

Abstract
Average intracellular calcium concentration ([Ca2+]i) and ciliary beat frequency (CBF) were simultaneously measured in rabbit airway ciliated cells in order to elucidate the molecular events that lead to ciliary activation by purinergic stimulation.
Extracellular ATP and extracellular UTP caused a rapid increase in both [Ca2+]i and CBF. These effects were practically abolished by a phospholipase C inhibitor (U-73122) or by suramin.
The effects of extracellular ATP were not altered: when protein kinase C (PKC) was inhibited by either GF 109203X or chelerythrine chloride, or when protein kinase A (PKA) was inhibited by RP-adenosine 3′, 5′-cyclic monophosphothioate triethylamine (Rp-cAMPS).
Activation of PKC by phorbol 12-myristate, 13-acetate (TPA) had little effect on CBF or on [Ca2+]i, while activation of PKA by forskolin or by dibutyryl-cAMP led to a small rise in CBF without affecting [Ca2+]i.
Direct activation of protein kinase G (PKG) with dibutyryl-cGMP had a negligible effect on CBF when [Ca2+]i was at basal level. However, dibutyryl-cGMP strongly elevated CBF when [Ca2+]i was elevated either by extracellular ATP or by ionomycin.
The findings suggest that the initial rise in [Ca2+]i induced by extracellular ATP activates the NO pathway, thus leading to PKG activation. In the continuous presence of elevated [Ca2+]i the stimulated PKG then induces a robust enhancement in CBF. In parallel, activated PKG plays a central role in Ca2+ influx via a still unidentified mechanism, and thus, through positive feedback, maintains CBF close to its maximal level in the continuous presence of ATP.
The primary function of the cilia of airway epithelium is to transport mucus over the cell, thereby contributing to mucociliary clearance. For this task to be performed efficiently, cilia beat in an approximately periodic pattern in space and time, called metachronal wave. To enable response to varying environmental conditions, ciliary cells possess a relatively large variety of different receptors capable of inducing intracellular events that lead to ciliary enhancement. Among these stimulators, extracellular ATP is the most potent activator of ciliary function, stimulating a variety of ciliary cells, including frog palate and oesophagus (Ovadyahu et al. 1988; Weiss et al. 1992), human nasal polyps (Mason et al. 1991), rabbit oviduct (Villalon et al. 1989), dog trachea (Wong & Yeates, 1992), and rabbit trachea (Korngreen & Priel, 1996).
It is well known that the cellular event most closely implicated in ciliary stimulation is a change in [Ca2+]i (Verdugo, 1980; Villalon et al. 1989; Di Benedetto et al. 1991; Lansley et al. 1992; Korngreen & Priel, 1994; Salathe & Bookman, 1995; Tarasiuk et al. 1995). Recently, the correlation between ciliary beat frequency (CBF) and [Ca2+]i enhancement was systematically examined in ciliary cells from rabbit airway epithelia (Korngreen & Priel, 1996). It was found that extracellular ATP caused a rapid and large rise in [Ca2+]i by mobilizing Ca2+ from intracellular stores, accompanied by a correlated enhancement of CBF. Following the maximum rise, [Ca2+]i decays to an elevated plateau while CBF remains close to its maximum value. The elevated plateau of [Ca2+]i and the sustained enhancement of CBF were found to require Ca2+ influx.
It is widely accepted that cAMP is also an important modulator of ciliary activity in a variety of ciliary systems. β-Adrenergic agonists known to elevate the concentration of cAMP stimulate CBF of respiratory epithelia (Verdugo et al. 1980). cAMP-dependent modulation of CBF has been reported in human and rabbit ciliary cells (Tamaoki et al. 1989; Di Benedetto et al. 1991; Yang et al. 1996). cAMP-dependent protein kinase (PKA) has been shown to phosphorylate specific axonemal targets that increase the forward swimming speed in Paramecium (Hamasaki et al. 1991). Similar axonemal targets for protein kinase A (PKA) phosphorylation have been identified in mammalian respiratory cilia (Salathe et al. 1993).
