

Abstract
The relationship between nitric oxide (NO) concentration measured with an NO-specific microelectrode and endothelium-dependent relaxation was investigated in isolated rat superior mesenteric artery contracted with 1 μM noradrenaline.
Acetylcholine (10 μM) induced endothelium-dependent simultaneous increases in luminal NO concentration of 21 ± 6 nM, and relaxations with pD2 values and maximum of 6.95 ± 0.32 and 97.5 ± 0.7 % (n = 7), respectively. An inhibitor of NO synthase, NG-nitro-L-arginine (L-NOARG, 100 μM) inhibited the relaxations and increases in NO concentration induced by acetylcholine.
Oxyhaemoglobin (10 μM) reversed the relaxations and increases in NO concentrations induced by acetylcholine, S-nitroso-N-acetylpenicillamine (SNAP) and S-morpholino-sydnonimine (SIN-1), but not the relaxations induced with forskolin. Oxyhaemoglobin also decreased the NO concentration below baseline level.
In the presence of L-NOARG (100 μM), a small relaxation to acetylcholine (10 μM) of noradrenaline-contracted segments was still seen; oxyhaemogobin inhibited this relaxation and decreased the NO concentration by 14 ± 4 nM (n = 4).
The NO concentration-relaxation relationship for acetylcholine resembled that for SNAP and SIN-1 more than for authentic NO. Thus while 7-17 nM NO induced half-maximal relaxations in response to SNAP or SIN-1, 378 ± 129 nM NO (n = 4) was needed for half-maximal relaxation to authentic NO.
The present study provides direct evidence that the relaxation of the rat superior mesenteric artery with the endothelium-dependent vasodilator acetylcholine is correlated to the endogeneous release of NO. The study also suggests that NO mediates the L-NOARG-resistant relaxations in this artery, and that there is a basal NO release.
Acetylcholine stimulates endothelial cells leading to relaxation of the underlying smooth muscle cells either through gap junction transmission or release of various diffusible substances. The identification of nitric oxide (NO) as an endothelium-derived relaxing factor (EDRF) rests on observations interfering with the L-arginine-NO pathway, as well as the physiological similarities between the endogenous substance and authentic NO. Thus pharmacological inhibition of the L-arginine-NO pathway in vivo and knockout of the gene for the endothelial cell constitutive nitric oxide synthase (NOS) raises the blood pressure (Huang et al. 1995), reduces acetylcholine-induced relaxations in vitro (Cohen & Vanhoutte, 1995; Huang et al. 1995), and reduces the production of the end-products of NO metabolism, nitrite (NO2−) and nitrate (NO3−) (Ignarro et al. 1993). EDRF and NO appear to cause comparable relaxations in bioassays (Palmer et al. 1987; Feelisch et al. 1994). Moreover, NO release has been measured with chemiluminescence (Palmer et al. 1987), by conversion of oxyhaemoglobin to methaemoglobin (Kelm & Schrader, 1990), and recently by the use of either membrane-covered electrodes (Goligorsky et al. 1994) or polarographic electrodes (Shibuki & Okada, 1991), which allow direct measurements of NO released from cell cultures (Malinski & Taha, 1992) and unmounted arteries (Cohen et al. 1997). Thus the evidence appears incontrovertible that NO participates in endothelium-dependent relaxations, but other endothelium-derived relaxing factors could be more important, since evidence for a direct relationship between endothelium-derived NO and acetylcholine-induced relaxation is lacking.
Endothelium-dependent relaxation of vascular smooth muscle involves in some cases hyperpolarization of the cell membrane, and has been attributed to a diffusible endothelium-derived hyperpolarizing factor (EDHF) (Murphy & Brayden, 1995; Cohen & Vanhoutte, 1995). EDHF may encompass more than one factor, since it displays properties similar to those of a cytochrome P-450-derived metabolite of arachidonic acid in porcine coronary arteries (Hecker et al. 1994), while P450 mono-oxygenase derivatives in rat mesenteric arteries were not involved in endothelium-dependent hyperpolarization and relaxation of the rat superior mesenteric artery (Van de Voorde & Vanheel, 1997; Vanheel & Van de Voorde, 1997). NO can also induce hyperpolarization (Tare et al. 1990; Bolotina et al. 1994), and inhibition of NOS with L-arginine analogues revealed that persisting hyperpolarization could be ascribed to the release of NO (Cohen et al. 1997), suggesting that persisting relaxation in the presence of an inhibitor of NOS might be due to an incomplete inhibition of NOS. Simultaneous measurements of the NO concentration and relaxations may clarify whether this is the case.
The aim of the present study was to gain evidence for a direct relationship between endothelium-derived NO and acetylcholine-induced relaxation. For this purpose a polarographic NO-selective electrode was introduced into the lumen of the mounted arterial segment, allowing simultaneous measurements of increases in NO concentration and force. Furthermore, we investigated whether relaxations persisting in the presence of an inhibitor of NOS, NG-nitro-L-arginine (L-NOARG), can be ascribed to NO in the rat superior mesenteric artery.
METHODS
Tissue preparation
Adult male Wistar rats (12-16 weeks old) were killed by inhalation of carbon dioxide gas in an increasing concentration and then exsanguinated. The procedure was in accordance with Danish animal law and regulations. The mesenteric vascular bed was removed and placed in physiological salt solution (PSS; see composition below). Segments (length 2 mm) of the rat superior mesenteric artery were mounted on 100 μm wires in a small vessel myograph for isometric tension recording (Mulvany & Halpern, 1977). The vessels were allowed to equilibrate for about 30 min in PSS of the following composition (mmol l−1): 119 NaCl, 25 NaHCO3, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4, 2.5 CaCl2, 0.026 EDTA and 5.5 glucose. The solution was gassed with 5 % CO2 in O2 to maintain pH at 7.4. The relation between resting wall tension and internal circumference was determined, and the internal circumference, L100, corresponding to a transmural pressure of 100 mmHg for a relaxed vessel in situ was calculated (Mulvany & Halpern, 1977). The vessels were set to the internal circumference L1, given by L1 = 0.9 L100. The effective internal lumen diameter was determined as l1 =L1/π, and was between 0.9 and 1.2 mm for the segments of the rat superior mesenteric artery.
Calibration of NO-sensitive electrodes
Two types of electrodes, membrane types and microelectrodes, were applied to monitor the concentration of NO electrochemically. The NO detection is the same for both sensors: a potential is applied to the measuring electrode relative to the reference electrode and the resulting current due to the electrochemical oxidation of NO is monitored.
The membrane-type NO-sensitive electrode is in many respects similar to the Clark electrode for oxygen tension. It consists of a working electrode covered by a gas-permeable membrane (World Precision Instruments, Stevenage, UK; see also Goligorsky et al. 1994). NO diffuses across the membrane and is oxidized on the surface of the prepolarized electrode, thus resulting in electrical current. The magnitude of the current is proportional to the concentration of NO in the sample and is amplified by an NO meter and registered on a chart recorder. The membrane-type electrode can be calibrated either by chemical titration based on the following equation:
or by using a known amount of gaseous NO in deoxygenated water. However, the membrane-type electrode is 2 mm in diameter and therefore does not permit measurements close to the endothelial surface in ring segments; it was applied to control NO concentrations in gaseous NO diluted in deoxygenated water to compare the two types of calibration.
