.
The chemoreceptor discharge response to adenosine (1 μM-1 mM) was recorded in six preparations as the mean discharge during the final 2 min of a 10 min exposure to each concentration, at which time discharge was steady. Adenosine significantly increased steady-state chemodischarge (P < 0.01, ANOVA). Basal discharge in the absence of adenosine was 0.12 ± 0.03 impulses s−1 and was 0.11 ± 0.02 and 0.17 ± 0.06 impulses s−1 at 1 and 10 μM adenosine, respectively. Discharge was elevated almost 5-fold to 0.55 ± 0.16 impulses s−1 at 100 μM adenosine and fell slightly but not significantly at 1 mM to 0.40 ± 0.13 impulses s−1. Post hoc testing (Scheffé‘s F test) revealed that this effect of adenosine was significant only at 100 μM and 1 mM.
The effect of adenosine in the absence of extracellular Ca2+ was determined in five preparations (Fig. 1). Chemoreceptor discharge was reduced considerably, but never totally abolished, to 0.03 ± 0.02 impulses s−1 (P < 0.01, ANOVA), in the continued presence of 100 μM adenosine, by the exclusion of CaCl2 from the superfusate and addition of 4 mM MgSO4 and 1 mM EGTA. This discharge level was significantly less than the adenosine stimulated (0.95 ± 0.28 impulses s−1; P < 0.05 post hoc Student's paired t test) and basal (0.23 ± 0.07 impulses s−1; P < 0.05 post hoc Student's paired t test) discharge levels in this series of experiments and was fully reversible upon return to normal Ca2+.
Figure 1. The stimulatory effect of adenosine is Ca2+ dependent.
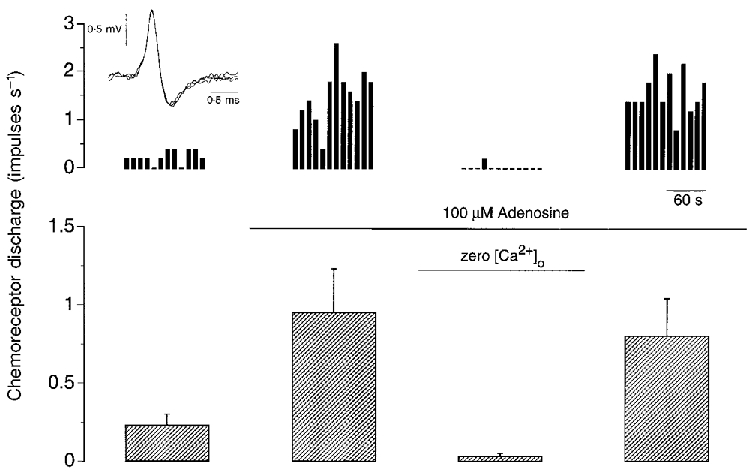
Single-fibre chemoreceptor activity recorded during steady-state control (left panel) and during exposure to 100 μM adenosine in the presence and absence of extracellular Ca2+ (middle panels) and after re-addition of Ca2+ (right panel). An example from one preparation is shown at the top, binned every 10 s and expressed in impulses s−1. The inset at top left shows three superimposed action potentials. The mean +s.e.m. of five preparations is shown below. Superfusate PO2 > 400 mmHg and PCO2≈35 mmHg throughout.
The source of Ca2+ entry in adenosine-mediated chemoexcitation was further determined by use of the non-selective Ca2+ channel antagonist, nickel chloride (Ni2+, 2 mM; n = 5) and the L-type Ca2+ channel blocker, methoxyverapamil (D600, 100 μM; n = 8). D600 was used in preference to dihydropyridines, such as nifedipine, as these compounds have been shown to decrease the uptake of adenosine through the nucleoside transporter (Bartrup & Stone, 1990). Both Ni2+ and D600 significantly (P < 0.001, ANOVA) reduced chemodischarge in the presence of 100 μM adenosine (Fig. 2) with Ni2+ reducing chemodischarge by 97 ± 1% from 0.27 ± 0.04 to 0.01 ± 0.004 impulses s−1 (P < 0.05, Scheffé‘s F test) and D600 reducing it by 68 ± 7% from 0.70 ± 0.17 to 0.23 ± 0.07 impulses s−1 (P < 0.05, Scheffé‘s F test). Whilst the adenosine-stimulated absolute discharges, in the absence of antagonists, were not significantly different between both groups of experiments (P > 0.05, unpaired t test), the absolute discharge in the presence of Ni2+ and D600 was significantly different (P < 0.05, unpaired t test) as was the normalized percentage inhibition between both groups (P < 0.02, unpaired t test).
Figure 2. Effect of Ni2+ and D600 upon adenosine stimulated chemoreceptor discharge.