Although the role of Ca2+ and cAMP as modulators of ciliary activity has been widely investigated, little is known about possible cross-talk between these two pathways in ciliary cells. Satir & Sleigh (1990) have suggested that there is dual control of ciliary activity by Ca2+ and by cAMP acting through independent pathways that interact or possibly converge at an axonemal or a subaxonemal level. In frog oesophagus and palate ciliated cells, we have recently shown that PKA releases Ca2+ from intracellular stores and enhances CBF in a Ca2+-dependent and -independent manner, suggesting a dual mechanism of action of PKA in this system (Braiman et al. 1998).
There is increasing evidence that the nitric oxide (NO) pathway also plays an important role in CBF stimulation. Expression of nitric oxide synthase (NOS) was demonstrated in human airway epithelium (Guo et al. 1997; Robbins et al. 1997), release of NO in rabbit tracheal mucosa (Tamaoki et al. 1995b), and synthesis of cGMP in human airway epithelium (Geary et al. 1993). Moreover, inhibition of NOS was shown to attenuate CBF stimulation induced by isoprenaline (Jain et al. 1993), cytokines (Jain et al. 1995), β-adrenergic stimulants (Tamaoki et al. 1995a), and methacholine (Yang et al. 1996). Direct application of 8-bromoguanosine 3′, 5′ cyclic monophosphate (8-Br-cGMP; a permeable analogue of cGMP) enhanced CBF in nasal airway epithelial cell culture (Geary et al. 1995). On the other hand, it was reported that cGMP inhibits ciliary motility in tissue cultures from rabbit trachea (Tamaoki et al. 1991).
The present study was designed to elucidate the pathway(s) underlying the strong and sustained enhancement in CBF induced by extracellular ATP in rabbit airway epithelium, based on simultaneous measurement of CBF and [Ca2+]i. Answers were sought to the following basic questions regarding CBF enhancement: (a) is a rise in [Ca2+]i necessary and sufficient to induce a rise in CBF, or are additional factors involved?; (b) are protein kinase C (PKC), PKA and protein kinase G (PKG) involved in CBF enhancement?; and (c) is there cross-talk between [Ca2+]i and the various protein kinases?
Part of this work has been published in abstract form (Uzlaner & Priel, 1998).
METHODS
Tissue culture
Experiments were carried out on monolayer tissue cultures grown from rabbit trachea using a procedure described previously (Korngreen & Priel, 1996). Briefly, adult New Zealand White rabbits (at least 1.5 kg) were killed according to the guidelines laid down by the animal welfare committee of Ben-Gurion University, by gradual exposure to carbon dioxide followed by exsanguination. Care was taken to slowly increase the gas flow over several minutes to prevent any visual signs of distress. Tracheas were removed, and the ciliary epithelium was peeled off the cartilage rings and cut into small pieces. Two or three pieces of epithelium were placed on a glass coverslip and were incubated in RPMI-1640 growth medium supplemented with 7.5 % fetal calf serum, 20 units ml−1 penicillin, 2.5 units ml−1 nystatin and 20 μg ml−1 streptomycin at 37°C with 5 % CO2. Prior to use, the glass coverslips were sterilized with ethanol, placed in plastic Petri dishes (Nunk, Denmark) and incubated with a small amount of medium for 24 h. The medium was replaced every 2 days. Measurements were performed on 8- to 20-day-old tissue cultures; this was the time period at which the epithelial monolayers were big enough to be used in the experiments.
Chemicals and solutions
Fetal calf serum, RPMI-1640 culture medium and antibiotics were from Biological Industries (Bet-Haemek, Israel). Fura-2 AM was from either Molecular Probes (Eugene, OR, USA) or from Teflabs (Austin, TX, USA). The dye was stored in solid form at -20°C and fresh solutions were made before each experiment in dry dimethyl sulphoxide (DMSO). Phorbol 12-myristate, 13-acetate (TPA), 8,8′-[carbonylbis [imino-3,1-phenylenecarbonylimino (4-methyl-3,1-phenylene) carbonylimino]] bis-1, 3, 5-naphthalenetrisulphonic acid hexasodium (Suramin hexasodium) and RP-adenosine 3′, 5′-cyclic monophosphothioate triethylamine (Rp-cAMPS) were from RBI (Natick, MA, USA). 1-(6-((17β-3-Methoxyestra-1, 3, 5 (10)-trien-17-yl) amino) hexyl)-1H-pyrrole-2, 5-dione (U-73122), 6-anilino-5, 8-quinolinedione (LY-83583), bisindolylmaleimide I (GF 109203X) and KT-5823 were from Calbiochem (La Jolla, CA, USA). L-NAME was from Biomol (Plymouth Meeting, PA, USA). All other materials were obtained from Sigma (USA). The standard Ringer solution was composed of (mM): 150 NaCl, 2.5 KCl, 1.5 CaCl2, 1.5 MgCl2, 5 D-glucose and 5 Hepes. The pH of all the solutions was set to 7.4 with NaOH and HCl.
