

Abstract
Single fibres were enzymatically isolated from interosseus muscles of dystrophic MDX mice, myotonic-dystrophic double mutant ADR-MDX mice and C57BL/10 controls. The fibres were kept in cell culture for up to 2 weeks for the study of Ca2+ homeostasis and sarcolemmal Ca2+ permeability.
Resting levels of intracellular free Ca2+, determined with the fluorescent Ca2+ indicator fura-2, were slightly higher in MDX (63 ± 20 nm; means ±s.d.; n = 454 analysed fibres) and ADR-MDX (65 ± 12 nm; n = 87) fibres than in controls (51 ± 20 nm; n = 265).
The amplitudes of electrically induced Ca2+ transients did not differ between MDX fibres and controls. Decay time constants of Ca2+ transients ranged between 10 and 55 ms in both genotypes. In 50% of MDX fibres (n = 68), but in only 20% of controls (n = 54), the decay time constants were > 35 ms.
Bath application of Mn2+ resulted in a progressive quench of fura-2 fluorescence emitted from the fibres. The quench rate was about 2 times higher in MDX fibres (3.98 ± 1.9% min−1; n = 275) than in controls (2.03 ± 1.4% min−1; n = 204). The quench rate in ADR-MDX fibres (2.49 ± 1.4% min−1; n = 87) was closer to that of controls.
The Mn2+ influx into MDX fibres was reduced to 10% by Gd3+, to 19% by La3+ and to 47% by Ni2+ (all at 50 μm). Bath application of 50 μm amiloride inhibited the Mn2+ influx to 37%.
We conclude that in isolated, resting MDX muscle fibres the membrane permeability for divalent cations is increased. The presumed additional influx of Ca2+ occurs through ion channels, but is well compensated for by effective cellular Ca2+ transport systems. The milder dystrophic phenotype of ADR-MDX mice is correlated with a smaller increase of their sarcolemmal Ca2+ permeability.
Dystrophin is a submembranous cytoskeletal protein that is vital for the long-term function of skeletal muscle (Hoffman et al. 1987; Straub & Campbell, 1997). Although it has been established that dystrophin anchors nitric oxide synthase to the inner surface of the sarcolemma (Brenman et al. 1996), other functions of this protein are still being discussed. For example, dystrophin is presumed to protect the sarcolemma against mechanical stress during muscle activity (Petrof et al. 1993; Pasternak et al. 1995; Menke & Jockusch, 1995). Deficiency of dystrophin, as in the X-linked hereditary disease of Duchenne muscular dystrophy (DMD) or its animal model, murine muscular dystrophy (MDX), leads to muscle fibre necrosis and muscle tissue fibrosis resulting in progressive weakness. MDX mice show a much milder phenotype than DMD patients although their muscle fibres also undergo cycles of necrosis and regeneration (Hoffman & Gorospe, 1991).
Even though the gene defects causing MDX and DMD were discovered more than a decade ago, neither the precise function of dystrophin nor the mechanism of the dystrophic process has been completely elucidated (McArdle et al. 1995; Gillis, 1996; Straub & Campbell, 1997). There is overwhelming evidence that in their final stages dystrophin-deficient muscle fibres are overloaded with calcium (Jackson et al. 1985; Glesby et al. 1988; Gillis, 1996) and that secondary calcium-mediated degradative effects are responsible for their degeneration. However, it is not clear whether the excess of calcium enters the fibres via transient membrane lesions (Petrof et al. 1993; Menke & Jockusch, 1995), through ion channels (Fong et al. 1990; Leijendekker et al. 1996; Hopf et al. 1996) or via both pathways.
The first alternative is suggested by the fact that the sarcolemma of dystrophin-deficient fibres is leaky for cytoplasmic proteins such as the muscle-specific enzyme creatine kinase (CK; Florence et al. 1985; Hoffman & Gorospe, 1991). The loss of CK occurs even prior to the onset of muscle fibre degeneration and may reach a degree such that plasma CK levels are elevated more than 100 times in both DMD patients and MDX mice. Extracellularly applied labelled albumin was taken up by MDX fibres in vivo thus demonstrating the leakiness of the dystrophin-deficient sarcolemma for macromolecules (Matsuda et al. 1995). The second possible explanation for the Ca2+ overload of dystrophin-deficient muscle fibres is an increased Ca2+ entry through sarcolemmal ion channels. Increased activity of voltage-independent calcium leak channels has been shown in MDX myotubes (Fong et al. 1990; Hopf et al. 1996) and the activity of mechano-sensitive Ca2+ channels may also contribute to elevated Ca2+ influx (Haws & Lansman, 1991). Nevertheless, the question as to whether extra calcium enters the fibres predominantly through lesions or channels is still open.
The major aim of our study was to address this important question again. Experimental approaches to determine intracellular calcium levels in DMD and MDX muscle have so far mostly been performed with myotubes (Fong et al. 1990; Hopf et al. 1996; Leijendekker et al. 1996) as these are easier to cultivate than adult muscle fibres. In this study, we used large numbers of enzymatically isolated intact muscle fibres from adult mice to test their resting calcium levels. Then we determined the entry of divalent cations into these fibres using the manganese quench technique. This method is good for events occurring in the time range from minutes to hours. Finally, using channel blockers we were also able to distinguish between the two mentioned possibilities for calcium entry, i.e. membrane lesions and overactive ion channels.
By breeding myotonic-dystrophic (ADR-MDX) double mutant mice we and others have been able to show that increased mechanical muscle activity may reduce the dystrophic process (Krämer et al. 1998; Heimann et al. 1998). The double mutant mice are viable for more than 100 days and at any age their muscles, though completely lacking dystrophin, show fewer signs of fibre necrosis and fibrosis than age-matched MDX muscles. Plasma creatine kinase levels are significantly lower than in MDX mice. Sections of ADR-MDX muscles show a uniform pattern of oxidative muscle fibres indicating a complete fibre type II B to II A transformation. Another aim of this work was therefore to test whether in the dystrophin-less ADR-MDX fibres the low degree of dystrophy could be explained by a more normal Ca2+ homeostasis.