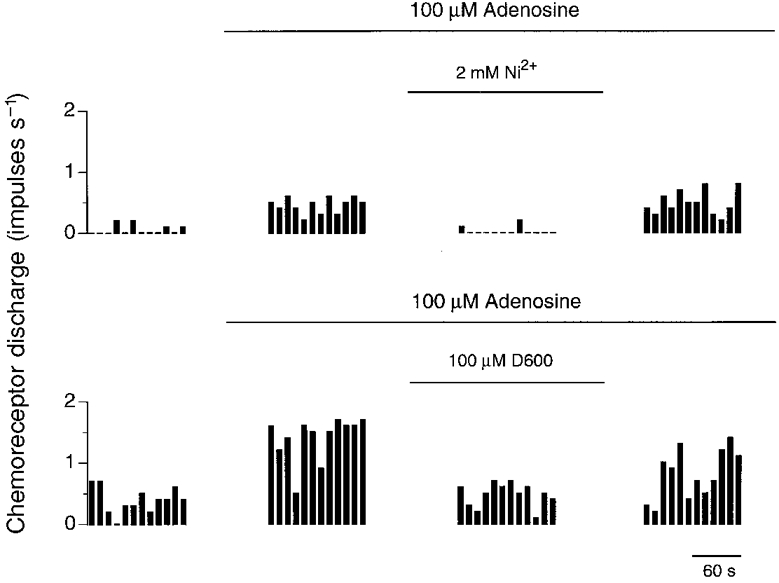
Single-fibre chemoreceptor activity recorded from two preparations during steady-state control (left panel) and during exposure to 100 μM adenosine in the presence and absence of either 2 mM Ni2+ or 100 μM D600 (middle panels) and after removal of antagonist (right panel). Discharge was binned every 10 s.
Dose-dependent effect of adenosine in isolated cells
Step depolarizations to +30 mV from a holding potential of -70 mV were applied for 300 ms every 6 s, firstly in the absence and then in the presence of various concentrations of adenosine in six type I cells. Example currents are shown in Fig. 3 where it can be seen that these currents were outward and sustained with inactivation not apparent within the pulse period. Increasing adenosine concentration decreased the amplitude of the outward current by 21.00 ± 2.40, 29.17 ± 3.26 and 32.92 ± 4.43% of the control current at 1, 10 and 100 μM respectively (P < 0.05, ANOVA) and in a reversible manner (Fig. 3). Post hoc testing (paired t test) revealed that this effect of adenosine was significantly different between 1 μM and 10 μM adenosine (P < 0.001) and between 1 μM and 100 μM (P < 0.001) but not between 10 μM and 100 μM adenosine (P > 0.2) suggesting that the maximal effect of adenosine was obtained between 10 and 100 μM. Further experiments were therefore performed with 10 μM adenosine as the minimum concentration able to induce the maximal effect.
Figure 3. Dose-dependent effect of adenosine on outward current.
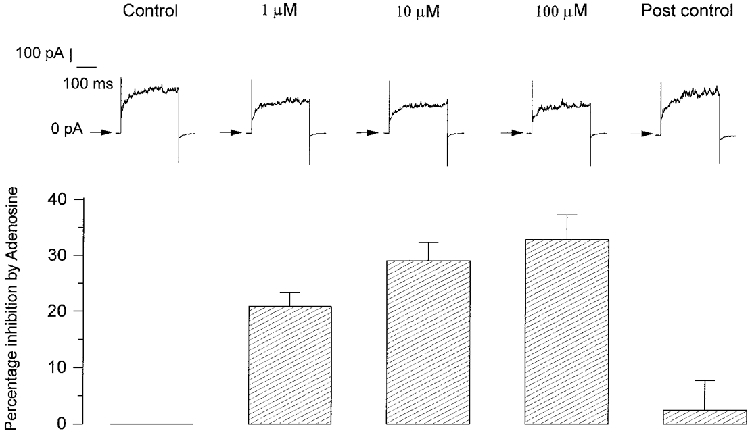
Top, example of outward currents elicited in one type I cell during 300 ms pulses to +30 mV from a holding potential of -70 mV in control conditions, at three different concentrations of adenosine and during post control conditions subsequent to the removal of adenosine. Currents during adenosine infusion were recorded 1 min after the calculated new solution equilibria would have been reached and 4 min after post control wash of adenosine. Bottom, means +s.e.m. (n = 6) of the percentage inhibition of outward currents at +30 mV induced by the three concentrations of adenosine.
Adenosine decreases the amplitude of outward current above -40 mV
Depolarization of the membrane between -90 and +30 mV, from a holding potential of -70 mV, elicited currents which were outward above -50 mV and which increased in amplitude with increasing depolarization. The application of 10 μM adenosine decreased the amplitude of these outward currents. Example currents from one cell are shown in Fig. 4A. The current density-voltage relations of this cell averaged with six other cells are shown in Fig. 4B. The current density-voltage relation was linear between -90 and -50 mV before adenosine and between -90 and -40 mV in the presence of adenosine and showed outward rectification at more positive potentials. Adenosine significantly decreased the amplitude of the current (P < 0.002, ANOVA). This effect of adenosine was significant only above -40 mV (P < 0.05, post hoc paired t test). Near the resting membrane potential of ∼-55 mV (corresponding to the membrane potential where the current density was 0 pA pF−1), adenosine was without effect on currents (Fig. 4B, inset) with no significant differences observed at -60 mV (P > 0.40) or -50 mV (P > 0.80). The current density-voltage relation of this adenosine-sensitive current (given as the difference between control and adenosine) is represented in Fig. 4C and shows an activation threshold near -50 mV.
