

Abstract
We have previously reported that brain angiotensin II type 2 receptors (AT2) contribute to immunological stress-induced hyperthermia (fever) in rats. Now, in mice, we report the effect of AT2 gene disruption on the hyperthermia induced by immunological (interleukin-1 (IL-1) injection) and non-immunological (saline injection or cage switch) stress.
AT2-deficient and control mice both showed typical circadian rhythmicity in body temperature and physical activity. During the latter half of the dark period, AT2-deficient mice exhibited a lower body temperature than the controls.
By comparison with the controls, AT2-deficient mice exhibited: (i) a significantly smaller hyperthermia after intraperitoneal (i.p.) injection of IL-1β; (ii) significantly greater increases in body temperature and physical activity after i.p. saline; and (iii) a significantly greater hyperthermia (but a similar increase in activity) during cage-switch stress.
These results suggest that AT2, presumably in the brain, plays important roles in stress-induced hyperthermia in mice.
It is widely accepted that the brain has its own renin- angiotensin system that is distinct from that in the periphery (Wright & Harding, 1994). There are two major types of angiotensin II (AII) receptors in the brain, AT1 and AT2 (Wright & Harding, 1994; Saavedra, 1994; Aguilera et al. 1995). AT1 mediates all the well-known central actions of AII, including those involved in blood pressure regulation. By contrast, the physiological roles played by the AT2 receptor in the brain are as yet poorly defined. Recently, two laboratories developed mice deficient in AT2. These mice showed (i) increased pressor responses to AII and a lower resting body temperature (Hein et al. 1995; Ichiki et al. 1995) and (ii) attenuated exploratory behaviour and a reduction in spontaneous movements. These findings suggest that endogenous AII regulates central nervous functions via AT2.
Animals exposed to stressors usually show activation of the sympathetic nervous system and the hypothalamo- pituitary adrenocortical axis, with consequent increases in body temperature, blood pressure, heart rate and the plasma concentration of adrenocorticotrophic hormone (McCubbin et al. 1991; Stanford & Samon, 1993). Recently, brain AII and its receptors have been shown to contribute to some stress-induced responses in rats. For example, we found that central administration of an AT2 antagonist, but not of an AT1 antagonist, attenuated the interleukin-1 (IL-1)-induced hyperthermic response (Watanabe et al. 1997). This suggests that AT2 in the brain may modulate (or mediate) the hyperthermia induced by immunological stress. On the other hand, to demonstrate inhibition of the hyperthermic, pressor and tachycardic responses to restraint stress required central injection of an AT1 antagonist (Saiki et al. 1997). Inhibition by an AT1 antagonist of the pressor response to heat or footshock stress has also been reported (Cierco & Israel, 1994; Kregel et al. 1994). Taken together, these findings suggest that brain AII and its receptors (AT1 and AT2) may play important roles in physiological reactions (such as body temperature increases) to several stress protocols in rats and they indicate the separate involvement of AT1 and AT2 in the effects induced by particular forms of stress.
The main aim of the present study was to investigate the role of AT2 in various types of stress-induced hyperthermia in mice. To that end, AT2-deficient mice and controls were exposed to both immunological (administration of IL-1) and non-immunological (saline injection or cage switch) stress. The results suggest that AT2, presumably in the brain, plays important roles in some types of stress-induced hyperthermia in mice.
METHODS
Animals
All the mice used in this study belonged to the C57BL/6 strain and were 19 weeks old. The generation of the AT2 gene-targeted mouse and its genetic background have been described in detail by Ichiki et al. (1995). The AT2-deficient mice and the controls were housed in individual plastic cages (28 cm × 16 cm × 13 cm; length × width × depth) with wood-chip bedding in a room maintained at 28 ± 1°C. They experienced a photoperiod of 12 h light : 12 h dark, lights coming on at 06.00 h. All animals had ad libitum access to drink and standard laboratory mouse chow. This experiment was reviewed by the Committee of the Ethics on Animal Experiment in Yamaguchi University School of Medicine and carried out under the control of the Guideline for Animal Experiments at Yamaguchi University School of Medicine and in accordance with Japanese Federal Law (no. 105) and Notification (no. 6) of the Japanese Government. At the end of the experiment the mice were killed by an overdose of the anaesthetic.
