

Abstract
We studied the effect of overexpression of the β2-adrenergic receptor (β2-AR) in the heart on ion channel currents in single cells isolated from hearts of fetal and neonatal transgenic and wild-type mice. The β2-AR transgene construct was under the control of the murine α-myosin heavy chain (α-MHC) promoter, and ion channel activity was measured at distinct developmental stages using whole-cell and perforated patch clamp techniques.
We found no change in L-type Ca2+ channel current (ICa) density in early embryonic stages (E11-13) of β2-AR transgenic positive (TG+) mice, but significant increases in ICa density in intermediate (E14-16, 152 %) and late (E17-19, 173.7 %) fetal and neonatal (1 day post partum, 161 %) TG+ compared with transgenic negative (TG-) mice. This increase in ICa was accompanied by a negative shift in the peak of the current-voltage relationship in TG+ mice.
Transient (< 3 min) or prolonged (16-24 h) exposure of TG- neonatal stage myocytes to 8-Br-cAMP (300 μM) increased ICa density and caused a shift in the current-voltage relationship to a similar extent to that seen in TG+ mice. In TG+ myocytes, 8-Br-cAMP had no effect. Exposure of TG+ cells to Rp-cAMPS reversed both the shift in voltage dependence and reduced the peak current density observed in these myocytes. We concluded from these results that the L-type Ca2+ channel is maximally modulated by cAMP-dependent protein kinase (PKA) in TG+ mice and that the α-MHC promoter is functional in the ventricle as early as embryonic day 14.
In contrast, we found that slow delayed rectifier K+ channels were not changed significantly at any of the developmental stages studied by the overexpression of β2-ARs compared with TG- mice. The sensitivity of murine slow delayed rectifier K+ channels to cAMP was tested by both transient and prolonged exposure to 8-Br-cAMP (300 μM), which increased the slow delayed rectifier K+ channel current (IK,s) density to a similar extent in both TG- and TG+ neonatal myocytes. In addition, we found that there was no difference in the concentration dependence of the response of ICa and IK,s to 8-Br-cAMP.
Thus, overexpression of the β2-AR in the heart results in distinct modulation of ICa, but not IK,s, and this is not due to differences in the 8-Br-cAMP sensitivity of the two channels. Instead, these results are consistent with both compartmentalization of β2-AR-controlled cAMP and distinct localization of L-type Ca2+ and slow delayed rectifier K+ channels. This cAMP is targeted preferentially to the L-type Ca2+ channel and is not accessible to the slow delayed rectifier K+ channel.
During the development of the murine heart, a variety of electrophysiological changes takes place: both the levels of expression of ion channels and their regulatory properties are altered (Kojima et al. 1990; Maki et al. 1996). During early murine embryogenesis, it has been shown that the L-type Ca2+ channel current (ICa) plays a dominant role in excitation, whereas the slow component of the delayed rectifier K+ channel current (IK,s) is not so apparent until the later stages of embryonic development, or even after birth (Honore et al. 1991; Davies et al. 1995). Also, during development of the mouse heart, the β-adrenergic signalling pathway has been found to be functionally incomplete until the later stages of embryogenesis suggesting a possible link between this pathway and expression of IK,s (Chen et al. 1982; Kojima et al. 1990; An et al. 1996).
The influence of catecholamines acting through β-adrenergic receptors (β-ARs) on the activity and expression of ion channels is of particular interest in the heart as it is well known that in chronic heart failure the myocardial β-AR system is defective. This functional impairment is associated with a decrease in agonist-induced inotropy, and is thought to be caused by a receptor defect, since the adenylyl cyclase response remains intact (Bristow et al. 1982). Specifically, it has been found that there is a selective downregulation of β1-ARs which increases substantially the percentage of total β-ARs that are of the β2-subtype (Bristow et al. 1986; Ungerer et al. 1993). Additionally, a population of remaining receptors (both β1- and β2-ARs) is functionally uncoupled, possibly due to increased homologous desensitization as the levels of β-adrenergic receptor kinase are increased in heart failure (Ungerer et al. 1993). Thus, β-AR agonists used to treat heart failure are not effective chronically and patients are at a higher risk of mortality as a result of the elevated levels of catecholamines (Ginsburg et al. 1983).
Recently, a transgenic mouse model has been developed in which β2-ARs are overexpressed specifically in the heart (Milano et al. 1994). In the hearts of the adult transgenic mice, there is a > 100-fold increase in β2-AR density accompanied by apparent maximal heart rate and cardiac contractility. The physiological changes in heart rate and contractility are not believed to arise simply as a result of stimulation of the increased number of β2-ARs by circulating catecholamines. Instead, they are thought to be due specifically to an increase in the number of β2-ARs present in the active conformation, which are able to activate adenylyl cyclase in the absence of agonist (Milano et al. 1994). This novel transgenic model provides a unique opportunity to investigate the effects of the β-AR pathway on the expression of ionic channels in the heart during development and to determine directly whether increased numbers of receptors by themselves activate functionally relevant steps in the β-AR signal cascade. In addition, it affords an opportunity to study modulatory properties that may be unique to the β2-AR and hence relevant to heart failure when the relative importance of this receptor subtype increases.
Here, we used this mouse model to study the effects of overexpression of the β2-AR on ICa and IK,s in the developing mouse heart. This study had multiple goals. First, we wanted to determine directly whether β2-AR overexpression modulates ion channel activity in the developing mouse heart in the absence of exogenous agonist and whether the effects of β2-AR overexpression could be detected functionally during embryonic development. Second, we used this model to test directly for an inter-relationship between the β2-AR signalling pathway and expression of IK,s. Our results indicate that β2-AR overexpression enhances ICa in a cAMP-dependent manner as early as day 14 of embryogenesis (E14). Surprisingly however, despite a robust sensitivity to exogenous 8-bromoadenosine 3′,5′-cyclic monophosphate (8-Br-cAMP), IK,s was not enhanced at any stage of development in β2-AR transgenic positive (TG+) animals. These results clearly indicate unique electrophysiological consequences of β2-AR-induced liberation of cAMP, and are consistent both with compartmentalization of β2-AR-controlled cAMP and distinct localization of L-type Ca2+ and slow delayed rectifier K+ channels.
METHODS
Embryonic cardiomyocyte isolation and culture
Pregnant female mice were killed by cervical dislocation and embryos, at different stages of development (11-13 days post coitus: early stage; 14-16 days: intermediate stage; or 17-20 days: late stage), were removed. For neonatal stage myocytes, mice at 1 day post partum were killed by decapitation, just prior to the removal of cardiac tissue as described by Sturm & Tam (1993) and in accordance with the guidelines of the local ethics committee (Institutional Animal Care and Use Committee, Columbia University College of Physicians and Surgeons). Cardiac myocytes were isolated from embryonic and neonatal hearts as described previously (Kubalak et al. 1995). In brief, hearts were dissected from embryos and neonates and placed in normal Tyrode solution (Kass et al. 1989). Atrial and ventricular tissues were separated under a dissecting microscope and placed in an Eppendorf tube with 0.5 ml Tyrode solution containing 0.5 mg ml−1 collagenase Type II (Worthington) and 1.0 mg ml−1 pancreatin (Gibco) for a 15 min digestion at 37°C. Cells from the enzymatic digestion were placed in culture medium (modified Eagle's medium; Gibco) containing 10 % fetal bovine serum, plated into plastic Petri dishes and cultured in a 10 % CO2 incubator at 37°C. Electrophysiological recordings were carried out approximately 18-24 h after plating of the cells and could be carried out for periods up to 48 h following plating. Unless specified for individual experiments, cells were maintained in agonist-free media for this entire period.