Figure 4. Adenosine decreases outward currents above -40 mV.

A, example of outward currents obtained during 300 ms pulses to membrane potentials between -90 and +30 mV from a holding potential of -70 mV in control conditions and with 10 μM adenosine in the bath. B, mean ±s.e.m. (n = 7) current density-voltage relation obtained in control conditions (•) and in the presence of 10 μM adenosine (○). The inset shows an expanded scale of the same current density-voltage relation between -75 and -45 mV which highlights the negligible effect of adenosine at membrane potentials where current density is equal to 0 pA pF−1. C, means ±s.e.m. (n = 7) of the adenosine-sensitive component of the total current density-voltage relation determined as the difference between the current density amplitudes in control conditions and in the presence of 10 μM adenosine.
The effect of adenosine is voltage independent and largely Ca2+ independent
To investigate the role of external Ca2+ in the effect of adenosine, the bathing solution was changed to a Ca2+-free solution for 2 min (to attain a steady state) in seven cells. In four of these cells we then applied 10 μM adenosine. Figure 5A shows, in one cell, the effect of removal of external Ca2+ and the effect of adenosine upon this residual current. The mean current density-voltage relation obtained in these three conditions is shown in Fig. 5B. Removal of external Ca2+ significantly decreased the current density (P < 0.001, ANOVA) and, in this condition, adenosine could still significantly decrease the amplitude of the remaining current (P < 0.001, ANOVA).
Figure 5. Adenosine decreases outward currents in the absence of external Ca2+.
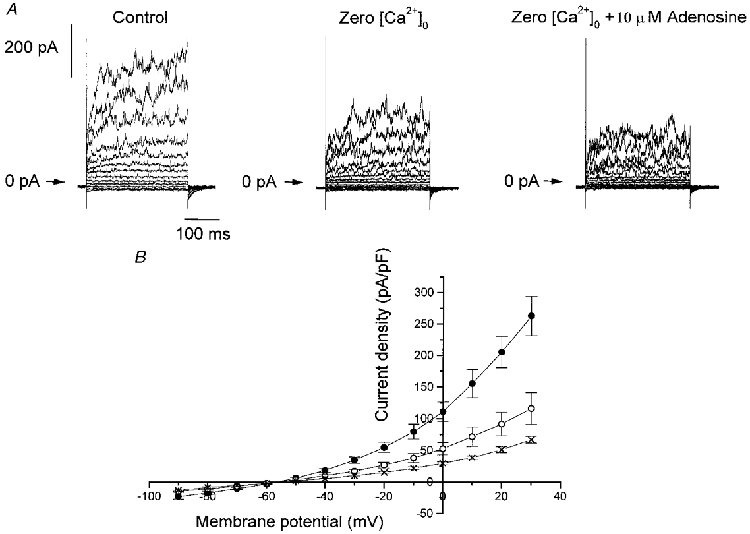
A, example of outward currents obtained during 300 ms pulses to membrane potentials between -90 and +30 mV from a holding potential of -70 mV in control conditions, in the absence of external Ca2+ (Zero [Ca2+]o) and in the presence of 10 μM adenosine. B, means ±s.e.m. of the current density-voltage relation obtained in control conditions (•, n = 7), in the absence of external Ca2+ (○, n = 7) and in the presence of 10 μM adenosine (×, n = 4).
The inhibition produced by adenosine in the presence, or absence, of external Ca2+ (Figs 4B and 5B) did not appear to be voltage dependent at potentials greater than -40 mV. This was confirmed by determining the degree of inhibition (difference in current density/control current density) produced by adenosine in both conditions. In the presence of Ca2+, adenosine decreased the amplitude of the control current by 42.68 ± 8.77% at -40 mV and by 47.60 ± 5.88% at +30 mV. This percentage of inhibition, induced by adenosine, was not significantly different across this potential range (P > 0.90, ANOVA) with a mean inhibition of 45.39 ± 2.55%. In the absence of external Ca2+, the inhibition of current by adenosine remained voltage independent (P > 0.90, ANOVA) with a mean inhibition of 34.20 ± 2.91% across the voltage range examined (P < 0.01, ANOVA compared with that when Ca2+ is present), suggesting that only approximately one-quarter (1 - (34.20/45.39)) of the inhibitory effect of adenosine was Ca2+ dependent.
4-AP abolishes the effect of adenosine
Figure 6A shows a family of outward currents obtained in one cell and from a holding potential of -70 mV to -90 mV and up to +30 mV and Fig. 6B represents the mean current density-voltage relation in control conditions, in the presence of 5 mM 4-AP in the absence or presence of 10 μM adenosine. In 10 cells, 4-AP significantly decreased the amplitude of the current density below that of control (P < 0.001, ANOVA) with a decrease of 80.60 ± 3.67% at +30 mV. In the continued presence of 5 mM 4-AP, adenosine had no effect on the remaining current (P > 0.70, ANOVA). At membrane potentials close to the reversal potential of the net whole-cell current, 4-AP did not decrease the current density. For example, at -60 mV, control current density was -2.84 ± 0.97 pA pF−1 and was 0.37 ± 0.32 pA pF−1 in the presence of 4-AP. This suggested, as was found for adenosine, that 4-AP would not depolarize isolated type I cells from their resting membrane potential.