TPA, GF 109203X, U-73122 and KT-5823 were dissolved in DMSO, chelerythrine chloride was dissolved in DMSO/H2O (1:1), and LY-83583 was dissolved in CH3OH/C2H5OH (1:1). Stock solutions of ATP and UTP were prepared in 0.05 M Hepes buffer (pH 7.4) and diluted into the Ringer solutions just prior to use. All other chemicals were dissolved in water. The final concentration of DMSO was not more than 0.1 %, and that of the alcohol not more than 0.5 %.
Simultaneous measurement of [Ca2+]i and ciliary beating
We used the method of simultaneous measurement of [Ca2+]i and ciliary beating that has been extensively described previously (Korngreen & Priel, 1994). Briefly, [Ca2+]i was measured with the fluorescent indicator fura-2. The dye-loaded cells were epi-illuminated with light from a 150 W xenon lamp (Oriel Corp., Stamford, CT, USA) filtered by 340 and 380 nm interference filters (Oriel) mounted on a four position rotating filter wheel. The fluorescence, emitted at 510 nm, was detected by a photon-counting photomultiplier (H3460-53, Hamamatsu, Japan). The 340 nm/380 nm fluorescence ratio, averaged over a period of 1 s, was stored on computer (IBM 486). CBF was measured by trans-illuminating the same cells with light at 600 nm (so as not to interfere with the fura-2 fluorescence at 510 nm). The light scattering from the beating cilia created amplitude modulations of the 600 nm light that were detected by a photomultiplier (R2014, Hamamatsu, Japan).
The [Ca2+]i was calculated using a calibration curve that correlates fura-2 fluorescence ratio with calcium concentration. The calibration curve was constructed from measurements of the fluorescence ratio obtained from calibration solutions composed of 115 mM KCl, 20 mM NaCl, 5 mM MgCl2, 5 mM D-glucose, 5 mM Hepes, 10 mM EGTA, 1 μM K2fura-2 and CaCl2 at a range of concentrations (Grynkiewicz et al. 1985). The [Ca2+]i concentration was calculated directly from the calibration curve using a table look-up algorithm. The frequency response was calibrated by: (1) an oscillating lamp in the range of 2-40 Hz; (2) a vibrating needle between 2 and 40 Hz. A linear response was revealed in these dynamic ranges.
Experimental procedure and data presentation
Cells were pre-loaded with fura-2 by incubating the tissue in growth medium containing 8.5 μM fura-2 AM for 60 min at 37°C in a rotating water bath. The cells were then rinsed with Ringer solution and maintained at room temperature (22-24°C) for 30 min to allow full de-esterification and equilibration of the dye in the cytoplasm. Only evenly fluorescent cells that did not display bright spots and had a steady CBF were used. In each experiment, basal CBF (F0) and basal [Ca2+]i ([Ca2+]i,0) were measured for 2-5 min in the appropriate solution. These were taken as reference values. Then the test chemical was introduced by exchanging the bath solution, and the frequency (F) and [Ca2+]i were monitored in the same ciliary area for 10-15 min. Each experimental condition was tested on naive cultures to avoid possible bias arising from previous manipulations. Beat frequencies are presented as the fold increase in CBF (F/F0). Changes in [Ca2+]i are presented as the difference between the observed [Ca2+]i and the basal [Ca2+]i ([Ca2+]i - [Ca2+]i,0). Each experiment was carried out on 3-15 tissue cultures from at least 2-3 animals. Results were averaged and are displayed as means ±s.e.m. Statistical significance was determined using Student's t test, with P < 0.05 being considered significant.
