

Abstract
The role of the cyclic AMP (cAMP) transduction cascade in mediating the prostaglandin E2 (PGE2)-induced decrease in potassium current (IK) was investigated in isolated embryonic rat sensory neurones using the whole-cell patch-clamp recording technique.
Exposure to 100 μM chlorophenylthio-adenosine cyclic 3′,5′-monophosphate (cpt-cAMP) or 1 μM PGE2 caused a slow suppression of the whole-cell IK by 34 and 36 %, respectively (measured after 20 min), without a shift in the voltage dependence of activation for this current. Neither of these agents altered the shape of the voltage-dependent inactivation curve indicating that the suppression of IK did not result from alterations in the inactivation properties.
To determine whether the PGE2-mediated suppression of IK depended on activation of the cAMP pathway, cells were exposed to this prostanoid in the presence of the protein kinase A (PKA) inhibitor, PKI. The PGE2-induced suppression of IK was prevented by PKI. In the absence of PGE2, PKI had no significant effect on the magnitude of IK.
Results obtained from protocols using different conditioning prepulse voltages indicated that the extent of cpt-cAMP- and PGE2-mediated suppression of IK was independent of the prepulse voltage. The subtraction of control and treated currents revealed that the cpt-cAMP- and PGE2-sensitive currents exhibited little time-dependent inactivation. Taken together, these results suggest that the modulated currents may be delayed rectifier-like IK.
Exposure to the inhibitors of IK, tetraethylammonium (TEA) or 4-aminopyridine (4-AP), reduced the control current elicited by a voltage step to +60 mV by 40-50 %. In the presence of 10 mM TEA, treatment with cpt-cAMP did not result in any further inhibition of IK. In contrast, cpt-cAMP reduced IK by an additional 25-30 % in the presence of 1 mM 4-AP. This effect was independent of the conditioning prepulse voltage.
These results establish that PGE2 inhibits an outward IK in sensory neurones via activation of PKA and are consistent with the idea that the PGE2-mediated sensitization of sensory neurones results, in part, from an inhibition of delayed rectifier-like IK.
Enhanced sensitivity to noxious stimuli is one of the characteristics of the inflammatory response and may result from an increase in the excitability of small-diameter sensory neurones that conduct nociceptive signals. Indeed, a number of pro-inflammatory agents that are synthesized and released in response to trauma can stimulate directly or augment the activation of sensory neurones (Treede et al. 1992). In this regard, prostaglandins represent an important group of agents that sensitize sensory neurones. Exposing various preparations of sensory neurones to prostaglandin E2 (PGE2) or prostaglandin I2 (PGI2) increases the number of action potentials elicited by noxious thermal, chemical and/or mechanical stimuli (Handwerker, 1976; Mense, 1981; Baccaglini & Hogan, 1983; Martin et al. 1987; Nicol & Cui, 1994; Nicol et al. 1997). These prostanoids also facilitate neurotransmitter release evoked by exposing sensory neurones to bradykinin, capsaicin or high extracellular potassium (Franco-Cereceda, 1989; Andreeva & Rang, 1993; Hingtgen & Vasko, 1994; Vasko et al. 1994).
Although the cellular mechanisms underlying the actions of these prostaglandins are not well understood, recent evidence suggests that these eicosanoids modulate ion channel activity that could enhance the excitability of sensory neurones. For example, PGE2 increases the amplitude of a tetrodotoxin (TTX)-resistant sodium current in adult and neonatal rat dorsal root ganglion (DRG) cells (Gold et al. 1996a; England et al. 1996). This prostanoid also suppresses a calcium-dependent slow after-hyperpolarization in adult rat nodose ganglia cells and DRG neurones (Fowler et al. 1985; Gold et al. 1996B). Furthermore, we demonstrated recently that PGI2 and PGE2 attenuate whole-cell potassium currents (IK), whereas the non-sensitizing prostanoid PGF2α is ineffective in sensory neurones (Nicol et al. 1997).
The question remains as to whether the prostaglandin-induced modulation of ion channels results in sensitization of sensory neurones. To initially address this issue, it is important to assess whether prostaglandin-induced sensitization and alterations in ion channels are mediated by the same transduction mechanisms. Because the sensitizing actions of PGE2 or PGI2 on sensory neurones are mediated by the cyclic AMP (cAMP) transduction cascade (Ferreira & Nakamura, 1979; Taiwo et al. 1989; Hingtgen et al. 1995; Cui & Nicol, 1995), modulatory effects of prostanoids on ion channels regulating membrane excitability should also be dependent on the cAMP pathway. We hypothesize that the sensitizing actions of PGE2 are also mediated by the suppression of IK. This notion is based, in part, on observations wherein cAMP modulates voltage-dependent IK in a variety of cell types. In the GH4C1 pituitary cell line, dibutyryl cAMP suppresses a delayed rectifier IK (Chung & Kaczmarek, 1995). Similarly, the current that arises from the expression of Kv3.2 (delayed rectifier potassium) channels in Chinese hamster ovary cells is inhibited by a membrane-permeant analogue of cAMP (Moreno et al. 1995). Activation of the cAMP pathway in mouse neurones isolated from the colliculus leads to a long-term (2-4 h) enhancement of excitability as exhibited by an increased duration of the action potential and a greater number of action potentials evoked by a depolarizing current pulse (Ansanay et al. 1995). These authors found that stimulation of protein kinase A (PKA) inhibits a delayed rectifier-like IK. Lastly, PGE2, through activation of the cAMP-PKA cascade, may play a role in the regulation of vascular smooth muscle tone. In smooth muscle cells isolated from the tail artery of the rat, PGE2 suppresses a non-inactivating IK (Ren et al. 1996). Therefore, taken together, the sensitivity of excitable cells and its modulation by different mediators will play an important role in the regulation of the physiological function of the cell.
To ascertain whether suppression of IK could also be a mechanism for prostaglandin-induced sensitization of sensory neurones, we examined the hypothesis that the cAMP pathway mediates the PGE2-induced decrease in IK in isolated rat embryonic sensory neurones. Our results demonstrate that the cAMP analogue, chlorophenylthio-adenosine cyclic 3′,5′-monophosphate (cpt-cAMP) inhibits a delayed rectifier-like IK in sensory neurones in a manner analogous to PGE2 and that the inhibitory effects of PGE2 on the whole-cell IK are blocked by the inhibition of PKA. Preliminary findings from this study have been reported in abstract form (Evans et al. 1996B).
METHODS
Isolation and culture of embryonic rat sensory neurones
The procedures for isolation and culture of rat sensory neurones have been described previously (Vasko et al. 1994). All procedures were approved by the Animal Care and Use Committee at Indiana University School of Medicine. Briefly, timed-pregnant rats were rendered unconscious with CO2, and killed by cervical dislocation. Embryos (embryonic day (E) 15-E17) were removed from the uterus and placed in a dish containing calcium- and magnesium-free Hanks’ balanced salt solution (Life Technologies, Grand Island, NY, USA). The dorsal root ganglia were dissected from each embryo and sensory neurones were dissociated from the ganglia with 0.025 % trypsin (37°C, 25 min) and mechanical agitation. The cells were grown in Dulbecco's modified Eagle's medium (Life Technologies, Grand Island, NY, USA) supplemented with 2 mM glutamine, 50 μg ml−1 penicillin, 50 μg ml−1 streptomycin, 10 % (v/v) heat-inactivated fetal bovine serum, 50 μM 5-fluoro-2′-deoxyuridine, 150 μM uridine and 250 ng ml−1 7S-nerve growth factor (Harlan Bioproducts for Science, Indianapolis, IN, USA). Approximately 150 000 cells ml−1 were plated in a collagen-coated culture dish containing small plastic coverslips. Cultures were maintained at 37°C in a 5 % CO2 atmosphere and the medium was changed every second day.
Recording procedures
Recordings were made using the whole-cell patch-clamp technique as described previously (Hamill et al. 1981; Nicol et al. 1997). Briefly, a coverslip with the sensory neurones (4-6 days in culture) was placed in a recording chamber where the neurones were bathed in normal Ringer solution of the following composition (mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes and 10 glucose; pH 7.4, adjusted with NaOH. Recording pipettes were pulled from disposable borosilicate glass tubing and typically had resistances of 2-5 MΩ when filled with the following solution (mM): 140 KCl, 5 MgCl2, 4 ATP, 0.3 GTP, 2.5 CaCl2, 5 EGTA (calculated free Ca2+ concentration of ∼100 nM) and 10 Hepes; pH 7.3, adjusted with KOH. For these solutions, a junction potential of 3.7 mV was calculated using the approach described by Barry (1994). We have not corrected for this potential and expect that the actual membrane potentials are 3-4 mV more negative than those listed.
Whole-cell currents were recorded from sensory neurones with either an Axopatch 200 (Axon Instruments, Foster City, CA, USA) or an EPC-7 (List Electronic, Darmstadt, Germany) patch-clamp amplifier; the data were acquired and analysed using pCLAMP 6 (Axon Instruments). The whole-cell recording configuration was established in normal Ringer solution. Both capacitance and series resistance compensation were used; however, no compensation was made for leak currents. The remaining uncompensated series resistance was 1.9 ± 0.3 MΩ (mean ±s.e.m.; range, 0.16-5.4 MΩ; n = 21). The maximum voltage error resulting from the uncompensated series resistance was calculated for each cell and averaged 9.8 ± 1.8 mV (n = 21).