Figure 6. 4-AP decreases outward currents and abolishes the effect of adenosine.
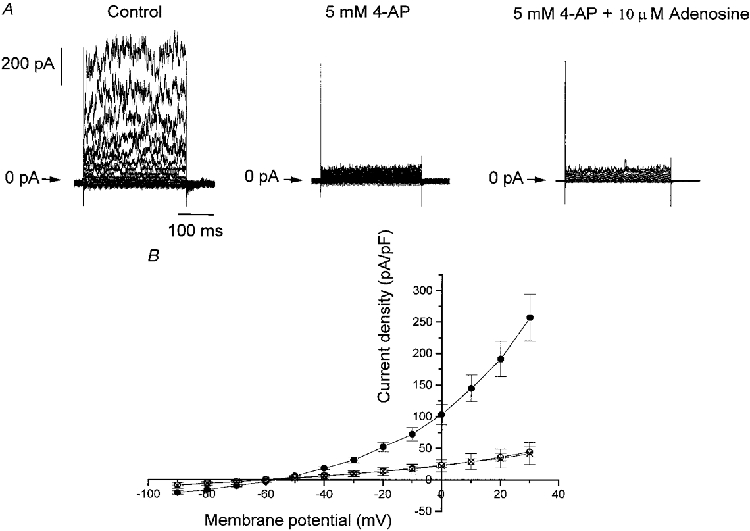
A, an example of outward currents obtained during 300 ms pulses to membrane potentials between -90 and +30 mV from a holding potential of -70 mV in control conditions, and in the presence of 5 mM 4-AP both in the absence and presence of 10 μM adenosine. B, mean ±s.e.m. current density-voltage relations obtained in control conditions (•, n = 10), in the presence of 5 mM 4-AP (○, n = 10) and in the last condition but with 10 μM adenosine (×, n = 3).
In the presence of 4-AP, the degree of inhibition observed in the presence of adenosine remained voltage independent with no significant difference between -40 and +30 mV (P > 0.90, ANOVA) with a mean inhibition of -2.29 ± 3.21%. This was significantly different (P < 0.0001, ANOVA) from the degree of inhibition induced by adenosine in the absence of 4-AP and further demonstrates that the current inhibited by adenosine is 4-AP sensitive.
Adenosine has no effect at the resting membrane potential
As adenosine was without significant effect on the amplitude of currents near the resting membrane potential, this suggested that it would not be able to modify this potential in isolated type I cells. We have addressed this suggestion more directly by testing the action of adenosine on the resting membrane potential using current clamp. Figure 7A shows an example obtained in one cell in which the resting membrane potential of -55 mV was not modified by 10 μM adenosine applied for 2 min. In six cells studied, we found the resting membrane potential to be -57.86 ± 1.45 mV which is close to the value found from the current density-voltage relation of Fig. 4B. In the presence of 10 μM adenosine, the resting membrane potential was -57.17 ± 4.36 mV, corresponding to a depolarization of just 0.50 ± 0.55 mV. This new resting membrane potential was not significantly different from control conditions (P > 0.05, paired t test). To test whether these cells could show stimulus-induced depolarization, we tested the effect of external (respiratory) acidosis, a known inducer of depolarization (Buckler & Vaughan-Jones, 1994b), on the resting membrane potential. In five cells, during control conditions (PCO2, 32.0 ± 0.5 mmHg; pHo, 7.39 ± 0.01) in which the resting membrane potential was -59.20 ± 1.68 mV, external acidosis (PCO2, 52.0 ± 2.6 mmHg; pHo, 7.25 ± 0.02) induced a significant depolarization of 4.80 ± 0.66 to -54.40 ± 1.03 mV (P < 0.002, paired t test).
Figure 7. Adenosine has no effect at the resting membrane potential.
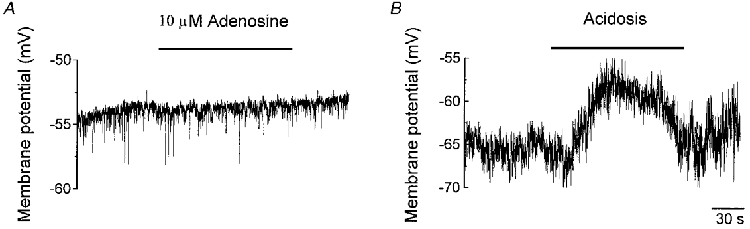
A, an example of the negligible effect of 10 μM adenosine in one cell on the resting membrane potential during current clamp. B, an example of the depolarizing effect of acidosis (pHo 7.30) in one cell on the resting membrane potential during current clamp.