RESULTS
Phospholipase C mediates the effects of ATP
The effect of extracellular ATP (10 μM) on [Ca2+]i is demonstrated in Fig. 1A. Following the addition of ATP, [Ca2+]i increased within 6 s by 350 nM, and then declined to an elevated plateau 100 nM higher than basal level [Ca2+]i. On average, the peak increase in [Ca2+]i in response to 10 μM ATP was 315 ± 39 nM (n = 13 coverslips from 10 rabbits), and in the continuous presence of ATP [Ca2+]i declined to 68 ± 15 nM after 8 min of exposure to the ATP (Fig. 1A, □). Figure 1B shows the changes in CBF measured simultaneously in the same cell. Following the addition of ATP, CBF increased 2.2-fold from a basal value of 10-22 Hz and then declined only slightly over the next 8 min of recording. On average, the maximal increase in CBF in response to 10 μM ATP was a factor of 2.0 ± 0.1 (n = 13 coverslips from 10 rabbits) (Fig. 1A, □).
Figure 1. Involvement of phospholipase C in purinergically induced ciliary activation.
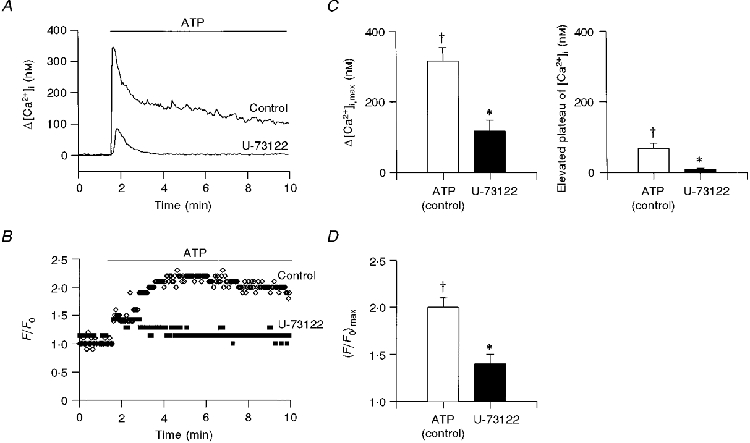
Time course of the effect of extracellular ATP (10 μM) on [Ca2+]i (A) and CBF enhancement (B) in the presence and absence of U-73122 (15 μM). In this and all subsequent figures, [Ca2+]i and CBF were sampled every second, but only each third data point is displayed for purposes of clarity. The average maximal value of Δ[Ca2+]i (C, left panel) and elevated [Ca2+]i plateau (C, right panel), and CBF enhancement (D) induced by extracellular ATP (10 μM) in the absence (□) and presence (▪) of U-73122 (15 μM) are presented. In all experiments, the tissue culture was incubated for 5 min with U-73122 prior to the addition of ATP. Each column represents the mean ±s.e.m. averaged over 3-13 experiments; † significantly different from basal values (Student's t test, P < 0.05); * significantly different from the effect at normal conditions (Student's t test, P < 0.05).
Recently we showed that the initial rapid rise in [Ca2+]i results from the release of Ca2+ from internal stores, while the sustained elevation in [Ca2+]i requires Ca2+ influx (Korngreen & Priel, 1996). The release of Ca2+ from internal stores strongly suggests that extracellular ATP activates phospholipase C (PLC), thus leading to the generation of inositol 1, 4, 5-trisphosphate (IP3). To test this possibility, the PLC pathway was inhibited by pre-incubating the cells with U-73122 and then applying ATP. The effect of extracellular ATP on [Ca2+]i and on CBF in a cell pre-treated with U-73122 (15 μM) is superimposed for purposes of comparison on the control responses in Fig. 1A and B. Clearly, U-73122 almost completely abolished the rise in [Ca2+]i and CBF. In nine experiments of this type, the peak increase in [Ca2+]i following addition of 10 μM ATP in cells pretreated with U-73122 (15 μM) was 117 ± 31 nM (n = 9 coverslips from 2 rabbits), and after 8 min of exposure to the ATP, [Ca2+]i was 9 ± 3 nM (Fig. 1A, ▪). In the same cells, CBF increased by only 1.4 ± 0.1 (Fig. 1A, ▪).
Surprisingly, no inhibitory effects were detected when the cells were exposed to 5 μM U-73122, while 10 μM U-73122 only partially inhibited the effects of extracellular ATP. This apparently strong dependence on the concentration of U-73122 may be related to the unstable nature of the compound (Bleasdale et al. 1989), and the actual effective concentrations of the inhibitor are not known. Nonetheless, the results strongly suggest that most, if not all, of the effect of extracellular ATP is mediated by the PLC pathway.