The microelectrode (ISONOP30, World Precision Instruments) is constructed from a carbon fibre with a polymer coating, giving selectivity for NO. This is also a combination electrode where measuring and reference electrodes have been combined within a Faraday shield designed to minimize susceptibility to environmental noise. The electrode has high selectivity for NO and a detection sensitivity of 1 nM (Broderick & Taha, 1995). Calibration of the coated carbon electrode was carried out with NO solution. NO solution was prepared from deoxygenated (argon-bubbled) distilled water, contained in glass vials with a septum closure. The argon gas was led through a 10 mM pyrogallol solution to remove traces of oxygen. Pure NO gas, bubbled through sodium hydroxide (NaOH, 10 mM) to remove higher nitrogen oxides, was then bubbled through the deoxygenated water for 10 min at room temperature (20°C), giving a solution of 1.9 mM (Gevantman, 1995), which was then diluted to 0.19 mM. The dissolved NO gas solution was withdrawn from the stock solution using gas-tight Hamilton microlitre syringes. The NO-sensitive microelectrode was calibrated in a glass vial with a septum and stirring just prior to the introduction of the electrode into the myograph bath, as described below.
To check the concentrations in stock solutions of gaseous NO dissolved in water, the stock was used to calibrate the membrane-type electrode and these calibrations were compared with those obtained with chemical titration as described above. The myograph experiments were performed bubbling with 5 % CO2 in oxygen, and therefore the calibrations were also conducted under these conditions.
Selectivity of NO-sensitive electrodes
The selectivity of the ISONOP30 electrodes was tested in connection with the calibration, where a lack of response to sodium nitrite (NaNO2) up to 100 μM was taken as evidence for an intact coating of the electrode. The electrodes did not respond to acetylcholine (10 μM) added in the calibration glass vial, but both 100 μM nicotine base and 1 μM calcium ionophore (A23187) increased the current of three new electrodes. Therefore, the latter substances were not applied in the present study. Oxyhaemoglobin was used to evaluate whether an increase in electrode current in response to L-NOARG was due to a direct effect or NO release. Noradrenaline is oxidized on Nafion-coated carbon fibres (CFN 30-1000, World Precision Instruments) and caused a pronounced increase in current (100 pA with 1 μM noradrenaline) when the polymer coating of an ISONOP30 electrode was damaged. Therefore, measurement of electrode current in response to noradrenaline (0.5-1 μM) was used to evaluate whether the coating, and hence the electrode selectivity, was changed by the procedure of placing the electrode in the artery lumen or during the experiment. If this was the case, the experiment was stopped and the electrode was discarded.
Simultaneous measurements of NO concentration and force
After calibration, the NO-sensitive microelectrode was inserted in the myograph through a hole drilled in one side, and then sealed with grease (high vacuum grease, Dow Corning GMBH, München, Germany). The microelectrodes were prepolarized with a steady poise voltage (0.9 V) allowing the oxidation of NO. The microelectrode was placed within the artery lumen by means of a micromanipulator. The electrode (length 0.5-2 mm, diameter of 30-60 μm) was inserted into the artery lumen and placed close to the endothelial surface by touching the vessel lumen until force increased 0.1 mN, and then retracting the electrode until the force decreased to 0 mN. The NO electrode was connected to an amplifier (NO meter, World Precision Instruments), and the amplified signal was registered on a recorder permitting simultaneous measurements of NO and force. The measurements were performed with mechanical stirring in one corner of the myograph, allowing a rapid distribution of the drugs in the myograph bath.
Experimental protocol
To investigate the effect of mechanical endothelial cell removal on the relaxations induced by acetylcholine, a stable contraction was induced in the endothelium-intact segments with 0.5-1 μM noradrenaline, and a single concentration of acetylcholine (10 μM) was added. The preparation was washed several times and the procedure repeated, but this time for 10 μM S-nitroso-N-acetyl-penicillamine (SNAP). The endothelial cells were removed by passing a human scalp hair through the vessel lumen and then rubbing it back and forth across the lumen several times, as described previously (Osol et al. 1989; Simonsen et al. 1997). The preparations were allowed to equilibrate for 45 min and the effect of acetylcholine and SNAP was evaluated in the noradrenaline-contracted segments.
The effect of the NO synthase inhibitor, NG-nitro-L-arginine (L-NOARG) was evaluated on the concentration-response curves for acetylcholine. The segments were contracted with noradrenaline (0.5-1 μM), and a cumulative concentration-response curve for acetylcholine (0.01-10 μM) was obtained. These curves served as controls. After washing, the vessels were incubated with 100 μM L-NOARG for 30 min, before a second concentration-response curve was constructed in the presence of the inhibitor.
The effect of the NO scavenger, oxyhaemoglobin, on the simultaneous changes in force and NO concentrations induced by single concentrations of either acetylcholine (10 μM), SNAP (10 μM), S-morpholinosydnonimine (SIN-1, 10 μM), or forskolin (1 μM) was investigated by evaluating the capacity of 10 μM oxyhaemoglobin to reverse increases in NO concentration and relaxations in noradrenaline-contracted preparations. Control responses without adding oxyhaemoglobin were also obtained.
Relaxations induced by acetylcholine in the presence of a NOS inhibitor are more prominent when precontraction is lower (Hatake et al. 1995), and therefore the vessels in these experiments were activated with 0.1-0.5 μM noradrenaline, relaxed with 10 μM acetylcholine and oxyhaemoglobin (10 μM) was added.
Concentration-response curves were also obtained for the NO donors, SNAP and SIN-1, and NO. The vessel was contracted with noradrenaline (0.5-1 μM) and relaxed by increasing concentrations of SNAP (0.01-10 μM), SIN-1 (0.01-10 μM), and NO (0.01-10 μM). NO was withdrawn from the stock solution using gas-tight Hamilton microlitre syringes and added to the myograph bath. The method of injecting small volumes of authentic NO into the myograph only gave reproducible responses when the solution was injected at precisely the same place of the myograph bath. This was most pronounced for the high concentrations (> 100 nM) of authentic NO.
To establish time relationships for relaxation and increase in NO concentration, the vessel was activated with noradrenaline (0.5-1 μM), and when the contraction was stable, paper speed was increased, and one concentration of acetylcholine, SNAP or SIN-1 was added. In other experiments, NO was infused continuously with a pump permitting the establishment of a steady-state concentration. As a control for the NO concentrations measured by use of the microelectrodes, a set of experiments using the same protocol was performed, but with the microelectrode exchanged for the membrane-type electrode, which was placed as close as possible to the vessel segment in the myograph.
Drugs
Acetylcholine HCl, (-)-noradrenaline HCl, L-NOARG, potassium nitrite (KNO2), pyrogallol, NaNO2, potassium iodide (KI) and forskolin were obtained from Sigma (USA), and SNAP and SIN-1 were obtained from GEA Ltd (Copenhagen, Denmark). The drugs were prepared in distilled water, which in the case of SNAP and SIN-1 was deoxygenated previously with nitrogen.
Oxyhaemoglobin was prepared from a 1 mM solution of commercial haemoglobin (bovine haemoglobin, Sigma) by addition of 10 mM sodium dithionite (Na2S2O4, Sigma). The reducing agent converting methaemoglobin to oxyhaemoglobin was removed by dialysis in 2 l of calcium-free PSS and bubbling with nitrogen at 4°C. The purity and concentration of oxyhaemoglobin was determined spectrophotometrically by measuring the difference in absorbence at 577 nm with methaemoglobin in the reference path.