Surgery
Body temperature and physical activity were measured using a biotelemetry system (Data Science Inc., St Paul, MN, USA) (Lange et al. 1991). Under sodium pentobarbitone anaesthesia (50 mg kg−1, i.p.), a battery-operated transmitter (model TA10TA-F20) was implanted i.p. into each mouse. The transmitter included a sensor and a radio-frequency transmitter, and its output was monitored by antennae mounted on a receiver board (model RA1010) placed under the cage of each animal. The data were fed into a peripheral processor (matrix model BCM100) connected to a Sanyo MBC-17J AX computer (IBM compatible). At least 10 days were allowed to elapse between the surgery and the start of the experiment.
Drugs
The human recombinant IL-1β preparation, supplied by Otsuka Pharmaceutical (Tokushima, Japan), was produced from recombinant strains of Escherichia coli. Its activity was found to be 2 × 104 units μg−1 (thymocyte co-proliferation assay) and it was shown to be free of significant endotoxin contamination (< 0.05 pg μg−1 protein; Limulus amoebocyte assay).
Experimental protocols
All recordings were made from freely moving mice. Each mouse remained in its home cage (except in the cage-switch stress experiment). On the day of the experiment, each mouse was gently picked up and its transmitter switched on using a magnet. The body temperature was then allowed to stabilize for a period of 90 min before experimentation.
Experiment 1
After the stabilizing period of 90 min, diurnal changes in resting body temperature were recorded in mutant and control mice for 5 days.
Experiment 2
The effects of a single i.p. injection of IL-1β (10 μg kg−1) or saline on body temperature were investigated in control and mutant mice. To minimize the confusing effects of the circadian rhythm, IL-1β or saline was always given between 10.00 h and 11.00 h.
Experiment 3
The effect on the resting body temperature of a single exposure to cage-switch stress was examined in the two groups of mice. The procedures for body temperature recording were the same as in Experiment 2. The cage-switch stress was evoked by removing a given mouse from its home cage and placing it in another plastic cage of a larger size (40 cm × 25 cm × 25 cm; length × width × depth) without wood-chip bedding. The mouse remained in this cage for 60 min. This experiment was performed between 10.00 h and 12.00 h.
Statistical analysis
All results are expressed as means ±s.e.m. (except the circadian data, which are given as mean values). Data were analysed for statistical significance by a repeated-measures ANOVA followed by Scheffé‘s post hoc test; (Macintosh, StatView 4.0). Differences were considered significant at P < 0.05.
RESULTS
Diurnal changes in body temperature and physical activity
Figure 1 shows that control and AT2-deficient groups each showed typical circadian rhythmicity in body temperature and activity levels, both parameters increasing during the dark period. In AT2-deficient mice, body temperature was slightly (but not statistically) higher than in control mice during the first half of the dark period, but significantly (P < 0.05) lower than in control mice during the latter half. It should be noted that the activity of the AT2-deficient mice increased markedly just after the onset of the dark period. This increased activity during the first half (or third) of the dark period tended to be higher than that seen in the control mice, although the difference did not reach significance.
Figure 1. Diurnal changes in body temperature and physical activity in control and AT2-deficient mice.
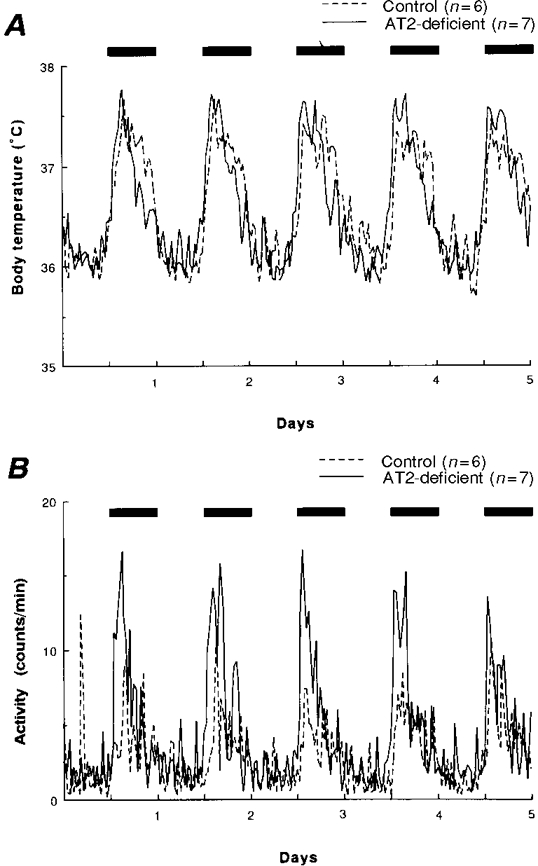
Mean changes in resting body temperature (A) and physical activity (B) in control and AT2-deficient mice over a 5 day period. The bars at the top of each panel indicate the periods of darkness. Each parameter was measured at 30 min intervals and the data for each group are presented as mean values.