Protein kinase C is not involved in ciliary activation by ATP
Activation of the PLC pathway results in the generation of IP3, which releases Ca2+ from internal stores, and the production of diacylglycerol (DAG), the natural activator of PKC. The possible involvement of PKC in ATP-induced ciliary activation in rabbit airway epithelium was therefore examined.
Two experimental approaches were used to assess whether PKC is involved in the enhancement of [Ca2+]i and CBF induced by extracellular ATP. First, the cells were exposed to TPA (16 nM), a direct activator of PKC. Second, the cells were pre-incubated with chelerythrine chloride (10 μM) or GF 109203X (10 μM), PKC inhibitors that operate via different mechanisms. Chelerythrine chloride (IC50 660 nM) competes with the protein substrate (Herbert et al. 1990), whereas GF 109230X (IC50 5-70 nM) competes selectively with ATP for binding to PKC (Toullec et al. 1991). Direct activation of PKC by TPA (n = 8 coverslips from 3 rabbits) failed to induce any change in CBF or [Ca2+]i. Furthermore, inhibition of PKC by chelerythrine chloride (n = 3 coverslips from 2 rabbits) or by GF 109203X (n = 5 coverslips from 2 rabbits) had no obvious effect on the ATP-induced changes in CBF and [Ca2+]i. The efficiency of TPA, chelerythrine chloride and GF 109203X was verified using frog palate or oesophagus. In the case of the frog cells, TPA induced Ca2+ influx and enhanced CBF 2.5-fold, while the two inhibitors, at the same concentrations as used earlier, cancelled both the elevated plateau of [Ca2+]i and the sustained enhancement of CBF induced by extracellular ATP (Levin et al. 1997).
The absence of any ciliary response to TPA and the inability of the two PKC inhibitors to alter the effects of extracellular ATP suggest that PKC is not involved in the signal transduction pathway induced by ATP in rabbit airway epithelium.
PKG and/or guanylate cyclase (GC) are involved in ciliary activation by ATP
The possible involvement of PKG in ATP-induced CBF enhancement was tested by pre-treating the cells with KT-5823 (1 μM), a well-known inhibitor of PKG (IC50 0.234 μM). Moreover, a potent inhibitor of GC, LY-83583 (IC50 2 μM), was tested at a concentration of 100 μM. Figure 2 shows the effects of extracellular ATP on [Ca2+]i and on CBF in a cell pretreated with KT-5823 (Fig. 2A and B), and on a cell pretreated with LY-83583 (Fig. 2A and D). Both inhibitors drastically reduced the CBF enhancement effect and lowered the elevated plateau of [Ca2+]i induced by extracellular ATP, while having no apparent effect on the initial rapid rise in [Ca2+]i. Under the control conditions, the peak rise in [Ca2+]i induced by extracellular ATP (100 μM) ranged from 190 to 650 nM with a mean of 373 ± 7 nM (n = 40 coverslips from 33 rabbits). The peak increase in [Ca2+]i in response to 100 μM ATP in cells pretreated with 1 μM KT-5823 (294 ± 39 nM; n = 5 coverslips from 3 rabbits) and for cells pretreated with 100 μM LY-83583 (388 ± 41 nM; n = 12 coverslips from 5 rabbits) was not significantly different (P = 0.25) from the control response (Fig. 3A). In contrast, the elevated plateau of [Ca2+]i was lowered from 111 ± 3 nM (n = 35 coverslips from 31 rabbits) in control cells to 38 ± 13 nM (n = 5 coverslips from 3 rabbits) in cells pretreated with KT-5823 and to 53 ± 11 nM (n = 11 coverslips from 5 rabbits) in cells pretreated with LY-83583 (Fig. 3A). Similarly, ATP-induced CBF enhancement was reduced from 2.2 ± 0.1 (n = 40 coverslips from 33 rabbits) in control cells to 1.2 ± 0.1 (n = 5 coverslips from 3 rabbits) with KT-5823, and to 1.4 ± 0.1 (n = 12 coverslips from 5 rabbits) with LY-83583 (Fig. 3A). Taken together, these results strongly suggest that PKG is indeed involved in sustaining the elevated plateau of [Ca2+]i and in ciliary activation induced by extracellular ATP.
Figure 2. Role of PKG and guanylate cyclase in the purinergically induced elevation of [Ca2+]i and CBF.