Calculations
The mechanical responses of the vessels were measured as force and expressed as active wall tension, ΔT, which is the increase in measured force, ΔF, divided by twice the segment length (Mulvany & Halpern, 1977). By using a computer program (GraphPad, Institute for Scientific Information, San Diego, CA, USA), the concentration-response curves were fitted to the classical Hill equation, as described earlier (Simonsen et al. 1997). Sensitivity to the agonists is expressed in terms of pD2 = -log (EC50), EC50 being the concentration (M) of agonist required to give half-maximal relaxation.
The results are expressed as means ±s.e.m., and the response curves are presented on a semilogarithmic scale. Differences between means were analysed using either one-way analysis of variance followed by a Bonferroni t test, Student's t test or paired t test as indicated. The Wilcoxon one-sample test was applied to evaluate whether there were significant increases in NO concentration and decreases in precontraction level. Probability levels less than 5 % were considered significant.
RESULTS
Calibrations
The ISONOP30 microelectrodes responded with increases in current to nanomolar concentrations of NO (Fig. 1A). The electrode was calibrated with dilutions of NO stock solution (approximately 0.19 mM), the output current of the probes correlating linearly with the concentration of NO (Fig. 1A). However, the sensitivity of different probes varied between 0.55 and 6.0 nM pA−1. Also, the sensitivity to NO of the electrodes declined with time: on the first day it was often 4.0 pA nM−1, and by the third to fourth day had fallen to 1-3 pA nM−1. In four experiments where calibrations of the NO electrode were performed before and after the experimental protocol, the sensitivity was not changed (results not shown). The electrodes were therefore calibrated daily. To check the stock solutions (1.9 and 0.19 mM) of authentic NO, the calibration of the membrane-type electrode with authentic NO was compared with those obtained with chemical titration. In four experiments, comparable calibrations of the membrane-type electrode were obtained with the two methods (Fig. 1A).
Figure 1. Calibration of NO-sensitive microelectrode.
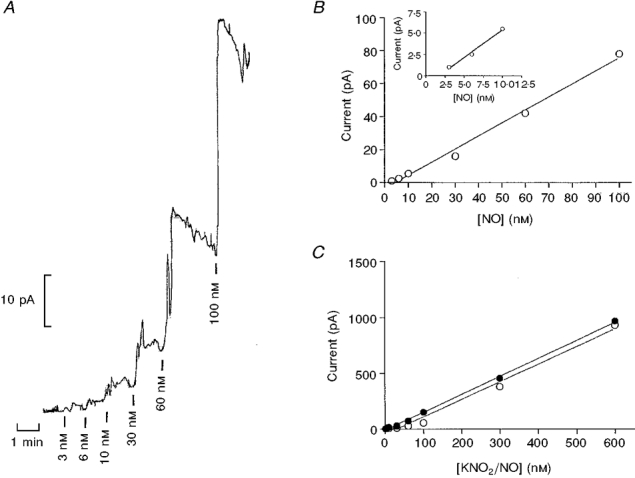
A, representative recording of the NO microelectrode calibration performed in a glass vial containing PSS at 37 °C with constant stirring. The vertical bars below the trace indicate injection of increasing concentrations of NO. The horizontal bar indicates time. B, linear regression analysis of the relationship between amount of NO added and output of the electrode for one electrode tip (ISONOP30), r2 = 0.9928, 1 pA = 3.0 nM. The inset shows the relationship for the lowest concentrations with this electrode. C, linear regression analyses of relationship for authentic NO (○) and chemical titration (•) versus current output for a membrane-type electrode, showing that both calibrations give the same result.
Mechanical stress influenced the current response of the electrode. Therefore, an artifact arose if the electrode touched the vessel wall when noradrenaline was added to contract the preparation. However, in a correct position of 10-35 μm from the endothelial surface, the increases in NO concentration induced with 3 μM acetylcholine appeared with the preparation at baseline tension or contracted with 1 μM noradrenaline (Fig. 2). Thus acetylcholine (3 μM) increased the NO concentration by 10 ± 2 nM (n = 4) and 16 ± 2 nM (n = 4) in the same preparations at baseline tension and contracted with 1 μM noradrenaline, respectively, and it relaxed the contracted preparations by 91 ± 2 % (n = 4).
Figure 2. Increases in measured NO concentrations are independent of force measurements.
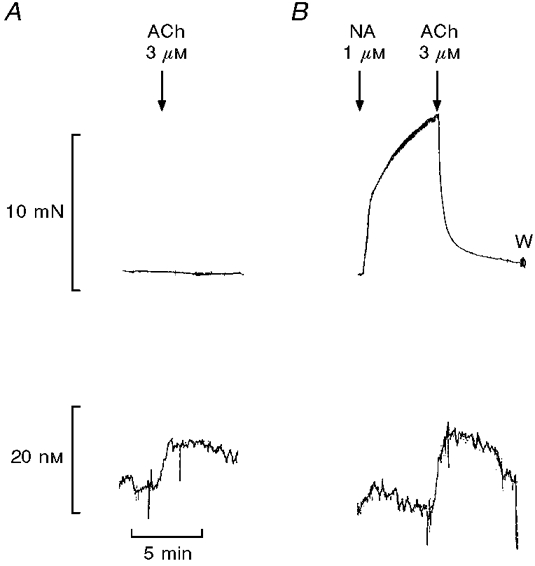
Simultaneous measurements of changes in isometric force (upper traces) and NO concentration (lower traces) in an endothelium-intact segment of rat superior mesenteric artery. A, acetylcholine (ACh, 3 μM) was added with the preparation at resting tension; B, ACh was added with the preparation contracted with 1 μM noradrenaline (NA). W, washout. The traces are representative of 4 experiments.
Effect of endothelial cell removal, L-NOARG and oxyhaemoglobin
In noradrenaline-contracted preparations, acetylcholine-induced relaxation was associated with NO release as detected by the NO sensor. Mechanical endothelial cell removal abolished the acetylcholine (10 μM)-induced relaxations and increases in NO concentration in preparations contracted with noradrenaline (n = 4, Fig. 3). In contrast, SNAP (10 μM) induced relaxations and increased the NO concentration in both endothelium-intact and -denuded preparations. Indeed, the SNAP-induced NO concentration was enhanced from 5.9 ± 1.2 nM (n = 4) in endothelium-intact to 10.8 ± 0.7 nM (n = 4) in endothelium-denuded preparations (Fig. 3).
Figure 3. The effect of mechanical endothelial cell removal on simultaneously obtained relaxations and increases in NO concentration.