Overall, the body temperature and activity levels were lower and more stable during the light period. Therefore, subsequent experiments (Figs 2–4) were performed during this period.
Figure 2. Body temperature and behavioural responses induced in control and AT2-deficient mice by intraperitoneal injection of interleukin-1 (IL-1).
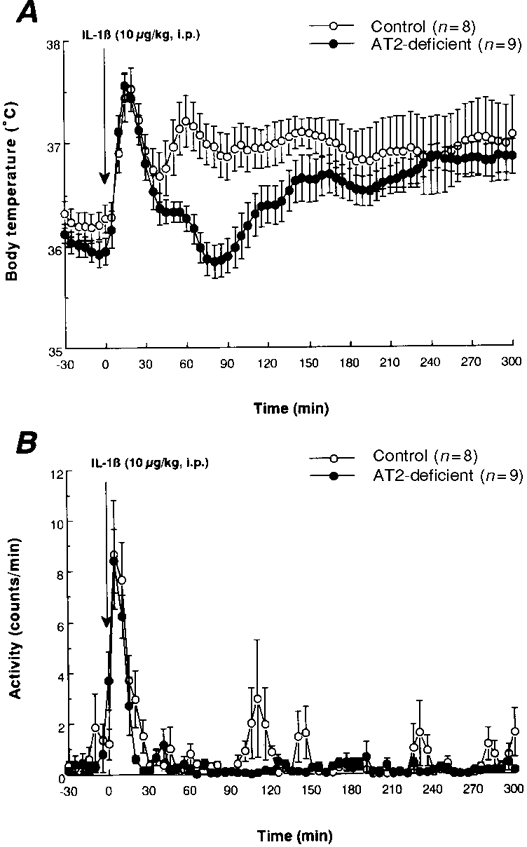
Mean values (±s.e.m.) obtained for body temperature (A) and physical activity (B) in control and AT2-deficient mice after i.p. injection of IL-1β (10 μg kg−1). Each parameter was measured at 5 min intervals.
Figure 4. Body temperature and behavioural responses induced in control and AT2-deficient mice by cage- switch stress.
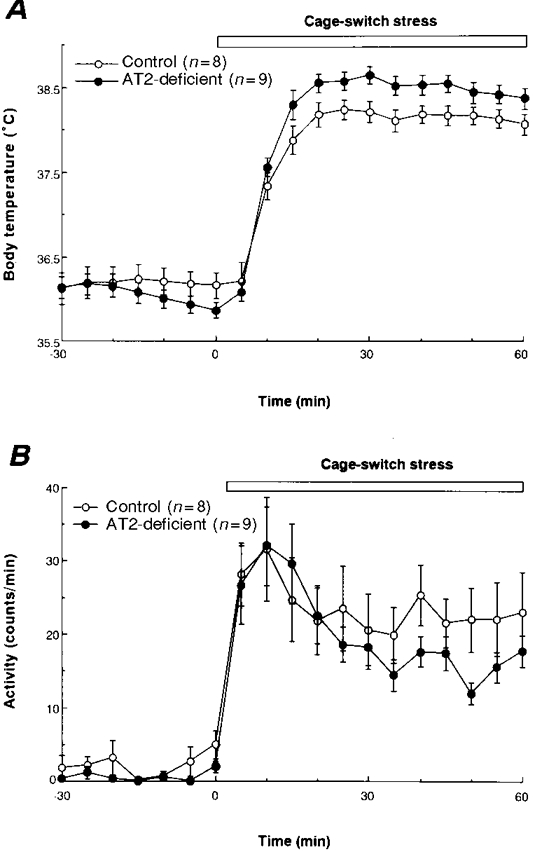
Mean values (±s.e.m.) obtained for body temperature (A) and physical activity (B) in control and AT2-deficient mice during exposure to cage-switch stress. Each parameter was measured at 5 min intervals.
