

Abstract
When intracellular recordings were made from preparations of rat anococcygeus muscle, transmural nerve stimulation evoked noradrenergic excitatory junction potentials (EJPs) made up of two distinct components. Both components were abolished by either guanethidine or α-adrenoceptor antagonists, indicating that they resulted from the release of transmitter from sympathetic nerves and the subsequent activation of α-adrenoceptors.
The first component was associated with a transient increase in the intracellular concentration of calcium ions ([Ca2+]i) and a contraction. Although the second component was often associated with a long lasting increase in [Ca2+]i it was not associated with a contraction unless the second component initiated an action potential.
The increase in [Ca2+]i associated with the first component resulted from Ca2+ release from an intracellular store and from entry of Ca2+ through voltage-dependent Ca2+ channels. The increase in [Ca2+]i associated with the second component resulted only from the entry of Ca2+ through L-type Ca2+ channels (CaL channels). The depolarization associated with the initial increase in [Ca2+]i was abolished by reducing the external concentration of chloride ions ([Cl−]o), suggesting that it involved the activation of a Cl− conductance.
When the relationships between changes in [Ca2+]i, membrane depolarization and contraction produced by an increasing number of sympathetic nerve stimuli were determined in control, and caffeine- and nifedipine-containing solutions, it was found that an increase in [Ca2+]i recorded in nifedipine produced a larger contraction and larger membrane depolarization than did a similar increase in [Ca2+]i recorded in either control or caffeine-containing solutions. These observations indicate that Ca2+ released from stores more readily triggers contraction and membrane depolarization than does Ca2+ entry via CaL channels.
The anococcygeus muscle receives a dense sympathetic innervation which when stimulated causes a contraction (Gillespie & Maxwell, 1971). Using electrophysiological recording techniques, it has been shown that transmural nerve stimulation evokes a noradrenergic excitatory junction potential (EJP) which has a latency of several hundred milliseconds and a duration of about 1 s (Creed et al. 1975). EJPs were abolished by guanethidine and by α-adrenoceptor antagonists indicating that they result from sympathetic nerve stimulation and involve the activation of α-adrenoceptors (Creed et al. 1975; Large, 1982). The noradrenergic EJPs evoked by sympathetic nerve stimulation can be mimicked by the ionophoretic application of noradrenaline (Large, 1982). These depolarizations were abolished when the external concentration of chloride ions ([Cl−]o) was reduced (Large, 1984). Since contractions to noradrenaline could still be detected after the associated depolarizations had been abolished, it was suggested that noradrenaline releases Ca2+ from intracellular stores (Large, 1982, 1984). In control solutions the resulting increase in the intracellular concentration of Ca2+ ([Ca2+]i) will trigger a contraction and at the same time activate Ca2+-activated Cl− channels to give a depolarization (Byrne & Large, 1987). It was also found that bath-applied noradrenaline produced a two-component depolarization, with both components resulting from the activation of α-adrenoceptors (Byrne & Large, 1985). Whereas the initial component was abolished when [Cl−]o was reduced to low levels, the second component was unaffected by these changes in [Cl−]o (Byrne & Large, 1985). Using isolated cells it has more recently been shown that as well as activating a Cl− conductance, applied catecholamines activate non-selective cation channels in several different types of smooth muscle (Wang & Large, 1991).
Frequently the α-adrenoceptors which trigger smooth muscle contractions activate phospholipase C, causing the formation of inositol 1,4,5 trisphosphate (IP3) (Berridge, 1993). IP3 interacts with receptors on the sarcoplasmic reticulum and releases Ca2+ from intracellular stores. The same metabolic pathway also leads to the formation of diacylglycerol which activates protein kinase C leading to phosphorylation of L-type Ca2+ channels (CaL channels) (McDonald et al. 1994). These channels then open more readily at negative membrane potentials and allow the entry of Ca2+ (McDonald et al. 1994). Thus the increase in [Ca2+]i that triggers contraction may arise from the release of Ca2+ from intracellular stores or from the entry of Ca2+ from the extracellular fluid.
The present study has re-examined the responses to sympathetic nerve stimulation in the rat anococcygeus muscle and found that sympathetic nerve stimulation initiates an EJP with two distinct components that depend upon the activation of α1A-adrenoceptors. The first component was closely associated with an increase in [Ca2+]i and a contraction whereas the second, although sometimes associated with an increase in [Ca2+]i, was not associated with a contraction unless an action potential occurred. The initial increase in [Ca2+]i resulted from the release of Ca2+ from an intracellular store and from the opening of CaL channels. However, Ca2+ released from the intracellular store was more effective at producing both a contraction and a membrane depolarization than was Ca2+ entering via CaL channels.
METHODS
Male rats (age 4-6 weeks, weight 100-140 g) were stunned and exsanguinated according to the guidelines laid down by Melbourne University Animal Ethics Committee. One of the anococcygeus muscles was rapidly removed and pinned in a shallow recording chamber, volume 0.5 ml. The base of the chamber consisted of a microscope cover slip coated with Sylgard silicone resin (Dow Corning Corp., Midland, MI, USA) that could be viewed using an inverted compound microscope. Preparations were superfused with physiological saline, composition (mM): NaCl, 120; NaHCO3, 25; NaH2PO4, 0.1; KCl, 5; MgCl2, 2; CaCl2, 2.5; glucose, 11.1; that had been gassed with 95 % O2-5 % CO2 and warmed to 35°C. In ten experiments [Cl−]o was reduced by substituting NaCl with equimolar amounts of either sodium isethionate (Sigma) or sodium methyl sulphonate (Aldrich Chemical Co., Milwaukee, WI, USA); to compensate for the resulting changes in free [Ca2+]o, CaCl2 was increased to 3.2 mM.
In the initial experiments, membrane potential changes and contractile responses were measured simultaneously. Subsequently, preparations were also loaded with fura-2, so allowing changes in [Ca2+]i to be measured concurrently (Fig. 1). In each experiment, intracellular recordings were made from the muscle layer using high resistance (100-200 MΩ) glass microelectrodes filled with 0.5 M KCl. Membrane potential changes were measured using a conventional amplifier (Axoclamp-2, Axon Instruments), all recordings were low-pass filtered, cut-off frequency 1 kHz, digitized at 500 Hz and stored for later analysis (Fig. 1A). Contractile responses were measured by using a transducer located in the anal end of the muscle; the output was low-pass filtered, cut-off frequency 10 Hz, digitized at 500 Hz and stored for later analysis (Fig. 1A). Transmural nerve stimulation was achieved by use of a pair of platinum stimulating electrodes, one set into the base of the recording chamber and the other positioned over the coccygeal end of the muscle. At the start of each experiment, the preparations were stimulated with 2 impulses (pulse duration 0.1 ms, stimulus frequency 10 Hz) and the stimulus strength increased until a maximal membrane potential change was detected. The stimulus strength was increased by a further 10 % and then left unchanged throughout the experiment. In experiments where contractile responses were measured, it was found that stimuli, applied in this way, provided they were brief (< 0.2 ms) and had strengths not more than double the maximal for the sympathetic nerves, failed to excite the nitrergic inhibitory nerves found in this tissue (Dail et al. 1993). Changes in [Ca2+]i were determined using a ratiometric method (Cousins et al. 1993, 1995). Preparations, loaded with fura-2, were illuminated with two periods of light, wavelengths 340 and 380 nm, alternating at a frequency of 30 Hz. Photons, emitted at a wavelength of 510 nm, were counted during each period of illumination to give separate outputs reflecting the increase in concentration of the fura-Ca2+ complex (Fig. 1Da) and the decrease in free-fura concentration (Fig. 1Db). A measure of changes in [Ca2+]i was obtained by taking a ratio of these values (Fig. 1Dc). In all subsequent illustrations only the ratio of emitted light intensities produced by illuminating with 340 and 380 nm light will be given. These conditions allowed changes in [Ca2+]i to be detected with a temporal accuracy of 33 ms. In a few early experiments, the switching rate between 340 and 380 nm wavelength light was increased to 100 Hz; this did not alter the time course of the recorded change in [Ca2+]i, indicating that the lower sampling rate provided an accurate measure of the time course of the change in [Ca2+]i evoked by sympathetic nerve stimulation. No attempt has been made to correlate the ratio values with absolute changes in [Ca2+]i. With this method, an area (50 μm2) of smooth muscle syncytium was illuminated and it was not possible to differentiate between uniform and non-uniform changes in [Ca2+]i occurring within that area.