METHODS
Isolation and cultivation of muscle fibres
The experiments were part of a project approved by the Reviewing Committee of the Ulm Interdisciplinary Centre for Clinical Research (IZKF) and were performed with muscle fibres isolated from a total of 124 pairs of interosseus muscles. These came from 52 MDX mice (obtained from Harlan Winkelmann, Germany), 56 animals of the C57BL/10 strain used as controls and 16 double mutant ADR-MDX mice. The latter were bred in the Central Animal Research Unit of the University of Ulm (Krämer et al. 1998) and had the presence of both mutations verified by PCR. All animals were killed by neck dislocation performed by authorized persons according to the regulations of the University of Ulm. Other preparations from these animals were used for different experiments not to be reported here.
The excised muscles were treated for 1 h at 37°C with collagenase (1.5 mg ml−1) and protease (2 mg ml−1) dissolved in Ham's F-12 medium (pH 7.2), supplemented with 2 mM Hepes and 25 μg ml−1 gentamicin. Dissociation into single fibres was achieved by trituration. The debris was removed by centrifugation (for 2 min at 40 g). Intact isolated muscle fibres were seeded on glass coverslips (42 mm diameter) that had been coated by overnight incubation at 37°C with 3 ml of 15 μg ml−1 poly-L-ornithine. The fibres were kept under cell culture conditions at 37°C in a medium based on a 1 : 1 mixture of Ham's F-12 medium and CMRL, supplemented with 0.5 % fetal calf serum, 0.5 % horse serum, 2 mM L-glutamine and 25 μg ml−1 gentamicin C (5 % CO2). The fibres attached themselves to the coverslips after 1–2 days. They were used for experimentation 2–10 days later.
Fluorescence measurements
Measurements of resting intracellular free Ca2+ and of Ca2+ transients during excitation as well as determinations of the membrane permeability to divalent cations were performed using the fluorescent calcium indicator fura-2. The dye was loaded by incubating the fibres for 30–45 min at 37°C in standard external solution (see below) containing 2 μM fura-2 AM. Fluorescence measurements were carried out at room temperature (20–24°C) using a fast fluorescence photometry system (MPM-FFP; Zeiss, Oberkochen, Germany). The excitation wavelength was switched at 400 Hz between 340 and 380 nm using appropriate interference filters (bandwidth, 10 nm) alternately mounted in a filter wheel. The system allows the emitted light (505–530 nm) to be monitored with a time resolution of 5 ms for one pair of excitation wavelengths. A computer-controlled motor-driven stage was available for repetitive cycles of observations on different individual fibres attached to the same coverslip. To reduce dye bleaching during long-term experiments, the illumination was shut off between recording cycles. Ca2+ levels are given in the figures as fluorescence ratios obtained from alternating excitation at 340 and 380 nm. Autofluorescence was determined to contribute less than 2 % to the fluorescence signals. No correction for it was considered necessary. Also, no corrections were made for errors induced by the kinetics of fura-2, as others, using similar experimental conditions, have found that the decay of the corrected signals largely follows the decay phase of the measured Ca2+ transients (Bakker et al. 1997). Rough estimates of the dynamic range of Ca2+ changes were provided by means of an in situ calibration of the fura-2 signals (Mayerhofer et al. 1992) using a Kd of 224 nM (Grynkiewicz et al. 1985).
For estimation of the permeability of the sarcolemma to divalent cations the manganese quench technique was used. Mn2+ enters cells via the same routes as Ca2+ and accumulates inside over time since it is poorly accepted by the cellular transport systems. As Mn2+ quenches the fluorescence of fura-2, the reduction of the intensity of fura-2 fluorescence can be used as an indicator of the time integral of Mn2+ influx. The quench rate of the fluorescence intensity was estimated using linear regression analysis and expressed as the decline per minute of the initial fluorescence intensity of the sum of the signals obtained with excitation at 340 and 380 nm. This method does not allow calculation of quench rates during transient changes of Ca2+ concentration. In such cases the calcium-independent fluorescence intensity at the isosbestic wavelength of 360 nm was used (Zhou & Neher, 1993).
Application of drugs
For short-term investigations drugs were locally administered using an L/M-SPS-8 superfusion system (List, Darmstadt, Germany) which allows application of drugs to a single cell or a small group of cells in a repetitive and highly reproducible manner (Collatz et al. 1997). For long-term experiments the contents of the bath were completely exchanged.
Electrical stimulation of muscle fibres
The muscle fibres were excited by field stimulation using an HP 33120 A frequency generator (Hewlett Packard, Germany) in combination with a home-made voltage amplifier. Two electrodes were placed in close proximity (100–200 μm) to the fibres. Stimulus duration was set to 0.5 ms and the amplitude was adjusted about 10 % above the threshold for visible contractions.
Solutions and chemicals
The standard bathing fluid contained (mM): 140 NaCl, 2.7 KCl, 1.5 CaCl2, 1 MgCl2, 6 glucose and 12 Hepes, adjusted to pH 7.3. This solution has an osmolarity of 300 mosmol l−1. Experiments with Mn2+ were performed in the absence of Ca2+. MnCl2 was from Merck (Darmstadt, Germany); Gd2O3, LaCl3, NiCl2, Hepes, protamine sulphate and protease were from Sigma; fetal calf serum, Ham's F-12, CMRL, L-glutamine and gentamicin were from Biochrom (Berlin, Germany); collagenase was from Serva (Heidelberg, Germany); horse serum and CEE were from Gibco (Eggenstein, Germany); and fura-2 and fura-2 AM were from Calbiochem (Bad Soden, Germany).
Results are given as means ± standard deviation of n fibres. For determination of the significance of the difference between mean values, Student's t test was applied. Correlations of data were analysed using Pearson's test.