After establishing the whole-cell configuration, IK was isolated by superfusing the cells with 140 mM N-methyl-glucamine chloride (NMG)-Ringer solution (equimolar substitution of NMG for NaCl), adjusted to pH 7.4 with KOH. The membrane voltage was held at -60 mV and two voltage-step protocols were then used to examine the activation and inactivation of the current. To determine the voltage dependence of activation, voltage steps of 175 ms were applied at 5 s intervals in +10 mV increments to a maximum of +60 mV. To determine the voltage dependence of inactivation, conditioning prepulses ranging from -100 to +20 mV were applied at 6 s intervals in 10 mV increments for 500 ms followed by a voltage step to +60 mV (200 ms duration). Another series of experiments examined the effects of a conditioning prepulse on the activation of the outward IK. Prepulses to either -100 or -30 mV were applied for 1 s; from this level incremental voltage steps (450 ms duration) depolarized the neurone to a maximum of +60 mV. After obtaining the control response, the superfusate was changed to NMG-Ringer solution containing either PGE2 or cpt-cAMP and cells were superfused continuously for the appropriate time. In experiments investigating the role of PKA in the PGE2-mediated decrease in IK, 20 μM of the PKA inhibitor, peptide protein kinase inhibitor (PKI14-24), was added to the whole-cell patch pipette and allowed to diffuse intracellularly for 10 min prior to recording potassium currents. In those experiments using 10 mM tetraethylammonium (TEA), the extracellular NaCl concentration in the Ringer solution was reduced to 130 mM, whereas 1 mM 4-aminopyridine (4-AP) was added directly to the Ringer solution. These agents were bath applied for 3-4 min prior to the acquisition of additional current recordings. All experiments were performed at room temperature (∼23°C).
Only the results obtained from neurones that satisfied the following criteria are presented in this report. First, after establishing the whole-cell configuration, neurones had to maintain zero-current potentials more hyperpolarized than -45 mV for at least 4-5 min, otherwise the recording was terminated. Second, over the initial 10 min period of the control recording, the peak amplitude of IK could not change by greater than 10 % (compared with the recording at time zero) or the recording was terminated. Third, neurones had to be sensitive to capsaicin. At the conclusion of each recording period, each neurone was exposed to normal Ringer solution containing 100 nM capsaicin. Neurones responsive to capsaicin exhibited an inward current that reversed following washout of capsaicin with normal Ringer solution. This neurotoxin was used to distinguish small-diameter sensory neurones as these neurones are believed to transmit nociceptive information (Holzer, 1991).
Data analysis
All values represent the mean ± standard error of the mean (s.e.m.). The voltage dependence of activation and inactivation of the outward IK was fitted with the Boltzmann relation (McFarlane & Cooper, 1991; Akins & McCleskey, 1993). For activation, the relation: G/Gmax =δ/[1 + exp(V0.5 - Vm)/k] was used, where G is the conductance, Gmax is the maximal conductance obtained at +60 mV, δ is a factor to account for the inhibition by PGE2 and cpt-cAMP, V0.5 is the voltage for half-maximal activation, Vm is the membrane voltage and k is a factor which describes the steepness of the voltage-conductance relation. Conductance was determined from the relation: G =I/(Vm - EK), where I is the measured membrane current, Vm is the voltage step and EK is the potassium equilibrium potential (calculated to be -84 mV). For inactivation, the relation: G/Gmax =c+ {(1 - c)/[1 + exp(V0.5 - Vm)/k]} was used, where c is the fraction of non-inactivating current (defined as the peak current obtained at +60 mV for the +20 mV prepulse) and the other parameters are as defined above. Fits were obtained using the curve fitting protocols in SigmaPlot 5.0 (Jandel Scientific, San Rafael, CA, USA). Statistical differences between the control recordings and those obtained under various treatment conditions were determined by using either Student's paired t test or a one-way analysis of variance (ANOVA) with repeated measures (whenever appropriate). When a significant difference was obtained with an ANOVA, post hoc analyses were performed using Student-Newman-Keuls test. Values of P < 0.05 were judged to be statistically significant.
Chemicals
PGE2 was obtained from Cayman Chemical Co. (Ann Arbor, MI, USA); cpt-cAMP was purchased from Boehringer Mannheim Corp. (Indianapolis, IN, USA); PKI was purchased from Peninsula Laboratories, Inc. (Belmont, CA, USA). All other chemicals were obtained from Sigma Chemical Corp. Prostaglandins and capsaicin were dissolved in 1-methyl-2-pyrrolidinone (HPLC grade, Aldrich Chemical Co., Milwaukee, WI, USA) to obtain concentrated stock solutions. These stock solutions were then diluted with Ringer solution to yield the appropriate concentration. We have demonstrated previously that the vehicle, 1-methyl-2-pyrrolidinone, had no effect on either the activation or steady-state inactivation curves obtained for potassium currents (Nicol et al. 1997).
RESULTS
cpt-cAMP suppresses an outward potassium current
We demonstrated previously that 1 μM PGE2 decreased whole-cell IK in isolated sensory neurones (Nicol et al. 1997). To determine whether the cAMP transduction cascade mediates this action, we first examined whether an analogue of cAMP could suppress IK in a manner analogous to PGE2. The peak amplitudes of IK for a series of voltage steps were measured in the absence and presence of 100 μM cpt-cAMP, a membrane-permeant analogue of cAMP. Consistent with our previous work, we found that the outward IK in these neurones ranged from a sustained type of current to current that exhibited varying degrees of time-dependent inactivation (Nicol et al. 1997). As can be seen in a representative neurone (Fig. 1, top left panel) depolarization of the membrane, in 20 mV steps, from a holding potential of -60 mV elicited outward currents that exhibited a small degree of time-dependent inactivation. An inactivation protocol further illustrated the degree of inactivation (Fig. 1, bottom left panel). This same neurone was then exposed to 100 μM cpt-cAMP. After a 20 min exposure, the outward current at all voltages was reduced (Fig. 1, top right panel); the maximum current (+60 mV) was lowered from 6.6 to 4.4 nA (23 % inhibition). Similar reductions in the test currents were observed using the inactivation protocol (Fig. 1, bottom right panel).
Figure 1. cpt-cAMP inhibits an outward potassium current in sensory neurones.

Outward potassium currents are shown from a representative sensory neurone in the absence (left panels) and presence (right panels) of 100 μM cpt-cAMP. In the top panels, IK was activated by incremental 20 mV steps from a holding potential of -60 mV (see inset). The bottom panels represent the currents obtained from this neurone using the steady-state inactivation protocol (see inset) before (left) and 20 min after (right) application of cpt-cAMP. In the bottom panels, the beginnings of the traces are the last 300 ms of the conditioning prepulses.
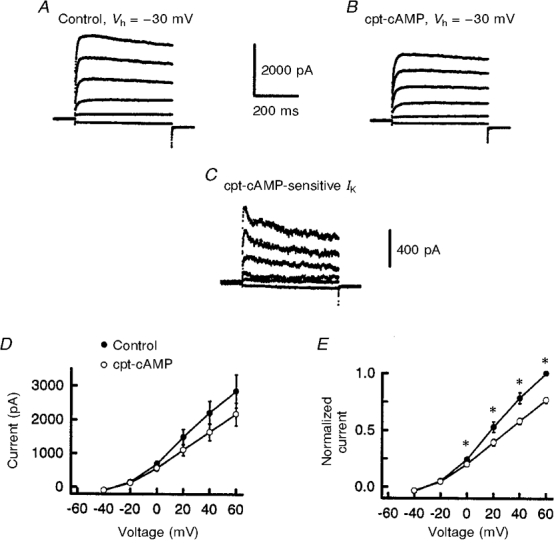
The capacity of cpt-cAMP to suppress the outward IK in sensory neurones is summarized in Fig. 2. Under control conditions, the peak amplitude of IK (at +60 mV) was 4.3 ± 0.6 nA (n = 10). After a 20 min exposure to 100 μM cpt-cAMP, the current was reduced to 2.9 ± 0.4 nA. Normalization of the current values to those obtained for the control recordings at +60 mV indicated that only 66 ± 2 % of the total current remained after 20 min (data not shown). Treatment with cpt-cAMP significantly reduced all the current values compared with the controls except for those currents obtained after the 2 min exposure for voltages between -50 and -20 mV (ANOVA with repeated measures). In another series of experiments (sensory neurones from other harvests), a 20 min treatment with 1 μM PGE2 significantly suppressed IK by ∼36 % (see below). These results indicate that both cpt-cAMP and PGE2 reduced IK by very similar amounts.
Figure 2. cpt-cAMP reduces the activation of an outward potassium current.