Figure 1. Simultaneous recording of changes in [Ca2+]i, membrane potential and contraction from rat anococcygeus muscle in response to sympathetic nerve stimulation.
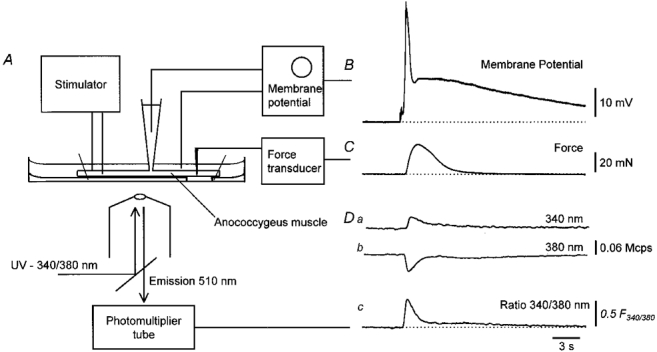
A schematic diagram of the experimental set-up is shown in A. An isolated muscle loaded with fura-2 was pinned out and viewed with an inverted compound microscope. Sympathetic nerves were stimulated with a pair of stimulating electrodes and the changes in membrane potential recorded with an intracellular recording electrode. Contractile responses were measured by attaching a transducer near to the anal end of the muscle. Changes in [Ca2+]i were determined ratiometrically. The response to three stimuli, delivered to the sympathetic nerves at 10 Hz, is shown in the right hand section of the figure. It can be seen that the train of stimuli evokes a biphasic depolarization (B) and a contraction (C). At the same time as the first component of membrane potential occurs, light emitted at wavelength 510 nm, when illuminating a small region of the preparation with light of wavelength 340 nm, increases (Da) and light emitted at wavelength 510 nm, when illuminating with light of wavelength 380 nm, falls (Db). The resting membrane potential in B was -74 mV. The time course of the change in [Ca2+]i (Dc) is given by taking the ratio of Da and Db in units of millions of counts per second. The time calibration bar applies to all traces.
Preparations to be loaded with fura-2 were again pinned out in the recording chamber. They were then incubated in physiological saline which contained fura-2 AM (5 μM), solubilized with the detergent pluronic F-127 (0.01 %), but otherwise had the same composition as given above except that CaCl2 was reduced to 0.5 mM. Preparations were incubated for 1 h at 20°C with the pH of the solution being kept constant by blowing 95 % O2-5 % CO2 over the surface of the recording chamber. Attempts to load the preparation with solutions using Hepes as a buffer, with or without NaHCO3, were unsuccessful. Although adequate loading was invariably achieved, the preparations had depolarized membrane potentials: similar depolarizations were observed when tissues were superfused with fura-2 AM/pluronic acid-free Hepes-buffered solutions. After loading, preparations were warmed to 30°C for 15 min in the fura-2 AM solution and then superfused with normal physiological saline, bath temperature 35°C, for 1 h before recordings were commenced.
Drugs used in this study were guanethidine, prazosin hydrochloride, phentolamine mesylate, WB4101, ryanodine, nifedipine hydrochloride, Hepes buffer and caffeine (Sigma Chemicals). Preparations were loaded using fura-2 AM and pluronic F-127 obtained from Molecular Probes Inc., Eugene, OR, USA.
RESULTS
General observations
When intracellular recordings were made from preparations of rat anococcygeus muscle, the membrane potential was invariably stable. The resting membrane potentials of non-fura-loaded preparations lay in the range -69 to -82 mV (-76.1 ± 0.6 mV, n = 37; mean ±s.e.m.; where n is the number of observations, and each n value in this and every other case represents an observation made on a separate preparation). Preparations which had been loaded with fura-2 had stable resting membrane potentials in the range -55 to -79 mV (-69.5 ± 1.4 mV, n = 20). Transmural stimulation with supramaximal stimuli triggered a series of membrane depolarizations which were abolished by adding guanethidine (10 μM) to the perfusion fluid. Most frequently the stimuli triggered a two-component EJP; however, the form of the EJP changed with the number of stimuli presented. In general, EJPs consisted of an initial phase of depolarization, which reached its peak amplitude after about 1 s and had a total duration of some 2 s; this depolarization will be referred to as the first component (Fig. 1A). The first component was invariably followed by a secondary sustained component of depolarization which had a duration of about 60 s; this depolarization will be referred to as the second component (Fig. 1A). With a single stimulus, the second component was seen to reach its peak amplitude after 6 to 8 s (Fig. 2Aa) and decayed over the next 20-30 s with a time constant of about 7 s (7.4 ± 0.7 s, n = 6). With more stimuli, its time course became more complex and the secondary component merged into the first component (Fig. 2Ca). Occasionally these components were preceded by EJPs which had amplitudes, when triggered by single stimuli, of 2-5 mV, rapid rising phases of 50-80 ms and total durations of about 400 ms. These EJPs, which appear to result from the release of ATP (Byrne & Large, 1984), were detected in less than half (22 out of 57) of the preparations examined (see also Byrne & Large, 1984) and were never associated with contraction or a change in [Ca2+]i. They have not been considered further in this study.
Figure 2. Changes in [Ca2+]i, membrane potential and contraction evoked by increasing numbers of sympathetic nerve stimuli in the rat anococcygeus muscle.
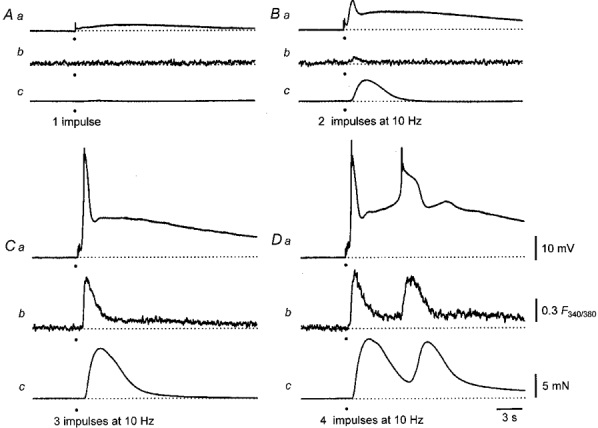
The responses to a single impulse are shown in A. A small long lasting depolarization (Aa) is associated with neither a change in [Ca2+]i (Ab) nor with a contraction (Ac). A pair of impulses triggers a two-component EJP (Ba), a small transient increase in [Ca2+]i (Bb) and a contraction (Bc). Increasing the number of stimuli to three triggers a larger two-component EJP (Ca), a large transient and a small persistent increase in [Ca2+]i (Cb) but only the transient increase in [Ca2+]i is associated with a contraction (Cc). A train of four sympathetic impulses again initiates a large two-component EJP and a muscle action potential (Da). Both the initial component and the muscle action potential are associated with large increases in [Ca2+]i (Db) and both trigger contractions (Dc). The resting membrane potential in all traces was -74 mV. The time calibration bar applies to all traces. The voltage calibration bar refers to all membrane potential recordings, the fluorescence calibration bar applies to all ratiometric measurements and the force calibration bar applies to all recordings of contractions.
Trains of stimuli, delivered to the sympathetic nerves at 10 Hz, also evoked increases in [Ca2+]i and associated contractions. An experiment is illustrated in Fig. 2. A single stimulus initiated little or no first component, no detectable change in [Ca2+]i and no contraction. A clearly defined second component was associated with neither an increase in tension nor a change in [Ca2+]i (Fig. 2A). Two stimuli initiated a first component with an amplitude of about 10 mV which was associated with a small increase in [Ca2+]i and a contraction. Although the amplitude of the second component was also increased it was not associated with a contraction nor a change in [Ca2+]i (Fig. 2A). When three stimuli were delivered, the peak amplitudes of the first component and the change in [Ca2+]i increased to their maximum values (Fig. 2A). The amplitude of the second component also increased and it was now accompanied by a small sustained increase in [Ca2+]i which did not trigger a contraction (Fig. 2A). With four stimuli, the peak amplitude of the first component was much the same as that produced by three stimuli. In this experiment the amplitude of the second component was further increased to such a level that it initiated an action potential (Fig. 2Da). Both the initial component and the action potential were associated with large increases in [Ca2+]i (Fig. 2Db). It should also be noted that even though the sustained increase in [Ca2+]i detected during the second component in response to three stimuli was larger than that detected during the first component in response to two stimuli, it failed to produce a contraction (Fig. 2).