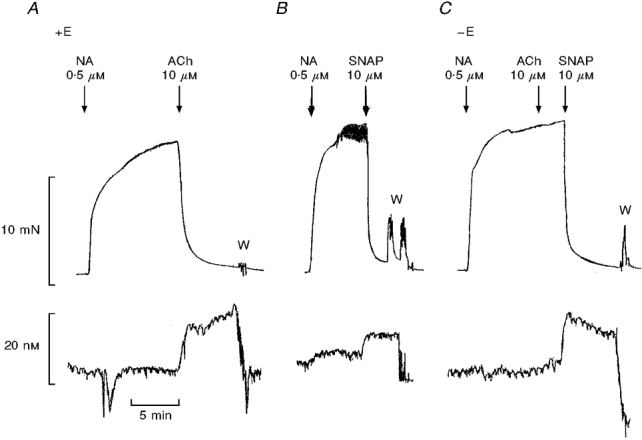
Simultaneous measurements of force (upper traces) and NO concentration (lower traces) in an endothelium-intact (+E) segment of rat superior mesenteric artery contracted with 0.5 μM noradrenaline (NA) and relaxed with either 10 μM acetylcholine (ACh) (A), or 10 μM SNAP (B), and the lack of relaxation to ACh, but relaxation and increases in NO induced by SNAP in the same segment after mechanical endothelial cell removal (-E) (C). The traces are representative of 4 experiments. W, washout.
The acetylcholine-induced relaxation and increase in measured NO was concentration dependent. Thus acetylcholine induced relaxations with pD2 6.95 ± 0.32 and maximal relaxations of 97.5 ± 0.7 % (n = 7), and increases in NO concentration that reached 21 ± 6 nM (n = 7) at 10 μM acetylcholine. More NO was released with 10 μM acetylcholine than with 3 μM acetylcholine, although relaxation was maximal at 3 μM acetylcholine (Fig. 4).
Figure 4. L-NOARG (100 μM) inhibits simultaneously measured relaxations and NO concentrations.

Average relaxations (circles) and increases in NO concentration (squares) induced by increasing concentrations of ACh in rat superior mesenteric arterial segments in the absence (open symbols) and the presence (filled symbols) of the nitric oxide synthase inhibitor, L-NOARG (100 μM). Relaxations are relative to the initial contraction induced by noradrenaline (NA, 0.5-1 μM) and NO concentrations are given as increases above the level in the absence of acetylcholine. The points are means ±s.e.m. of 6 experiments. First concentration of acetylcholine causing significant relaxation or increase in NO concentration: *, P < 0.05.
In the presence of L-NOARG (100 μM) and contracted with 0.5-1 μM noradrenaline, the concentration-response curves for acetylcholine-induced relaxations and increases in NO were almost abolished, although transient relaxations were still observed at the highest concentrations (3-10 μM) of acetylcholine (Fig. 4). Thus acetylcholine (10 μM) induced maximal relaxations of 96.3 ± 0.8 % and increases in NO concentrations of 17 ± 5 nM (n = 6) in the absence of L-NOARG, and transient maximal relaxations of 40 ± 13 % (n = 6,P < 0.05, paired t test) and increases in NO concentration of 4 ± 1 nM (n = 6,P < 0.05, paired t test), in the presence of 100 μM L-NOARG.
Unexpectedly, incubation with L-NOARG (100 μM) increased the measured NO concentration. This could be due to tissue degradation of the L-arginine analogue. Therefore, in control experiments, L-NOARG (1 μM to 1 mM) was added to the calibration glass vial, and it increased the measured current concentration dependently, reaching 6 ± 1 pA (n = 3). These increases corresponding to 11 ± 1 nM NO were inhibited by oxyhaemoglobin (10 μM), suggesting that L-NOARG does release NO.
Oxyhaemoglobin (10 μM), a scavenger of NO, reversed acetylcholine (10 μM) and SNAP-induced relaxations and increases in NO concentration (Fig. 5, Table 1), while the SIN-1-induced relaxations and NO concentrations were only partially reversed (Fig. 5, Table 1). Forskolin (1 μM), which is an activator of adenylate cyclase, caused relaxations of noradrenaline-contracted preparations without significant changes in NO concentration, and oxyhaemoglobin did not change the relaxations. Oxyhaemoglobin (10 μM) lowered the measured NO concentrations below baseline in the case of acetylcholine, SNAP and forskolin (Table 1). When oxyhaemoglobin (10 μM) was added at baseline tension, it induced small contractions corresponding to 11 ± 2 % (0.38 ± 0.05 N m−1, n = 4) of the contractions induced by 5 μM noradrenaline. Moreover, the NO concentration was decreased 6 ± 3 nM (n = 4) below baseline.
Figure 5. Oxyhaemoglobin reverses relaxations and increases in NO concentration.
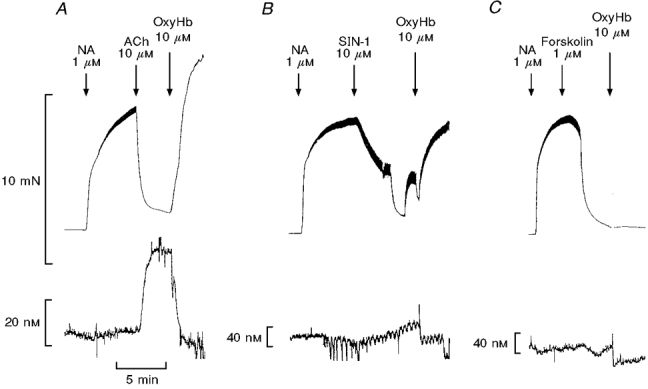
Records showing simultaneous measurements of force (upper traces) and NO concentration (lower traces) in segments of the superior mesenteric artery. The vessels were contracted with 1 μM noradrenaline (NA) and relaxed with either 10 μM ACh (A), 10 μM SIN-1 (B), or 1 μM forskolin (C), and when a plateau was reached, oxyhaemoglobin (OxyHb, 10 μM) was added. The average results are summarized in Table 1.
Table 1.
The effect of oxyhaemoglobin (10 μM) on the simultaneously measured relaxations and changes from baseline in NO concentrations induced by ACh (10 μM), SIN-1 (10 μM), SNAP (10 μM) and forskolin (1 μM) in segments of the rat superior mesenteric artery contracted with noradrenaline (0.5-1 μM)
| Control | Oxyhaemoglobin | |||||
|---|---|---|---|---|---|---|
| n | Preconstriction (N m−1) | Relaxation (%) | Δ[NO] (nM) | Relaxation (%) | Δ[NO] (nM) | |
| Ach | 7 | 2.3 ± 0.2 | 93.3 ± 2.1 | 33 ± 5 | −15.6 ± 16.0 | −13 ± 6 |
| SIN-1 | 5 | 2.5 ± 0.2 | 83.6 ± 6.5 | 16 ± 4 | 40.6 ± 15.3 | −2 ± 3 |
| SNAP | 9 | 3.5 ± 0.3 | 72.2 ± 8.0 | 40 ± 8 | −1.2 ± 6.9 | −15 ± 6 |
| Forskolin | 6 | 3.0 ± 0.2 | 85.4 ± 5.8 | 1 ± 1 | 71.7 ± 10.6 | −14 ± 4 |
Values are means ± S.E.M., and n is the number of arteries (one from each animal). The protocol is shown in Fig. 5. The increases and decreases in NO concentration (Δ[NO]) are represented with respect to baseline before adding the relaxant agonist.
In preparations incubated with 100 μM L-NOARG, contracted with a lower concentration of noradrenaline (0.1-0.5 μM), and relaxed by 72 ± 5 % (n = 4) with 10 μM acetylcholine, 10 μM oxyhaemoglobin added after 2 min reduced the relaxations to 6 ± 5 % and decreased the NO concentrations by 14 ± 4 nM (n = 4). In a set of control experiments performed in the presence of L-NOARG (100 μM), 10 μM acetylcholine induced transient relaxations of 71 ± 4 % after 2 min and with a duration of 13 ± 3 min (n = 6) to 50 % reversal of the relaxations in noradrenaline- contracted preparations.