Effect of i.p. injection of IL-1β on body temperature and physical activity
In the controls, a single injection of IL-1β (10 μg kg−1, i.p.) resulted in a biphasic hyperthermia which began after 5 min and reached its first peak at 15–20 min (Fig. 2A). The second peak occurred at around 60 min. The first phase involves an injection stress-induced hyperthermia and the second phase is an IL-1-induced hyperthermia (fever), because injection of saline (a vehicle for IL-1) evoked only the early increase in body temperature (see Fig. 3A). The IL-1-induced hyperthermia was significantly smaller in AT2-deficient mice (P < 0.05 for the period 30–150 min). By contrast, the first phase of the hyperthermia did not differ between the two groups of mice. The body temperature of the mutant mice was similar to that of the controls from 155 min onwards.
Figure 3. Body temperature and behavioural responses induced in control and AT2-deficient mice by intraperitoneal injection of saline.
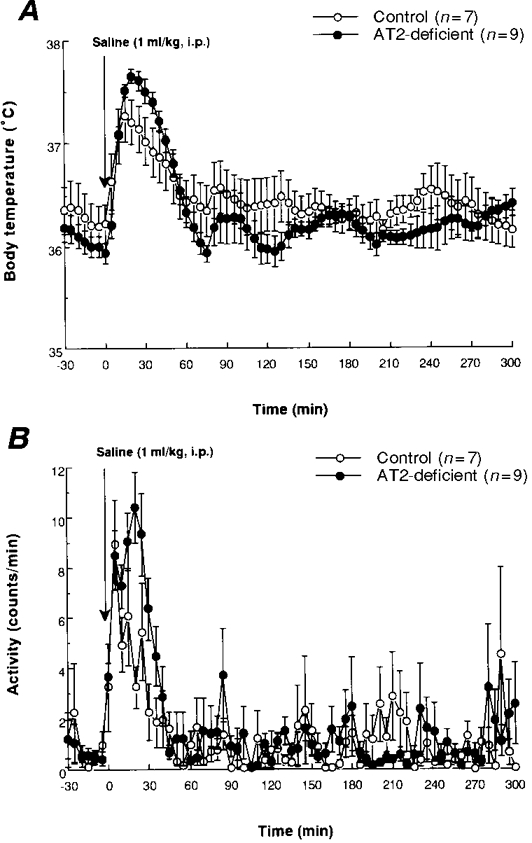
Mean values (±s.e.m.) obtained for body temperature (A) and physical activity (B) in control and AT2-deficient mice after i.p. injection of saline (1 ml kg−1). Each parameter was measured at 5 min intervals.
Immediately after the injection of IL-1, there was a marked increase in physical activity in each group (Fig. 2B). This change in activity might have been partly responsible for the first phase of the hyperthermia. There was no significant difference in physical activity between the groups after the injection of IL-1, except for the greater activity in the control mice seen at 20–25 min and 95–105 min.
Effect of i.p. injection of saline on body temperature and physical activity
As shown in Fig. 3A, i.p. injection of saline produced a monophasic increase in body temperature that was significantly greater in the mutant mice than in the controls (15–30 min).
A monophasic increase in physical activity was observed just after the saline injection in each group of mice (Fig. 3B). This early increase in activity was greater in the mutant mice than in the controls (P < 0.05 for the period 15–20 min).
Effect of cage-switch stress on body temperature and physical activity
Marked increases in body temperature and physical activity were observed during cage-switch stress in each group of mice (Fig. 4). The mutant mice showed a significantly greater body temperature response than the controls between 10 and 60 min (the end of the recording period), while there was no significant difference between the groups in terms of the increases in physical activity seen during this period.
DISCUSSION
The present results show that the AT2-deficient mice had a lower hyperthermia (fever) than the controls after an i.p. injection of IL-1β. These results suggest that AT2 contributes to the development of IL-1-induced hyperthermia in mice. In rats, central injection of an AT2 antagonist attenuates IL-1-induced hyperthermia (Watanabe et al. 1997), so it is likely that brain AT2 is important in inducing such hyperthermia in both mice and rats. In fact, the reduced response in AT2-deficient mice was observed only during the early stage of the hyperthermia (30–150 min). Similarly we previously found that only the early stage of the IL-1-induced hyperthermia in rats was attenuated by central injection of an AT2 antagonist (Watanabe et al. 1997). Thus, it may be that activation of AT2 is one of a number of factors contributing to immunological stress-induced hyperthermia and that its role lies mainly in the early stage of the hyperthermia.