Electrophysiology
Experimental results shown in this paper were obtained using patch clamp procedures in conventional whole-cell (Hamill et al. 1981) or in the perforated patch configuration (Horn & Marty, 1988). In experiments to study IK,s, E4031 (5 μM; obtained as a gift from Eisai Limited, Tokyo, Japan) was added to the external solution to block the rapid component of the delayed rectifier K+ channel current (Sanguinetti & Jurkiewicz, 1990) and nisoldipine (1 μM) was added to block the L-type Ca2+ channel current. For the measurement of IK,s, the external solution contained (mM): KCl, 5; N-methyl-glucamine, 125; MgCl2, 1; CaCl2, 1; Hepes, 10; glucose, 5 (pH 7.4 with KCl). The internal solution for recording whole-cell IK,s contained (mM): potassium aspartate, 110; CaCl2, 1; Hepes, 10; EGTA, 11; MgCl2, 1; K2ATP, 5; pH 7.3 (KOH). Whole-cell ICa was recorded using external solution containing (mM): CsCl, 5; Hepes, 10; N-methyl-glucamine, 125; glucose, 5; MgCl2, 1. BaCl2 (20 mM) was added to this solution as charge carrier and the internal solution contained (mM): aspartic acid, 50; K2ATP, 5; CsCl, 60; EGTA, 11; Hepes, 10; CaCl2, 1 (pH 7.2 with CsOH). For perforated patch recordings of L-type Ca2+ channel currents, amphotericin B was dissolved in DMSO at a concentration of 30 mg ml−1, and then added to the above internal solution to yield a final concentration of 120-250 μg ml−1. Both the amphotericin B stock solution and the amphotericin B-containing pipette solution were subjected to 5-10 min of ultrasonication before use. Capacity transients were monitored as a function of time after attaining a high resistance seal with the surface membrane. Electrical access to the cell was judged by the time course of the capacity transient, and adequate access was usually attained within 10 min of seal formation.
In order to measure the time course of regulatory responses, Ca2+ channel currents were measured during test pulses (40 ms) to +10 mV applied once every 10 s. Holding potentials of -40 mV were used for both Ca2+ and K+ channel current recordings, and isochronal (2 s pulses applied at 10 s intervals) activation curves were used to measure the activation of IK,s. Similarly, ICa activation was measured by 40 ms test pulses to a series of potentials (10 mV increments) applied at 10 s intervals from a holding potential of -40 mV to +60 mV. ICa inactivation curves were obtained by measuring peak current at a test potential of +10 mV after application of a series of 5 s conditioning pules (-80 to +30 mV, 10 mV increments). A 10 ms return to the holding potential (-40 mV) separated each test and conditioning pulse. Patch pipettes (Clay Adams glass) were pulled to resistances of 2.5-5.0 MΩ when filled with internal solution. Total cell membrane capacitance was used as a measure of membrane area and was determined either by analog capacity compensation or by integration of current transients in response to 10 mV test pulses. Electrophysiological recordings were carried out at room temperature (20-22°C) except for the experiments in which the slow delayed rectifier K+ channel was transiently exposed to 8-Br-cAMP, which were conducted at 30-32°C.
Chemicals were obtained from the following suppliers: amphotericin B (Sigma); 8-chlorophenylthio-cAMP (8-CPT-cAMP; Boehringer Mannheim); 8-Br-cAMP (Sigma); adenosine 3′,5′-cyclic monophosphothioate, RP-isomer, triethylammonium salt (Rp-cAMPS; CalBiochem). Stock (20 mM) solutions of 8-CPT-cAMP or 8-Br-cAMP (dissolved in H2O) were mixed and diluted to 300 μM for each experiment. Stock Rp-cAMPS solution (10 mM dissolved in H2O) was mixed and diluted daily to 300 μM.
Data were collected, stored and analysed on IBM (486)-compatible computers interfaced to Axopatch (200A) amplifiers (Axon Instruments) under the control of pCLAMP (version 6.0) software (Axon Instruments). Graphics and statistical data analysis were carried out using Origin software (Microcal, Northampton, MA, USA). Averaged data are shown as means ±s.e.m. and were compared using Student's t test with a P value of < 0.05 taken to indicate statistical significance.
Cell capacitance was measured and compared for TG+ and transgenic negative (TG-) cells as a function of developmental stage. For each stage there was no significant difference at the 0.05 level between TG+ and TG- cell capacitance; data obtained were (TG-, TG+, means ±s.e.m.): early stage: 25.4 ± 3.3 pF (n = 10), 29.9 ± 6.5 pF (n = 8); intermediate stage: 23.4 ± 7.5 pF (n = 26), 26.0 ± 9 pF (n = 34); late stage: 26.5 ± 11.5 pF (n = 32), 31.4 ± 3.2 pF (n = 15); neonatal: 25.7 ± 1.8 pF (n = 10), 28.0 ± 3.2 pF (n = 20).
Transgenic mice
The transgenic mice line utilized (TG4) has been described in detail (Milano et al. 1994). These mice (TG4) possess cardiac overexpression of the human β2-AR at > 100-fold over endogenous myocardial levels. Myocardial specificity was targeted by the use of the murine α-myosin heavy chain (α-MHC) gene promoter (Milano et al. 1994). Breeding pairs used were heterozygous for the transgene as determined by Southern blotting (Milano et al. 1994) and pregnancies were timed for the purpose of these studies in order to isolate embryonic myocytes. Embryos and neonatal pups were genotyped by PCR analysis with primers specific for the α-MHC gene promoter and the β2-AR (Milano et al. 1994).
RESULTS
L-type Ca2+ channel activity is enhanced in TG+ mice
We first focused on possible modulation of ICa by β2-AR overexpression because we had previously found that the L-type Ca2+ channel was expressed at robust levels and could be modulated in a cAMP-dependent manner at early stages in the developing mouse heart (Davies et al. 1995; An et al. 1996). As in our previous studies, we focused on distinct stages of development and compared ICa density at each stage between cells isolated from TG- and TG+ hearts. It should be pointed out that we also compared data obtained in cells isolated from wild-type (WT) and TG- hearts, and found no difference in stage-dependent current densities or responses to 8-Br-cAMP for either ICa or IK,s (data not shown). Figure 1 shows that ICa was increased by β2-AR overexpression, and that the increase in channel activity could be detected clearly during embryogenesis. As seen in Fig. 1, at the earliest stage studied (days 11-13), no effect of the transgene was observed. However, by the intermediate embryonic period studied (days 14-16), we were able to detect significantly larger currents in TG+ compared with TG- hearts and this difference was maintained throughout development. In the remainder of this study, we focused on channel activity in cells isolated from neonatal hearts because the effect of the transgene was clear at that stage, and baseline channel activity was more pronounced than at earlier developmental stages.
Figure 1. β2-AR overexpression increases ICa in the developing mouse heart.