This general pattern of behaviour was noted in each of the 57 preparations examined. However, the number of stimuli required to initiate threshold responses varied from preparation to preparation. In some preparations a single stimulus initiated a well-defined first component and a contraction; in these preparations a pair of stimuli gave a maximal depolarization and a large contraction. In other preparations, a pair of stimuli gave barely detectable first components and four stimuli were required to initiate a maximal depolarization. Whereas the amplitude of the first component of the EJP (see also Large, 1982) and associated contraction increased dramatically with increasing numbers of stimuli, the increase in the peak amplitude of the second component of the EJP was more modest (Fig. 2). Thus when the number of stimuli was increased, the amplitude of the first component increased rapidly towards a maximum of about 50 mV whereas the amplitude of the second component increased more linearly with the number of stimuli presented.
Together the observations indicate that sympathetic nerve stimulation initiates a two-component membrane depolarization but that the two components show different patterns of facilitation following repetitive sympathetic nerve stimulation. Furthermore only the first component was consistently associated with an increase in [Ca2+]i and contraction.
Effect of nifedipine on responses to sympathetic nerve stimulation
Since the membrane potential changed from a resting value of -75 mV to about -25 mV during the peak depolarization of the EJP triggered by repetitive nerve stimulation, it seemed likely that voltage-dependent CaL channels would be activated and contribute to both depolarization and contraction. Moreover in view of the abrupt way in which the initial component increased in amplitude, it was considered possible that the increase in amplitude of the first component largely reflected the recruitment of CaL channels. To examine this possibility the effect of nifedipine on the responses to sympathetic nerve stimulation was examined. In preliminary experiments it was found that nifedipine, applied at a concentration of 0.3 μM, abolished the contractile responses produced by increasing the external concentration of potassium ions ([K+]o). Thus in experiments where CaL channels were to be blocked a supramaximal concentration of nifedipine (1 μM) was routinely added to the physiological saline. When this was done, nifedipine reduced the amplitude of the first component along with the associated increase in [Ca2+]i and contraction (Fig. 3A and B). Nevertheless when the number of stimuli was increased further, it was possible to partly restore the amplitude of the first component (see Fig. 8). Nifedipine abolished any increases in [Ca2+]i associated with the second component (Fig. 3A). If the second component was sufficiently large to initiate an action potential, nifedipine readily abolished the action potential. Under these conditions the peak amplitude of the second component increased linearly with the number of stimuli present over a wide range.
Figure 3. Effect of nifedipine on changes in [Ca2+]i, membrane potential and contraction evoked by sympathetic nerve stimulation in the rat anococcygeus muscle.
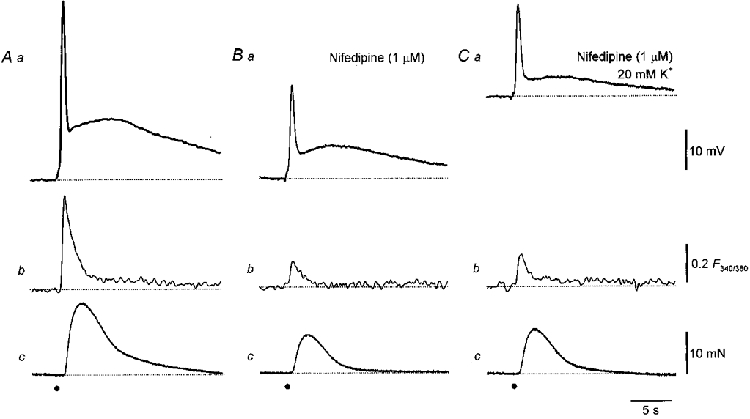
Three impulses triggered a biphasic EJP (Aa), a transient and persistent increase in [Ca2+]i (Ab) and a contraction (Ac). Nifedipine (1 μM) reduced the amplitudes of the initial component (Ba), the change in [Ca2+]i (Bb) and of the contraction (Bc) and abolished the persistent increase in [Ca2+]i associated with the second component. The resting membrane potential in traces A and B was -71 mV. When [K+]o was increased to 20 mM, the membrane potential depolarized by about 20 mV (C). Even though nifedipine was still present, the stimuli continued to initiate a biphasic EJP (Ca), a transient increase in [Ca2+]i (Cb) and a contraction (Cc). The time calibration bar applies to all traces. The voltage calibration bar refers to all membrane potential recordings, the fluorescence calibration bar applies to all ratiometric measurements and the force calibration bar applies to all recordings of contractions.
Figure 8. Relationships between changes in [Ca2+]i, membrane potential and contraction and number of sympathetic nerve stimuli delivered in control solution and after preventing Ca2+ entry.
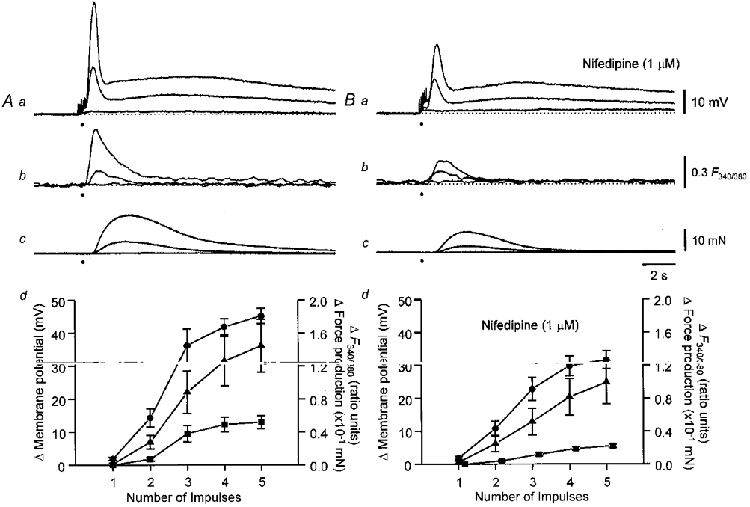
Recordings obtained in control solution are shown in A. The upper three families of traces show membrane potential changes (Aa), changes in [Ca2+]i (Ab), and contractions (Ac) initiated by 1, 2 and 5 stimuli. The graph (Ad) shows the dependence of peak amplitude of the initial phase of depolarization (0), peak contraction (8), and peak increase in [Ca2+]i (▪) upon number of stimuli from the experimental series of six. Comparable recordings obtained in nifedipine- (1 μM) containing solutions are shown in B. The graph (Bd) shows the dependence of peak amplitude of the initial phase of depolarization (0), peak contraction (8) and peak increase in [Ca2+]i (▪) upon the number of stimuli, from the same experimental series recorded in the presence of nifedipine. In both graphs the vertical bars represent ±s.e.m.; n = 6. The resting membrane potential in all traces was -71 mV. The time calibration bar refers to all traces. The voltage calibration bar refers to all membrane potential recordings, the fluorescence calibration bar applies to all ratiometric measurements and the force calibration bar applies to all recordings of contractions.
It has been shown that the block of CaL channels by nifedipine is voltage dependent (Nelson & Worley, 1989). To check whether the residual responses recorded in the presence of nifedipine occurred because nifedipine was unable to block CaL channels at negative membrane potentials, responses were recorded with nifedipine added to the physiological saline and after the membrane potential had been depolarized by increasing [K+]o to 20 mM. In five experiments the resting membrane potential in control solution was -72.6 ± 1.3 mV; with [K+]o increased, the resting membrane potential was -53.2 ± 3.2 mV. In each experiment, after [K+]o had been increased, sympathetic nerve stimulation continued to initiate a biphasic EJP, an increase in [Ca2+]i and a contraction (Fig. 3A). In control solution the peak increase in [Ca2+]i initiated by three stimuli in the presence of nifedipine was 0.19 ± 0.03 F340/F380; with [K+]o increased, the same trains of stimuli caused an increase in [Ca2+]i of 0.21 ± 0.05 F340/F380.