Simultaneous measurements of relaxations and NO concentrations induced by exogenously added NO
In noradrenaline-contracted preparations, SNAP, SIN-1 and injected NO solution caused concentration-dependent decreases in force and increases in NO concentration (Fig. 6). Thus SNAP induced relaxations with pD2 values and maximal relaxations of 5.68 ± 0.16 and 83 ± 6 %, respectively, while increases in NO concentration were 19 ± 9 nM NO (n = 6) at the highest concentration of SNAP applied (10 μM) (Fig. 6A). SIN-1 induced relaxations with pD2 values and maximal relaxations of 5.42 ± 0.09 and 97 ± 2 % (n = 5), and increased the NO concentration by 12 ± 5 nM (n = 5) at the highest concentration of SIN-1 (10 μM) (Fig. 6A). Higher concentrations of authentic NO had to be applied to obtain relaxations that reached a maximum of 75 ± 8 % (n = 4) with a pD2 value of 6.41 ± 0.39 (n = 4), and the NO concentration measured in the myograph bath reached 3.1 ± 0.8 μM at the highest concentration of authentic NO applied (10 μM). This was not due to a loss of NO, since the measured NO concentration at the electrode corresponded to the injected NO concentration at low, but not at high concentrations of injected authentic NO. Thus when 10 nM and 1 μM authentic NO was added, 98 ± 18 and 27 ± 11 % (n = 4), respectively, was measured at the electrode.
Figure 6. Concentration-dependent relaxations and increases in NO concentration induced by exogenously added NO.
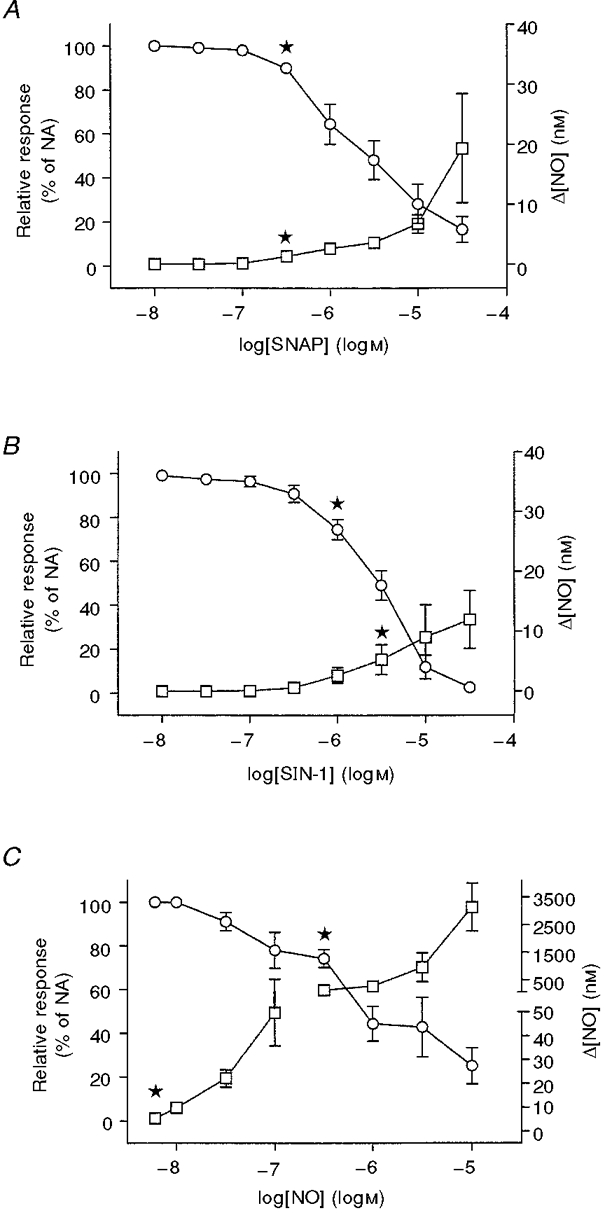
Graphs showing average relaxations (○) and increases in NO concentrations (□) with increasing concentrations of SNAP (A), SIN-1 (B) and dissolved authentic NO (C) in the rat superior mesenteric artery precontracted with noradrenaline (NA, 0.5-1 μM). NO concentrations are given as increases above the level in the absence of NO donor. The results are means ±s.e.m. of 4-6 experiments. First concentration of NO donor causing significant relaxation or increase in NO concentration: *,P < 0.05.
SNAP- (10 μM) and SIN-1 (10 μM)-induced increases in NO concentration preceded the relaxations by 8 ± 2 and 20 ± 10 s, respectively, and increased for both NO donors during the observation period (Fig. 7). For half-maximal relaxation, the estimated increases in NO concentration were 17 ± 7 nM (n = 6) and 7 ± 2 nM (n = 5) for SNAP and SIN-1, respectively. These responses were compared with the response to infused NO. The increases in NO concentration induced by infusion of dissolved NO gas (50 nmol l−1 min−1) accompanied with relaxation were much higher (Fig. 7A), and the increase in NO concentration associated with half-maximal relaxation when authentic NO was infused was 378 ± 129 nM (P < 0.05, n = 4, compared with SNAP and SIN-1, ANOVA followed by Bonferroni t test). After stopping the infusion of authentic NO, the measured NO concentration fell exponentially with time, and it was associated with recovery of the noradrenaline-induced contraction (Fig. 7A). When NO infusion was stopped, the initial NO concentration was 1.0 ± 0.3 μM (n = 4), and the fall in measured NO concentration fitted with an exponential decay yielded a half-life of 115 ± 19 s (n = 4).
Figure 7. Time relationship for simultaneously measured relaxations and increases in NO.

Averaged relaxations (○) and increases in NO (□) at various times after the addition of 10 μM SNAP (A), 10 μM SIN-1 (B), and 50 nmol l−1 min−1 infused NO (C) in rat superior mesenteric arteries contracted with 0.5 μM noradrenaline (NA). The NO infusion (C) was stopped after 10 min (arrow), and this was followed by a fall in NO concentration and recovery of the noradrenaline-induced contraction. NO concentrations are given as increases above the level in the absence of acetylcholine. Each point represents the mean ±s.e.m. of 4-6 preparations.
Experiments with the membrane-type NO-sensitive electrode placed close to the vessel segment in the myograph gave increases in NO concentration similar to the ISONOP30 microelectrode when 50 nmol l−1 min−1 authentic NO was infused (n = 3, results not shown).
Time relationship for simultaneous measurements of relaxations and endogenous NO release
In arteries contracted with noradrenaline, acetylcholine (10 μM) induced an increase in the concentration of NO, which reached a maximum of 22 ± 9 nM (n = 6) after 2 min and was accompanied by a simultaneous reduction of vascular tone averaging 87 ± 1 % (n = 6) (Fig. 8A and B). The acetylcholine-induced relaxation and increase in NO concentration occurred simultaneously (difference between the two events was 2 ± 4 s (n = 6)). After the addition of acetylcholine, the measured NO concentration after 10 min had decreased to 15 ± 4 nM above baseline, but the relaxation was unchanged (88 ± 7 %, n = 6). Plotting the 10 μM acetylcholine-induced relaxation against measured NO concentration indicated that an increase in NO concentration of 9 ± 2 nM (n = 6) was sufficient to cause 50 % relaxation of the noradrenaline-contracted segments (Fig. 8A).