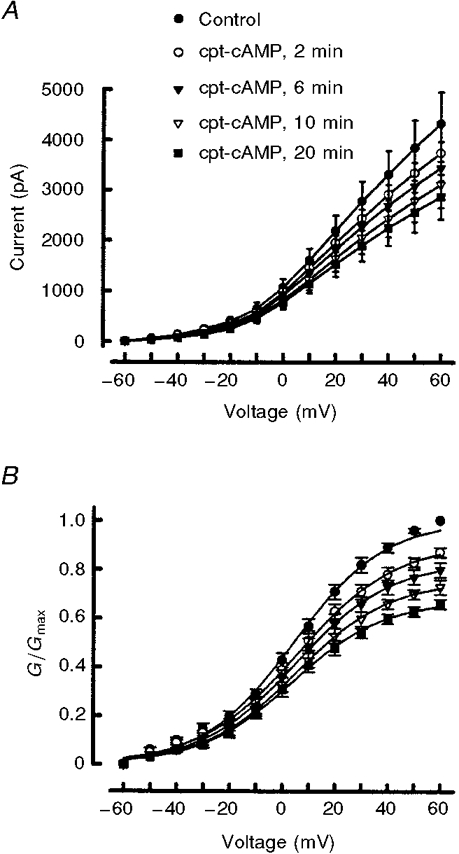
A, time-dependent suppression of IK after exposure to 100 μM cpt-cAMP. Currents were obtained in response to incremental 10 mV steps from a holding potential of -60 mV. B illustrates the currents shown in A after they were converted to conductance (G) values and fitted by the Boltzmann relation as described in the Methods section. In A and B, each point represents the mean ±s.e.m. from ten neurones. For those points appearing to lack error bars, the size of the bar is smaller than the symbol.
To ascertain the effects of cpt-cAMP on the voltage dependence for the activation of IK, the conductances were calculated and fitted with the Boltzmann equation to determine the voltage of half-maximal activation (V0.5), the steepness of the voltage-conductance relation (k) and G/Gmax (see Fig. 2A and Table 1). The value of G/Gmax (obtained at +60 mV) was reduced significantly from a control value of 1.0 to 0.69 ± 0.02 after a 20 min exposure to 100 μM cpt-cAMP (n = 10, ANOVA with repeated measures). Neither V0.5 nor k were altered significantly after exposure to cpt-cAMP (see Table 1). This lack of change in V0.5 and k indicates that cpt-cAMP does not cause a shift in the voltage dependence of activation for IK in these neurones. There was, however, a significant decrease in the value of the Boltzmann fitting parameter, δ, at all time points following exposure to cpt-cAMP and this is reflected in the decreased G/Gmax values obtained at +60 mV (ANOVA with repeated measures).
Table 1.
Effects of cpt-cAMP, PGE2 and PKI on the voltage dependence of IK activation
| V0.5 (mV) | k (mV) | G/Gmax | ||
|---|---|---|---|---|
| Control† | 4 ± 2 | 17 ± 1 | 1.0 | |
| cpt-cAMP | 2 min | 7 ± 2 | 18 ± 1 | 0.92 ± 0.02* |
| 6 min | 7 ± 3 | 18 ± 1 | 0.85 ± 0.04* | |
| 10 min | 6 ± 2 | 17 ± 1 | 0.76 ± 0.03* | |
| 20 min | 5 ± 2 | 17 ± 1 | 0.69 ± 0.02* | |
| Control† | 1 ± 2 | 14 ± 1 | 1.0 | |
| PGE2 | 2 min | 1 ± 2 | 15 ± 1 | 0.90 ± 0.02* |
| 6 min | 1 ± 3 | 15 ± 1 | 0.81 ± 0.03* | |
| 10 min | 2 ± 3 | 15 ± 1 | 0.73 ± 0.03* | |
| 20 min | −1 ± 3 | 16 ± 1 | 0.64 ± 0.05* | |
| Control + PKI‡ | 5 ± 2 | 15 ± 1 | 1.0 | |
| PGE2+ PKI | 20 min | 5 ± 2 | 17 ± 1 | 0.97 ± 0.05 |
Values are means ± S.E.M.
Significant difference from respective controls
n = 10
n = 7.
The potassium currents inhibited by cpt-cAMP and PGE2 are similar
Increasing intracellular cAMP levels appeared to suppress IK in a manner analogous to that observed with PGE2. These findings suggest that cpt-cAMP and PGE2 may inhibit similar types of IK. To address this idea, we used a subtraction protocol to obtain the currents inhibited by cpt-cAMP (the cpt-cAMP-sensitive current) for comparison with the PGE2-sensitive current. The currents recorded for the different voltage steps after a 20 min exposure to these agents were subtracted from their respective control recordings. As illustrated in Fig. 3A and B, the cpt-cAMP- and PGE2-sensitive currents in representative neurones displayed little time-dependent inactivation. Similar results were found in 20 neurones exposed to either cpt-cAMP (n = 10) or PGE2 (n = 10), suggesting that the subtracted currents were a sustained type of IK rather than a fast-inactivating type (e.g. an IA-like potassium current). Comparison of the current-voltage relations for the cpt-cAMP- and PGE2-sensitive currents revealed no significant differences (see Fig. 3A). Also, Boltzmann fits to individual cpt-cAMP- or PGE2-sensitive conductances demonstrated no significant differences between either V0.5 (6 ± 3 and 9 ± 2 mV, respectively) or k (18 ± 1 and 12 ± 1 mV, respectively).
Figure 3. The cpt-cAMP- and PGE2-sensitive currents are similar.
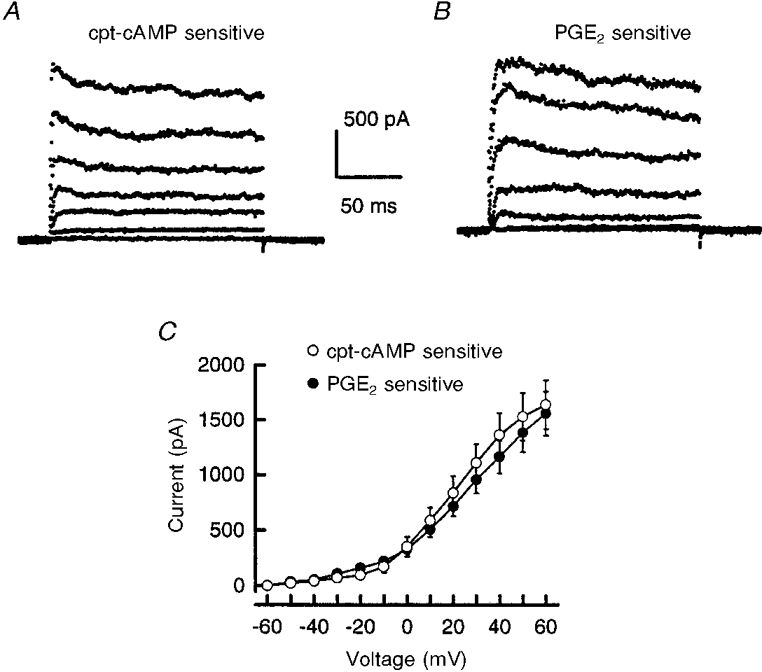
The currents inhibited by 100 μM cpt-cAMP (A) or 1 μM PGE2 (B) were obtained from representative neurones by subtracting the currents remaining after a 20 min exposure to these agents from those currents recorded under control conditions. The currents were obtained from incremental 20 mV steps from a holding potential of -60 mV. C shows the current-voltage curves for the cpt-cAMP- and PGE2-sensitive currents. Each data point represents the mean ±s.e.m. from ten neurones.
cpt-cAMP and PGE2 increase the resting input resistance
We examined the resting membrane resistance by assessing the amplitudes of the currents just prior to the termination of 500 ms conditioning prepulses (-100 to -40 mV) before and after treatment with cpt-cAMP or PGE2. The slope of the linear regression line was used to calculate the input resistance between -100 and -40 mV. The resting input resistance was increased significantly after a 20 min exposure to 100 μM cpt-cAMP (paired t test, data not shown). Under control conditions, the resistance was 180 ± 26 MΩ whereas after treatment with cpt-cAMP the resistance increased to 324 ± 33 MΩ (n = 10). Similar results were observed for PGE2. Treatment with 1 μM PGE2 (20 min exposure) increased the resistance from a control value of 158 ± 13 to 258 ± 38 MΩ (n = 10). These results show that the activation of the cAMP signalling pathway increases the resting input resistance of sensory neurones. This modulation may, in part, give rise to the enhanced excitability observed after treatment with PGE2 (Cui & Nicol, 1995; Nicol et al. 1997).
Inhibition of protein kinase A blocks PGE2-mediated suppression of outward potassium currents
To establish a causal relationship between activation of the cAMP transduction cascade and the PGE2-induced suppression of potassium currents, we examined the effects of PKI, a peptide inhibitor of PKA (Cheng et al. 1986), on the PGE2-mediated decrease in potassium current. As shown in Fig. 4A, 1 μM PGE2 elicited a time-dependent suppression of the total outward current. Under control conditions, the mean peak IK obtained at +60 mV was 4.8 ± 0.6 nA (n = 10). After exposure times to PGE2 of 10 and 20 min, the peak current was reduced to 3.6 ± 0.5 and 3.2 ± 0.5 nA, respectively. The current values after PGE2 treatment for 10 and 20 min were significantly different from the controls at all voltage steps (ANOVA with repeated measures). Activation curves for IK were fitted with the Boltzmann relation; the fitting parameters are shown in Table 1. After exposure to PGE2, there was a significant decrease in the value of the Boltzmann fitting parameter, δ, which reflects the decrease in G/Gmax obtained at +60 mV (ANOVA with repeated measures). Neither V0.5 nor k were altered significantly following exposure to this prostanoid suggesting that PGE2 did not alter the voltage-dependent activation profile of the potassium current.