DISCUSSION
Our results show that rat carotid body chemoafferent discharge is increased by adenosine via a non-vascular mechanism and thus confirm previous reports from which recordings were made from multi-fibre or whole sinus nerve recordings from cat carotid bodies (McQueen & Ribeiro, 1981; Runold et al. 1990). As we have demonstrated this action of adenosine occurs at the level of single fibres we can, in addition, discount fibre recruitment as the mechanism underlying these previous reports of chemoexcitation. We cannot compare directly the concentrations of adenosine used in our study with those used in the in vivo study of McQueen & Ribeiro (1981) but do note that the concentrations required to cause significant chemoexcitation in our rat in vitro preparation do exceed those required in the cat in vitro (Runold et al. 1990). At present we have no explanation for this difference which may reflect a genuine species difference or may be related to methodological differences.
The generation of chemoafferent discharge is now widely accepted to result from Ca2+-dependent neurotransmission from the type I cell onto postsynaptically located receptors on the afferent nerve terminals. In accordance with this hypothesis, the excitatory actions of natural stimuli such as hypoxia and hypercapnia are considerably reduced by the absence of external Ca2+ from the superfusion/perfusion media of isolated carotid body preparations (Roy et al. 1997) or in situ preparations (Shirahata & Fitzgerald, 1991). In their series of studies on the cat carotid body, McQueen & Ribeiro (1981, 1983, 1986) suggested that adenosine most probably acted at the presynaptically located type I cell, basing their argument upon evidence from other cell types, but they did not test this more directly. In the present study, we therefore demonstrated that a Ca2+-free superfusate could significantly reduce the chemodischarge induced by adenosine and take this to be more direct evidence of an action of adenosine on type I cells.
The Ca2+ dependence of the excitatory effect of adenosine implies an absolute requirement for receptor-mediated influx of Ca2+ into type I cells. This concept is supported by our finding that Ni2+ could abolish the adenosine-mediated discharge to almost the same degree as Ca2+-free solutions. Part, but not all, of this Ca2+ influx appears to be through L-type Ca2+ channels as D600 was also able to reduce discharge significantly, suggesting that membrane depolarization is an essential step for the full organ response to adenosine. These data are in close agreement with those reported for intracellular Ca2+ measurements made in type I cells in response to anoxic (Buckler & Vaughan-Jones, 1994a) or hypercapnic (Buckler & Vaughan-Jones, 1994b) stimulation.
We have demonstrated that adenosine can decrease the amplitude of the rat type I cell outward current, at potentials in excess of -40 mV and therefore having an effect similar to hypoxia in these cells (Peers, 1990). To our knowledge, this is the first time that this effect has been described in type I cells and is observable at a concentration known also to affect K+ currents in smooth muscle, cardiac and PC12 cells (Dart & Standen, 1993; Hiraoka & Furakawa, 1998; Kobayashi et al. 1998). In smooth and cardiac muscle, hypoxia/ischaemia and adenosine both increase the amplitude of the K+ current. In contrast, adenosine does not mimic hypoxia in PC12 cells (Kobayashi et al. 1998) as, in these cells, the increase in K+ current induced by adenosine acts to attenuate the decrease in this current induced by hypoxia (Zhu et al. 1996). Thus, adenosine appears protective against hypoxia in PC12 cells, whilst it may mediate, in part, the action of hypoxia in smooth muscle, cardiac and type I cells of the carotid body.
The K+ current of type I cells contains both Ca2+-sensitive and -insensitive components (Peers & O'Donnell, 1990; Lopez-Lopez et al. 1997). As a large part of the effect of adenosine, in the present study, was still observed in the absence of external Ca2+ and 4-AP significantly reduced the amplitude of the outward current and prevented any subsequent action of adenosine, this suggests that adenosine acts primarily to decrease the amplitude of a delayed rectifier K+ current in rat type I cells. A similar Ca2+-insensitive effect has been reported for the action of hypoxia upon isolated type I cells in rabbits (Lopez-Barneo et al. 1988) and in PC12 cells (Zhu et al. 1996) and a similar preventative action of 4-AP upon adenosine-mediated effects has been described in other neuronal tissue (Scholfield & Steel, 1988). However, a small but significant component of the current blocked by adenosine was Ca2+ dependent. We cannot therefore exclude the existence of an additional action of adenosine upon Ca2+-activated K+ currents as has been described for the action of hypoxia in rat type I cells (Peers, 1990) and/or an adenosine-mediated increase in Ca2+ currents. The action of adenosine upon Ca2+ currents in the rat type I cell has not been reported, but it has recently been shown that adenosine can decrease the amplitude of L-type Ca2+ currents in PC12 cells (Kobayashi et al. 1998).