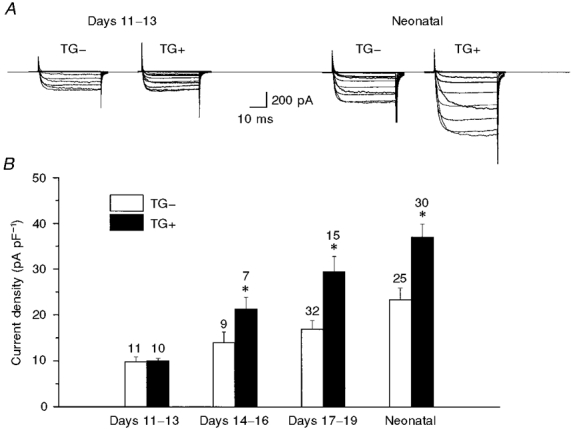
A, current traces illustrating families of currents (for voltages, see Methods) measured in cells from TG- and TG+ early stage embryonic (left) and neonatal (right) hearts. B, bar graph summarizing peak ICa density recorded at +10 mV from a large number of cells (indicated above each bar) for three embryonic stages and for neonatal cells. * Significant difference compared with TG-, P < 0.01.
Functional consequences of β2-AR overexpression: a role for elevated cAMP
In order to assess the functional consequences of β2-AR overexpression we compared the voltage dependence of activation and inactivation in cells isolated from TG+ and TG- mice. Figure 2 summarizes these experiments and shows that there were clear differences in the voltage dependence of L-type Ca2+ channel activation that were reflected both in the peak current-voltage relationships (Fig. 2A) and in the conductance-voltage relationships extracted from these data (Fig. 2A). These effects occurred with no change in the voltage dependence of steady-state inactivation (Fig. 2A).
Figure 2. Voltage-dependent changes in ICa in TG+ cells.
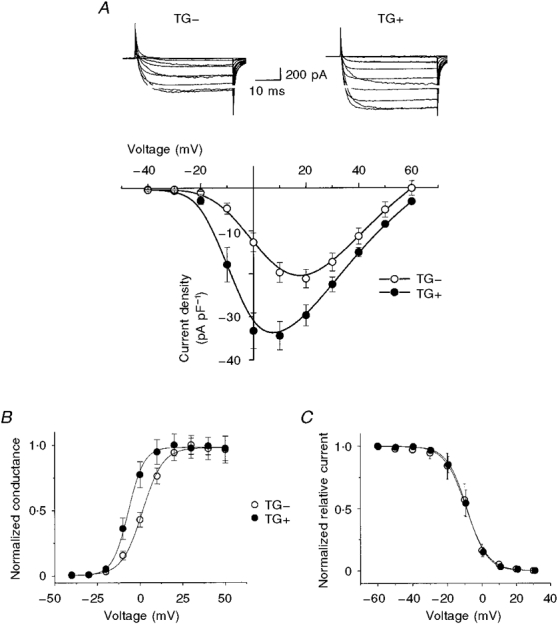
A, mean peak current-voltage relationships for TG+ ( n = 25) and TG- ( n = 20) cells. Note that the peak of the relationship occurred near +5 mV for TG+ and near +15 mV for TG- cells. The current traces (top) illustrate typical TG- (left) and TG+ (right) recordings (see Methods for voltage protocol). B, conductance- voltage relationships determined from A by normalizing currents to driving force. The smooth curves are Boltzmann fits to the data with the following parameters: potential at half-maximal activation (V½) = -7 mV, slope factor (VK) = 5.2 mV (TG+); V½ = 1.8 mV, VK = 6.5 mV (TG-). C, β2-AR overexpression did not affect ICa steady-state inactivation. Steady-state inactivation was measured using 5 s conditioning pulses for TG+ and TG- cells. The smooth curves are Boltzmann relationships: potential at half-maximal inactivation (V½) = -8.6 mV; VK = 5.8 mV.
The shift in the voltage dependence of activation and the increase in peak current density observed in TG+ cells are consistent with well-known modulatory effects of cAMP-dependent protein kinase A (PKA) on cardiac L-type Ca2+ channels (reviewed by McDonald et al. 1994; Hove-Madsen et al. 1996). The results are consistent with the view that increased cAMP production caused by β2-AR overexpression-induced stimulation of adenylyl cyclase, even in the absence of agonist, modulates L-type Ca2+ channel activity in these cells (Rockman et al. 1996). In order to test more directly for this possibility, we studied the effects of 8-Br-cAMP on ICa recorded in TG- cells to determine whether or not cAMP causes similar changes in the voltage dependence and density of ICa in these cells.
We found that 8-Br-cAMP did increase peak current density and shifted the peak of the current-voltage relationship in the negative direction in TG- cells in a manner that strikingly resembled the effects of overexpression of the β2-AR in the absence of exogenous 8-Br-cAMP application. Figure 3A illustrates the effects of transient exposure of a TG- cell to 8-Br-cAMP, which is a cAMP analogue. In this and the following experiments, the perforated patch arrangement of the patch clamp technique was used to minimize disruption of key intracellular signalling molecules. The response to 8-Br-cAMP reached a steady state within 1 min, was maintained in the presence of 8-Br-cAMP and was reduced completely within minutes of return to 8-Br-cAMP-free solution (Fig. 3A). We found similar results with 8-CPT-cAMP, another membrane-permeant cAMP analogue (data not shown). Figure 3B summarizes the effects of 8-Br-cAMP on the current-voltage relationship of several cells. These data were obtained after steady state had been reached in the presence of 8-Br-cAMP. Peak current was increased and the peak of the current-voltage relationship was shifted in the negative direction. The increase in peak current in TG- myocytes measured in response to 8-Br-cAMP was remarkably similar to the increase in current density measured as a result of overexpression of the transgene. It was also of interest to study the effect of 8-Br-cAMP on ICa in TG+ cells since it is possible that the increased ICa observed in TG+ cells was due to an influence other than cAMP, such as a direct membrane-delimited pathway that caused functional changes in channels which simply resemble those induced by cAMP (Brown & Birnbaumer, 1988, 1990). Such experiments would also help to establish whether ICa was maximally modulated in the TG+ animals. Therefore, TG+ cells were exposed overnight to 8-Br-cAMP and Ca2+ current density was measured. In contrast to ICa in TG- cells, TG+ICa was not enhanced by exposure to exogenous 8-Br-cAMP (Fig. 3A), providing further evidence that not only is [cAMP] elevated in the vicinity of L-type Ca2+ channels in TG+ cells but also that it is sufficiently high to saturate the modulatory response of L-type Ca2+ channels.
Figure 3. Influence of 8-Br-cAMP on ICa in TG- and TG+ cells.