Together these experiments indicate that nifedipine is effective at blocking the entry of Ca2+ through voltage-dependent channels in this tissue. They confirm that the EJP consists of two distinct components and also show that the contraction triggered by the first component results only in part from Ca2+ entry through CaL channels. In contrast any increase in [Ca2+]i associated with the second component resulted entirely from Ca2+ entry through CaL channels.
Effect of α-adrenoceptor antagonists on responses to sympathetic nerve stimulation
The effects of a series of α-adrenoceptor antagonists on the responses to sympathetic nerve stimulation were examined in tissues where voltage-dependent Ca2+ entry had been abolished by adding nifedipine (1 μM), to the physiological saline. Both components of the response to sympathetic nerve stimulation depended upon the activation of α-adrenoceptors. Phentolamine (1 μM) rapidly abolished both the EJP and the associated contraction in the three preparations examined (Fig. 4A). When present, purinergic EJPs persisted in the presence of phentolamine (Fig. 4A; see also Byrne & Large, 1984). Both the noradrenergic components of the EJP and the associated contractions resulted from the activation of an α1-adrenoceptor as the amplitudes of each were reduced by very low concentrations of prazosin (0.3 to 1 nM; Fig. 4A). The first component of the EJP and associated contraction was reduced by a greater proportion than the second component (Fig. 4A). Thus prazosin (0.3 nM) reduced the peak amplitude of the first component to 10 ± 5 % (n = 4) of control, the peak amplitude of the contraction to 7 ± 3 % but that of the second component only to 35 ± 9 % of control. When prazosin (1 nM) was added to the physiological saline the first component could not be detected whereas a part of the second component was invariably present; even a concentration of 10 nM prazosin failed to abolish this component (Fig. 4A). The idea that the adrenoceptors activated by sympathetic nerve stimulation were of the α1-subtype was confirmed when the effects of yohimbine on the two components of the EJP and associated contractions were examined. Concentrations of yohimbine less than 10 nM failed to reduce the amplitudes of the first and second component. Concentrations in the range 10-100 nM reduced both components and the associated contractions (Fig. 4A). At these concentrations, yohimbine has been shown to block α1-adrenoceptors (Lomasney et al. 1991). Again, when high concentrations of yohimbine were applied, the first component and associated contractions were reduced to a greater extent than was the second component (Fig. 4A).
Figure 4. The effect of phentolamine, prazosin and yohimbine on EJPs and associated contractions evoked by sympathetic nerve stimulation.
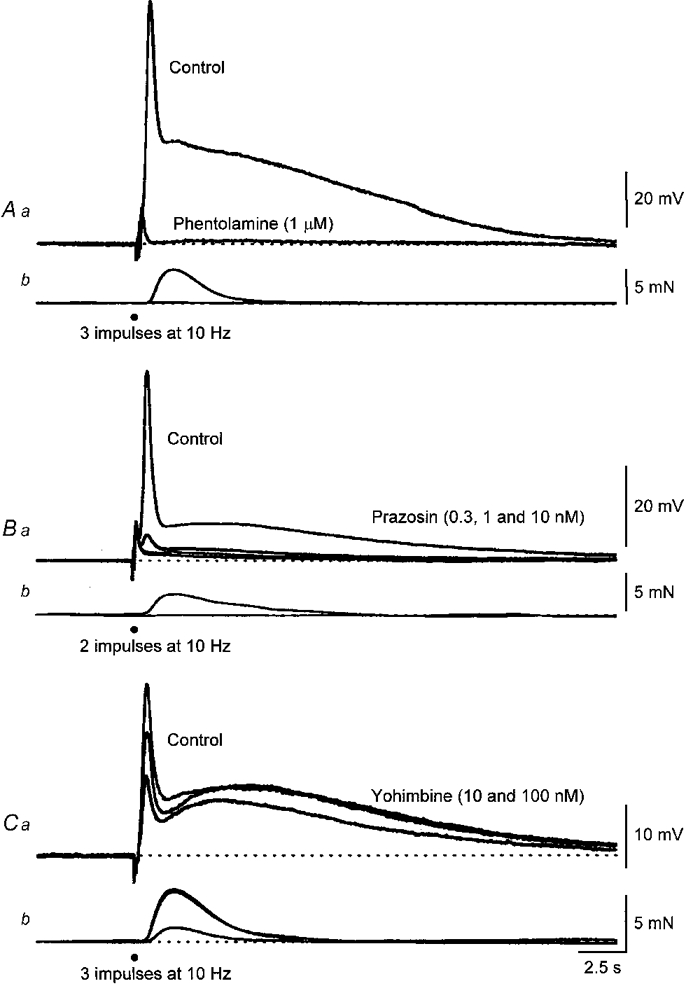
Both the first and second components of the EJP, along with the contraction, were abolished by phentolamine (1 μm; A). The resting membrane potential was -72 mV. In this preparation the stimuli initiated purinergic EJPs and these persisted in the presence of phentolamine. The middle pair of traces (B) show control responses and the effect of increasing concentrations of prazosin (0.3, 1 and 10 nM) on the membrane potential changes and contractions triggered by a pair of sympathetic stimuli. It can be seen that the lowest concentration applied (0.3 nM) abolished the contraction and reduced the amplitudes of both the first and second components of the EJP. Increasing the concentration of prazosin to 1 and 10 nM abolished the first but failed to abolish the second component. The resting membrane potential of this cell was -74 mV. The lower pair of traces (C) show control responses and the effect of increasing concentrations of yohimbine (10 and 100 nM) on the membrane potential changes and contractions triggered by three sympathetic stimuli. It can be seen that yohimbine (10 nM) reduced the amplitude of the first component but not that of the second component. Increasing the concentration of yohimbine to 100 nM reduced the amplitudes of both components but that of the first component more dramatically. The resting membrane potential of this cell was -75 mV. The time calibration applies to all recordings. Nifedipine (1 μM) was present throughout.
The finding that concentrations of prazosin of less than 1 nM are very effective at reducing the amplitudes of the first and second components of the EJP suggests that the adrenoceptors responsible for triggering both may be of the α1A-subtype (Lomasney et al. 1991). This view was supported by the effects of WB4101 on EJPs and associated contractions. A concentration of 1 nM reduced the mean peak amplitude of the first component to 30 ± 8 % (n = 3) of control, that of the associated contraction to 15 ± 4 % and that of the second component to 52 ± 8 %. At a concentration of 3 nM, the first component and associated contraction were abolished; the peak amplitude of the second component was reduced to 23 ± 6 % of control. Together the observations indicate that both components of the EJP are reduced in amplitude by very low concentrations of α1A-adrenoceptor antagonists suggesting that the activation of a single set of adrenoceptors triggers both components. Conversely, part of the second component persisted in high concentrations of several antagonists.
Involvement of intracellular stores in the responses to sympathetic nerve stimulation
When ryanodine (10 μM) was added to the physiological saline (n = 3) and preparations were stimulated regularly with brief trains of stimuli, one to five stimuli at 10 Hz delivered at 1 min intervals, the amplitude of the first component was slowly reduced over a period of 90 min (Fig. 5A and B). Concurrently the amplitudes of the associated contractions were reduced. When more sympathetic stimuli were applied, although the peak amplitude of the first component was somewhat restored, a response as large as the maximal responses detected in control could not be detected (Fig. 5A); ryanodine did not affect the second component (Fig. 5). In two experiments, ryanodine (10 μM) was added to the physiological saline and the preparations were not stimulated for 1 h. At the end of this time, trains of stimuli produced responses very similar to those detected in control solution. Clearly ryanodine only affected the first component of the EJP if the preparations were stimulated regularly, suggesting its action was use dependent.
Figure 5. The effect of ryanodine on EJPs and associated contractions evoked by sympathetic nerve stimulation.