Figure 4. PKA mediates the PGE2-induced suppression of IK.
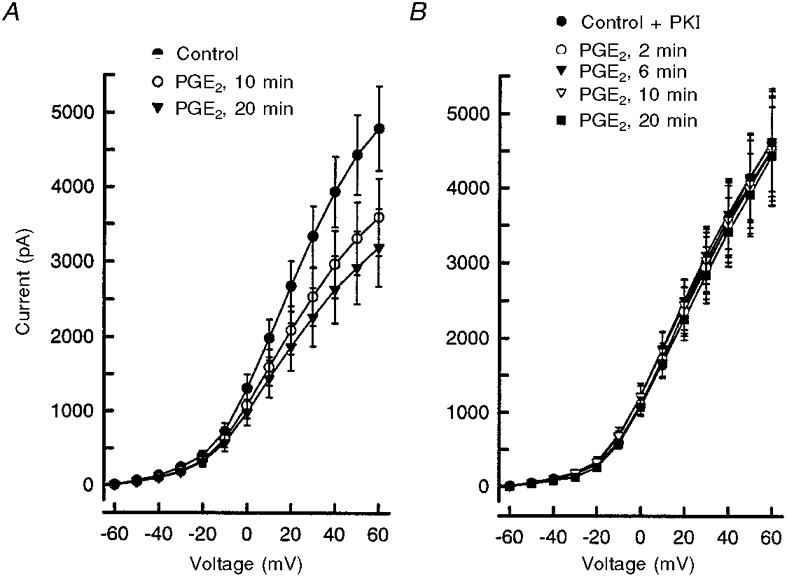
A, treatment with 1 μM PGE2 elicited a time-dependent suppression of IK. Currents were elicited by incremental 10 mV steps from a holding potential of -60 mV. Each point represents the mean ±s.e.m. from ten neurones. B shows that 20 μM PKI abolished the inhibition of IK produced by PGE2. Data points represent means ±s.e.m. from seven neurones. For those data points appearing to lack error bars, the size of the bars is smaller than the symbol.
To determine whether the PGE2-mediated suppression of IK depends on activation of the cAMP pathway, cells were exposed to this prostanoid in the presence of PKI. The recording pipette solution contained 20 μM PKI, which was allowed to diffuse into the cell for at least 10 min prior to obtaining the control IK. As illustrated in Fig. 4A, PKI completely abolished the PGE2-induced suppression of IK; after a 20 min exposure to 1 μM PGE2 the peak current at +60 mV (4.4 ± 0.6 nA, n = 7) was not changed from the mean control value of 4.6 ± 0.7 nA. For this IK, a G/Gmax value of 0.97 ± 0.05 was calculated; this was significantly different from the G/Gmax value (0.64 ± 0.05) obtained for neurones exposed to PGE2 in the absence of PKI, but was not significantly different from control values (ANOVA, compare Fig. 4A with B). Elimination of the PGE2-mediated decrease in IK by PKI was not a direct effect of PKI on the current because, in the absence of PGE2, PKI had no significant effect on the magnitude of the outward currents. The mean amplitude of the peak current (obtained at +60 mV) in the presence of PKI was 4.6 ± 0.7 nA (n = 7), whereas, in those neurones that did not receive PKI, the peak current was 4.8 ± 0.5 nA (n = 10). Furthermore, the values of the Boltzmann parameters obtained from neurones exposed to PKI alone were not different from the values obtained under control conditions in either the PGE2 or the cpt-cAMP experiments (Table 1). These results establish that PGE2 inhibits an outward potassium current(s) in sensory neurones via activation of PKA.
The nature of IK suppressed by cpt-cAMP
The effects of cpt-cAMP on the voltage dependence of inactivation of IK were examined by measuring the amplitudes of the peak currents obtained at +60 mV following the conditioning prepulse voltage step before and after the application of cpt-cAMP (see Fig. 5). The peak amplitude of IK was reduced significantly by cpt-cAMP treatment at all prepulse voltages (see Fig. 5A; n = 10, paired t test). These currents were then normalized to the value obtained for the -100 mV conditioning prepulse step and fitted with the Boltzmann equation (Fig. 5A). Under control conditions, V0.5, k and the fraction of non-inactivating current (c) were -39 ± 6 mV, 21 ± 2 mV and 0.70 ± 0.03, respectively. These values were not altered significantly following 20 min exposure to 100 μM cpt-cAMP, where V0.5, k and c were -39 ± 6 mV, 24 ± 2 mV and 0.62 ± 0.02, respectively. Although the peak current was reduced by cpt-cAMP at all prepulse voltages, there was little change in the shape of the inactivation curves indicating that the cpt-cAMP-mediated suppression did not result from consequential alterations in the inactivation properties of IK. Taken together, these results suggest that cpt-cAMP reduced the total amount of IK capable of activation and that the extent of this reduction did not depend on the conditioning prepulse.
Figure 5. cpt-cAMP reduces IK but does not alter the shape of the steady-state inactivation profile.

A, the peak IK obtained for the +60 mV step in the steady-state inactivation protocol was suppressed after 20 min treatment with 100 μM cpt-cAMP. The current values obtained after treatment with cpt-cAMP were significantly different from the controls at all voltage steps (paired t test). These values for IK were then used to determine the Boltzmann parameters that described the inactivation of this current and are shown in B. The lines drawn through the data points represent the Boltzmann fits to the control data and and data obtained after 20 min treatment with cpt-cAMP. In A and B, data points represent means ±s.e.m. from ten neurones.
The notion that cpt-cAMP led to the suppression of IK without having a significant effect on the inactivation of this current was examined in further detail. The results obtained with the inactivation protocol were used to determine the extent of inhibition by cpt-cAMP on the peak amplitude (Ipeak) compared with the current just prior to the end of the +60 mV step (steady-state current; Iss) for different prepulse voltages. These different conditioning prepulses could be used to activate/inactivate different populations of potassium currents (e.g. fast-inactivating IAvs. delayed rectifier types). Subtraction of Iss from Ipeak for different prepulse voltages should reveal any selective inhibition produced by 100 μM cpt-cAMP. As shown in Fig. 6A, the difference (Ipeak - Iss) became smaller for more depolarizing prepulse voltages and was consistent with the loss of an IA-like potassium current. However, after a 20 min exposure to cpt-cAMP the extent of (Ipeak - Iss) was not significantly different from the control values. Similarly, a comparison of the percentage of inhibition produced by cpt-cAMP for either Ipeak or Iss as a function of the prepulse voltage demonstrated that, regardless of the amplitude of the conditioning prepulse, cpt-cAMP produced an approximately 35 % suppression of IK (data not shown). Our results for cpt-cAMP are in contrast to those obtained by Chung & Kaczmarek (1995) in GH4C1 cells. They observed that dibutyryl cAMP reduced IK to a greater extent at the end of the pulse compared with the beginning, suggesting that cAMP increased the rate of inactivation. Analysis of the effects of 1 μM PGE2 and their dependence on the prepulse voltage demonstrated that PGE2, like cpt-cAMP, had no significant action on the value of (Ipeak - Iss) compared with the control condition (see Fig. 6A) and that the inhibition of both Ipeak and Iss ranged between 35 and 40 %. This observation supports the notion that cpt-cAMP and PGE2 led to the suppression of the same IK by reducing the number of channels capable of activation rather than by affecting their rates of inactivation.
Figure 6. Inhibition of IK by cpt-cAMP or PGE2 does not depend on the conditioning prepulse voltage.
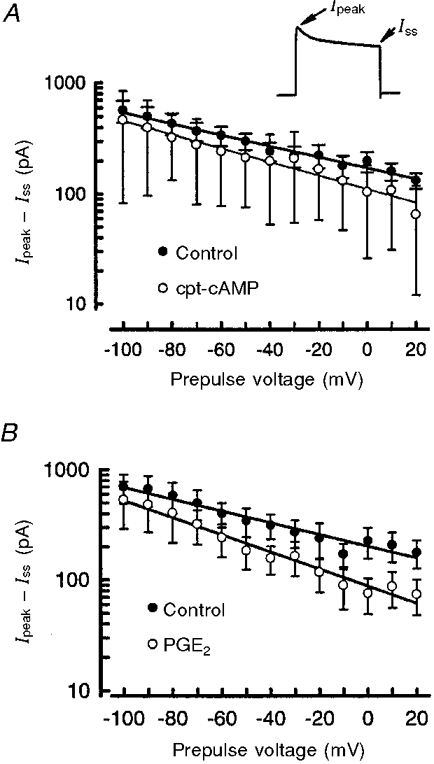
The current (Ipeak - Iss) was measured as the difference between the peak (Ipeak) and steady-state (Iss) currents for the voltage step to +60 mV using the steady-state inactivation protocol and is shown in the inset in A. A, the value of (Ipeak - Iss) was determined in the absence and presence of 100 μM cpt-cAMP (20 min treatment) as a function of the conditioning prepulse voltage. The points represent means ±s.e.m. from ten neurones. Note that the ordinate is a logarithmic scale. The lines drawn through the data points are linear regression lines wherein the correlation coefficients (r2) for the control and cpt-cAMP treatments were 0.98 and 0.99, respectively. B shows the value of (Ipeak - Iss) in the absence and presence of 1 μM PGE2 (20 min treatment) as a function of the conditioning prepulse voltage. The points represent means ±s.e.m. from ten neurones. The lines drawn through the data points are linear regression lines; the correlation coefficients (r2) for the control and PGE2 treatments were 0.94 and 0.97, respectively.