Figure 8. Time relationship for simultaneously measured relaxations and endogenous NO concentrations.

A, average of simultaneously obtained force (○) and NO concentration (□) curves for acetylcholine (10 μM) in segments of the rat mesenteric superior artery contracted with noradrenaline (0.5 μM). B, the same results as A with relaxation plotted against NO concentration. The results are means of 6 experiments and horizontal and vertical bars represent s.e.m.
DISCUSSION
The main finding of this study is the demonstration of simultaneous changes in nanomolar NO concentrations and contractility in isolated arteries exposed to either exogenous NO or to NO released upon stimulation with the endothelium-dependent vasodilator, acetylcholine. The real time analysis of associated increases in NO concentration and relaxation also suggests that the transient responses still induced by acetylcholine in the presence of the NOS inhibitor, L-NOARG, are mediated by NO. In addition to agonist-induced release of NO, there appears to be an endogenous basal release of NO.
Evaluation of increases in NO concentration
The NO concentration measured with the microelectrode in the vessel lumen reflects the NO release minus clearance by degradation and diffusion, and as expected in the present investigation, the NO concentration measured in the myograph was substantially less than the quantity of authentic NO added, although that was most pronounced for concentrations above 100 nM (Figs 6 and 7). Both calibration and experiments were performed in the presence of oxygen, since the endogenous NO production is oxygen dependent (Hashimoto et al. 1993). Therefore, degradation of NO could play a role. NO reacts with oxygen by a second-order reaction in physiological buffer, and the half-life of NO in this reaction is inversely proportional to the concentration of NO, i.e. the half-life of NO would be 560 s for 1 μM and 5600 s for 100 nM in water saturated with room air (Beckman & Koppenol, 1996). Superoxide formed in oxygenated glucose-rich buffer in bioassays using a perfusion-cascade system can probably explain the short half-life (5-6 s) of EDRF and NO, which has been reported earlier (Palmer et al. 1987; Feelisch et al. 1994; Beckman & Koppenol, 1996). In the present study where the preparation was exposed to 95 % oxygen, the exponential decay in NO concentration had an estimated half-life of 115 s after stopping the infusion of authentic NO. This suggests that diffusion and tissue degradation of NO, rather than destruction in the buffer, could contribute to the NO concentrations measured in the present study. Therefore, mainly cellular diffusion barriers or the presence of oxyhaemoglobin would create a difference in NO concentration at the production site and that measured with the NO electrode.
The NO concentrations measured in response to acetylcholine in the present study were in the low nanomolar range. In contrast, other studies have reported that endothelium-dependent vasodilators are associated with much higher NO concentrations (Goligorsky et al. 1994; Tschudi et al. 1996). However, there are differences with respect to preparation examined, species and the conditions under which the experiments were performed. The NO concentration is much higher in studies performed with the arteries kept at 24°C (Tschudi et al. 1996; Cohen et al. 1997) than in experiments performed at physiological temperature like the present investigation, where the concentrations of NO would be expected to be lower, since less NO is dissolved at higher temperature and will disappear before reaching the electrode. The different structure of the porphyrinic microelectrode used in other studies (Tschudi et al. 1996; Cohen et al. 1997) and the ISONOP30 microelectrode applied in the present study can also influence the measured NO concentrations. Thus the Nafion-covered nickel porphyrin-type NO sensors were found to be less selective against ascorbate, dopamine and nitrite than polymer-coated electrodes (Friedemann et al. 1996). Detection of NO with either chemiluminescence (Palmer et al. 1987) or the oxyhaemoglobin technique (Kelm & Schrader, 1990) have suggested that nanomolar concentrations of NO are released from endothelial cells, and the apparent Km of NO for guanylate cyclase is in the low nanomolar range (Ignarro et al. 1993). In the present study, the nanomolar increases in NO concentration observed with acetylcholine were sustained for a long period, which is in agreement with the experiments with infused NO suggesting a long half-life for low concentrations of NO. Thus there are several lines of evidence supporting the present finding that nanomolar concentrations of endogenously released NO initiate and maintain the acetylcholine-induced relaxation of the rat superior mesenteric artery.
One finding of the present study was that the NO concentration associated with relaxation was different for authentic NO compared with other NO donors. Injecting small volumes or infusion of authentic NO at a constant rate suggested that concentrations of 300-400 nM were needed to cause 50 % relaxation of the rat superior mesenteric artery. In contrast, when SNAP or SIN-1 was added, concentrations of only 7-17 nM induced half-maximal relaxation. Authentic NO may arrive at the tissue at much lower concentrations than the calibrated concentration, but this is an unlikely explanation for the concentration range below 100 nM where most of the added NO reached the electrode that was situated close to the vessel wall. Therefore, other mechanisms leading to relaxation, in addition to NO release from SNAP and SIN-1, might explain the different NO release-relaxation relationship for these NO donors compared with authentic NO. SNAP is a nitrosothiol, and in addition to NO release, nitrosothiols have been suggested to cause relaxations through activation of stereoselective S-nitrosothiol recognition sites (Davisson et al. 1996), cyclic GMP-independent direct inhibition of Ca2+ channels (Hu et al. 1997) or activation of K+ channels (Koh et al. 1995). The NO release-relaxation relationships for SNAP and SIN-1 appear to be alike, but other mechanisms can probably explain the different NO release-relaxation relationship for SIN-1 compared with authentic NO. SIN-1, in addition to NO, releases superoxide, which can inactivate NO (Feelisch & Stamler, 1996), and SIN-1 was also reported to cause relaxation through a direct activation of K+ channels in mesenteric small arteries (Plane et al. 1996). SIN-1-induced relaxations were only partially reversed by oxyhaemoglobin in the present study, which would be compatible with mechanisms apart from NO release mediating the relaxations. In contrast to SIN-1, the SNAP-induced responses were abolished by oxyhaemoglobin in the present study. Oxyhaemoglobin can create a gradient for NO out of the cell (Lancaster, 1994). Therefore, an alternative explanation could be a different location in the vascular wall of the NO release from SNAP compared with authentic NO. Indeed, SNAP releases NO catalysed by the vascular membrane or by heterolytic transfer, a transnitrosation reaction, to intracellular glutathione in the smooth muscle cells (Butler & Rhodes, 1997). Thus either additional mechanisms apart from NO release or a different location of NO release can explain that nanomolar concentrations of NO released from SNAP and SIN-1 are sufficient to cause relaxation of the mesenteric superior artery, while submicromolar concentrations of authentic NO are necessary to cause full relaxation of the artery.