RESULTS
Cultivation and attachment of muscle fibres to glass coverslips
The best attachment of muscle fibres to glass surfaces, a prerequisite for fluorescence measurements on single cells, was achieved by coating the glass with poly-L-ornithine. Uncoated or poly-L-lysine-coated coverslips did not support the attachment of fibres. Muscle fibres from young mice attached themselves faster than those from older ones. Up to the age of 50 days attachment was firm within 1 day. Fibres from animals older than 90 days required 2–3 days for firm attachment. During establishment of conditions for long-term cultivation we routinely observed that fibres from MDX mice attached themselves much better to poly-L-ornithine-coated glass than controls. Fibres from the double mutant ADR-MDX animals were similar to those from MDX mice. Experiments were performed with fibres from animals older than 90 days (range, 90–220 days). Using an eyepiece micrometer we estimated the diameters of MDX, ADR-MDX and control fibres to vary between 10 and 60 μm. The mean diameters ±s.d. were 30 ± 8 μm (n = 409) for MDX, 28 ± 7 μm (n = 87) for ADR-MDX and 38 ± 9 μm (n = 242) for controls. The results from MDX and ADR-MDX fibres did not significantly differ (P > 0.08) whereas they both significantly differed from controls (P < 0.0001).
Resting Ca2+ levels and electrically induced Ca2+ transients
Resting Ca2+ levels of muscle fibres were slightly but significantly higher in MDX (63 ± 20 nM; n = 454) and ADR-MDX (65 ± 12; n = 87) fibres than in controls (51 ± 20 nM; n = 265; P < 0.0001; Fig. 1). We concluded that Ca2+ entry into MDX fibres at rest is either marginally increased or, if markedly increased, compensated for by effective cellular Ca2+ transport systems.
Figure 1. Intracellular free Ca2+ concentrations in dystrophin-deficient and control muscle fibres.
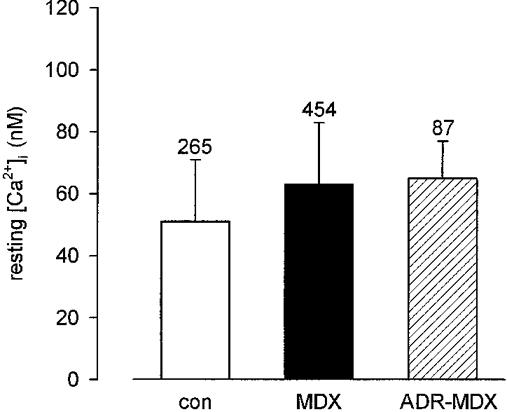
Ca2+ concentrations in resting mouse interosseus muscle fibres from control (con), dystrophic (MDX) and myotonic-dystrophic (ADR-MDX) animals. Number of analysed fibres indicated for each genotype above bar (means ±s.d.).
To test the influence of activity on the Ca2+ homeostasis of MDX fibres we induced visible contractions by electrical stimulation. At a stimulus frequency of 1 Hz the Ca2+ transients in MDX (Fig. 2A) and control preparations showed no gross differences in shape and size. Although our recording system was not fast enough to follow exactly the rise of the signal, the peak size from an individual fibre varied by no more than ±10 %. Between individual fibres the mean peak size varied between 0.8 and 1.4 (in terms of the fluorescence ratio). This was true for both MDX and control fibres, i.e. the size of the calcium signals was independent of the genotype. The decay phase of the signals was well fitted with a single exponential (Fig. 2B). Analysis of the variation of the decay time constant revealed a range from 10 to 65 ms for both genotypes. The fraction of fibres with large time constants was higher for MDX than for controls. In only 20 % of controls (n = 54), but in about 50 % of MDX fibres (n = 68) time constants of decay were > 35 ms (Fig. 3). The larger time constants measured in MDX fibres were not a consequence of smaller fibre diameters, as the two parameters were not correlated in either MDX or control fibres.
Figure 2. Sarcoplasmic Ca2+ transients of an MDX muscle fibre.
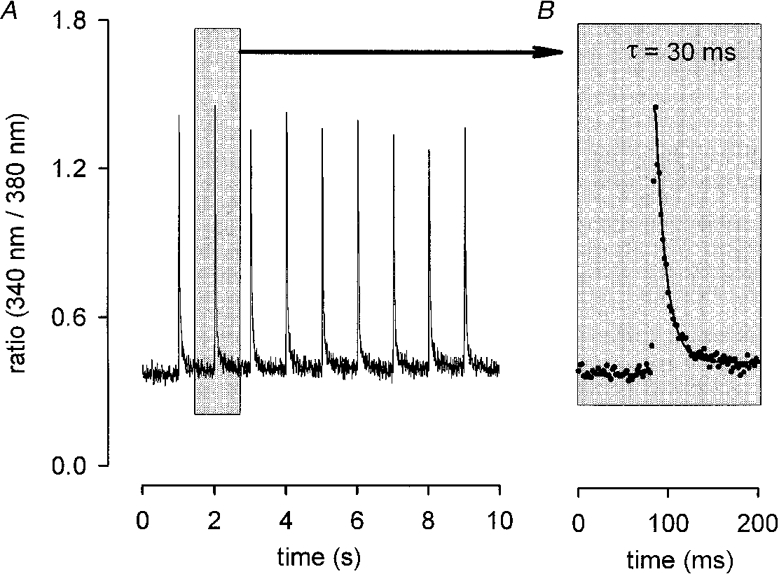
A, ratio of emission intensities (340 nm/380 nm, sampled every 5 ms) vs. recording time. Stimulus frequency, 1 Hz. B, single transient plotted on expanded time scale. Ascribed decay time constant (τ) obtained by fitting single exponential to falling phase (continuous line).
Figure 3. Decay time constants of Ca2+ transients of electrically stimulated muscle fibres.
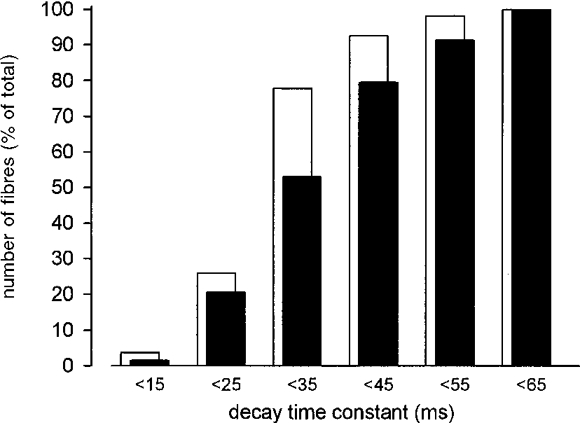
Data determined from 49 MDX and 36 control fibres grouped into 10 ms intervals. Percentages of fibres are plotted for each interval for MDX (▪) and control fibres (□).