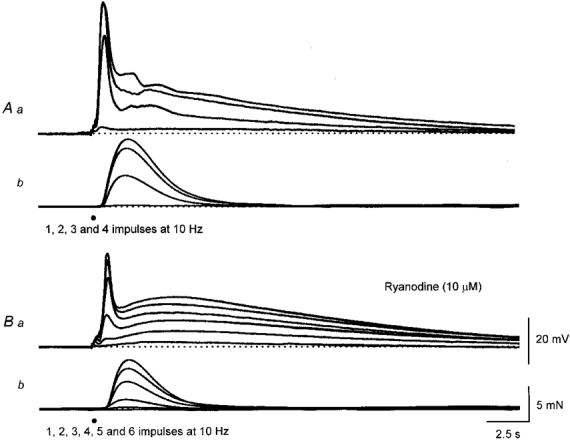
The upper sets of traces show EJPs and associated contractions evoked by 1, 2, 3 and 4 sympathetic nerve stimuli delivered at 10 Hz (A). The lower sets of traces show EJPs and associated contractions evoked by 1, 2, 3, 4, 5 and 6 sympathetic nerve stimuli, recorded 90 min after adding ryanodine (10 μM) to the physiological saline (B). It can be seen that the amplitude of the second component was unchanged whereas the amplitudes of the first component and associated contraction, triggered by a given number of stimuli, were much reduced. The sets of calibration bars refer to both sets of traces.
Caffeine (3 mM) reduced the amplitude of the first component but not that of the second component (n = 11; see as example Fig. 6). In each experiment, caffeine produced a small transient contraction and a membrane depolarization. These were associated with an increase in [Ca2+]i; each of these changes could not be detected 10-15 min after changing to the caffeine-containing solutions. The subsequent addition of nifedipine (1 μM) abolished the remaining part of the first component but again the second component was unaffected (Fig. 6). Higher concentrations of caffeine are often employed to deplete intracellular Ca2+ stores (Bolton & Lim, 1989). When applied at a higher concentration, caffeine (10 mM) produced a transient contraction and reduced the amplitude of both the first and second components; in preparations where purinergic EJPs were detected these were concurrently abolished. This suggests that high concentrations of caffeine, as well as causing the release of Ca2+ from intracellular stores, inhibit the release of transmitter from sympathetic nerves. All the effects of caffeine were readily reversed by washing with drug-free solutions.
Figure 6. Effect of caffeine and nifedipine on changes in [Ca2+]i, membrane potential and contraction evoked by sympathetic nerve stimulation in the rat anococcygeus muscle.
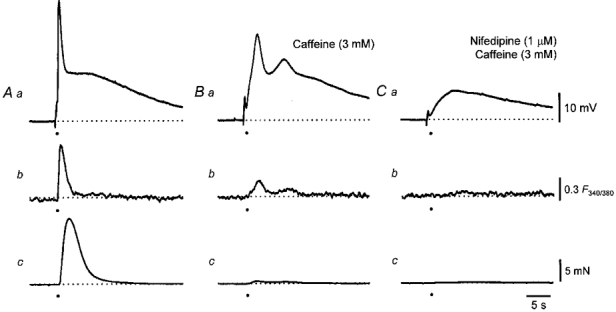
A train of stimuli triggered a biphasic EJP (Aa), a transient increase in [Ca2+]i (Ab) and a contraction (Ac). Caffeine (3 mM) reduced the amplitudes of the first component of the EJP (Ba), the increase in [Ca2+]i (Bb) and almost abolished the contraction (Bc). The further addition of nifedipine (1 μM) abolished the increase in [Ca2+]i (Cb), the associated contraction (Cc) and the remaining part of the first component but again left the second component of the EJP intact (Ca). The resting membrane potential in all traces was -74 mV. The time calibration bar refers to all traces. The voltage calibration bar refers to all membrane potential recordings, the fluorescence calibration bar applies to all ratiometric measurements and the force calibration bar applies to all recordings of contractions.
Together the observations indicate that the first and second components were differentially affected by ryanodine and by moderate concentrations of caffeine. The observations also suggest that the nifedipine-resistant increases in [Ca2+]i and associated contractions result from the release of Ca2+ from intracellular stores. Either caffeine or ryanodine deplete these stores, with the response to ryanodine being use dependent.
Effect of changing the external concentration of Cl− on the responses to sympathetic nerve stimulation
It has been shown that smooth muscle cells of the rat anococcygeus muscle possess sets of Ca2+-activated Cl− channels which are activated by ionophoretically applied noradrenaline (Large, 1984). In an attempt to differentiate between the two components of EJPs the effect of substituting Cl− in the physiological saline with other impermeant ions was examined. When the [Cl−]o was reduced from control (134 mM) to 14 mM by substituting isethionate ions (three experiments) or methylsulphonate ions (three experiments) for Cl− the first component was rapidly abolished whereas the second component was little changed (Fig. 7). These effects were readily reversed by restoring the control solution. With either substitute, although the amplitudes of the contractile responses were reduced, a contraction invariably persisted after the first component had been abolished (Fig. 7A). In a further four preparations loaded with fura-2, sympathetic nerve stimulation evoked a transient early increase in [Ca2+]i in isethionate-containing solutions. Clearly since a contraction and an increase in [Ca2+]i occurred in the absence of a membrane potential change much of the increase in [Ca2+]i must result from the release of Ca2+ from intracellular stores. An unexpected observation made during this series of experiments was that, with either Cl− substitute, even when the experiments were repeated in preparations where voltage-dependent Ca2+ channels were first blocked by adding nifedipine (1 μM) to the bathing solution, the subsequent reduction in [Cl−]o continued to reduce the amplitude of the contractile responses. This effect developed over a period of 10-20 min and reversed over a similar period of time.
Figure 7. The effect of changing [Cl−]o on EJPs and associated contractions evoked by sympathetic nerve stimulation.
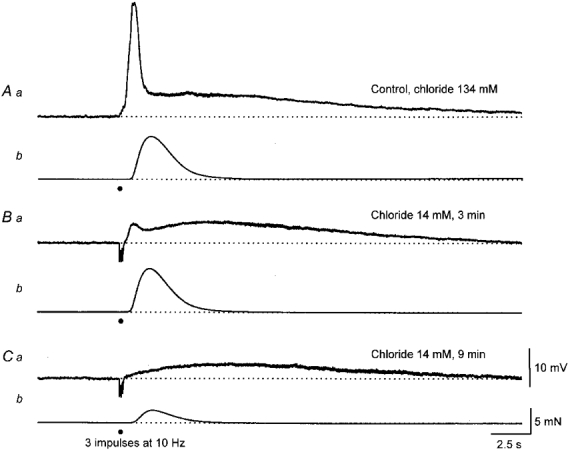
The upper pair of traces (A) show the EJP and associated contraction initiated by three stimuli, recorded in the presence of nifedipine (1 μM). Three minutes after a 90 % replacement of [Cl−]o with isethionate ions, the first component was dramatically reduced in amplitude but the second component and contraction were little changed (B). Some 9 min after reducing the [Cl−]o, the first component was abolished, the second component persisted and the contraction was reduced in amplitude (C). The calibration bars refer to each set of traces.
Relationships between changes in membrane potential, [Ca2+]i and contraction produced by sympathetic nerve stimulation
The relationships between peak membrane potential change, peak increase in [Ca2+]i, peak contraction and number of sympathetic stimuli were determined in control solution (Fig. 8A) and after entry of Ca2+ via CaL channels had been blocked with nifedipine (1 μM; Fig. 8A). A similar series of experiments was carried out using control solutions and solutions in which the intracellular storage of Ca2+ had been disrupted by caffeine (3 mM; Fig. 9A and B). It can be seen that nifedipine (Fig. 8Ab and Bb) or caffeine (Fig. 9Ab and Bb) reduced the increase in [Ca2+]i produced by each train of sympathetic stimuli. However, even quite large increases in [Ca2+]i recorded in the presence of caffeine, produced quite small contractions and depolarizations (Fig. 9Bd). In contrast similar or smaller increases in [Ca2+]i recorded in the presence of nifedipine produced quite substantial contractions and depolarizations (Fig. 8Bd).
Figure 9. Relationships between changes in [Ca2+]i, membrane potential and contraction and number of sympathetic nerve stimuli delivered in control solution and after preventing Ca2+ release from intracellular stores.