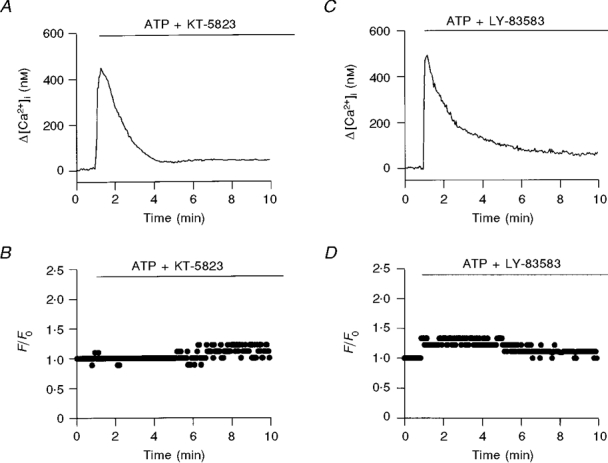
The time course of the effect of extracellular ATP (100 μM) on [Ca2+]i and CBF in the presence of KT-5823 (A and B, respectively) and LY-83583 (C and D, respectively) is shown. Both KT-5823 (1 μM) and LY-83583 (100 μM) failed to reduce the initial rise in [Ca2+]i, but strongly reduced the elevated plateau of [Ca2+]i and CBF enhancement caused by ATP. In each experiment, cells were pre-incubated with inhibitor for 10 min.
Figure 3. Effect of blockers of the NO pathway on the CBF and the [Ca2+]i induced by extracellular ATP.
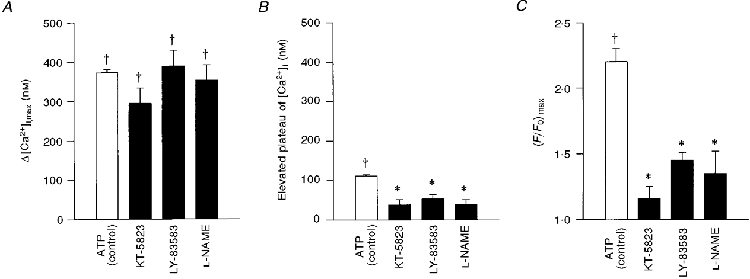
Average maximal values of Δ[Ca2+]i (A), elevated [Ca2+]i plateau (B) and normalized CBF enhancement (C), induced by extracellular ATP (100 μM) in the presence of KT-5823 (1 μM), LY-83583 (100 μM) and L-NAME (50 μM), inhibitors of PKG, GC and NOS, respectively. Each column represents the mean ±s.e.m. averaged over 5-14 experiments; † significantly different from basal values (Student's t test, P < 0.05); * significantly different from the effect at normal conditions (Student's t test, P < 0.05).
PKG is activated via the NO pathway
It is well known that activation of GC produces cGMP, which in turn activates PKG. Recently, it was reported that NO activates GC in airway epithelium (Geary et al. 1993). In order to explore whether the NO pathway is involved in the activation of PKG by extracellular ATP, the cells were pre-treated with L-NAME (50 μM), a potent inhibitor of NOS (IC50 50 nM). Indeed, inhibiting NOS mimicked the effects of the PKG and GC inhibitors. In experiments of this type, the elevated plateau of [Ca2+]i in cells pretreated with L-NAME was only 39 ± 12 nM (n = 14 coverslips from 6 rabbits) (Fig. 3A), while the increase in CBF was only 1.3 ± 0.1-fold (n = 13 coverslips from 6 rabbits) (Fig. 3A).
The three experimental manipulations that blocked PKG (KT-5823), GC (LY-83583) and NOS (L-NAME) drastically reduced the extracellular ATP-induced enhancement of CBF and the elevated [Ca2+]i plateau, but did not abolish them completely (Fig. 3A and C). It is presently not known whether the residual response to ATP was an outcome of incomplete inhibition of enzyme activity, or whether an additional pathway was activated by extracellular ATP. These possibilities were not investigated further. In any case, the results suggest that activation of PKG via the NO pathway underlies most, if not all, of the extracellular ATP-induced ciliary activation.