Different holding potentials and their effects on the capacity of cpt-cAMP to reduce IK
Analogous to the inactivation protocol, two specific conditioning prepulses of longer duration were used to manipulate selectively various components of the total whole-cell IK. A prepulse voltage step to -100 mV (1 s duration) was used to remove most of the inactivation associated with the rapidly inactivating potassium currents, such as IA, thereby revealing currents that are a combination of non-inactivating and inactivating currents (Connor & Stevens, 1971; Neher, 1971). Conversely, a prepulse voltage step to -30 mV (1 s duration) was used to inactivate rapidly inactivating currents and should result in the generation of currents dominated by sustained or delayed rectifier-type currents. From these long conditioning prepulses, an incremental series of depolarizing voltage steps (450 ms duration) was used to establish the current- voltage relation for both the peak current and the steady-state current.
The current-voltage relations for these two different holding potentials represent the compilation of all control studies for the experiments involving the effects of either the different prepulses alone or treatments with TEA or 4-AP (the pharmacology is shown below). For the 1 s prepulse to -100 mV, the mean peak current obtained at +60 mV was 5.6 ± 0.6 nA whereas for the prepulse to -30 mV the mean peak current was reduced significantly to 3.8 ± 0.4 nA (n = 15, data not shown). For all voltage steps, the peak IK was smaller for the -30 mV prepulse compared with the -100 mV prepulse. Similarly, the mean steady-state currents obtained for all voltage steps were reduced significantly for the -30 mV prepulse compared with those for the -100 mV prepulse (e.g. at +60 mV, IK was 3.1 ± 0.3 vs. 4.1 ± 0.4 nA, respectively). When the current values were normalized to their respective values obtained at +60 mV, all current-voltage relations exhibited a ‘half-maximal’ activation at 20 mV (data not shown).
As described above, treatment with cpt-cAMP for 20 min produced a suppression of IK that was similar for each conditioning prepulse. The representative currents obtained in the absence and presence of 100 μM cpt-cAMP for the conditioning prepulse to -100 mV are illustrated in Fig. 7. Under control conditions, the currents elicited by the depolarizing steps (450 ms duration) exhibited some time-dependent inactivation (Fig. 7A) and were similar to the traces shown in Fig. 1. Treatment with cpt-cAMP (Fig. 7A) reduced the peak current from 4.6 to 3.5 nA and the steady-state current from 3.8 to 3.2 nA, inhibitions of 23 and 20 %, respectively. Subtraction of these traces yielded the cpt-cAMP-sensitive current exhibited by this sensory neurone (Fig. 7A). This current demonstrated a small amount of time-dependent inactivation wherein the peak current at +60 mV was 850 pA compared with a steady-state current of 580 pA. The cpt-cAMP-mediated inhibition of IK for the -100 mV prepulse is summarized in Fig. 7A. When the effects of cpt-cAMP on the absolute values of IK were examined, cpt-cAMP did not produce a significant suppression of IK. This probably occurred because of the variability in the different amplitudes of IK. This idea was corroborated when the currents obtained after application of cpt-cAMP were normalized to their respective control values obtained at +60 mV. As shown in Fig. 7A, cpt-cAMP caused a significant inhibition of IK (paired t test). For a prepulse of -100 mV, a mean suppression of 22 ± 6 % (n = 4) was observed for the voltage step to +60 mV.
Figure 7. The cpt-cAMP-sensitive current obtained for a -100 mV conditioning prepulse.

A illustrates representative currents obtained in response to 20 mV incremental steps from a prepulse voltage of -100 mV (Vh, holding potential). B shows these currents after a 20 min exposure to 100 μM cpt-cAMP. Subtraction of the respective traces in B from those in A yield the cpt-cAMP-sensitive currents for these different voltage steps (C). The calibration bar for time is the same for all three panels. D shows the effect of 100 μM cpt-cAMP on the current-voltage relation of IK for the prepulse voltage of -100 mV. In E, the currents have been normalized to their respective control values obtained for the step to +60 mV. In D and E, the data points represent means ±s.e.m. obtained from four neurones. For those points appearing to lack error bars, the size of the bar is smaller than the symbol. * P < 0.05 vs. cpt-cAMP.
The results obtained from a representative neurone before and after exposure to cpt-cAMP at a prepulse voltage of -30 mV are shown in Fig. 8A and B, respectively. Exposure to cpt-cAMP produced a similar extent of suppression of IK for both the peak and steady-state currents, 27 and 23 %, respectively, as observed at -100 mV. The cpt-cAMP-sensitive current obtained for the -30 mV prepulse is shown in Fig. 8A. The peak and the steady-state currents at +60 mV were 800 and 480 pA, respectively. The inhibition of IK produced by cpt-cAMP for the -30 mV prepulse is summarized in Fig. 8A. When the effects of cpt-cAMP on the absolute values of IK were examined, cpt-cAMP did not produce a significant suppression of IK. As shown in Fig. 8A, when the cpt-cAMP currents were normalized to their control values obtained at +60 mV, cpt-cAMP caused a significant inhibition of IK (paired t test). For a prepulse of -30 mV, a mean suppression of 23 ± 3 % (n = 4) was observed for the voltage step to +60 mV. Therefore, the results obtained for the cpt-cAMP-sensitive currents at both -100 and -30 mV were similar to those illustrated in Fig. 3 for a holding potential of -60 mV.
Figure 8. The cpt-cAMP-sensitive current obtained for a -30 mV conditioning prepulse.
A illustrates representative currents elicited from the -30 mV prepulse voltage. These currents were obtained from the same neurone shown in Fig. 7. B shows those currents resulting after a 20 min exposure to 100 μM cpt-cAMP. The cpt-cAMP-sensitive current (C) was then determined by subtracting IK remaining after a 20 min exposure to 100 μM cpt-cAMP (B) from its respective control IK(A). The calibration bar for time is the same for all three panels. D shows the effect of 100 μM cpt-cAMP on the current-voltage relation of IK for the prepulse voltage of -100 mV. In E, the currents have been normalized to their respective control values obtained for the step to +60 mV. In D and E, the data points represent means ±s.e.m. obtained from four neurones. For those points appearing to lack error bars, the size of the bar is smaller than the symbol. * P < 0.05 vs. cpt-cAMP.
To determine whether the inhibition produced by cpt-cAMP was reversible, neurones were then superfused with the NMG-Ringer solution for 20 min and IK was determined for the two prepulse voltages. For the -100 mV prepulse, neither the peak nor the steady-state IK recovered to control values (data not shown). For example, the peak IK (at +60 mV) after a 20 min washout was 3.5 ± 0.8 nA (n = 3) compared with 3.3 ± 0.5 nA (n = 4) for the cpt-cAMP treatment. Similar results for the lack of reversibility were obtained for the peak and steady-state IK at the -30 mV prepulse (data not shown). Akins & McCleskey (1993) also reported that the inhibitory actions of membrane-permeant analogues of cAMP were irreversible. These results suggest that the extent of cpt-cAMP-induced suppression of IK was long-lived (at least for 20 min). Thus, our findings suggest that the IK suppressed by cpt-cAMP is analogous to a delayed rectifier-like current rather than a rapidly inactivating-like current such as that described as IA (or that current exhibited by the expression of Kv1.4).
Pharmacological characterization of IK inhibited by cpt-cAMP
The inhibitors of IK, TEA and 4-AP, were used in an attempt to distinguish further the nature of the current reduced by cpt-cAMP (Thompson, 1977). Conditioning prepulses to -100 and -30 mV were used in combination with the pharmacological intervention to isolate delayed rectifier-like currents from rapidly inactivating currents. The effects of 10 mM TEA on IK elicited from a prepulse of -30 mV in a representative sensory neurone are shown in Fig. 9A. Because it was established that the prepulse voltage did not influence the extent of IK suppression by either PGE2 or cpt-cAMP, only the results obtained for -30 mV will be represented. Under control conditions, a voltage step to +60 mV from the -30 mV prepulse evoked a peak IK of 6.2 nA whereas the steady-state IK was 5.3 nA (Fig. 9A, top panel). For the -100 mV prepulse, the peak and steady-state IK were 8.2 and 6.3 nA, respectively (data not shown). Treatment with 10 mM TEA-Ringer solution caused an approximately 60 % reduction in the peak and steady-state IK for both conditioning prepulses (Fig. 9A, middle panel). After exposure to TEA, the time-dependent inactivation exhibited by IK was still apparent for the -100 mV prepulse whereas it was reduced for the -30 mV prepulse. This sensory neurone was then treated with 100 μM cpt-cAMP and the results obtained after 20 min are illustrated in the bottom panel of Fig. 9A. Interestingly, after TEA, cpt-cAMP did not produce a further inhibition of the peak or the steady-state IK for either prepulse voltage.