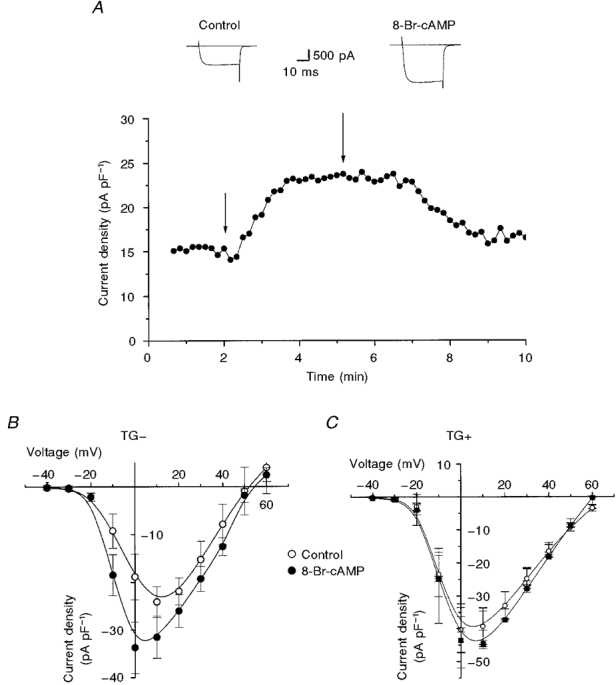
A, transient exposure to 8-Br-cAMP (300 μM) reversibly increased the peak ICa (activated by repeated 40 ms depolarizations to +10 mV) in a TG- cell. Typical Ca2+ channel currents shown above the plot were sampled at the points indicated by the arrows. B, mean current-voltage relationships for ICa in TG- cells before (Control) and after overnight (16-24 h) exposure to 8-Br-cAMP (n = 4-6). C, mean current-voltage relationships for ICa in TG+ cells in which overnight exposure to 8-Br-cAMP had no significant effect (P > 0.05; n = 6-18).
A summary of the Ca2+ current density in TG- and TG+ myocytes, a comparison of these data with the effect of 8-Br-cAMP on ICa density in TG- myocytes (both transient and overnight exposure), and finally data on the lack of effect of 8-Br-cAMP (overnight exposure) on the Ca2+ current density in TG+ cells are illustrated in Fig. 4. When the increased ICa density found in TG+ myocytes was compared with the effect of transient exposure of TG- myocytes to 8-Br-cAMP on ICa density in a large number of cells, no significant difference between the two groups was found (unpaired t test, P > 0.05; Fig. 4). Further, we found that sustained exposure to 8-Br-cAMP (16-24 h) produced changes in current density and ICa voltage dependence that were not significantly different from changes caused by transient exposure to 8-Br-cAMP or overexpression of β2-ARs (Fig. 4). Lastly, Fig. 4 also shows that not only did 8-Br-cAMP fail to enhance ICa further in TG+ cells, but also that the current density in these cells was of a similar level to that observed in TG- cells only after they were exposed to 8-Br-cAMP.
Figure 4. Comparison of the effects of β2-AR overexpression and 8-Br-cAMP (300 μM) on peak ICa.
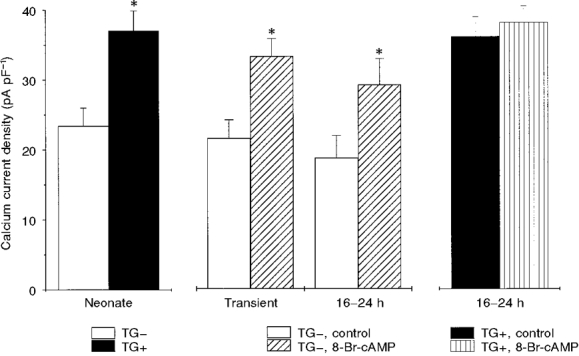
Bar graphs summarizing peak current densities recorded at +10 mV. Left, peak currents in TG- (n = 26) vs. TG+ (n = 30) cells. Middle, peak current density in TG- cells before and after transient (< 3 min; middle left, n = 6 each) and prolonged (16-24 h; middle right, n = 8 each) exposure to 8-Br-cAMP. * There was no significant difference between the enhancement of peak ICa in the TG+ cells (left) and that produced by 8-Br-cAMP in TG- cells (middle; unpaired t test, P > 0.05). Right, peak current density before and after prolonged exposure of TG+ cells to the same concentration of 8-Br-cAMP.
Although these results strongly suggest that the functional changes in ICa caused by β2-AR overexpression are due to increased cAMP, it is still possible that the effects are due to a direct membrane-delimited pathway that causes functional changes in channels that simply resemble those induced by increased cAMP (Brown & Birnbaumer, 1988, 1990). In order to distinguish between these possibilities, we exposed TG+ cells to the inhibitor of PKA, Rp-cAMPS (Rothermel & Parker, 1988). Rp-cAMPS was applied to TG+ cells, in which ICa was again measured in the perforated patch configuration. We reasoned that, if the voltage-dependent changes in current density caused by β2-AR overexpression in TG+ cells were due to a membrane-delimited pathway, this inhibitor would have no effect on the measured channel activity. Instead, we found that Rp-cAMPS reduced ICa density and shifted the peak of the current-voltage relationship in the positive direction such that it now occurred over the same voltage range as that observed for ICa in TG- cells (Fig. 5; compare with the TG- current-voltage relationship shown in Fig. 2A). The data summarized in Fig. 5 were obtained after steady state was reached in the presence of Rp-cAMPS (300 μM), which typically took 5 min. The properties of currents measured in the presence of Rp-cAMPS strongly resembled those of ICa in TG- cells, consistent with the conclusion that the effects of the transgene on ICa are due to elevation of cAMP.
Figure 5. Effect of Rp-cAMPS on ICa recorded from TG+ cells.
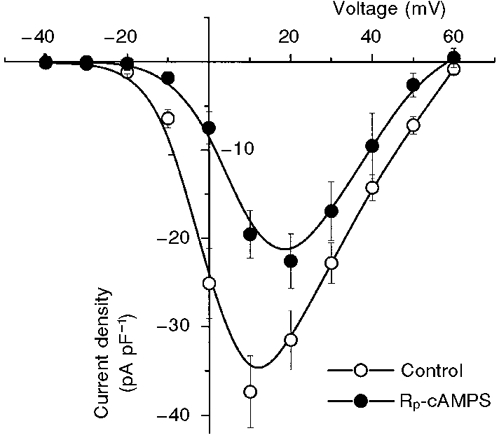
Mean ( n = 6) peak current-voltage relationships for currents measured in TG+ cells before (Control) and after exposure to Rp-cAMPS (300 μM). Steady-state effects which were obtained within 3-5 min of exposure to the PKA inhibitor are shown. Note that the peak of the current-voltage relationship was shifted to positive voltages and reduced in amplitude.
Delayed K+ current (IK,s) is not enhanced by β2-AR overexpression
Our experiments up to this point have provided strong evidence that ICa is enhanced in the TG+ cells by elevation of cAMP. Since IK,s is also known to be modulated by cAMP in the heart, we investigated the effects of β2-AR overexpression on IK,s by measuring its activation as a function of voltage at each developmental stage. In contrast to ICa, we found no statistically significant difference in the expression of IK,s or its voltage dependence at any developmental stage between cells isolated from TG+ and TG- mice. Figure 6 shows the results obtained from the neonatal stage of development. As in the case of our study of ICa, we focused on currents in cells isolated from neonatal hearts because expression of IK,s is most pronounced at this stage (Davies et al. 1995).
Figure 6. β2-AR overexpression did not affect IK,s.