At 1 Hz stimulation frequency the Ca2+ transients returned to baseline levels between single stimuli for MDX and controls (see Fig. 2A). This was true for the longest trains tested (10 min). At higher stimulation frequencies (10 and 30 Hz) again the amplitudes of the Ca2+ transients in MDX and control fibres were not different. Estimation of decay times was hampered by the fact that the Ca2+ transients did not always return to baseline. In some cases complete Ca2+ deregulation was observed after stimulation with 30 pulses at 30 Hz (Fig. 4). Deregulation was seen in control as well as in MDX fibres.
Figure 4. Sarcoplasmic Ca2+ transients in control muscle.
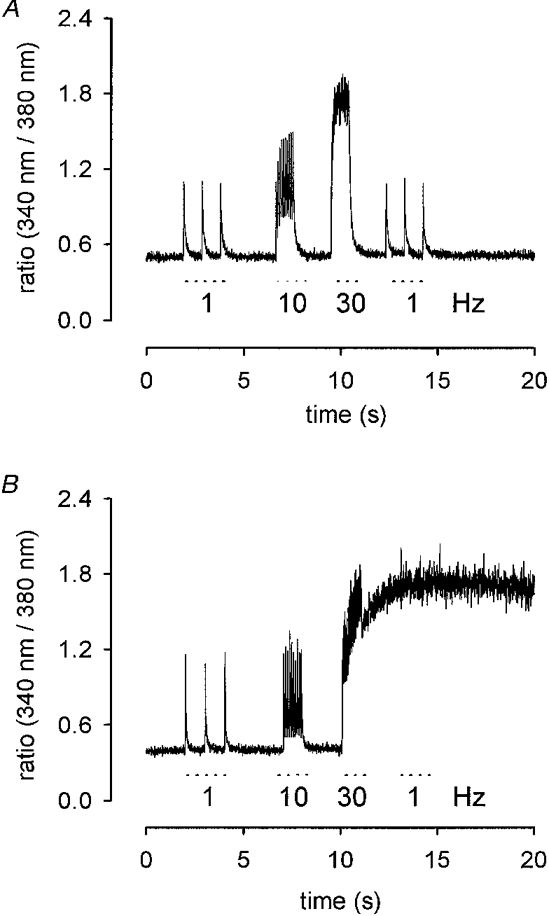
A, ratio of emission intensities (sampled every 5 ms) vs. recording time. Stimulus frequency: 1 Hz (3 pulses), 10 Hz (10 pulses) and 30 Hz (30 pulses). B, Ca2+ deregulation after 30 Hz stimulation.
Membrane permeability of muscle fibres for divalent cations
Extracellular Mn2+ enters cells via the same routes as Ca2+ and quenches the fluorescence signal of fura-2 at both excitation wavelengths. The speed of the method can be demonstrated with the fast Ca2+ entry effected by application of 10 μM acetylcholine (ACh): the fluorescence dropped to a low value within less than a minute (Fig. 5). This rapid drop was not observed when the fibres had been incubated with the ACh antagonist α-bungarotoxin (0.1 μM for 15 min, Fig. 5). Since Mn2+ is poorly accepted by cellular transport systems, the fluorescence signal remains low after removal of Mn2+ and ACh, thus providing an estimate of the integral ion influx.
Figure 5. Quench of fura-2 fluorescence generated by entry of Mn2+ into MDX muscle fibres.
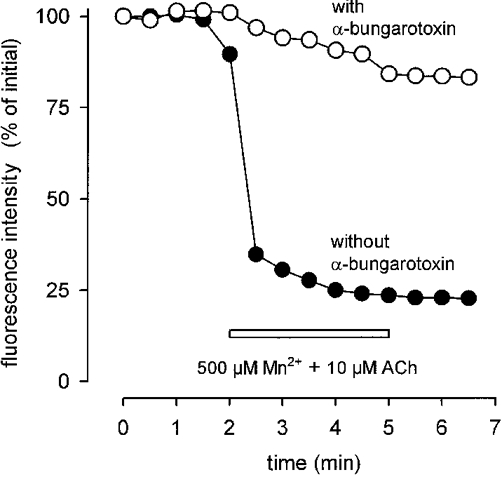
Sudden drop of fluorescence intensity upon application of 10 μM ACh in the presence of 500 μM Mn2+ (•). Removal of Mn2+ and ACh does not restore original fluorescence intensity. After preincubation with α-bungarotoxin (0.1 μM, 15 min; ^) quench rate is as small (4.8 % min−1) as in the absence of ACh (for control see Fig. 7).
During short-term application of 500 μM Mn2+ no quench was observed in either preparation independently of whether the cells were at rest or stimulated at 1 or 10 Hz (Fig. 6). During long-term application the signal slowly decreased and this quench ceased upon removal of Mn2+ (Fig. 7A). For quantification we used linear regression analysis to define a quench rate given by the percentage reduction of the initial fluorescence signal per minute.
Figure 6. Absence of Mn2+ entry during short-term muscle activity.
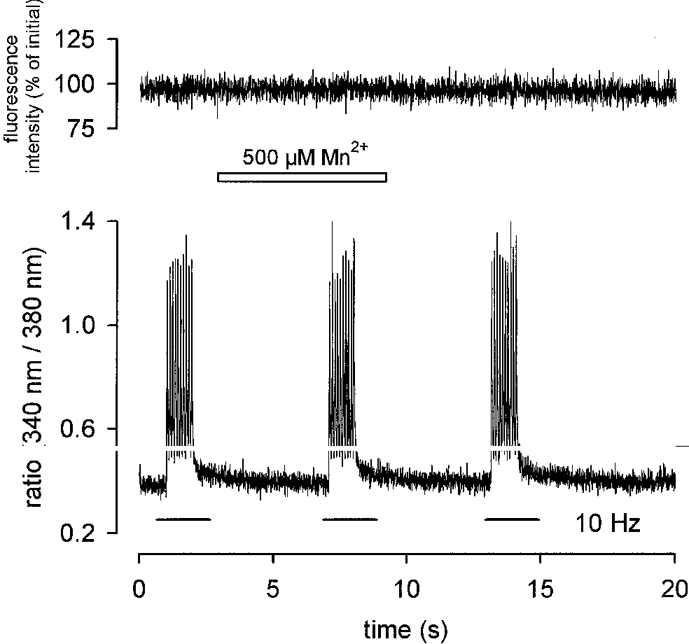
Application of 500 μM Mn2+ to an MDX fibre during electrically induced contractions (10 Hz, 10 pulses, lower trace) does not reduce fluorescence intensity (upper trace). Ca2+-independent fluorescence intensity calculated as described in Methods.