By contrast, in the present study the mutant mice showed a greater increase in body temperature than the control mice following either a single i.p. injection of saline or exposure to cage-switch stress. This suggests that AT2 mediates attenuation of non-immunological stress-induced hyperthermia in mice. Alternatively, for the following reasons the hyperthermic response may be dependent on the balance between AT1- and AT2-mediated effects. In rats, restraint-stress-induced hyperthermia was reduced following a central injection of an AT1 antagonist (Saiki et al. 1997), indicating a contribution of brain AT1 to non-immunological stress-induced hyperthermia. If this is so, our AT2-deficient mice might have experienced a greater hyperthermia because the action of AII on AT1 within the brain was not opposed by its action on AT2. Ichiki et al. (1995) and Hein et al. (1995) demonstrated an increased vasopressor response to AII in AT2-deficient mice. Their finding is similar to the present one in that it suggests that AII may act through two receptors with opposing effects, the pressor response mediated through AT1 activation being unopposed in AT2-deficient individuals. Further, it has recently been suggested that there is an interaction or cross-talk between AT1 and AT2 during their activation (Gelband et al. 1997, 1998). Thus the response actually evoked by AII may reflect the net effect of AT1 and AT2 activation.
In the present study an i.p. injection of saline produced a greater increase in physical activity in AT2-deficient mice than in the controls, an effect that might in part be responsible for the greater evoked hyperthermia in the mutant group. However, when mice were exposed to cage-switch stress, AT2-deficient mice exhibited a greater hyperthermia than the controls, although there was no difference in physical activity between the two groups. To judge from these results, the central mechanisms regulating physical activity may act in concert with other mechanisms to determine the magnitude of the hyperthermia seen in AT2-deficient mice during non-immunological stress.
It was reported in the 1980s that central injection of ‘exogenous’ AII induced hypothermia in sheep and rats (Huang et al. 1985; Shido et al. 1985; Wilson & Fregly, 1985). However, in the study by Shido et al. (1985) the hypothermia induced by intracerebroventricular injection of a large dose of AII was reduced by sinoaortic denervation. This indicates that the large dose of AII markedly increased blood pressure and thus produced a baroreceptor reflex response (including an inhibition of sympathetic nervous activity) which resulted in the hypothermia. Thus, the role of ‘endogenous’ brain AII in thermoregulation remained unclear. In the present study the diurnal rhythm of the mutant mice led to an increased body temperature just after the onset of darkness. This might have been secondary to the increase in physical activity consequent upon the arousal caused by the lights being switched off. However, the AT2-deficient mice had a significantly lower body temperature than the controls during the latter half of the dark period, a time at which there was no difference in physical activity between the two groups. This result suggests that AT2 may participate directly in the regulation of normal body temperature in mice. Future studies should examine the mechanism underlying this presumed role of AT2.
The present results represent the first evidence in mice that AT2 participates in the induction of some types of stress-related hyperthermia. However, we should keep in mind the possibility that some unknown compensatory reactions might occur in mice with a targeted disruption of a certain gene. In future, we should examine the role of AT2 in stress-related hyperthermia using transgenic mice that, for example, over-express the AT2 gene.
Acknowledgments
We are grateful to Dr Robert J. Timms for his critical reading of the English manuscript. This work was partly supported by the Ministry of Education, Science and Culture with a Grant-in-Aid for Scientific Research (C09670071).