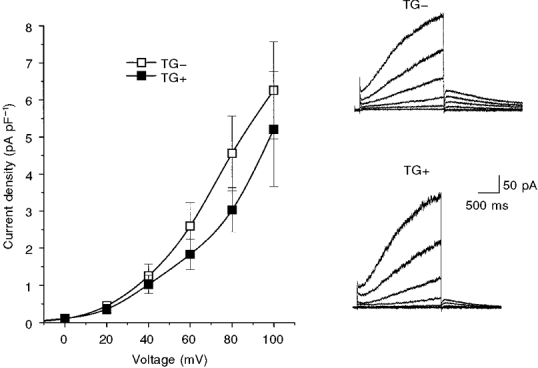
Right, representative traces of IK,s (activated by isochronal 2 s depolarizations from -40 to +60 mV in 20 mV increments from a holding potential of -40 mV) recorded from TG- (top) and TG+ (bottom) cells. Left, the mean current-voltage relationships for time-dependent IK,s activated during depolarization for TG- ( n = 19) and TG+ ( n = 20) cells (neonatal stage). There was no significant difference between TG- and TG+ currents at any voltage.
Because there was no difference in the properties of IK,s measured in TG+ and TG- cells, as summarized in Fig. 6, and because we have demonstrated that β2-AR overexpression increases [cAMP] in the vicinity of L-type Ca2+ channels, the results in Fig. 6 raise the possibility that murine IK,s is insensitive to intracellular cAMP. In fact, species differences in modulatory properties of recombinant IK,s have been reported (Varnum et al. 1993). In order to test for this possibility, we studied the effects of 8-Br-cAMP on IK,s measured in cells from neonatal hearts using the same protocols previously applied to the measurement of ICa. We incubated TG- cells for 16-24 h in the presence 8-Br-cAMP in order to study the effects of maintained cAMP elevation, similar to maintained elevation of cAMP caused by overexpression of β2-AR as detected in our studies of ICa. Under these conditions, we found that the slow delayed rectifier K+ channel activity in TG- myocytes was enhanced by 8-Br-cAMP and that the effect was statistically significant (P < 0.01; Fig. 7A). Thus, as is the case for many other species, IK,s in neonatal murine heart is potentiated by cAMP.
Figure 7. Effects of 8-Br-cAMP on IK,s in TG+ and TG- cells.
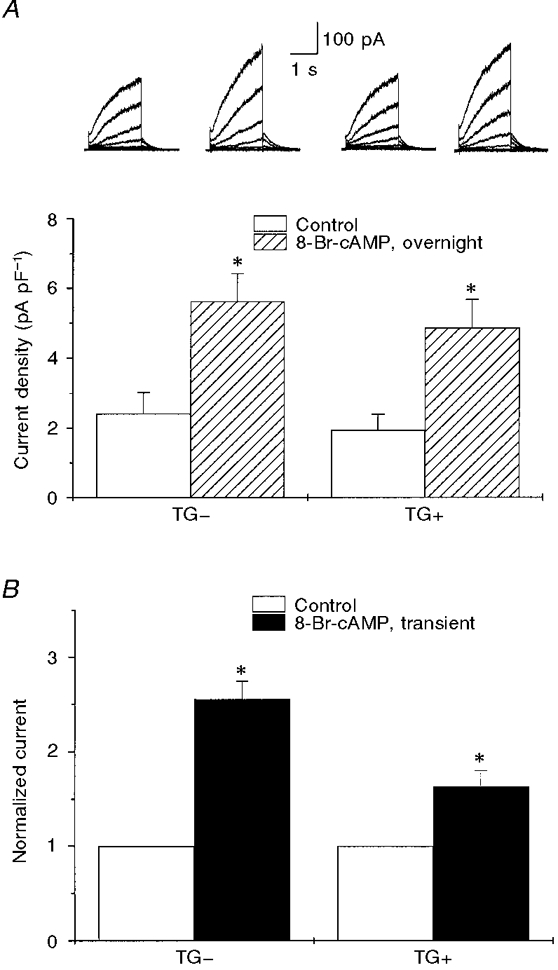
A shows mean IK,s density measured at +40 mV in TG- and TG+ cells in the absence of 8-Br-cAMP (Control) and after 16-24 h incubation in the presence of 8-Br-cAMP (300 μM), n = 5-18. Representative IK,s traces (at +60 mV) before and after exposure to cAMP in TG- and TG+ cells are shown above the bars. * Significant difference compared with control, P < 0.01. B shows the relative IK,s amplitude before (Control) and after transient (< 3 min) exposure to 300 μM 8-Br-cAMP in TG- and TG+ cells. Currents were normalized to those in control (n = 2-6). * Significant difference compared with control, P < 0.01.
If prolonged exposure to elevated intracellular 8-Br-cAMP enhances IK,s in TG- (and WT) hearts, then why is the current not affected by β2-AR overexpression in the TG+ cells in which we have shown that ICa is enhanced in a cAMP-dependent manner? One possibility is that β2-AR overexpression also stimulates an inhibitory pathway that is specific for IK,s and not ICa. If this were the case, then exposure of cells obtained from TG+ hearts to 8-Br-cAMP might not be expected to enhance IK,s. We tested for this possibility by incubating TG+ cells in 300 μM 8-Br-cAMP but found that IK,s was enhanced to the same extent as currents in TG- cells (P < 0.01; Fig. 7A). This finding is consistent with the hypothesis that there is no inhibitory factor preventing the cAMP-dependent modulation of IK,s.
It is possible, however, that the increases in IK,s observed following prolonged exposure of the TG- and TG+ cells to 8-Br-cAMP might have resulted from cAMP-mediated changes in channel expression rather than a direct effect of cAMP on the channels themselves. Therefore, we also transiently exposed both TG- and TG+ cells to 8-Br-cAMP and found that, under these conditions, IK,s was also enhanced in both TG- and TG+ cells (Fig. 7A). These results provide further evidence to support the hypothesis that IK,s in TG- and TG+ cells is sensitive to cAMP and are consistent with the lack of an inhibitory pathway or factor in the TG+ cells preventing a cAMP-dependent regulation of IK,s.
Finally, it might be possible that L-type Ca2+ channels are significantly more sensitive to cAMP than slow delayed rectifier K+ channels. If this were the case, then perhaps β2-AR overexpression might increase cAMP in regions near both slow delayed rectifier K+ and L-type Ca2+ channels, but to a concentration that saturates the response of ICa but which is not sufficiently high to increase IK,s. To test this, the concentration dependence of both IK,s and ICa to cAMP was investigated by exposure of TG- myocytes to 8-Br-cAMP at different concentrations. In Fig. 8A, the relative increase in ICa and IK,s at different 8-Br-cAMP concentrations is shown. There was no significant difference between the effect of 8-Br-cAMP on ICa and IK,s at any 8-Br-cAMP concentration tested. This can be seen more clearly in Fig. 8A, where the concentration- response curves for ICa and IK,s to [8-Br-cAMP] are shown. The EC50 values for 8-Br-cAMP for IK,s and ICa were determined: for IK,s (overnight exposure), 1.52 μM; ICa (transient exposure), 1.47 μM; and ICa (overnight exposure), 1.56 μM. Thus, the EC50 values were broadly similar and therefore a difference in the cAMP sensitivity is unlikely to explain the observed distinction between IK,s and ICa enhancement in TG+ cells.