Figure 7. Quench rates obtained from different muscle fibres.
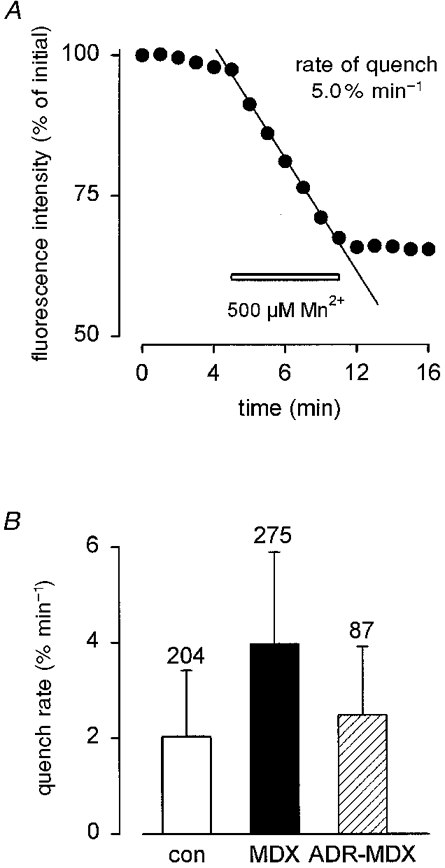
A, determination of quench rate (5 % min−1) in an MDX fibre using linear regression analysis. Calibration of both axes different from those in Fig. 5. The decline of the fura-2 fluorescence is expressed as percentage per minute (initial fluorescence intensity = 100 %). Bar indicates presence of 500 μM Mn2+. B, overall results from control (□), MDX (▪) and ADR-MDX ( ) fibres.
) fibres.
The quench rate in resting MDX fibres (3.98 ± 1.9 % min−1; n = 275) was about twice that of control (2.03 ± 1.4 % min−1; n = 204) (Fig. 7B). For ADR-MDX fibres the rate was close to control (2.49 ± 1.4 % min−1; n = 87). Statistical analysis of these data revealed that the difference in quench rates between control and MDX as well as between MDX and ADR-MDX was highly significant (P < 0.0001). Differences in quench rates between control and ADR-MDX were also significant (P < 0.001). These findings indicate that the membrane of MDX muscle fibres is more permeable to divalent cations than that of control or ADR-MDX muscle.
Effects of ion channel blockers on the Mn2+ quench rate
First we looked for Ca2+ entry via the dihydropyridine receptor Ca2+ channel by applying 50 μM nifedipine. Mn2+ entry was not inhibited (not shown). Analysis of other possible Ca2+ entry sites included the test for leak and store-mediated ion channels. The test involving the leak channel blocker protamine was not feasible with our experimental set-up since, according to our routinely performed control experiments, protamine reduces the fluorescence signal even in the absence of Mn2+. Another possible pathway for Ca2+ entry could be provided by store-mediated Ca2+ channels. Although there are no such channels described for adult skeletal muscle we investigated this possibility using carboxyamidotriazole. At 3 μM, this putative blocker of store-mediated Ca2+ channels was ineffective in reducing Mn2+ entry. Furthermore, preincubation with α-methylprednisolone (1 or 4 days, 10 μM), supposed to reduce stress-induced Ca2+ entry in MDX myotubes (Metzinger et al. 1995), did not prevent Mn2+ entry in resting muscle fibres from either MDX or control mice.
However, application of 50 μM Gd3+ immediately stopped fluorescence quench in MDX fibres (Fig. 8). On average the quench rate was reduced from about 4 % min−1 to 0.38 ± 0.37 % min−1 (n = 70). This value is very close to the reduced rate that Gd3+ produced in controls (0.36 ± 0.29 % min−1, n = 63; Fig. 9A) and is close to the reduction rate of basal fluorescence seen in the absence of Mn2+ (about 0.2 % min−1). Block of Mn2+ entry by Gd3+ was reversible since fluorescence quench continued after removal of Gd3+ (not illustrated). The persisting quench rates in the presence of Gd3+ were about the same for control and MDX or ADR-MDX, and for MDX and ADR-MDX (differences not statistically significant).
Figure 8. Block of Mn2+ entry into an MDX fibre by Gd3+.
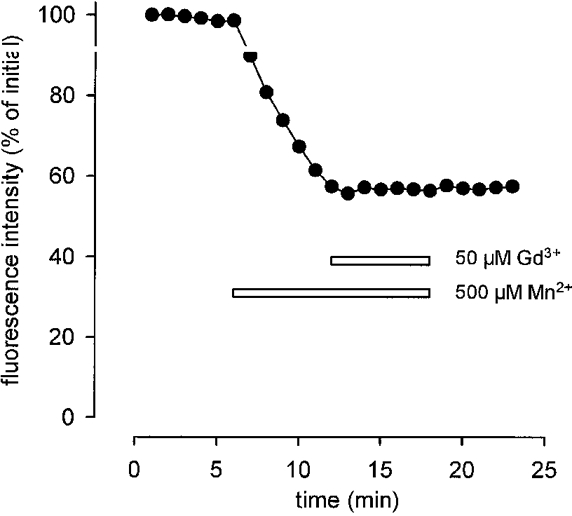
Application of 50 μM Gd3+ immediately prevents entry of Mn2+. Presence of Mn2+ and Gd3+ indicated by bars.
Figure 9. Block of Mn2+ entry into MDX, ADR-MDX and control fibres by various ion channel blockers.