Pharmacological evidence using specific adenosine-receptor agonists suggested that the action of adenosine at the cat carotid body was mediated via activation of A2 receptors (McQueen & Ribeiro, 1986) and given that in situ hybridization has revealed intense A2A mRNA in rat carotid bodies (Weaver, 1993) and low doses of an A2A agonist stimulated ventilation in rats (Sebastiao & Ribeiro, 1996) this appears the most likely receptor subtype expressed in this organ. This receptor is widely accepted as being coupled to stimulatory guanine nucleotide binding (Gs) proteins which stimulate adenylate cyclase in many cell types (Fredholm, 1995; Sebastiao & Ribeiro, 1996) including those of the carotid body (Chen et al. 1997). This action on adenylate cyclase activity could explain our observed responses, as elevations in cAMP levels have been shown to decrease the amplitude of 4-AP-sensitive outward currents in isolated rabbit type I cells (Lopez-Lopez et al. 1993). Moreover, the decrease of the outward current by cAMP in type I cells is voltage independent (Lopez-Lopez et al. 1993) as we have also observed with the effect of adenosine. The suggestion that this occurred via a phosphorylation of the K+ channels (Lopez-Lopez et al. 1993) was confirmed in PC12 cells in which adenosine A2A receptor activation could activate the cAMP-PKA pathway (Kobayashi et al. 1998). As we have shown that the effect of adenosine on type I cells is largely Ca2+ independent, as is the hypoxia-induced elevation in cAMP in these cells (Perez-Garcia et al. 1990; Wang et al. 1991), the simplest explanation for the action of adenosine in type I cells would therefore be via an increase in cAMP. Our data could therefore support the contention of Chen et al. (1997) that the elevation of cAMP during hypoxia is mediated by the release of endogenous adenosine. However, in another study, no effects of cAMP or PKA were observed on currents recorded from neonatal rat type I cells (Hatton & Peers, 1996) and these authors argued against a role for cyclic nucleotides in chemotransduction. Whether this reflects differences in species, age, temperature or in the choice of pH buffers remains an issue to be resolved. We cannot, however, exclude an action of adenosine upon the βδ subunit of Gs protein acting to decrease K+ currents (Peers & Carpenter, 1998) via activated PKC (Fredholm, 1995).
Hypoxia-induced neurosecretion from type I cells is Ca2+-dependent (Gonzalez et al. 1994) with the most compelling evidence suggesting that this elevation in intracellular Ca2+ occurs via influx through L-type Ca2+ channels (Buckler & Vaughan-Jones, 1994a). This implies cell depolarization as a prerequisite for hypoxic chemotransduction which necessitates a component of the current contributing to the resting membrane potential to be hypoxia sensitive. Of the three types of K+ currents inhibited by hypoxia that have been described (Ganfornina & Lopez-Barneo, 1992; Wyatt & Peers, 1995; Buckler, 1997), a voltage-, Ca2+- and 4-AP-insensitive background (leak) current (Buckler, 1997) appears the most likely to mediate cell depolarization as it is activated at resting membrane potentials. Additionally, pharmacological block of the other channel types does not increase chemoreceptor discharge (Doyle & Donnelly, 1994; Pepper et al. 1995). Not surprisingly therefore, our results showed that adenosine had no effect on the current near the resting membrane potential and was not able to depolarize isolated type I cells. The observation that a slight external acidosis of around 0.15 pH units could induce depolarization, as previously described (Buckler & Vaughan-Jones, 1994b), demonstrates that any small effect of adenosine would have been observed if it were present.
How then can we explain the excitatory effect of adenosine observed in the whole carotid body when we have no evidence for membrane depolarization in isolated type I cells? It is possible that the resting membrane potential of cells in the whole organ is held more depolarized than that of isolated cells and therefore at potentials at which a 4-AP-sensitive current is activated. Qualitative differences in the electrophysiological responses to natural stimuli between single cells and small clusters of cells have been reported (Pang & Eyzaguirre, 1992) which may arise from inter-cell chemical and/or electrical synaptic contact between type I cells (McDonald & Mitchell, 1975). The membrane resistance of an individual cell could thus be affected by its neighbours either directly through gap junctions or through the release of neurotransmitters/modulators acting in an autocrine or paracrine fashion. Another possibility is that type I cell depolarization, subsequent to K+ channel inhibition, is not a requirement for chemoexcitation. Such a suggestion has previously been made as the membrane resistance of type I cells recorded from within the whole carotid body was not changed by a level of hypoxia which increased chemoafferent discharge and 4-AP was unable to excite chemoafferent discharge from the whole carotid body, even in the presence of hypoxia (Donnelly, 1995). A more recent study (Lahiri et al. 1998) has come to a similar conclusion, although a role for the leak K+ current could not be excluded due to the lack of specific blockers of this current.
In summary, we have confirmed an excitatory action of adenosine upon carotid body chemoafferent discharge in vitro and have demonstrated an inhibitory action of adenosine on a 4-AP-sensitive component of the total outward current in isolated type I cells. The causal link between these two actions remains to be established.
Acknowledgments
We thank Dr David Davies for the LabView programming and Dr David Pepper for technical advice. This work was supported by The Wellcome Trust. P. K. is a Lister Institute Research Fellow.