Figure 8. Concentration dependence of IK,s and ICa response in TG- cells to 8-Br-cAMP.
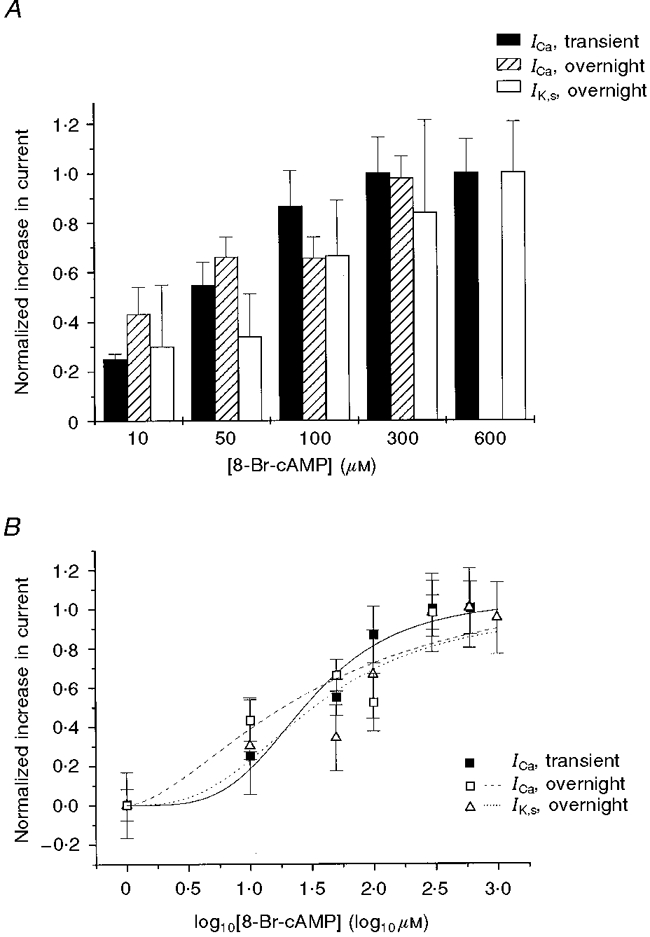
A, bar graph summarizing the normalized change in ICa (measured at +10 mV) following transient and 16-24 h (overnight) exposures to 8-Br-cAMP and the change in IK,s following 16-24 h exposure to 8-Br-cAMP (measured at +40 mV) as a function of [8-Br-cAMP]. There was no significant difference between groups at each 8-Br-cAMP concentration (P > 0.3). B, log concentration-response curves for ICa and IK,s (normalized change in peak ICa for transient and 16-24 h exposures and in peak IK,s for 16-24 h exposure plotted against [8-Br-cAMP]).
DISCUSSION
In this paper we report the first cellular measurements of ion channel activity and its modulation by the β-AR signalling cascade in developing hearts of mice engineered to overexpress β2-AR. We found that β2-AR overexpression enhanced L-type Ca2+ channel currents in a cAMP-dependent manner, but that the slow delayed rectifier K+ channel current was not affected by the transgene. Our results, based on functional assays of expressed channel activity, provide new insights into mechanisms by which receptor activation can direct modulatory enzymes to distinct targets (ion channels).
Evidence for embryonic activity of the α-MHC promoter
In cells isolated from TG+ hearts, we found significant effects of the β2-AR transgene at very early developmental stages. ICa was enhanced in TG+ cells as early as day 14 of embryogenesis indicating that the α-MHC promoter must be active in the ventricle at this stage of development, in contrast to previous reports suggesting activation after birth (Milano et al. 1994). Transgene activity in the ventricle is thus likely to be much more important during embryogenesis than previously estimated.
cAMP-dependent modulation of ICa in TG+ mice in the absence of exogenous agonist
In their studies of in vivo heart function in β2-AR TG+ mice, Milano et al. (1994) suggested that changes in heart rate and contractility detected in these hearts were not due to circulating catecholamines, but instead were due specifically to an increase in the number of β2-ARs present in the active conformation. This increase in active receptors is thought subsequently to activate adenylyl cyclase in the absence of agonist (Bond et al. 1995; Bond, 1997). Our data confirm this interpretation. Cells in our experiments were enzymatically isolated and then plated and kept under cell culture conditions in agonist-free media for 24 to 36 h before experiments were carried out. Before measuring ion channel activity, plates of cells were washed with agonist-free Tyrode solution and then cells which were studied with patch clamp procedures were continuously perfused with agonist-free solutions before and during the measurement of channel activity. Hence it is very unlikely that any of the effects on channel activity that we report caused by the transgene are due to stimulation of expressed receptor by exogenous catecholamines.
The observed functional differences in Ca2+ channel activity between TG- and TG+ cells as well as the effects of Rp-cAMPS on the voltage dependence and current density of ICa in TG+ cells presents strong evidence in favour of a cytoplasmic cAMP-dependent pathway for modulation of ICa in TG+ cells, consistent with activation of adenylyl cyclase and subsequent production of cAMP caused directly by an increased number of β2-ARs in the active conformation (Bond et al. 1995; Bond, 1997). This hypothesis is consistent with regulation of the L-type Ca2+ channel current by cytoplasmic pathways in the presence of β-AR stimulation in the heart previously shown by Hartzell et al. 1991.
cAMP discrimination between IK,s and ICa in TG+ mice: evidence for cAMP compartmentalization
Despite the cAMP-induced enhancement of ICa we found no effect of β2-AR overexpression on IK,s activity, either in its voltage dependence or in the level of expression. This suggested to us that the β2-AR-induced change in intracellular cAMP was not uniform throughout the cell, but was localized to regions near the L-type Ca2+ channel. This was supported by the finding that in both TG- and TG+ cells, exogenous 8-Br-cAMP application (both transient and prolonged exposure) increased peak IK,s density to a similar extent in both TG- and TG+ cells, demonstrating that IK,s was sensitive to cAMP in the mouse cells and that in the TG+ cells, this is consistent with the absence of any inhibitory pathway that might have prevented cAMP-dependent regulation. A comparison of the concentration dependence of ICa and IK,s for 8-Br-cAMP-mediated changes in current showed that the two channel types share a similar sensitivity to 8-Br-cAMP. We measured the change in each current caused by exposure to 8-Br-cAMP and the results were consistent with previous reports in other cell types, which have shown similar cAMP sensitivities for the two channel types (Kass & Wiegers, 1982; Giles et al. 1989; Iijima et al. 1990; Yazawa & Kameyama, 1990). Taken together, these results support the view that in the TG+ cells, the basal cAMP concentrations differ in regions near these two channel types consistent with targeting or compartmentalization of the β2-AR-induced increases in cAMP.