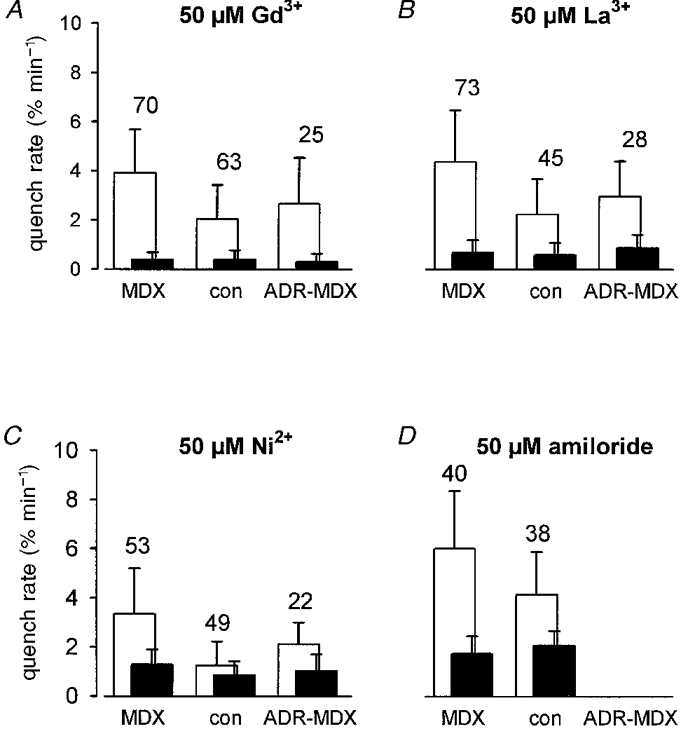
Quench rates determined in the presence of 500 μM Mn2+ and the agent indicated (□, before; ▪, after application of channel blocker). A, Gd3+; B, La3+; C, Ni2+; D, amiloride.
Lanthanum and nickel ions (each at 50 μM) as well as the organic blocker amiloride (50 μM) were also able to reduce the quench rate. In comparison to Gd3+, La3+, Ni2+ and amiloride were less effective blockers. Relative to its value in the absence of blockers, the rate of Mn2+ influx in MDX fibres was reduced to 10 % by Gd3+, to 19 % by La3+ and to 47 % by Ni2+. Amiloride reduced the rate of Mn2+ influx to 37 % (Fig. 9A-D).
DISCUSSION
Most studies analysing Ca2+ regulation in MDX muscle have been performed with cultivated myotubes which may not be representative of the situation in adult fibres. In only a few cases were freshly dissociated muscle fibres used (see Gillis, 1996). Cultivation has the advantage that many fibres from an individual muscle can be used for experimentation over an extended period of time. Thus, we were able to analyse Ca2+ homeostasis and membrane permeability of more than 1000 adult mouse muscle fibres of three different genotypes. During the cultivation period of 2–10 days we never observed substantial changes of the resting intracellular free Ca2+ levels in our mouse interosseus fibres (MDX and controls). This same result was reported for cultivated fibres from the mouse flexor digitorum brevis (Liu et al. 1996). Therefore it can be concluded that the process of cultivation does not harm the cellular mechanisms regulating the resting Ca2+ level in mouse muscle fibres.
It has been reported that cultivated myotubes, especially those from MDX muscle, have normal (Pressmar et al. 1994) or slightly increased Ca2+ levels (Head et al. 1990). Our finding of nearly normal resting Ca2+ levels in cultivated MDX fibres is in accordance with the majority of findings obtained with freshly isolated fibres (Bakker et al. 1993; Rivet-Bastide et al. 1993; Gailly et al. 1993; Pressmar et al. 1994; Gillis, 1996), but disagrees with others (Turner et al. 1991; Hopf et al. 1996). The differences may be due to different procedures of dye loading or to different muscles used for analysis. Dye loading performed at 37°C (in contrast to 23°C) diminishes differences in resting Ca2+ levels between MDX and control fibres, most probably due to dye compartmentalization (Hopf et al. 1996). Different muscles may have different fibre-type compositions influencing the results. But even when independent groups used the same muscle, the results (reviewed by Gillis, 1996) were sometimes controversial.
Upon stimulation, the decay of the Ca2+ transients in cultivated interosseus fibres from MDX muscle was slower than in control. This contrasts with findings obtained with freshly isolated MDX fibres from the flexor digitorum brevis (Head, 1993). There, the transients showed a narrow range of decay time constants (around 40 ms), whereas in interosseus fibres the distribution ranged from 10 to 65 ms. The fraction of MDX fibres having large decay time constants was clearly higher than control. This might be explained by a defective regulation of energy metabolism (Even et al. 1994) or by functionally altered Ca2+ pumps of the sarcoplasmic reticulum (Kargacin & Kargacin, 1996). Since isolated muscle fibres did not tolerate high stimulation frequency in a long run, it was impossible to verify results reported from intact muscles which showed an increase in muscle Ca2+ content during long-term stimulation experiments (Everts et al. 1993).
Using the manganese quench technique we found that in MDX fibres the membrane permeability to divalent cations was twice the value of controls. The differences in quench rates between the genotypes were not due to differences in fibre calibre, since the calculated correlation coefficients (r2) between diameters and quench rates were extremely small (Pearson's r2 < 0.04 for each genotype). An increased Mn2+ entry into MDX muscle was also obtained with cultivated myotubes (Hopf et al. 1996). The increase might be based on an increased open probability of single mechanosensitive ion channels (Franco-Obrégon & Lansman, 1994) or on an increased mean open time of leak channels (Hopf et al. 1996). Both channel types seem to be clustered in MDX muscle fibres, and it is conceivable that an increased activity might initiate focal damage. These same types of ion channels are also found in myotubes at about 10-fold higher frequency, with similar or mildly changed gating properties (Franco-Obrégon & Lansman, 1994; Hopf et al. 1996). Together with the finding of nearly normal resting Ca2+ levels the increased membrane permeability to divalent cations means that in MDX fibres the Ca2+-sequestering mechanisms must be continually coping with an overload.