References
- Bartrup J, Stone T. Dihydropyridines alter adenosine sensitivity in rat hippocampal slice. British Journal of Pharmacology. 1990;101:97–102. doi: 10.1111/j.1476-5381.1990.tb12096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. A novel oxygen-sensitive potassium current in rat carotid body type I cells. The Journal of Physiology. 1997;498:649–662. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. The Journal of Physiology. 1994a;476:423–428. doi: 10.1113/jphysiol.1994.sp020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypercapnia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. The Journal of Physiology. 1994b;478:157–171. doi: 10.1113/jphysiol.1994.sp020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Dinger B, Fidone SJ. cAMP production in rabbit carotid body: Role of adenosine. Journal of Applied Physiology. 1997;82:1771–1775. doi: 10.1152/jappl.1997.82.6.1771. [DOI] [PubMed] [Google Scholar]
- Dart C, Standen NB. Adenosine-activated potassium current in smooth muscle cells isolated from the pig coronary artery. The Journal of Physiology. 1993;471:767–786. doi: 10.1113/jphysiol.1993.sp019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC, Surgeon JW. A methodological approach to rapid and sensitive monoamine histofluorescence using a modified glyoxylic acid technique: The SPG method. Histochemistry. 1976;49:81–93. doi: 10.1007/BF00495672. [DOI] [PubMed] [Google Scholar]
- Donnelly DF. Modulation of glomus cell membrane currents of intact rat carotid body. The Journal of Physiology. 1995;489:677–688. doi: 10.1113/jphysiol.1995.sp021082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle TP, Donnelly DF. Effect of Na+ and K+ channel blockade on base-line and anoxia-induced catecholamine release from rat carotid body. Journal of Applied Physiology. 1994;77:2606–2611. doi: 10.1152/jappl.1994.77.6.2606. [DOI] [PubMed] [Google Scholar]
- Fredholm BB. Purinoceptors in the nervous system. Pharmacology and Toxicology. 1995;76:228–239. doi: 10.1111/j.1600-0773.1995.tb00135.x. [DOI] [PubMed] [Google Scholar]
- Ganfornina MD, Lopez-Barneo J. Potassium channel types in arterial chemoreceptor cells and their selective modulation by oxygen. Journal of General Physiology. 1992;100:401–426. doi: 10.1085/jgp.100.3.401. 10.1085/jgp.100.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiological Reviews. 1994;74:829–899. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Hatton CJ, Peers C. Hypoxic inhibition of K+ currents in isolated rat type I carotid body cells - evidence against the involvement of cyclic-nucleotides. Pflügers Archiv. 1996;433:129–135. doi: 10.1007/s004240050258. [DOI] [PubMed] [Google Scholar]
- Hiraoka M, Furakawa T. Functional modulation of cardiac ATP-sensitive K+ channels. News in Physiological Sciences. 1998;13:131–137. doi: 10.1152/physiologyonline.1998.13.3.131. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Conforti L, Pun RYK, Millhorn DE. Adenosine modulates hypoxia-induced responses in rat PC12 cells via the A2A receptor. The Journal of Physiology. 1998;508:95–107. doi: 10.1111/j.1469-7793.1998.095br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S, Roy A, Rozanov C, Mokashi A. K+-current modulated by PO2 in type I cells in rat carotid body is not a chemosensor. Brain Research. 1998;794:162–165. doi: 10.1016/s0006-8993(98)00276-5. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Lopez-Lopez JR, Urena J, Gonzalez C. Chemotransduction in the carotid body - K+ current modulated by PO2 in type I chemoreceptor cells. Science. 1988;241:580–582. doi: 10.1126/science.2456613. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez JR, De Luis DA, Gonzalez C. Properties of a transient K+ current in chemoreceptor cells of rabbit carotid body. The Journal of Physiology. 1993;460:15–32. doi: 10.1113/jphysiol.1993.sp019456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lopez JR, Gonzalez C, Perez-Garcia MT. Properties of ionic currents from isolated adult rat carotid body chemoreceptor cells: effect of hypoxia. The Journal of Physiology. 1997;499:429–441. doi: 10.1113/jphysiol.1997.sp021939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DM, Mitchell RA. The innervation of glomus cells, ganglion cells and blood vessels in the rat carotid body: a quantitative ultrastructural analysis. Journal of Neurocytology. 1975;104:177–230. [Google Scholar]
- McQueen DS, Ribeiro JA. Effect of adenosine on carotid chemoreceptor activity in the cat. British Journal of Pharmacology. 1981;74:129–136. doi: 10.1111/j.1476-5381.1981.tb09964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueen DS, Ribeiro JA. On the specificity and type of receptor involved in carotid-body chemoreceptor activation by adenosine in the cat. British Journal of Pharmacology. 1983;80:347–354. doi: 10.1111/j.1476-5381.1983.tb10040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueen DS, Ribeiro JA. Pharmacological characterisation of the receptor involved in chemoexitation induced by adenosine. British Journal of Pharmacology. 1986;88:615–620. doi: 10.1111/j.1476-5381.1986.tb10242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro EC, Ribeiro JA. Ventilatory effects of adenosine mediated by carotid body chemoreceptors in the rat. Naunyn-Schmiedeberg's Archives of Pharmacology. 1987;335:143–148. doi: 10.1007/BF00177715. [DOI] [PubMed] [Google Scholar]