Although this is the first report of cAMP compartmentalization in cardiac cells in a manner that distinguishes target ion channel regulation by β2-AR overexpression, our results complement those of others who have focused on calcium entry and calcium transients in cardiac myocytes using pharmacological approaches (Xiao et al. 1994; Jurevicius & Fischmeister, 1996, 1997; Zhou et al. 1997). Previous studies have linked β2-AR-induced changes in contraction, cytosolic Ca2+, and L-type Ca2+ channel currents to activation of adenylyl cyclase and subsequent increases in intracellular cAMP (Skeberdis et al. 1997). Distinct functional differences have been reported in modulation of these parameters by stimulation of β1- or β2-receptors (Xiao & Lakatta, 1993), and these differences have been suggested to be due in part to β-receptor subtype-specific compartmentalization of cAMP (Aass et al. 1988; Hohl & Li, 1991; Xiao et al. 1994; Jurevicius & Fischmeister, 1996; Rockman et al. 1996; Skeberdis et al. 1997; Zhou et al. 1997). Our results support this view. The data presented in this paper strongly suggest that the modulation of ICa in the TG+ hearts is indeed due to increased cAMP activity in the vicinity of the L-type Ca2+ channels, and that the discrimination between modulation of ICa and IK,s is consistent with localization of this increased cAMP pool. Furthermore, the results of this study indicate that this mouse model in which β2-ARs are overexpressed will make an excellent system in which to study the molecular mechanism by which cAMP is targeted to L-type Ca2+ rather than slow delayed rectifier K+ channels. Future studies will be directed at determining whether distinct anchoring proteins are involved in this targeting.
Relationship to arrhythmias and heart failure
Electrical activity of the mammalian myocardium is maintained by a fine balance of a large number of ionic currents carried by ion channel and transport proteins (Kass, 1995). Sympathetic stimulation, a key regulatory pathway in the cardiovascular system, modulates contractile activity, but also, importantly, electrical activity at the single cell level. In order to ensure adequate diastolic filling time in the face of increased heart rate in the presence of elevated sympathetic tone, it is essential that the ventricular action potential duration be controlled by the sympathetic nervous system such that, concomitant with elevated heart rate, the ventricular action potential shortens. Key to this regulation is a balance between β-AR modulation of IK,s and ICa (Kass & Wiegers, 1982; Kass, 1994). This regulation occurs in a cAMP-dependent manner (Hove-Madsen et al. 1996). Dysfunction in slow delayed rectifier K+ channel activity has been shown to be linked to at least one inherited human cardiac arrhythmia, the long QT syndrome (Sanguinetti et al. 1996; Splawski et al. 1997). Interestingly, patients with defects in the genes encoding either of the two subunits of the slow delayed rectifier K+ channel, KvLQT1 or minK, are prone to arrhythmias in the face of elevated sympathetic tone, providing key clinical evidence for the importance of the regulation of this ion channel by the sympathetic nervous system for the maintenance of normal cardiac function (Moss, 1997). Because the relative contribution of β2-receptors to AR stimulation increases both in heart failure (Bristow et al. 1985, 1986) and in a recent experimental transgenic model in which nerve growth factor was overexpressed in a cardiac-specific manner (Heath et al. 1998), it is possible that cAMP-dependent regulation of the slow delayed rectifier K+ channel also may change in failure. If this were the case, action potential duration would not shorten sufficiently with increased levels of circulating catecholamines promoting conditions that resemble LQT1-type dysfunction. Investigation of conditions that promote arrhythmias in models of the failing heart should thus clearly include studies of the control of slow delayed rectifier K+ channels by β-AR stimulation.
Acknowledgments
This study was supported by USPHS grant 2R01 HL 44365-05 (R. S. K.). We thank Dr Xiao-Li Wang for excellent help in preparing myocytes and Mr John Crosby and Ms Sandy Duncan for ensuring co-ordination of mice between Durham, NC and New York, NY, USA.
References
- Aass H, Skomedal T, Osnes JB. Increase of cyclic AMP in subcellular fractions of rat heart muscle after β-adrenergic stimulation: prenalterol and isoprenaline caused different distribution of bound cyclic AMP. Journal of Molecular and Cellular Cardiology. 1988;20:847–860. doi: 10.1016/s0022-2828(88)80009-9. [DOI] [PubMed] [Google Scholar]
- An RH, Davies MP, Doevendans PA, Kubalak SW, Bangalore R, Chien KR, Kass RS. Developmental changes in β-adrenergic modulation of L-type calcium channels in embryonic mouse heart. Circulation Research. 1996;78:371–378. doi: 10.1161/01.res.78.3.371. [DOI] [PubMed] [Google Scholar]
- Bond RA. Do recent operational studies indicate that a single state model is no longer applicable to G protein-coupled receptors? Annals of the New York Academy of Sciences. 1997;812:92–97. doi: 10.1111/j.1749-6632.1997.tb48149.x. [DOI] [PubMed] [Google Scholar]
- Bond RA, Leff P, Johnson TD, Milano CA, Rockman HA, McMinn TR, Apparsundaram S, Hyek MF, Kenakin TP, Allen LF. Physiological effects of inverse agonists in transgenic mice with myocardial overexpression of the β2-adrenoceptor. Nature. 1995;374:272–276. doi: 10.1038/374272a0. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. New England Journal of Medicine. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S. β1- and β2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective β1-receptor downregulation in heart failure. Circulation Research. 1986;59:297–309. doi: 10.1161/01.res.59.3.297. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Kantrowitz NE, Ginsburg R, Fowler MB. β-Adrenergic function in heart muscle disease and heart failure. Journal of Molecular and Cellular Cardiology. 1985;17(suppl. 2):41–52. doi: 10.1016/0022-2828(85)90007-0. [DOI] [PubMed] [Google Scholar]
- Brown AM, Birnbaumer L. Direct G protein gating of ion channels. American Journal of Physiology. 1988;254:H401–410. doi: 10.1152/ajpheart.1988.254.3.H401. [DOI] [PubMed] [Google Scholar]
- Brown AM, Birnbaumer L. Ionic channels and their regulation by G protein subunits. Annual Review of Physiology. 1990;52:197–213. doi: 10.1146/annurev.ph.52.030190.001213. [DOI] [PubMed] [Google Scholar]
- Chen FC, Yamamura HI, Roeske WR. Adenylate cyclase and β adrenergic receptor development in the mouse heart. Journal of Pharmacology and Experimental Therapeutics. 1982;222:7–13. [PubMed] [Google Scholar]
- Davies MP, An RH, Doevendans P, Kubalak S, Chien KR, Kass RS. Developmental changes in ionic channel activity in the embryonic mouse heart. Circulation Research. 1995;78:15–25. doi: 10.1161/01.res.78.1.15. [DOI] [PubMed] [Google Scholar]
- Giles W, Nakajima T, Ono K, Shibata EF. Modulation of the delayed rectifier K+ current by isoprenaline in bull-frog atrial myocytes. The Journal of Physiology. 1989;415:233–249. doi: 10.1113/jphysiol.1989.sp017720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg R, Bristow MR, Billingham ME, Stinson EB, Schroeder JS, Harrison DC. Study of the normal and failing isolated human heart: decreased response of failing heart to isoproterenol. American Heart Journal. 1983;106:535–540. doi: 10.1016/0002-8703(83)90698-1. 10.1016/0002-8703(83)90698-1. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hartzell HC, Mery P-F, Fischmeister R, Szabo G. Sympathetic regulation of cardiac calcium current is due exclusively to cAMP-dependent phosphorylation. Nature. 1991;351:573–576. doi: 10.1038/351573a0. [DOI] [PubMed] [Google Scholar]