Investigation of the NO release-relaxation relationship for endogenous NO would clarify whether the location in the vessel wall of the NO release plays a role in the NO measurements. In the present study, the NO concentration- force relationships for acetylcholine and authentic NO are different. In contrast to authentic NO, which reaches the smooth muscle either from the adventitial side or by diffusion through the endothelial cell layer, acetylcholine acts on the endothelium followed by release of NO, which will diffuse both towards the sensor in the lumen and towards the smooth muscle, producing relaxation and increase in the NO signal. Thus the endothelial cell layer or adventitia might constitute a barrier to exogenously added authentic NO, since the cut ends of the vessel only constitute 1.5 % of the vascular segment in vitro. Indeed, the superoxide anion-generating enzyme NADPH oxidase located in the adventitia was recently shown to generate sufficient extracellular superoxide anion to constitute a barrier capable of inactivating NO (Wang et al. 1998). The existence of a barrier within the endothelium and adventitia of the vessel wall for authentic NO would imply a concentration difference and that low NO concentrations close to the smooth muscle should relax the artery. Recent studies where NO was released intracellularly from ruthenium nitrosyl chlorides or iron-sulphur- nitrosyls have actually demonstrated that 3.7-13 nM NO caused half-maximal relaxation of the rabbit aorta and rat isolated tail artery (Flitney et al. 1996; Carter et al. 1997). Therefore, in the present study the different NO concentration-force relationship for acetylcholine compared with authentic NO can probably be explained by a barrier for the latter. On the other hand, SNAP, which probably releases NO close to the smooth muscle cell membrane, has a NO concentration- force relationship similar to that of acetylcholine.
Time-associated increase in NO concentration and relaxation
The present study reveals that endogenous NO concentrations and relaxations induced by acetylcholine are temporally related. This is further supported by the observation that modulation of the acetylcholine-induced increases in NO concentration and relaxations by mechanical endothelial cell removal, incubation with the NOS inhibitor, L-NOARG, or the NO scavenger, oxyhaemoglobin, caused simultaneous changes in both NO concentration and relaxation. These results are in agreement with earlier studies in the rat superior mesenteric artery showing that inhibition of the NO-L-arginine pathway almost abolishes the relaxations induced by acetylcholine (Hwa et al. 1994; Van de Voorde & Vanheel, 1997), but in contrast to those in mesenteric small arteries where acetylcholine induces maximal relaxations in the presence of an inhibitor of NOS and oxyhaemoglobin (Hwa et al. 1994). However, the present study has for the first time related the increase in NO concentration and relaxation induced by acetylcholine of the rat superior mesenteric artery.
One of the unexpected findings in the present study was that L-NOARG did not decrease, but increased the NO concentration when it was added to the myograph bath. This could be due to biochemical conversion of L-NOARG in the tissue, but L-NOARG also caused an oxyhaemoglobin-sensitive increase in the NO concentration in a glass vial in the absence of vascular tissue, suggesting that it is a spontaneous process. NOS inhibitors, such as L-NOARG and L-NAME containing a nitro group, have previously been demonstrated to release NO by a photochemical reaction (Bauer & Fung, 1993). Thus the NO released by L-NOARG plus the residual NO released by acetylcholine in the presence of L-NOARG could provide in total sufficient NO to explain the transient relaxations observed with acetylcholine.
In the presence of L-NOARG, we still observed transient relaxations at the highest concentrations of acetylcholine, and these relaxations were associated temporally with small and transient increases in NO concentrations. These results agree with the observations in the rabbit carotid artery where endothelium-dependent relaxations in the presence of L-NOARG have previously been claimed to be due to an incomplete inhibition of NOS (Cohen et al. 1997). As pointed out in the Introduction, in the rat superior mesenteric artery cytochrome P450 metabolites do not appear to mediate the L-NOARG-resistant relaxations (Van de Voorde & Vanheel, 1997) or the transient endothelium-dependent hyperpolarizations (Vanheel & Van de Voorde, 1997) induced by acetylcholine. The observation that oxyhaemoglobin also reversed the L-NOARG resistant relaxations induced by acetylcholine in the present study provides further evidence for the suggestion that only NO is mediating the endothelium-dependent relaxations in the rat superior mesenteric artery. In rat aorta and main pulmonary artery, haemoglobin also inhibited the acetylcholine-induced relaxation, but did not change the transient hyperpolarization or increases in marked rubidium efflux (Chen et al. 1988). Therefore, the present results do not exclude the presence of an EDHF, which could play a modulatory role in the acetylcholine-induced relaxations of the rat superior mesenteric artery.
Basal NO release
In some vascular preparations, incubation with inhibitors of NOS induce contractions in endothelium-intact, but not in endothelium-denuded preparations, suggesting a basal release of NO (Chester et al. 1990; Simonsen et al. 1997), and evaluation of the contractile effect induced by NOS inhibitors has been used to evaluate endothelial cell dysfunction in cardiovascular disease (Chester et al. 1990). In the present study, oxyhaemoglobin, in addition to causing inhibition of the relaxations induced with acetylcholine and SNAP, caused a decrease in measured NO concentration below baseline. A noradrenaline-induced NO release cannot be exluded, but oxyhaemoglobin also reduced the NO signal below baseline in the absence of noradrenaline. Therefore these results suggests the presence of a continuous basal NO release in the preparation.
In conclusion, the present study demonstrates that the endogenous NO concentration correlates with relaxation of the rat superior mesenteric artery when it is stimulated with the endothelium-dependent vasodilator acetylcholine. It also suggests that NO is mediating the L-NOARG-resistant relaxations in the rat superior mesenteric artery, and that there is a basal NO release. Thus the study provides direct evidence that endothelium-derived NO is of major importance in the physiological regulation of the contractility of the rat superior mesenteric artery.
Acknowledgments
R. M. Wadsworth was supported by a grant from the Royal Society under the European Science Exchange Programme. This study was supported by grants from the NOVO Foundation, Aarhus Universitets Forskningsfond E-1996-SUN-1-172, and AP Møller fonden.