Figure 9. The effects of TEA or 4-AP and cpt-cAMP on IK at the -30 mV conditioning prepulse voltage.

These representative current traces illustrate the effects of either TEA or 4-AP and 100 μM cpt-cAMP in the presence of TEA or 4-AP. A shows currents obtained under control conditions from a prepulse of -30 mV (top). The middle panel represents the suppression of these currents by 10 mM TEA. The bottom panel shows the currents remaining after a 20 min exposure to cpt-cAMP in the presence of 10 mM TEA. B illustrates the effects of 4-AP, where the top panel shows the currents obtained under control conditions from a prepulse of -30 mV. The middle panel represents the suppression of these currents by 1 mM 4-AP. The bottom panel shows the currents remaining after a 20 min exposure to cpt-cAMP in the presence of 1 mM 4-AP. The calibration bar for time is the same for all six panels.
Different classes of potassium currents have varying affinities for particular inhibitors. Therefore, we examined the sensitivity of the cpt-cAMP-modulated IK to 4-AP. The representative effects of 1 mM 4-AP on IK obtained for the -30 mV prepulse are illustrated in Fig. 9B. For a -30 mV prepulse, the application of 4-AP reduced the peak and steady-state IK obtained at +60 mV by 50 % (compare top and middle panels in Fig. 9A). The results obtained for the -100 mV prepulse were similar to those for -30 mV wherein 4-AP reduced the peak and steady-state IK by 43 and 44 %, respectively (data not shown). After treatment with 4-AP, IK exhibited a time-dependent inactivation that was similar to that observed in the control recordings for the -100 mV prepulse. This observation, in combination with the similar reductions in peak and steady-state currents, suggests that the IK comprising the component(s) that regulates the time-dependent inactivation is not sensitive to this concentration of 4-AP. In contrast to TEA, treatment with 100 μM cpt-cAMP in the presence of 1 mM 4-AP produced additional inhibition of IK. In this representative neurone, the peak IK in the presence of 4-AP (-30 mV prepulse) was reduced from 1.8 to 1.4 nA (+60 mV step) after a 20 min exposure to cpt-cAMP; steady-state IK was decreased from 1.7 to 1.3 nA. At the more depolarized voltage steps, the current traces exhibited some time-dependent inactivation. For the -100 mV prepulse voltage, peak and steady-state IK were suppressed by 25 and 31 %, respectively, after a 20 min treatment with cpt-cAMP (data not shown).
The inhibitory effects of TEA, 4-AP and cpt-cAMP in the presence of either blocker are summarized in Fig. 10. For the prepulse to -100 mV, the mean peak IK obtained at +60 mV was 6.4 ± 1.5 nA (n = 5, see Fig. 10A). Exposure to TEA reduced this peak current to 3.7 ± 0.9 nA. The inhibition of IK by TEA was significantly different from the control for only the +20, +40 and +60 mV steps (ANOVA with repeated measures). Similar results were obtained for the current values normalized to their respective control values for the +60 mV step and indicated that TEA caused a significant decrease in IK for the steps to +20 mV (control, 0.52 ± 0.03 vs. TEA, 0.31 ± 0.08), +40 mV (0.78 ± 0.01 vs. 0.45 ± 0.08) and +60 mV (1.0 vs. 0.61 ± 0.09). The addition of 100 μM cpt-cAMP in the presence of TEA did not significantly alter the peak currents (or the normalized values) obtained for any voltage step (e.g. for +60 mV, IK was 3.4 ± 0.9 nA; ANOVA with repeated measures). The cpt-cAMP-sensitive current in the presence of TEA was determined in order to examine the inactivation kinetics of these traces. For a given prepulse, the currents obtained in TEA + cpt-cAMP were subtracted from the currents in TEA alone and are shown in the inset. The cpt-cAMP-sensitive currents shown in the insets to Fig. 10A and B were obtained for the voltage step to +60 mV from the neurone shown in Fig. 9A. These current traces indicate that in the presence of TEA there was very little, if any, cpt-cAMP-sensitive current. Figure 10B illustrates the effects of TEA and cpt-cAMP for the -30 mV prepulse. Here, the mean peak IK obtained at +60 mV was 4.4 ± 0.9 nA (n = 5). TEA significantly inhibited IK (both absolute and normalized current values) for the steps -20 to +60 mV. Similar to the results obtained with the -100 mV prepulse, normalization indicated that TEA significantly reduced IK to 0.45 ± 0.06 of the control value at +60 mV (1.0). Treatment with 100 μM cpt-cAMP did not produce any additional inhibition of IK for any voltage step. The cpt-cAMP-sensitive current is shown in the inset. Similar results were obtained for the effects of TEA and cpt-cAMP on the steady-state IK (data not shown). After exposure to TEA, the remaining steady-state IK was only 0.45 ± 0.03 and 0.44 ± 0.03 relative to the control values at +60 mV for prepulses to -100 and -30 mV, respectively. However, cpt-cAMP had no significant effect on steady-state IK in the presence of TEA. Taken together, these results demonstrate that TEA inhibits both the peak and steady-state IK by approximately 50 % for either prepulse voltage. In the presence of TEA, cpt-cAMP was incapable of causing further inhibition of IK, suggesting that the current modulated by cpt-cAMP is a potassium current sensitive to TEA.
Figure 10. The effects of TEA or 4-AP on the inhibition of IK by cpt-cAMP.

A and B summarize the inhibition produced by 10 mM TEA and 100 μM cpt-cAMP in the presence of TEA for the peak IK recorded for the -100 mV (A) and the -30 mV (B) prepulses. Values represent means ±s.e.m. from five neurones. C and D summarize the inhibition produced by 1 mM 4-AP and 100 μM cpt-cAMP in the presence of 4-AP for the peak IK recorded for the -100 mV (C) and the -30 mV (D) prepulses. Values represent means ±s.e.m. from six neurones. The cpt-cAMP-sensitive currents (voltage step to +60 mV) obtained under these different conditions are illustrated in the insets in each respective panel. The calibration bars apply to each trace. * Significant difference (ANOVA with repeated measures, P < 0.05) between the control and TEA or 4-AP treatments; † significant difference between the 4-AP and 4-AP + cpt-cAMP treatments.
The inhibitory effects of both 4-AP and cpt-cAMP are also summarized in Fig. 10. The peak IK obtained for both the -100 (Fig. 10A) and -30 mV (Fig. 10A) prepulses was decreased significantly by 1 mM 4-AP (ANOVA with repeated measures). For the voltage step to +60 mV, these reductions corresponded to inhibitions of 35 ± 4 and 45 ± 4 % (n = 6) for the -100 and -30 mV prepulses, respectively. The insets to Fig. 10A and D represent the cpt-cAMP-sensitive current obtained in the presence of 4-AP (same neurone as shown in Fig. 9A). Under these conditions, the cpt-cAMP-sensitive current exhibited little time-dependent inactivation. Suppression of the steady-state IK by 4-AP was identical to that observed for the peak current wherein the extent of inhibition for the +60 mV step was 33 ± 6 and 41 ± 5 % (n = 6) for the -100 and -30 mV prepulses, respectively (data not shown). In the presence of 4-AP, 20 min treatment with 100 μM cpt-cAMP further decreased the peak and steady-state IK (ANOVA with repeated measures) by values that ranged from 26 to 30 % for the more depolarized voltage steps (20-60 mV) obtained in 4-AP for either prepulse voltage. This amount of cpt-cAMP-induced inhibition was similar to that observed under control conditions (compare Figs 2, 7 and 8). Taken together, these results suggest that the IK modulated by cAMP was insensitive to the actions of 4-AP.
DISCUSSION
Our results demonstrate that cpt-cAMP and PGE2 suppress, in an analogous manner, potassium currents in embryonic rat sensory neurones. This suppression of IK is dependent on activation of the cAMP-PKA transduction cascade since pretreatment with the selective PKA inhibitor, PKI, completely abolished the suppressive effects of PGE2. Thus, these results support the growing evidence that the cAMP signalling pathway mediates the sensitizing actions of PGE2. This evidence includes findings wherein PGE2 elevates intracellular levels of cAMP in rat sensory neurones (Hingtgen et al. 1995). In addition, the PGE2-induced facilitation of both neuropeptide release and the number of action potentials evoked by bradykinin are attenuated by blocking the production of cAMP or inhibiting the activation of PKA (Hingtgen et al. 1995; Cui & Nicol, 1995). In behavioural assays measuring the effects of noxious stimulation in rats, the application of membrane-permeant analogues of cAMP produce a sensitization similar to the hyperalgesia evoked by PGE2 (Ferreira & Nakamura, 1979; Taiwo et al. 1989).