References
- Aguilera G, Kiss A, Luo X, Akbasak BS. The renin angiotensin system and the stress response. Annals of the New York Academy of Sciences. 1995;771:173–86. doi: 10.1111/j.1749-6632.1995.tb44679.x. [DOI] [PubMed] [Google Scholar]
- Cierco M, Israel A. Role of angiotensin AT1 receptor in the cardiovascular response to footshock. European Journal of Pharmacology. 1994;251:103–106. doi: 10.1016/0014-2999(94)90450-2. [DOI] [PubMed] [Google Scholar]
- Gelband CH, Sumners C, Lu D, Raizada MK. Angiotensin receptors and norepinephrine neuromodulation: implications of functional coupling. Regulatory Peptides. 1998;73:141–147. doi: 10.1016/s0167-0115(97)11050-3. 10.1016/S0167-0115(97)11050-3. [DOI] [PubMed] [Google Scholar]
- Gelband CH, Zhu M, Lu D, Reagan LP, Fluharty SJ, Posner P, Raizada MK, Sumners C. Functional interactions between neuronal AT1 and AT2 receptors. Endocrinology. 1997;138:2195–2198. doi: 10.1210/endo.138.5.5236. 10.1210/en.138.5.2195. [DOI] [PubMed] [Google Scholar]
- Hein L, Barsh GS, Pratt RE, Dzau VJ, Kobilka BK. Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor gene in mice. Nature. 1995;377:744–747. doi: 10.1038/377744a0. 10.1038/377744a0. [DOI] [PubMed] [Google Scholar]
- Huang BS, Kluger MJ, Malvin RL. Thermal responses to central angiotensin II, SQ 20881, and dopamine infusions in sheep. American Journal of Physiology. 1985;248:R371–377. doi: 10.1152/ajpregu.1985.248.3.R371. [DOI] [PubMed] [Google Scholar]
- Ichiki T, Labosky PA, Shiota C, Okuyama S, Imagawa Y, Fogo A, Niimura F, Ichikawa I, Hogan BLM, Inagami T. Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type-2 receptor. Nature. 1995;377:748–750. doi: 10.1038/377748a0. 10.1038/377748a0. [DOI] [PubMed] [Google Scholar]
- Kregel KC, Stauss H, Unger T. Modulation of autonomic nervous system adjustments to heat stress by central ANGII receptor antagonism. American Journal of Physiology. 1994;266:R1985–1991. doi: 10.1152/ajpregu.1994.266.6.R1985. [DOI] [PubMed] [Google Scholar]
- Lange J, Brockway B, Azar S. Telemetric monitoring of laboratory animals: an advanced technique that has come of age. Lab Animal – New York. 1991;20:28–34. [Google Scholar]
- McCubbin JA, Kaufmann PG, Nemeroff CB. Stress, Neuropeptides and Systemic Disease. 1. New York: Academic Press; 1991. [Google Scholar]
- Saavedra JM. Brain angiotensin II receptor subtypes. In: Saavedra JM, Timmermans PBMWM, editors. Angiotensin Receptors. New York: Plenum Press; 1994. pp. 151–175. [Google Scholar]
- Saiki Y, Watanabe T, Tan N, Matsuzaki M, Nakamura S. Role of central ANGII receptors in stress-induced cardiovascular and hyperthermic responses in rats. American Journal of Physiology. 1997;272:R26–33. doi: 10.1152/ajpregu.1997.272.1.R26. [DOI] [PubMed] [Google Scholar]
- Shido O, Kifune A, Nagasaka T. Contributions of baroreceptor reflex to the hypothermic effects of intraventricular angiotensin II in rats. Japanese The Journal of Physiology. 1985;35:301–309. doi: 10.2170/jjphysiol.35.301. [DOI] [PubMed] [Google Scholar]
- Stanford SC, Samon P. Stress (from Synapse to Syndrome) 1. New York: Academic Press; 1993. [Google Scholar]
- Watanabe T, Saiki Y, Sakata Y. The effect of central angiotensin II receptor blockade on interleukin-1β- and prostaglandin E-induced fevers in rats: possible involvement of brain angiotensin II receptor in fever induction. Journal of Pharmacology and Experimental Therapeutics. 1997;282:873–881. [PubMed] [Google Scholar]
- Wilson KM, Fregly MJ. Factors affecting angiotensin II-induced hypothermia in rats. Peptides. 1985;6:695–701. doi: 10.1016/0196-9781(85)90174-3. [DOI] [PubMed] [Google Scholar]
- Wright JW, Harding JW. Brain angiotensin receptor subtypes in the control of physiological and behavioral responses. Neuroscience and Biobehavioral Reviews. 1994;18:21–53. doi: 10.1016/0149-7634(94)90034-5. [DOI] [PubMed] [Google Scholar]