References
- Bauer JA, Fung H-L. Photochemical generation of nitric oxide from nitro-containing compounds: possible relation to vascular photorelaxation phenomena. Life Sciences. 1993;54:PL1–4. doi: 10.1016/0024-3205(94)00578-8. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. American Journal of Physiology. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- Broderick MP, Taha Z. Nitric oxide detection using a popular electrochemical sensor. World Precision Instruments, Satellite Symposium, 4th IBRO World Congress of Neuroscience, Kyoto, Japan. 1995:1–10. [Google Scholar]
- Butler AR, Rhodes P. Chemistry, analysis, and biological roles of S-nitrosothiols. Analytical Biochemistry. 1997;249:1–9. doi: 10.1006/abio.1997.2129. 10.1006/abio.1997.2129. [DOI] [PubMed] [Google Scholar]
- Carter TD, Bettache N, Ogden D. Potency and kinetics of nitric oxide-mediated vascular smooth muscle relaxation determined with flash photolysis of ruthenium nitrosyl chlorides. British Journal of Pharmacology. 1997;122:971–973. doi: 10.1038/sj.bjp.0701549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. British Journal of Pharmacology. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester AH, O'Neil GS, Moncada S, Tadjkarimi S, Yacoub MH. Low basal and stimulated release of nitric oxide in atherosclerotic epicardial coronary arteries. Lancet. 1990;336:897–900. doi: 10.1016/0140-6736(90)92269-n. [DOI] [PubMed] [Google Scholar]
- Cohen RA, Plane F, Najibi S, Huk I, Malinski T, Garland CJ. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery. Proceedings of the National Academy of Sciences of the USA. 1997;94:4193–4198. doi: 10.1073/pnas.94.8.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RA, Vanhoutte PM. Endothelium-dependent hyperpolarization. Beyond nitric oxide and cyclic GMP. Circulation. 1995;92:3337–3349. doi: 10.1161/01.cir.92.11.3337. [DOI] [PubMed] [Google Scholar]
- Davisson RL, Travis MD, Bates JN, Lewis SJ. Hemodynamic effects of L- and D-S-nitrosocysteine in the rat. Stereoselctive S-nitrosothiol recognition sites. Circulation Research. 1996;79:256–262. doi: 10.1161/01.res.79.2.256. [DOI] [PubMed] [Google Scholar]
- Feelisch M, Poel MT, Zamora R, Deussen A, Moncada S. Understanding the controversy over the identity of EDRF. Nature. 1994;368:62–65. doi: 10.1038/368062a0. 10.1038/368062a0. [DOI] [PubMed] [Google Scholar]
- Feelisch M, Stamler JS. Donors of nitric oxide. In: Feelisch M, Stamler JS, editors. Methods in Nitric Oxide Research. New York: Wiley & Sons; 1996. pp. 71–119. [Google Scholar]
- Flitney FW, Megson IL, Joanne LM, Thomson GD, Kennovin GD, Butler AR. Vasodilator responses of rat isolated tail artery enhanced by oxygen-dependent, photochemical release of nitric oxide from iron-sulphur-nitrosyls. British Journal of Pharmacology. 1996;117:1549–1557. doi: 10.1111/j.1476-5381.1996.tb15320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedemann MN, Robinson SW, Gerhardt GA. O-Phenylenediamine-modified carbon fiber electrodes for the detection of nitric oxide. Analytical Chemistry. 1996;68:2621–2628. doi: 10.1021/ac960093w. 10.1021/ac960093w. [DOI] [PubMed] [Google Scholar]
- Gevantman LH. In: Handbook of Chemistry and Physics. Lide D, editor. Boca Raton, FL, USA: CRC Press; 1995. pp. 6–3. [Google Scholar]
- Goligorsky MS, Tsukahara H, Magazine H, Andersen TT, Malik AB, Bahou WF. Termination of endothelin signaling: role of nitric oxide. Journal of Cellular Physiology. 1994;158:485–494. doi: 10.1002/jcp.1041580313. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Close LA, Ishida Y, Paul RJ. Dependence of endothelium-mediated relaxation on oxygen and metabolism in porcine coronary arteries. American Journal of Physiology. 1993;265:H299–306. doi: 10.1152/ajpheart.1993.265.1.H299. [DOI] [PubMed] [Google Scholar]
- Hatake K, Wakabayashi I, Hishida S. Endothelium-dependent relaxation resistant to NG-nitro-L-arginine in rat aorta. European Journal of Pharmacology. 1995;274:25–32. doi: 10.1016/0014-2999(94)00704-b. 10.1016/0014-2999(94)00704-B. [DOI] [PubMed] [Google Scholar]
- Hecker M, Bara AT, Bauersachs J, Busse R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. The Journal of Physiology. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Chaimvimonvat N, Yamagishi T, Marban E. Direct inhibition of expressed cardiac L-type Ca2+ channels by S-nitrosothiol nitric oxide donors. Circulation Research. 1997;81:742–752. doi: 10.1161/01.res.81.5.742. [DOI] [PubMed] [Google Scholar]
- Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking a gene for endothelial nitric oxide synthase. Nature. 1995;337:239–242. doi: 10.1038/377239a0. 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- Hwa JJ, Ghibaudi L, Williams P, Chatterjee M. Comparison of acetylcholine-dependent relaxation in large and small arteries of rat mesenteric vascular bed. American Journal of Physiology. 1994;266:H952–958. doi: 10.1152/ajpheart.1994.266.3.H952. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Fukuto JM, Crisgavage JM, Rogers NE, Byrns RE. Oxidation of nitric oxide in aqueous solution to nitrite but not nitrate: Comparison with enzymatically formed nitric oxide from L-arginine. Proceedings of the National Academy of Sciences of the USA. 1993;90:8103–8107. doi: 10.1073/pnas.90.17.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm M, Schrader J. Control of coronary vascular tone by nitric oxide. Circulation Research. 1990;66:1561–1575. doi: 10.1161/01.res.66.6.1561. [DOI] [PubMed] [Google Scholar]
- Koh SD, Campell JD, Carl A, Sanders KM. Nitric oxide activates multiple potassium channels in canine colonic smooth muscle. The Journal of Physiology. 1995;489:735–743. doi: 10.1113/jphysiol.1995.sp021087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster JR. Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proceedings of the National Academy of Sciences of the USA. 1994;91:8137–8141. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinski T, Taha Z. Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature. 1992;358:676–678. doi: 10.1038/358676a0. 10.1038/358676a0. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circulation Research. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Murphy ME, Brayden JE. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. The Journal of Physiology. 1995;486:47–58. doi: 10.1113/jphysiol.1995.sp020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osol G, Cipolla M, Knutson S. A new method for mechanically denuding the endothelium of small (50–150 microns) arteries with a human hair. Blood Vessels. 1989;26:320–324. doi: 10.1159/000158781. [DOI] [PubMed] [Google Scholar]
- Palmer RMJ, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- Plane F, Hurrell A, Jeremy JY, Garland CJ. Evidence that potassium channels make a major contribution to SIN-1-evoked relaxation of rat isolated mesenteric artery. British Journal of Pharmacology. 1996;119:1557–1562. doi: 10.1111/j.1476-5381.1996.tb16072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuki M, Okada D. Endogenous nitric oxide release required for long-term synaptic depression in the cerebellum. Nature. 1991;349:326–328. doi: 10.1038/349326a0. 10.1038/349326a0. [DOI] [PubMed] [Google Scholar]
- Simonsen U, Garcia-Sacristan A, Prieto D. Apamin-sensitive K+ channels involved in the inhibition of acetylcholine-induced contractions in lamb coronary small arteries. European Journal of Pharmacology. 1997;329:153–163. 10.1016/S0014-2999(97)10129-7. [PubMed] [Google Scholar]
- Tare N, Parkington HC, Coleman HA, Neild TO, Dusting GJ. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- Tschudi MR, Barton M, Bersinger NA, Moreau P, Cosentino F, Noll G, Malinski T, Lüscher TF. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. Journal of Clinical Investigation. 1996;98:899–905. doi: 10.1172/JCI118872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Voorde J, Vanheel B. Influence of cytochrome P-450 inhibitors on endothelium-dependent nitro-L-arginine-resistant relaxation and cromakalim-induced relaxation in rat mesenteric arteries. Journal of Cardiovascular Pharmacology. 1997;29:827–832. doi: 10.1097/00005344-199706000-00018. 10.1097/00005344-199706000-00018. [DOI] [PubMed] [Google Scholar]
- Vanheel B, Van de Voorde J. Evidence against the involvement of cytochrome P450 metabolites in endothelium-dependent hyperpolarization of the rat mesenteric artery. The Journal of Physiology. 1997;501:331–341. doi: 10.1111/j.1469-7793.1997.331bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HD, Pagano PJ, Du Y, Cayatte AJ, Quinn MT, Brecher P, Cohen RA. Superoxide anion from the adventitia of the rat thoracic aorta inactivates nitric oxide. Circulation Research. 1998;28:810–818. doi: 10.1161/01.res.82.7.810. [DOI] [PubMed] [Google Scholar]