- Heath BM, Xia J, Dong E, An RH, Brooks A, Liang C-S, Federoff HJ, Kass RS. Overexpression of nerve growth factor in the heart alters ion channel activity and β-adrenergic signalling in an adult transgenic mouse. The Journal of Physiology. 1998;512:779–791. doi: 10.1111/j.1469-7793.1998.779bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl CM, Li QA. Compartmentation of cAMP in adult canine ventricular myocytes. Relation to single-cell free Ca2+ transients. Circulation Research. 1991;69:1369–1379. doi: 10.1161/01.res.69.5.1369. [DOI] [PubMed] [Google Scholar]
- Honore E, Attali B, Romey G, Heurteaux C, Ricard P, Lesage F, Barhanin J. Cloning, expression, pharmacology and regulation of a delayed rectifier K+ channel in mouse heart. EMBO Journal. 1991;10:2805–2811. doi: 10.1002/j.1460-2075.1991.tb07829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. Journal of General Physiology. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove-Madsen L, Mery PF, Jurevicius J, Skeberdis AV, Fischmeister R. Regulation of myocardial calcium channels by cyclic AMP metabolism. Basic Research in Cardiology. 1996;91(suppl. 2):1–8. doi: 10.1007/BF00795355. [DOI] [PubMed] [Google Scholar]
- Iijima T, Imagawa JI, Taira N. Differential modulation by β adrenoceptors of inward calcium and delayed rectifier potassium current in single ventricular cells of guinea pig heart. Journal of Pharmacology and Experimental Therapeutics. 1990;254:142–146. [PubMed] [Google Scholar]
- Jurevicius J, Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by β-adrenergic agonists. Proceedings of the National Academy of Sciences of the USA. 1996;93:295–299. doi: 10.1073/pnas.93.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevicius J, Fischmeister R. Longitudinal distribution of Na+ and Ca2+ channels and β-adrenoceptors on the sarcolemmal membrane of frog cardiomyocytes. The Journal of Physiology. 1997;503:471–477. doi: 10.1111/j.1469-7793.1997.471bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RS. Genesis of cardiac arrhythmias: roles of calcium and delayed potassium channels in the heart. In: Andreoli T, Fambrough D, Hoffman J, Welsh M, Schultz S, Brown AM, editors. Molecular Biology of Membrane Transport Disorders. Plenum Press; 1994. pp. 595–604. [Google Scholar]
- Kass RS. Ionic basis of electrical activity in the heart. In: Sperelakis N, editor. Physiology and Pathophysiology of the Heart. Norwell, MA, USA: Kluwer Academic; 1995. pp. 77–90. [Google Scholar]
- Kass RS, Arena JP, Chin S. Modulation of calcium channels by charged and neutral dihydropyridines. Annals of the New York Academy of Sciences. 1989;560:189–197. doi: 10.1111/j.1749-6632.1989.tb24097.x. [DOI] [PubMed] [Google Scholar]
- Kass RS, Wiegers SE. The ionic basis of concentration-related effects of noradrenaline on the action potential of calf cardiac Purkinje fibres. The Journal of Physiology. 1982;322:541–558. doi: 10.1113/jphysiol.1982.sp014054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima M, Ishima T, Taniguchi N, Kimura K, Sada H, Sperelakis N. Developmental changes in β-adrenoceptors, muscarinic cholinoceptors and Ca2+ channels in rat ventricular muscles. British Journal of Pharmacology. 1990;99:334–339. doi: 10.1111/j.1476-5381.1990.tb14704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubalak S, Doevendans PA, Rockman H, Hunter JJ, Tanaka N, Ross J, Chien KR. Molecular analysis of cardiac muscle diseases via mouse genetics. In: Adolph KW, editor. Methods in Molecular Genetics. Orlando, FL, USA: Academic Press; 1995. pp. 470–487. [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Maki T, Gruver EJ, Davidoff AJ, Izzo N, Toupin D, Colucci W, Marks AR, Marsh JD. Regulation of calcium channel expression in neonatal myocytes by catecholamines. Journal of Clinical Investigation. 1996;97:656–663. doi: 10.1172/JCI118462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, Johnson TD, Bond RA, Lefkowitz RJ. Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science. 1994;264:582–586. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- Moss AJ. Clinical management of patients with the long QT syndrome: drugs, devices, and gene-specific therapy. Pacing and Clinical Electrophysiology. 1997;20:2058–2060. doi: 10.1111/j.1540-8159.1997.tb03627.x. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ, Milano CA, Lefkowitz RJ. Myocardial β-adrenergic receptor signalling in vivo: insights from transgenic mice. Journal of Molecular Medicine. 1996;74:489–495. doi: 10.1007/BF00204974. [DOI] [PubMed] [Google Scholar]
- Rothermel JD, Parker BL. A mechanistic and kinetic analysis of the interactions of the diastereoisomers of adenosine 3′, 5′-(cyclic)phosphorothioate with purified cyclic AMP-dependent protein kinase. Biochemical Journal. 1988;251:757–762. doi: 10.1042/bj2510757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector P S X A D, Keating MT. Coassembly of K(v)LQT1 and mink (Isk) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. Journal of General Physiology. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeberdis VA, Jurevicius J, Fischmeister R. β-2 Adrenergic activation of L-type Ca2+ current in cardiac myocytes. Journal of Pharmacology and Experimental Therapeutics. 1997;283:452–461. [PubMed] [Google Scholar]
- Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nature Genetics. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- Sturm K, Tam PP. Isolation and culture of whole post-implantation embryos and germ layer derivatives. Methods in Enzymology. 1993;225:164–190. doi: 10.1016/0076-6879(93)25013-r. [DOI] [PubMed] [Google Scholar]
- Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of β-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454–463. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- Varnum MD, Busch AE, Bond CT, Maylie J, Adelman JP. The min K channel underlies the cardiac potassium current IKs and mediates species-specific responses to protein kinase C. Proceedings of the National Academy of Sciences of the USA. 1993;90:11528–11532. doi: 10.1073/pnas.90.24.11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao RP, Hohl C, Altschuld R, Jones L, Livingston B, Ziman B, Tantini B, Lakatta EG. β2-Adrenergic receptor-stimulated increase in cAMP in rat heart cells is not coupled to changes in Ca2+ dynamics, contractility, or phospholamban phosphorylation. Journal of Biological Chemistry. 1994;269:19151–19156. [PubMed] [Google Scholar]
- Xiao RP, Lakatta EG. β1-Adrenoceptor stimulation and beta 2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circulation Research. 1993;73:286–300. doi: 10.1161/01.res.73.2.286. [DOI] [PubMed] [Google Scholar]
- Yazawa K, Kameyama M. Mechanism of receptor-mediated modulation of the delayed outward potassium current in guinea-pig ventricular myocytes. The Journal of Physiology. 1990;421:135–150. doi: 10.1113/jphysiol.1990.sp017937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YY, Cheng H, Bogdanov KY, Hohl C, Altschuld R, Lakatta EG, Xiao RP. Localized cAMP-dependent signalling mediates β2-adrenergic modulation of cardiac excitation-contraction coupling. American Journal of Physiology. 1997;273:H1611–1618. doi: 10.1152/ajpheart.1997.273.3.H1611. [DOI] [PubMed] [Google Scholar]