As to the main aim of this study, our evidence of several channel blockers being able to reduce the extra Mn2+ (i.e. Ca2+) entry in MDX fibres clearly favours the hypothesis that this entry occurs through channels and not through transient membrane lesions. A similar conclusion was drawn from findings of an increased Mn2+ influx into cultured myotubes effected by the leak channel activator nifedipine (Hopf et al. 1996). As nifedipine neither stimulated nor inhibited the Mn2+ quench in our adult MDX fibres we tend to believe that in this case mechanosensitive rather than store-mediated Ca2+ channels are responsible for the extra Ca2+ influx. This conclusion is corroborated by other reports of mechanosensitive channels being blockable by amiloride (Hamill et al. 1992) and Gd3+ (Imbert et al. 1996). As to the entry of Mn2+ into myotubes, this may well occur via different channels.
Judging from the nearly identical quench rates remaining in the three different genotypes after application of each of the four tested blocking agents, we assume that the passageway is identical in MDX and control fibres, but of different activity. The increase in channel activity might rise by a much greater factor than two when MDX muscle is stressed by isometric or eccentric contractions. Thus it is conceivable that this overactivity effects the late muscle damage in the course of Duchenne muscular dystrophy.
As already reported (Krämer et al. 1998), the muscles of the myotonic-dystrophic double mutant ADR-MDX, though also completely lacking dystrophin, show fewer signs of fibre necrosis and fibrosis than pure MDX muscles. We have now shown that fibres of ADR-MDX muscle, compared with those of MDX muscle, have a lower fura-2 quench rate during exposure to Mn2+. The quench rate for ADR-MDX was close to that of controls indicating nearly normal expression and activity of stretch-activated channels in the fibres of the double mutant. Thus, there is a correlation between sarcolemmal permeability for divalent cations and severity of muscular dystrophy in the three tested genotypes. From these results it may be concluded that stretch-activated sarcolemmal ion channels are of pathophysiological relevance for dystrophin-deficient muscle. They may be responsible for subsarcolemmally increased Ca2+ causing damage to the sarcolemma, disturbances in signalling pathways and alterations in structures of the cytoskeleton near the plasma membrane.
In conclusion, we would like to stress that our results do not exclude the possibility that dystrophin-deficient muscle fibres may also develop lesions under mechanical stress.
Acknowledgments
This study was supported by the IZKF Ulm and the Wilhelm-Sander-Stiftung. The technical assistance of Ms Margit Rudolf-Dauner and Ms Silvia Gabriel is gratefully acknowledged.
References
- Bakker AJ, Head SI, Stephenson DG. Time course of calcium transients derived from Fura-2 fluorescence measurements in single fast twitch fibres of adult mice and rat myotubes developing in primary culture. Cell Calcium. 1997;21:359–364. doi: 10.1016/s0143-4160(97)90029-4. [DOI] [PubMed] [Google Scholar]
- Bakker AJ, Head SI, Williams DA, Stephenson DG. Ca2+ levels in myotubes grown from the skeletal muscle of dystrophic (mdx) and normal mice. The Journal of Physiology. 1993;460:1–13. doi: 10.1113/jphysiol.1993.sp019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Collatz MB, Rüdel R, Brinkmeier H. The intracellular calcium chelator BAPTA protects cells against toxic calcium overload but also alters physiological calcium responses. Cell Calcium. 1997;21:453–459. doi: 10.1016/s0143-4160(97)90056-7. [DOI] [PubMed] [Google Scholar]
- Even PC, Decrouy A, Chinet A. Defective regulation of energy metabolism in mdx-mouse skeletal muscles. Biochemical Journal. 1994;304:649–654. doi: 10.1042/bj3040649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts ME, Lømo T, Clausen T. Changes in K+, Na+ and calcium contents during in vivo stimulation of rat skeletal muscle. Acta Physiologica Scandinavica. 1993;147:357–368. doi: 10.1111/j.1748-1716.1993.tb09512.x. [DOI] [PubMed] [Google Scholar]
- Florence JM, Fox PT, Planer GJ, Brooke MH. Activity, creatine kinase, and myoglobin in Duchenne muscular dystrophy: a clue to etiology? Neurology. 1985;35:758–761. doi: 10.1212/wnl.35.5.758. [DOI] [PubMed] [Google Scholar]
- Fong PY, Turner PR, Steinhardt RA. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science. 1990;250:673–676. doi: 10.1126/science.2173137. [DOI] [PubMed] [Google Scholar]
- Franco-Obrégon AJ, Lansman JB. Mechanosensitive ion channels in skeletal muscle from normal and dystrophic mice. The Journal of Physiology. 1994;481:299–309. doi: 10.1113/jphysiol.1994.sp020440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailly P, Boland B, Himpens B, Casteels R, Gillis JM. Critical evaluation of cytosolic calcium determination in resting muscle fibres from normal and dystrophic (mdx) mice. Cell Calcium. 1993;14:473–483. doi: 10.1016/0143-4160(93)90006-r. 10.1016/0143-4160(93)90006-R. [DOI] [PubMed] [Google Scholar]
- Gillis JM. Membrane abnormalities and Ca homeostasis in muscles of the mdx mouse, an animal model of the Duchenne muscular dystrophy: a review. Acta Physiologica Scandinavica. 1996;156:397–406. doi: 10.1046/j.1365-201X.1996.201000.x. 10.1046/j.1365-201X.1996.201000.x. [DOI] [PubMed] [Google Scholar]
- Glesby MJ, Rosenmann E, Nylen EG, Wrogemann K. Serum CK, calcium, magnesium, and oxidative phosphorylation in mdx mouse muscular dystrophy. Muscle and Nerve. 1988;11:852–856. doi: 10.1002/mus.880110809. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of calcium indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hamill OP, Lane JW, McBride DW., Jr Amiloride: a molecular probe for mechanosensitive channels. Trends in Pharmacological Sciences. 1992;13:373–376. doi: 10.1016/0165-6147(92)90115-m. 10.1016/0165-6147(92)90115-M. [DOI] [PubMed] [Google Scholar]