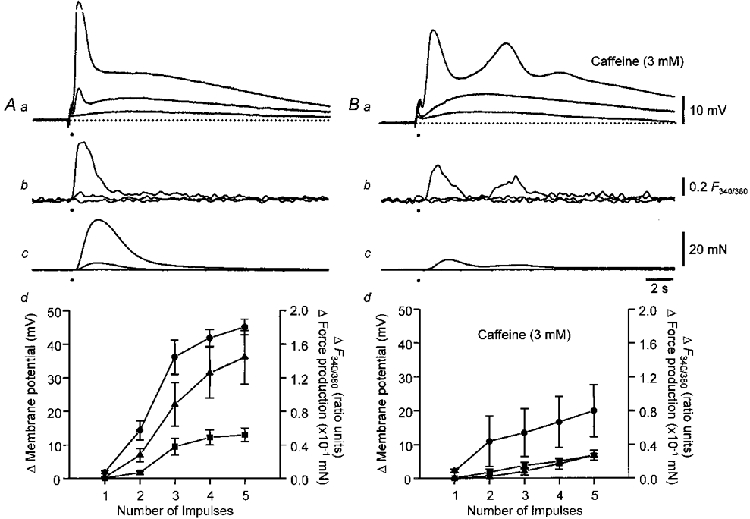
Control recordings are shown in A. The families of traces show changes in membrane potential (Aa), [Ca2+]i (Ab) and contractions (Ac) initiated by 1, 2 and 5 stimuli. The graph (Ad) shows the dependence of peak amplitude of the initial phase of depolarization (0), peak contraction (8) and peak increase in [Ca2+]i (▪) upon the number of stimuli. Recordings obtained in solutions containing caffeine (3 mM) are shown in B. The graph (Bd) shows the dependence of peak amplitude of the initial phase of depolarization (0), peak contraction (8) and peak increase in [Ca2+]i (▪) upon the number of stimuli, from the same experimental series recorded in the presence of caffeine. The vertical bars represent ±s.e.m.; n = 6. The resting membrane potential in all traces was -68 mV. The time calibration bar refers to all traces. The voltage calibration bar refers to all membrane potential recordings, the fluorescence calibration bar applies to all ratiometric measurements and the force calibration bar applies to all recordings of contractions.
Using the data shown in Figs 8 and 9 one can obtain the relationship between changes in [Ca2+]i and contraction when Ca2+ both enters by CaL channels and is released from a store (control responses), when the Ca2+ source is an intracellular store (responses in the presence of nifedipine) and when the Ca2+ source is Ca2+ entering via CaL channels (responses in the presence of caffeine). These relationships are shown in Fig. 10. In the upper graph (Fig. 10A), the relationships between peak contraction and the change in [Ca2+]i produced by different numbers of sympathetic stimuli are shown. The relationship between [Ca2+]i and contraction, recorded in the presence of nifedipine, was steeper than that determined either in control solution or in the presence of caffeine. Similarly the relationships between [Ca2+]i and membrane depolarization, recorded in the presence of nifedipine was steeper than those determined in either control or caffeine-containing solution (Fig. 10A). An analysis of covariance revealed significant interactions between the two treatments. For the relationship between Δ[Ca2+]i and Δforce, F1,51 = 6.44 (P < 0.05) and for the relationship between Δ[Ca2+]i and Δmembrane potential, F1,51 = 4.54 (P < 0.05). This indicates that slopes of the regression lines of Δ[Ca2+]i and Δforce obtained in nifedipine and caffeine are significantly different, as are those of Δ[Ca2+]i and Δmembrane potential. Thus Ca2+ released from stores is more effective at triggering contractions and membrane depolarizations than Ca2+ entering via CaL channels.
Figure 10. Relationships between changes in [Ca2+]i and contraction and between changes in [Ca2+]i and membrane potential.
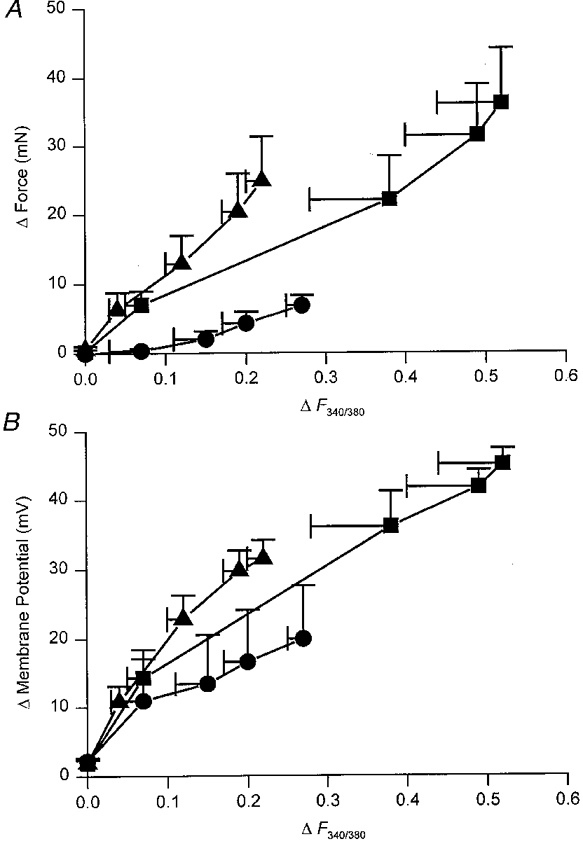
The upper graph (A) shows the relationship between the changes in [Ca2+]i and amplitudes of contraction displayed in Figs 8 and 9. As increasing numbers of sympathetic nerve stimuli were presented, both the change in [Ca2+]i and the amplitude of contractions increased giving rise to the control relationship plotted in A (▪). The relationship plotted (▴) represents data recorded in the presence of nifedipine and so reflects the ability of store-released Ca2+ to trigger a contraction. The relationship plotted (•) shows data recorded in the presence of caffeine and so reflects the ability of ‘extracellular’ Ca2+ to trigger a contraction. It can be seen that the relationship between store-released Ca2+ and contraction is much steeper than that between ‘external’ Ca2+ and contraction. The lower graph (B) shows the relationship between the changes in [Ca2+]i and amplitudes of depolarization obtained from Figs 8 and 9. Again it can be seen that the relationship between depolarization and Ca2+released from a store (8) is steeper than either that obtained in control (▪) or that obtained in the presence of caffeine (0). The vertical and horizontal bars represent ±s.e.m.; n = 6.
DISCUSSION
In the rat anococcygeus muscle, sympathetic nerve stimulation initiated a two-component response that resulted from the activation of α-adrenoceptors. A transient depolarization was followed by a more prolonged phase of depolarization. The two components could be distinguished on a number of grounds other than their distinct time courses. The first component was associated with a contraction whereas the second was not. Only the first component was associated with an increase in [Ca2+]i unless the amplitude of the second component was large enough to activate CaL channels. The first component involved the release of Ca2+ from intracellular stores whereas the second component was unchanged in the presence of agents which disrupt the internal storage of Ca2+. The first component was abolished when [Cl−]o was reduced whereas the second component was unaffected. The amplitudes of the two components varied in different ways as the number of stimuli presented was increased. The peak amplitude of the second component increased linearly with increasing number of stimuli whereas the increase in amplitude of the first component was non-linear: a single stimulus gave almost no response and three or four stimuli gave a maximal response. However, both components appeared to result from the activation of a single set of α-adrenoceptors. As both components were sensitive to very low concentrations of prazosin and WB4101 but were relatively insensitive to yohimbine, these are likely to be α1A-adrenoceptors (Minneman, 1988).
In many tissues α-adrenoceptors are linked to phospholipase C which triggers the formation of IP3 and other metabolic products (Minneman, 1988). The simplest explanation for the events underlying the first component of the EJP is that this pathway is activated, IP3 then triggers the release of Ca2+ from an intracellular store and the increase in [Ca2+]i activates Ca2+-activated Cl− channels. Since the chloride equilibrium potential (ECl) is positive to the resting membrane potential, depolarization occurs (Large & Wang, 1996). The depolarization then triggers the opening of CaL channels and the increase in [Ca2+]i is augmented. Although we could not show directly that the properties of CaL channels were changed by α-adrenoceptor activation in this tissue, as has been shown in other tissues (McDonald et al. 1994), it seems likely that this was the case. Nifedipine reduced the amplitude of the first component of the EJP if it had an amplitude greater than 10 mV in control solution. This represents a membrane potential of about -65 mV, considerably more negative than that associated with the normal activation of CaL channels (Bolton et al. 1988).