- Pang L, Eyzaguirre C. Different effects of hypoxia on the membrane potential and input resistance of isolated and clustered carotid body glomus cells. Brain Research. 1992;575:167–173. doi: 10.1016/0006-8993(92)90440-k. [DOI] [PubMed] [Google Scholar]
- Peers C. Hypoxic suppression of K+ currents in type I carotid body cells - selective effect on the Ca2+ -activated K+ current. Neuroscience Letters. 1990;119:253–256. doi: 10.1016/0304-3940(90)90846-2. [DOI] [PubMed] [Google Scholar]
- Peers C, Buckler KJ. Transduction of chemostimuli by the type I carotid body cell. Journal of Membrane Biology. 1995;144:1–9. doi: 10.1007/BF00238411. [DOI] [PubMed] [Google Scholar]
- Peers C, Carpenter E. Protein kinase C activation causes inhibition of maxi-K channels in isolated rat carotid body type I cells. The Journal of Physiology. 1998;506.P:50. doi: 10.1111/j.1469-7793.1998.743bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C, O'Donnell J. Potassium currents recorded in type I carotid body cells from the neonatal rat and their modulation by chemoexcitatory agents. Brain Research. 1990;522:259–266. doi: 10.1016/0006-8993(90)91470-2. [DOI] [PubMed] [Google Scholar]
- Pepper DR, Landauer RC, Kumar P. Effect of charybdotoxin on hypoxic chemosensitivity in the adult rat carotid body, in vitro. The Journal of Physiology. 1995;487.P:177P–178P. doi: 10.1113/jphysiol.1995.sp020750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper DR, Landauer RC, Kumar P. Postnatal development of CO2-O2 interaction in the rat carotid body in vitro. The Journal of Physiology. 1995;485:531–541. doi: 10.1113/jphysiol.1995.sp020749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia MT, Almaraz L, Gonzalez C. Effects of different types of stimulation on cyclic-AMP content in the rabbit carotid body: functional significance. Journal of Neurochemistry. 1990;55:1287–1293. doi: 10.1111/j.1471-4159.1990.tb03137.x. [DOI] [PubMed] [Google Scholar]
- Roy A, Rozanov C, Iturriaga R, Mokashi A, Lahiri S. Acid-sensing by carotid body is inhibited by blockers of voltage-sensitive Ca2+ channels. Brain Research. 1997;769:396–399. doi: 10.1016/s0006-8993(97)00820-2. [DOI] [PubMed] [Google Scholar]
- Runold M, Cherniack NS, Prabhakar NR. Effect of adenosine on isolated and superfused cat carotid body activity. Neuroscience Letters. 1990;113:111–114. doi: 10.1016/0304-3940(90)90504-3. [DOI] [PubMed] [Google Scholar]
- Scholfield CN, Steel L. Presynaptic K-channel blockade counteracts the depressant effect of adenosine in olfactory cortex. Neuroscience. 1988;24:81–91. doi: 10.1016/0306-4522(88)90313-2. [DOI] [PubMed] [Google Scholar]
- Sebastiao AM, Ribeiro JA. Adenosine A2 receptor-mediated excitatory actions on the nervous system. Progress in Neurobiology. 1996;48:167–189. doi: 10.1016/0301-0082(95)00035-6. [DOI] [PubMed] [Google Scholar]
- Shirahata M, Fitzgerald RS. Dependency of hypoxic chemotransduction in cat carotid body on voltage-gated calcium channels. Journal of Applied Physiology. 1991;71:1062–1069. doi: 10.1152/jappl.1991.71.3.1062. [DOI] [PubMed] [Google Scholar]
- Vandier C, Conway AF, Kumar P. Pre-synaptic action of adenosine on the rat carotid body in vitro. The Journal of Physiology. 1998;511.P:101P. doi: 10.1111/j.1469-7793.1999.419ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WJ, Cheng GF, Yoshizaki K, Dinger B, Fidone S. The role of cyclic-AMP in chemoreception in the rabbit carotid body. Brain Research. 1991;540:96–104. doi: 10.1016/0006-8993(91)90495-h. [DOI] [PubMed] [Google Scholar]
- Watt AH, Reid PG, Stephens MR, Routledge PA. Adenosine-induced respiratory stimulation in man depends on site of infusion - evidence for an action on the carotid body. British Journal of Clinical Pharmacology. 1987;23:486–490. doi: 10.1111/j.1365-2125.1987.tb03081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver DR. A2a adenosine receptor gene expression in developing rat brain. Molecular Brain Research. 1993;20:313–327. doi: 10.1016/0169-328x(93)90058-w. [DOI] [PubMed] [Google Scholar]
- Winn HR, Rubio R, Berne RM. Brain adenosine concentration during hypoxia in rats. American Journal of Physiology. 1981;241:H235–242. doi: 10.1152/ajpheart.1981.241.2.H235. [DOI] [PubMed] [Google Scholar]
- Wyatt CN, Peers C. Ca2+ -activated K+ channels in isolated type I cells of the neonatal rat carotid body. The Journal of Physiology. 1995;483:559–565. doi: 10.1113/jphysiol.1995.sp020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WH, Conforti L, Czyzyk-Krzeska MF, Millhorn DE. Membrane depolarization in PC-12 cells during hypoxia is regulated by an O2-sensitive K+ current. American Journal of Physiology. 1996;271:C658–665. doi: 10.1152/ajpcell.1996.271.2.C658. [DOI] [PubMed] [Google Scholar]