- Haws CM, Lansman JB. Developmental regulation of mechanosensitive calcium channels in skeletal muscle from normal and mdx mice. Proceedings of the Royal Society of London B. 1991;245:173–177. doi: 10.1098/rspb.1991.0105. [DOI] [PubMed] [Google Scholar]
- Head SI. Membrane potential, resting calcium and calcium transients in isolated muscle fibres from normal and dystrophic mice. The Journal of Physiology. 1993;469:11–19. doi: 10.1113/jphysiol.1993.sp019801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head SI, Stephenson DG, Williams DA. Properties of enzymatically isolated skeletal fibres from mice with muscular dystrophy. The Journal of Physiology. 1990;422:351–367. doi: 10.1113/jphysiol.1990.sp017988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimann P, Augustin M, Wienecke S, Heising S, Jockusch H. Mutual interference of myotonia and muscular dystrophy in the mouse: a study on ADR.MDX double mutants. Neuromuscular Disorders. 1998;8:551–560. doi: 10.1016/s0960-8966(98)00079-0. 10.1016/S0960-8966(98)00079-0. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Gorospe JRM. The animal models of Duchenne muscular dystrophy: windows on the pathophysiological consequences of dystrophin-deficiency. Current Topics in Membranes. 1991;38:113–154. [Google Scholar]
- Hopf FW, Reddy P, Hong J, Steinhardt RA. A capacitative calcium current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. Journal of Biological Chemistry. 1996;271:22358–22367. doi: 10.1074/jbc.271.37.22358. 10.1074/jbc.271.37.22358. [DOI] [PubMed] [Google Scholar]
- Imbert N, Vandebrouck C, Constantin B, Duport G, Guillou C, Cognard C, Raymond G. Hypoosmotic shocks induce elevation of resting calcium level in Duchenne muscular dystrophy myotubes contracting in vitro. Neuromuscular Disorders. 1996;6:351–360. doi: 10.1016/0960-8966(96)00351-3. 10.1016/0960-8966(96)00351-3. [DOI] [PubMed] [Google Scholar]
- Jackson MJ, Jones DA, Edwards RH. Measurements of calcium and other elements in muscle biopsy samples from patients with Duchenne muscular dystrophy. Clinica Chimica Acta. 1985;147:215–221. doi: 10.1016/0009-8981(85)90202-5. 10.1016/0009-8981(85)90202-5. [DOI] [PubMed] [Google Scholar]
- Kargacin ME, Kargacin GJ. The sarcoplasmic reticulum calcium pump is functionally altered in dystrophic muscle. Biochimica et Biophysica Acta. 1996;1290:4–8. doi: 10.1016/0304-4165(95)00180-8. [DOI] [PubMed] [Google Scholar]
- Krämer R, Lochmüller H, Abicht A, Rüdel R, Brinkmeier H. Myotonic ADR-MDX mutant mice show less severe muscular dystrophy than MDX mice. Neuromuscular Disorders. 1998;8:542–550. doi: 10.1016/s0960-8966(98)00078-9. 10.1016/S0960-8966(98)00078-9. [DOI] [PubMed] [Google Scholar]
- Leijendekker WJ, Passaquin AC, Metzinger L, Rüegg UT. Regulation of cytosolic calcium in skeletal muscle cells of the mdx mouse under conditions of stress. British Journal of Pharmacology. 1996;118:611–616. doi: 10.1111/j.1476-5381.1996.tb15445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Schrank B, Waterston RH. Interaction between a putative mechanosensory membrane channel and a collagen. Science. 1996;273:361–364. doi: 10.1126/science.273.5273.361. [DOI] [PubMed] [Google Scholar]
- Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. Journal of Biochemistry (Tokyo) 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- Mayerhofer A, Föhr KJ, Sterzik K, Gratzl M. Carbachol increases intracellular free calcium concentrations in human granulosa-lutein cells. Journal of Endocrinology. 1992;135:153–159. doi: 10.1677/joe.0.1350153. [DOI] [PubMed] [Google Scholar]
- McArdle A, Edwards RH, Jackson MJ. How does dystrophin deficiency lead to muscle degeneration?- evidence from the mdx mouse. Neuromuscular Disorders. 1995;5:445–456. doi: 10.1016/0960-8966(95)00001-4. 10.1016/0960-8966(95)00001-4. [DOI] [PubMed] [Google Scholar]
- Menke A, Jockusch H. Extent of shock-induced membrane leakage in human and mouse myotubes depends on dystrophin. Journal of Cell Science. 1995;108:727–733. doi: 10.1242/jcs.108.2.727. [DOI] [PubMed] [Google Scholar]
- Metzinger L, Passaquin AC, Leijendekker WJ, Poindron P, Rüegg UT. Modulation by prednisolone of calcium handling in skeletal muscle cells. British Journal of Pharmacology. 1995;116:2811–2816. doi: 10.1111/j.1476-5381.1995.tb15930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak C, Wong S, Elson EL. Mechanical function of dystrophin in muscle cells. Journal of Cell Biology. 1995;128:355–361. doi: 10.1083/jcb.128.3.355. 10.1083/jcb.128.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proceedings of the National Academy of Sciences of the USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressmar J, Brinkmeier H, Seewald MJ, Naumann T, Rüdel R. Intracellular Ca2+ concentrations are not elevated in resting cultured muscle from Duchenne (DMD) patients and in MDX mouse muscle fibres. Pflügers Archiv. 1994;426:499–505. doi: 10.1007/BF00378527. [DOI] [PubMed] [Google Scholar]
- Rivet-Bastide M, Imbert N, Cognard C, Duport G, Rideau Y, Raymond G. Changes in cytosolic resting ionized calcium level and in calcium transients during in vitro development of normal and Duchenne muscular dystrophy cultured skeletal muscle measured by laser cytofluorimetry using indo-1. Cell Calcium. 1993;14:563–571. doi: 10.1016/0143-4160(93)90077-j. [DOI] [PubMed] [Google Scholar]
- Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Current Opinion in Neurology. 1997;10:168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- Turner PR, Fong PY, Steinhardt RA. Increased calcium influx in dystrophic muscle. Journal of Cell Biology. 1991;115:1701–1712. doi: 10.1083/jcb.115.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Neher E. Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. The Journal of Physiology. 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]