Although the amplitudes of each component changed differently when different numbers of stimuli were presented, this could be explained if a common pool of receptors was activated but the amplitude of each component was a different function of the number of receptors activated. If the amplitude of the first component was a function of the number of receptors occupied, N, raised to a power (e.g. N4) and the amplitude of the second component was simply related to the number of receptors occupied (i.e. N1), doubling the number of occupied receptors would increase the amplitude of the first component by a factor of 16 and that of the second by a factor of 2. An antagonist will therefore reduce the amplitude of the first component more dramatically than that of the second component (Fig. 4). At a biochemical level, adrenoceptor activation could trigger the formation of IP3 and hence the release of Ca2+ from an intracellular store. The increase in [Ca2+]i might be further augmented by Ca2+-induced Ca2+ release (Fabiato & Fabiato, 1975) so that the responses increase non-linearly with increasing numbers of stimuli presented. Conversely, if the amplitude of the second component simply reflected the opening or closing of a set of channels directly in response to IP3 or to another biochemical product formed by the same neurally activated pathway, its amplitude might linearly increase with the number of stimuli presented.
These experiments have not identified the nature of the conductance change underlying the second component. In many other tissues, the activation of adrenoceptors leads to a long lasting decrease in potassium conductance (gK). Thus in the rat tail artery, a purinergic EJP is followed by a depolarization which peaks in 10 s and decays with a time constant of 7 s (Cheung, 1982). Similar EJPs are detected in spleen (Jobling, 1994) and in veins (Van Helden, 1988). In the anococcygeus, in preparations where purinergic EJPs were detected, their time constants of decay were slowed when initiated during a second component (authors’ unpublished observations). Since the rate of decay of purinergic EJPs frequently reflects the membrane time constant (Hirst & Edwards, 1989), it seems likely that the second component of the EJPs recorded in this tissue results from closing of membrane channels, i.e. a fall in gK. A fall in gK, following the activation of α1A-adrenoceptors, has been detected in dorsal Raphe neurones (Pan et al. 1994). Again in Raphe neurones, as in the anococcygeus muscle, the responses do not depend upon a change in [Ca2+]i and are not altered when the internal storage of Ca2+ is disrupted (Pan et al. 1994). The alternative possibility is that the second component reflects the opening of sets of cation-selective channels (Wang & Large, 1991). However, this seems unlikely as such channels readily allow Ca2+ entry (Large, 1991) and an increase in [Ca2+]i, independent of CaL channels was not detected during the second component.
It is not clear why the amplitudes of the changes in [Ca2+]i and contractile responses fell when [Cl−]o was reduced. The time course of the reduction was slower than that taken to reduce the amplitude of the first component of the EJP. The rate of recovery was also slower. In some smooth muscles a change in [Cl−]o leads to a change in [Cl−]i (Aicken & Brading, 1983). This also occurs over several minutes and might suggest that the release of Ca2+ from intracellular stores is reduced when [Cl−]i is reduced (Daniel et al. 1993). Alternatively it has been shown that changes in [Cl−]o lead to changes in the internal pH of cells (Aicken & Brading, 1984); such a change could perhaps also reduce the release of Ca2+ from intracellular stores.
The observations indicate that an increase in [Ca2+]i triggered by sympathetic nerve stimulation is more effective at triggering a contraction and a depolarization if recorded in the presence of nifedipine, than it is if recorded in control or caffeine-containing solutions (Fig. 10). This could arise if nifedipine and caffeine exerted their actions in ways that involved neither CaL channels nor depletion of intracellular stores, respectively. However, we are unaware of an action of nifedipine to sensitize contractile proteins; such a property would be required to explain the increased ability of Ca2+to trigger a contraction after nifedipine had been added to the physiological saline. Although it could be argued that caffeine was, perhaps, by inhibiting phosphodiesterases and causing the accumulation of cyclic nucleotides, interfering with contraction (Taylor et al. 1989) we think that this is an unlikely explanation. In control solutions it was apparent that an increase in [Ca2+]i occurring during the first component, which contains a contribution of Ca2+ from intracellular stores, was far more effective at triggering a contraction than was an increase in [Ca2+]i occurring during the second component which resulted from Ca2+ entry via CaL channels. Thus we suggest that Ca2+ released from an internal store is more effective at triggering either a contraction or a depolarization than Ca2+ entering via CaL channels.
There are several observations which could be taken to indicate that store-released Ca2+ is more effective at triggering a contraction than Ca2+ entering from the extracellular space. For example when vascular smooth muscle is depolarized by increasing [K+]o, large increases in [Ca2+]i are required to trigger a contraction when compared with those required to produce a contraction when triggered by applied catecholamines (Morgan & Morgan, 1984; Khalil & van Breemen, 1990). These observations have usually been attributed to an ability of catecholamines to change the sensitivity of contractile elements to Ca2+ (Morgan & Morgan, 1984). Whilst such changes in sensitivity undoubtedly occur (Gailly et al. 1997) they do so in this tissue with a time constant in excess of 10 s during sympathetic nerve stimulation (authors’ unpublished observations). Clearly such an explanation would not apply to our observations where the responses had finished within a few seconds.
It is possible that the increase in [Ca2+]i produced when neurally released transmitter causes the release of Ca2+ from an intracellular store is not uniform, whereas that resulting from Ca2+ entry is more uniform. We have pointed out that our method of detecting changes in [Ca2+]i does not allow a distinction to be made between several large local increases in [Ca2+]i and a more uniform, smaller increase in [Ca2+]i. At other sympathetic neuroeffector junctions, the probability of release of transmitter per nerve impulse is extremely low, with each varicosity releasing a quantum of transmitter less than once every hundred impulses (Hirst & Edwards, 1989). If this were the case in the anococcygeus muscle, even though it receives a dense sympathetic innervation, the likelihood that transmitter is released near a particular cell will be small, even during trains of stimuli. If the transmitter did not diffuse freely through the extracellular space, or the messenger through the syncytium, then only a few cells would be activated. These would display large changes in [Ca2+]i as Ca2+ was released from intracellular stores and contract. Hence only a few cells would contribute to a contractile response. Since the smooth muscle cells form an electrical syncytium, all cells will depolarize and Ca2+ entry via CaL channels will be widespread, hence every muscle would contract to a small extent. This idea could be tested if local changes in [Ca2+]i occurring throughout the syncytium could be monitored. However, this explanation seems unlikely as it implies that when contraction depends on release of Ca2+ from intracellular stores, most muscle cells will make only a minor contribution to the contractile response.
The most likely explanation is that Ca2+ from one source does not readily access all parts of the smooth muscle cell (see for examples Chen & van Breemen, 1993; Ganitkevich & Isenberg, 1996). Thus Ca2+ released from a store would have better access to the contractile elements than did Ca2+ entering from the extracellular fluid. A similar explanation for the observation that ‘internal’ Ca2+ is more effective than ‘external’ Ca2+ at triggering depolarization seems likely. It is generally agreed that an increase in [Ca2+]i activates Ca2+- activated Cl− channels in this and a number of other smooth muscle cells (Large & Wang, 1996). If Ca2+ entering via CaL channels had access to the Ca2+-activated Cl− channels a depolarization would result from the inward flow of Ca2+ across the membrane and the outward flow of Cl−. After blocking CaL channels, only internally released Ca2+ would activate Ca2+-activated Cl− channels. Consequently an increase in [Ca2+]i resulting from internally released Ca2+ would be expected to trigger a smaller depolarization than that triggered by a similar increase in [Ca2+]i following Ca2+ entry. This was clearly not the case (Fig. 10). The simplest explanation is that CaL channels and Ca2+-activated Cl− channels are spatially separated so that entering Ca2+ does not have access to Ca2+-activated Cl− channels. Conversely the Ca2+-store release channels must allow the ready access of Ca2+ to membrane-located Ca2+-activated Cl− channels.
In summary, these experiments have shown that neurally released catecholamines activate a set of adrenoceptors and produce a two-component EJP. The first component results from the release of Ca2+ from intracellular stores and activation of Ca2+-activated Cl− channels, along with CaL channel activation and further Ca2+ entry. Ca2+ from the two different sources has different abilities to trigger contraction or depolarization. Presumably Ca2+ released from intracellular stores have easier access to critical sites on the contractile proteins and on Ca2+-activated Cl− channels than Ca2+ entering via CaL channels. This idea is in accord with the view that distinct compartments exist within smooth muscles which prevent free movement of Ca2+ through the cytosol of individual smooth muscle cells (Chen & van Breemen, 1993).