The question remains as to the specific mechanism of action whereby pro-inflammatory prostaglandins give rise to this sensitization in sensory neurones. These prostaglandins are known to modulate multiple types of membrane currents in various model systems. Examples of this mediation are as follows: a non-selective inward current activated by hyperpolarization termed Ih (Ingram & Williams, 1994), a calcium-dependent current that gives rise to a slow after-hyperpolarization (AHPslow) (Fowler et al. 1985; Gold et al. 1996B), calcium currents (Miwa et al. 1988; Alloatti et al. 1991; Mochizuki-Oda et al. 1991; Nicol et al. 1992), a TTX-resistant sodium current (Gold et al. 1996a; England et al. 1996), and outward potassium current (Ren et al. 1996; Nicol et al. 1997). However, several lines of evidence suggest that alterations of some of these currents are not the mechanism(s) by which PGE2 enhances the excitability of small-diameter sensory neurones. First, Ih is expressed mainly in medium to large cells, but not in small-diameter Aδ and C-type sensory neurones (Tokimasa et al. 1990). Second, the inhibitory effects of PGE2 on AHPslow were observed in only about half of the neurones sensitized by this prostanoid (Gold et al. 1996B). Also, PGE2 increased the excitability of neurones not expressing AHPslow, thus modulation of this current was not critical for sensitization. Finally, we reported recently that blockade of N-, L- or P-type voltage-dependent calcium channels did not attenuate the PGE2-mediated facilitation of peptide release from rat sensory neurones, indicating that these channels were not involved in PGE2-induced sensitization (Evans et al. 1996a).
Our findings are consistent with and expand on previous work that has shown that increasing levels of intracellular cAMP led to the suppression of voltage-dependent potassium currents in adult and neonatal DRG cells. Indeed, previous studies indicated a role for the cAMP signalling cascade in the enhancement of membrane excitability. In both chick (Dunlap, 1985) and mouse (Grega & Macdonald, 1987) sensory neurones grown in culture, activation of the cAMP pathway increased the duration of the action potential and this lengthening of the action potential resulted from the inhibition of IK. In sensory neurones isolated from adult rats and grown in culture, the application of membrane-permeant analogues of cAMP produced an inhibition (∼15 %) of the outward IK (Akins & McCleskey, 1993). The modulation of potassium currents as a means of controlling excitability in sensory neurones may be a general scheme. In Aplysia sensory neurones, the serotonin-induced sensitization is analogous to the effects of PGE2 on mammalian sensory neurones and results, in part, from the cAMP-mediated suppression of multiple potassium currents (Klein et al. 1982; Baxter & Byrne, 1989; Goldsmith & Abrams, 1992). Furthermore, in another sensory system, increased levels of cAMP led to the suppression of a TEA-sensitive IK in fungiform taste bud cells (Cummings et al. 1996).
The capacity of cAMP-PKA to decrease IK has also been observed in other neuronal preparations. For example, the activity of the type 2 BK (high conductance calcium-sensitive) potassium channel in recordings from planar bilayers is reduced greatly after exposure to PKA (Reinhart et al. 1991). In addition, PKA reduced the open probability of the human BK channel (hSlo) when it was co-expressed with the β subunit (hSloβ) and exhibited properties that were similar to the native type 2 channel (Dworetzky et al. 1996). In mouse anterior pituitary cells (AtT20 cells), cpt-cAMP reduced an IK believed to be carried by BK potassium channels by about 30 % (Shipston et al. 1996). In the presence of TEA, cpt-cAMP was ineffective at reducing IK. Thus, activation of the cAMP-PKA transduction cascade may be an important signalling mechanism in the regulation of membrane excitability in a variety of neurones.
The exact nature of the IK modulated by activation of PKA is difficult to ascertain. At least two different types of voltage-dependent IK have been described consistently in adult, neonatal and embryonic sensory neurones (Kostyuk et al. 1981; Kameyama, 1983; Akins & McCleskey, 1993; Nicol et al. 1997): an inactivating IA-type and a delayed rectifier-type current. In addition, recent work of Gold and co-workers suggests that additional types of IK exist (Gold et al. 1996c). Although a more careful characterization of the IK modulated by PGE2 and cAMP remains to be determined, our results suggest that the PGE2-/cAMP-modulated IK in rat sensory neurones may be a delayed rectifier-like current. This conclusion is based upon several lines of evidence. First, the current-voltage relations for both the PGE2- and cpt-cAMP-sensitive currents were quite similar, both began to exhibit activation at approximately -20 mV (Fig. 3). Second, the cpt-cAMP-sensitive current observed under various experimental conditions exhibited slow inactivation kinetics, which are characteristic of delayed rectifier currents. Third, the extent of inhibition by either PGE2 or cpt-cAMP did not depend on the level of the conditioning prepulse voltage and was approximately the same for either the peak or steady-state values. Lastly, cpt-cAMP effectively suppressed IK in the presence of 4-AP, whereas no further inhibition of IK was observed in the presence of TEA, an established inhibitor of delayed rectifier-like currents.
Our present work and that of Gold et al. (1996a) and England et al. (1996) demonstrate that PGE2 may enhance neuronal excitability by suppressing potassium currents and/or augmenting a TTX-resistant sodium current. Additionally, both the sensitizing actions of PGE2 and the effects of this prostanoid on potassium and TTX-resistant sodium currents are dependent on the activation of the cAMP transduction cascade (Cui & Nicol, 1995; Hingtgen et al. 1995; England et al. 1996). Therefore, taken together, activation of the cAMP signalling pathway gives rise to PKA-mediated modulation of the activity of multiple ion channels, which, in turn, results in the enhanced excitability of sensory neurones. This prostaglandin-induced modification of ion channel activity may then account for the heightened sensitivity to noxious stimulation exhibited in behavioural observations.
Acknowledgments
We would like to thank Dr James Kenyon for his helpful comments and critique of this work. This work was supported by NIH grants NS09733 to A. R. Evans, NS34159 to M. R. Vasko and NS30527 to G. D. Nicol.
References
- Akins PT, McCleskey EW. Characterization of potassium currents in adult rat sensory neurons and modulation by opioids and cyclic AMP. Neuroscience. 1993;56:759–769. doi: 10.1016/0306-4522(93)90372-m. 10.1016/0306-4522(93)90372-M. [DOI] [PubMed] [Google Scholar]
- Alloatti G, Serazzi L, Levi RC. Prostaglandin I2 (PGI2) enhances calcium current in guinea-pig ventricular heart cells. Journal of Molecular and Cellular Cardiology. 1991;23:851–860. doi: 10.1016/0022-2828(91)90218-b. 10.1016/0022-2828(91)90218-B. [DOI] [PubMed] [Google Scholar]
- Andreeva L, Rang HP. Effect of bradykinin and prostaglandins on the release of calcitonin gene-related peptide-like immunoreactivity from the rat spinal cord in vitro. British Journal of Pharmacology. 1993;108:185–190. doi: 10.1111/j.1476-5381.1993.tb13460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansanay H, Dumuis A, Sebben M, Bockaert J, Fagni L. cAMP-dependent, long-lasting inhibition of a K+ current in mammalian neurons. Proceedings of the National Academy of Sciences of the USA. 1995;92:6635–6639. doi: 10.1073/pnas.92.14.6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccaglini PI, Hogan PG. Some rat sensory neurons in culture express characteristics of differentiated pain sensory cells. Proceedings of the National Academy of Sciences of the USA. 1983;80:594–598. doi: 10.1073/pnas.80.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry PH. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. Journal of Neuroscience Methods. 1994;51:107–116. doi: 10.1016/0165-0270(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Baxter DA, Byrne JH. Serotonergic modulation of two potassium currents in the pleural sensory neurons of Aplysia. Journal of Neurophysiology. 1989;62:665–679. doi: 10.1152/jn.1989.62.3.665. [DOI] [PubMed] [Google Scholar]
- Cheng H-C, Kemp BE, Pearson RB, Smith AJ, Misconi L, Van Patten SM, Walsh DA. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. Journal of Biological Chemistry. 1986;261:989–992. [PubMed] [Google Scholar]
- Chung S, Kaczmarek L. Modulation of the inactivation of voltage-dependent potassium channels by cAMP. Journal of Neuroscience. 1995;15:3927–3935. doi: 10.1523/JNEUROSCI.15-05-03927.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JA, Stevens CF. Voltage clamp studies of a transient outward current in gastropod neural somata. The Journal of Physiology. 1971;213:21–30. doi: 10.1113/jphysiol.1971.sp009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Nicol GD. Cyclic AMP mediates the prostaglandin E2-induced potentiation of bradykinin excitation in rat sensory neurons. Neuroscience. 1995;66:459–466. doi: 10.1016/0306-4522(94)00567-o. [DOI] [PubMed] [Google Scholar]