Acknowledgments
This project was supported by a research grant from the Australian NH & MRC. We wish to thank Dr Mark Elgar for his statistical advice and Dr Frank Edwards for his helpful comments on the manuscript.
References
- Aicken CC, Brading AF. Towards an estimate of chloride permeability in the smooth muscle of guinea-pig vas deferens. The Journal of Physiology. 1983;336:179–197. doi: 10.1113/jphysiol.1983.sp014575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aicken CC, Brading AF. The role of chloride- bicarbonate exchange in the regulation of intracellular chloride in guinea-pig vas deferens. The Journal of Physiology. 1984;349:587–606. doi: 10.1113/jphysiol.1984.sp015175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Bolton TB, Lim SP. Properties of calcium stores and transient outward currents in single smooth muscle cells of rabbit intestine. The Journal of Physiology. 1989;409:385–401. doi: 10.1113/jphysiol.1989.sp017504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton TB, Mackenzie I, Aaronson PI. Voltage dependent calcium channels in smooth muscle cells. Journal of Cardiovascular Pharmacology. 1988;12:S3–7. doi: 10.1097/00005344-198812006-00003. [DOI] [PubMed] [Google Scholar]
- Byrne NG, Large WA. Comparison of the biphasic excitatory junction potential with membrane responses to adenosine triphosphate and noradrenaline in the rat anococcygeus muscle. British Journal of Pharmacology. 1984;83:751–758. doi: 10.1111/j.1476-5381.1984.tb16229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne NG, Large WA. Evidence for two mechanisms of depolarization associated with alpha 1-adrenoceptor activation in the rat anococcygeus muscle. British Journal of Pharmacology. 1985;86:711–721. doi: 10.1111/j.1476-5381.1985.tb08950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne NG, Large WA. Action of noradrenaline on single smooth muscle cells freshly dispersed from the rat anococcygeus muscle. The Journal of Physiology. 1987;389:513–525. doi: 10.1113/jphysiol.1987.sp016669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, van Breemen C. The superficial buffer barrier in venous smooth muscle: sarcoplasmic reticulum refilling and unloading. British Journal of Pharmacology. 1993;109:336–343. doi: 10.1111/j.1476-5381.1993.tb13575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung DW. Two components in cellular response of rat tail arteries to nerve stimulation. The Journal of Physiology. 1982;328:461–468. doi: 10.1113/jphysiol.1982.sp014277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousins HM, Edwards FR, Hirst GDS. Neuronally released and applied acetylcholine on the longitudinal muscle of the guinea-pig ileum. Neuroscience. 1995;65:193–207. doi: 10.1016/0306-4522(94)00466-i. [DOI] [PubMed] [Google Scholar]
- Cousins HM, Edwards FR, Hirst GDS, Wendt IR. Cholinergic neuromuscular transmission in the longitudinal muscle of the guinea-pig ileum. The Journal of Physiology. 1993;471:61–86. doi: 10.1113/jphysiol.1993.sp019891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed KE, Gillespie JS, Muir TC. The electrical basis of excitation and inhibition in the rat anococcygeus muscle. The Journal of Physiology. 1975;245:33–47. doi: 10.1113/jphysiol.1975.sp010833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dail WG, Galloway B, Bordegaray J. NADPH diaphorase innervation of the rat anococcygeus and retractor penis muscles. Neuroscience Letters. 1993;160:17–20. doi: 10.1016/0304-3940(93)90906-2. 10.1016/0304-3940(93)90906-2. [DOI] [PubMed] [Google Scholar]
- Daniel EE, Jury J, Bourreau JP, Jager L. Chloride and depolarization by acetylcholine in canine airway smooth muscle. Canadian Journal of Physiology and Pharmacology. 1993;71:284–292. doi: 10.1139/y93-044. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. The Journal of Physiology. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailly P, Gong MC, Somlyo AV, Somlyo AP. Possible role of atypical protein kinase C activated by arachidonic acid in Ca2+ sensitization of rabbit smooth muscle. The Journal of Physiology. 1997;500:95–109. doi: 10.1113/jphysiol.1997.sp022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganitkevich VY, Isenberg G. Dissociation of subsarcolemmal from global cytosolic [Ca2+] in myocytes from guinea-pig coronary artery. The Journal of Physiology. 1996;490:305–318. doi: 10.1113/jphysiol.1996.sp021145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JS, Maxwell JD. Adrenergic innervation of sphincteric and nonsphincteric muscle in the rat intestine. Journal of Histochemistry and Cytochemistry. 1971;19:676–681. doi: 10.1177/19.11.676. [DOI] [PubMed] [Google Scholar]
- Hirst GDS, Edwards FR. Sympathetic neuroeffector transmission in arteries and arterioles. Physiological Reviews. 1989;69:546–604. doi: 10.1152/physrev.1989.69.2.546. [DOI] [PubMed] [Google Scholar]
- Jobling P. Electrophysiological events during neuroeffector transmission in the spleen of guinea-pigs and rats. The Journal of Physiology. 1994;476:153–165. [PMC free article] [PubMed] [Google Scholar]
- Khalil RA, van Breemen C. Intracellular free calcium concentration/force relationship in rabbit inferior vena cava activated by norepinepherine and high K+ Pflügers Archiv. 1990;416:727–734. doi: 10.1007/BF00370622. [DOI] [PubMed] [Google Scholar]
- Large WA. Membrane potential responses of the mouse anococcygeus muscle to ionophoretically applied noradrenaline. The Journal of Physiology. 1982;326:385–400. doi: 10.1113/jphysiol.1982.sp014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Large WA. The effect of chloride removal on the responses of the isolated rat anococcygeus muscle to alpha 1-adrenoceptor stimulation. The Journal of Physiology. 1984;352:17–29. doi: 10.1113/jphysiol.1984.sp015275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Large WA. Three membrane-conductance mechanisms activated by noradrenaline in vascular smooth muscle. Zeitschrift für Kardiologie. 1991;80(suppl. 7):55–57. [PubMed] [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. American Journal of Physiology. 1996;217:C435–454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Lomasney JW, Cotecchia S, Lefkowitz RJ, Caron MG. Molecular biology of alpha-adrenergic receptors: implications for receptor classification and for structure-function relationships. Biochimica et Biophysica Acta. 1991;1095:127–139. doi: 10.1016/0167-4889(91)90075-9. [DOI] [PubMed] [Google Scholar]
- Mcdonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Minneman KP. α1-Adrenergic receptor subtypes, inositol phosphates, and sources of cell Ca2+ Pharmacological Reviews. 1988;40:87–119. [PubMed] [Google Scholar]
- Morgan JP, Morgan KG. Stimulus-specific patterns of intracellular calcium levels in smooth muscle of ferret portal vein. The Journal of Physiology. 1984;351:155–167. doi: 10.1113/jphysiol.1984.sp015239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Worley JF. Dihydropyridine inhibition of single calcium channels and contraction in rabbit mesenteric artery depends on voltage. The Journal of Physiology. 1989;412:65–91. doi: 10.1113/jphysiol.1989.sp017604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZZ, Grudt TJ, Williams JT. α1-Adrenoceptors in rat dorsal raphe neurones: regulation of two potassium conductances. The Journal of Physiology. 1994;478:437–447. doi: 10.1113/jphysiol.1994.sp020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DA, Bowman BF, Stull JT. Cytoplasmic Ca2+ is a primary determinant for myosin phosphorylation in smooth muscle cells. Journal of Biological Chemistry. 1989;264:6207–6213. [PubMed] [Google Scholar]
- Van Helden DF. Electrophysiology of neuromuscular transmission in guinea-pig mesenteric veins. The Journal of Physiology. 1988;401:469–488. doi: 10.1113/jphysiol.1988.sp017173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Large WA. Noradrenaline-evoked cation conductance recorded with the nystatin whole-cell method in rabbit portal vein cells. The Journal of Physiology. 1991;435:21–39. doi: 10.1113/jphysiol.1991.sp018496. [DOI] [PMC free article] [PubMed] [Google Scholar]