- Cummings TA, Daniels C, Kinnamon SC. Sweet taste transduction in hamster: sweeteners and cyclic nucleotides depolarize taste cells by reducing a K+ current. Journal of Neurophysiology. 1996;75:1256–1263. doi: 10.1152/jn.1996.75.3.1256. [DOI] [PubMed] [Google Scholar]
- Dunlap K. Forskolin prolongs action potential duration and blocks potassium current in embryonic chick sensory neurons. Pflügers Archiv. 1985;403:170–174. doi: 10.1007/BF00584096. [DOI] [PubMed] [Google Scholar]
- Dworetzky SI, Boissard CG, Lum-Ragan JT, McKay MC, Post-Munson DJ, Trojnacki JT, Chang C-P, Gribkoff VK. Phenotypic alteration of a human BK (hSlo) channel by hSloβ subunit coexpression: changes in blocker sensitivity, activation/relaxation and inactivation kinetics, and protein kinase A modulation. Journal of Neuroscience. 1996;16:4543–4550. doi: 10.1523/JNEUROSCI.16-15-04543.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. The Journal of Physiology. 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AR, Nicol GD, Vasko MR. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Research. 1996a;712:265–273. doi: 10.1016/0006-8993(95)01447-0. [DOI] [PubMed] [Google Scholar]
- Evans AR, Vasko MR, Nicol GD. The cyclic AMP transduction cascade mediates the prostaglandin E2-induced inhibition of potassium currents in rat sensory neurons. Society for Neuroscience Abstracts. 1996b;22:1812. [Google Scholar]
- Ferreira SH, Nakamura M. Prostaglandin hyperalgesia, a cAMP/Ca2+ dependent process. Prostaglandins. 1979;18:179–190. doi: 10.1016/0090-6980(79)90103-5. [DOI] [PubMed] [Google Scholar]
- Fowler JC, Wonderlin WF, Weinreich D. Prostaglandins block a Ca2+-dependent slow afterhyperpolarization independent of effects on Ca2+ influx in visceral afferent neurons. Brain Research. 1985;345:345–349. doi: 10.1016/0006-8993(85)91014-5. [DOI] [PubMed] [Google Scholar]
- Franco-Cereceda A. Prostaglandins and CGRP release from cardiac sensory nerves. Naunyn-Schmiedeberg's Archives of Pharmacology. 1989;340:140–184. doi: 10.1007/BF00168966. [DOI] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proceedings of the National Academy of Sciences of the USA. 1996a;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Shuster MJ, Levine JD. Role of a Ca2+-dependent slow afterhyperpolarization in prostaglandin E2-induced sensitization of cultured rat sensory neurons. Neuroscience Letters. 1996b;205:161–164. doi: 10.1016/0304-3940(96)12401-0. [DOI] [PubMed] [Google Scholar]
- Gold MS, Shuster MJ, Levine JD. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. Journal of Neurophysiology. 1996c;75:2629–2646. doi: 10.1152/jn.1996.75.6.2629. [DOI] [PubMed] [Google Scholar]
- Goldsmith BA, Abrams TW. cAMP modulates multiple K+ currents, increasing spike duration and excitability in Aplysia sensory neurons. Proceedings of the National Academy of Sciences of the USA. 1992;89:11476–11480. doi: 10.1073/pnas.89.23.11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grega DS, Macdonald RL. Activators of adenylate cyclase and cyclic AMP prolong calcium-dependent action potentials of mouse sensory neurons in culture by reducing a voltage-dependent potassium conductance. Journal of Neuroscience. 1987;7:700–707. doi: 10.1523/JNEUROSCI.07-03-00700.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Handwerker HO. Influences of algogenic substances and prostaglandins on the discharges of unmyelinated cutaneous nerve fibers identified as nociceptors. Advances in Pain Research and Therapy. 1976;1:41–45. [Google Scholar]
- Hingtgen CM, Vasko MR. Prostacyclin enhances the evoked release of substance P and calcitonin gene-related peptide from rat sensory neurons. Brain Research. 1994;655:51–60. doi: 10.1016/0006-8993(94)91596-2. [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′,5′-cyclic monophosphate transduction cascade. Journal of Neuroscience. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacological Reviews. 1991;43:143–201. [PubMed] [Google Scholar]
- Ingram SL, Williams JT. Opioid inhibition of Ih via adenylyl cyclase. Neuron. 1994;13:179–186. doi: 10.1016/0896-6273(94)90468-5. [DOI] [PubMed] [Google Scholar]
- Kameyama M. Ionic currents in cultured dorsal root ganglion cells from adult guinea pigs. Journal of Membrane Biology. 1983;72:195–203. doi: 10.1007/BF01870586. [DOI] [PubMed] [Google Scholar]
- Klein M, Camardo J, Kandel ER. Serotonin modulates a specific potassium current in the sensory neurons that show presynaptic facilitation in Aplysia. Proceedings of the National Academy of Sciences of the USA. 1982;79:5713–5717. doi: 10.1073/pnas.79.18.5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyuk PG, Veselovsky NS, Fedulova SA, Tsyndrenko AY. Ionic currents in the somatic membrane of rat dorsal root ganglion neurons. III. Potassium currents. Neuroscience. 1981;6:2439–2444. doi: 10.1016/0306-4522(81)90090-7. [DOI] [PubMed] [Google Scholar]
- McFarlane S, Cooper E. Kinetics and voltage dependence of A-type currents on neonatal rat sensory neurons. Journal of Neurophysiology. 1991;66:1380–1391. doi: 10.1152/jn.1991.66.4.1380. [DOI] [PubMed] [Google Scholar]
- Martin HA, Basbaum AI, Kwiat GC, Goetzl EJ, Levine JD. Leukotriene and prostaglandin sensitization of cutaneous high-threshold C- and A-delta mechanonociceptors in the hairy skin of rat hindlimbs. Neuroscience. 1987;22:651–659. doi: 10.1016/0306-4522(87)90360-5. [DOI] [PubMed] [Google Scholar]
- Mense S. Sensitization of group IV muscle receptors to bradykinin by 5-hydroxytryptamine and prostaglandin E2. Brain Research. 1981;225:95–105. doi: 10.1016/0006-8993(81)90320-6. [DOI] [PubMed] [Google Scholar]
- Miwa N, Sugino H, Ueno R, Hayaishi O. Prostaglandin induces Ca2+ influx and cyclic GMP formation in mouse neuroblastoma × rat glioma hybrid NG 108–15 cells in culture. Journal of Neurochemistry. 1988;50:1418–1424. doi: 10.1111/j.1471-4159.1988.tb03025.x. [DOI] [PubMed] [Google Scholar]
- Mochizuki-Oda N, Mori K, Negishi M, Ito S. Prostaglandin E2 activates Ca2+ channels in bovine adrenal chromaffin cells. Journal of Neurochemistry. 1991;56:541–547. doi: 10.1111/j.1471-4159.1991.tb08183.x. [DOI] [PubMed] [Google Scholar]
- Moreno H, Kentros C, Bueno E, Weiser M, Hernandez A, Vega-Saenz De Miera E, Ponce A, Thornhill W, Rudy B. Thalamocortical projections have a K+ channel that is phosphorylated and modulated by a cAMP-dependent protein kinase. Journal of Neuroscience. 1995;15:5486–5501. doi: 10.1523/JNEUROSCI.15-08-05486.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Two fast transient current components during voltage clamp on snail neurons. Journal of General Physiology. 1971;58:36–53. doi: 10.1085/jgp.58.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol GD, Cui M. Enhancement by prostaglandin E2 of bradykinin activation of embryonic rat sensory neurons. The Journal of Physiology. 1994;480:485–492. doi: 10.1113/jphysiol.1994.sp020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol GD, Klingberg DK, Vasko MR. Prostaglandin E2 increases calcium conductance and stimulates release of substance P in avian sensory neurons. Journal of Neuroscience. 1992;12:1917–1927. doi: 10.1523/JNEUROSCI.12-05-01917.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol GD, Vasko MR, Evans AR. Prostaglandins suppress an outward potassium current in embryonic rat sensory neurons. Journal of Neurophysiology. 1997;77:167–176. doi: 10.1152/jn.1997.77.1.167. [DOI] [PubMed] [Google Scholar]
- Reinhart PH, Chung S, Martin BL, Brautigan DL, Levitan IB. Modulation of calcium-activated potassium channels from rat brain by protein kinase A and phosphatase 2A. Journal of Neuroscience. 1991;11:1627–1635. doi: 10.1523/JNEUROSCI.11-06-01627.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Karpinski E, Benishin CG. The actions of prostaglandin E2 on potassium currents in rat tail artery smooth muscle cells: regulation by protein kinase A and protein kinase C. Journal of Pharmacology and Experimental Therapeutics. 1996;277:394–402. [PubMed] [Google Scholar]
- Shipston MJ, Kelly JS, Antoni FA. Glucocorticoids block protein kinase A inhibition of calcium-activated potassium channels. Journal of Biological Chemistry. 1996;271:9197–9200. doi: 10.1074/jbc.271.16.9197. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD. Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience. 1989;32:577–580. doi: 10.1016/0306-4522(89)90280-7. [DOI] [PubMed] [Google Scholar]
- Thompson SH. Three pharmacologically distinct potassium channels in molluscan neurones. The Journal of Physiology. 1977;265:465–488. doi: 10.1113/jphysiol.1977.sp011725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokimasa T, Shiraishi M, Akasu T. Morphological and electrophysiological properties of C-cells in bullfrog dorsal root ganglia. Neuroscience Letters. 1990;116:304–308. doi: 10.1016/0304-3940(90)90091-m. [DOI] [PubMed] [Google Scholar]
- Treede RD, Meyer RA, Raja SN, Campbell JN. Peripheral and central mechanisms of cutaneous hyperalgesia. Progress in Neurobiology. 1992;38:397–421. doi: 10.1016/0301-0082(92)90027-c. [DOI] [PubMed] [Google Scholar]
- Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. Journal of Neuroscience. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]