NOS is activated by a rise in [Ca2+]i
In rabbit airway ciliated cells, the enhancement in CBF induced by extracellular ATP was completely inhibited when the intracellular Ca2+ stores were first discharged with ionomycin (Korngreen & Priel, 1996). This finding suggests that a rise in [Ca2+]i is a prerequisite for CBF stimulation by ATP. Consistent with this conclusion, preventing change in [Ca2+]i by loading the cells with the Ca2+ chelator BAPTA AM abolished CBF enhancement induced by extracellular ATP (data not shown).
It has been shown that NOS can be directly activated by a rise in [Ca2+]i (Motte et al. 1995), raising the possibility that the initial increase in [Ca2+]i is needed to activate the NO pathway. To test this possibility, the ATP-induced signal transduction pathway was bypassed by elevating [Ca2+]i with ionomycin in Ringer solution. Figure 4A and B shows that ionomycin (1 μM) increased CBF and [Ca2+]i to a similar degree as ATP. Similar effects of ionomycin were observed in 4 of 4 additional experiments from 3 different animals (Table 1, row 1). When NOS was inhibited with L-NAME (50 μM), the increase in CBF induced by ionomycin was attenuated (Fig. 4A and D). Similar results were obtained in 6 additional experiments (from 4 rabbits) of this type (Table 1, row 2). These results suggest that the NO pathway is activated by a rise in [Ca2+]i.
Figure 4. Inhibiting the NO pathway attenuates ciliary activation induced by ionomycin.
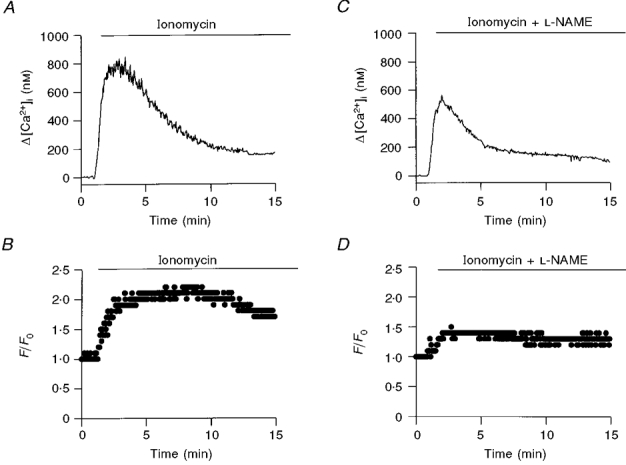
The time course of the effect of ionomycin on Δ[Ca2+]i and CBF in the absence (A and B) and presence of NOS inhibitor (C and D) is shown. Pretreatment of ciliary cells with L-NAME (50 μM) drastically reduced the enhancement in CBF and the sustained enhancement in [Ca2+]i.
Table 1.
ATP, UTP or ionomycin induced elevation in [Ca2+]i and CBF in the absence and presence of dB-cGMP and/or specific inhibitors
| Substance | Δ[Ca2+]i,max (nM) | (F/Fo)max | Elevated plateau (nM) | n: experiments (n: animals) | |
|---|---|---|---|---|---|
| 1 | Ionomycin, 1 μM | 782 ± 166 | 1.9 ± 0.04 | 176 ± 30 | 5 (4) |
| 2 | Ionomycin, 1 μM + L-NAME, 50 μM | 625 ± 57 | 1.4 ± 0.1* | 67 ± 17* | 7 (4) |
| 3 | db-cGMP, 100 μM | 25 ± 13 | 1.1 ± 0.1 | 25 ± 13 | 4 (2) |
| 4 | db-cGMP, 100 μM + L- NAME, 50 μM | 7 ± 3 | 1.0 ± 0.03 | — | 3 (2) |
| 5 | Ionomycin, 1 μM + db-cGMP, 100 μM + L-NAME, 50 μM | 727 ± 245 | 1.9 ± 0.1 | 110 ± 46 | 3 (2) |
| 6 | db-cGMP, 100 μM + ATP, 100 μM + LY-83583, 100 μM | 557 ± 155 | 2.1 ± 0.1 | 97 ± 35 | 3 (2) |
| 7 | ATP, 100 μM | 373 ± 7 | 2.2 ± 0.1 | 111 ± 3 | 40 (33) |
| 8 | UTP, 100 μM | 360 ± 37 | 2.2 ± 0.1 | 108 ± 18 | 15 (14) |
| 9 | UTP, 10 μM | 278 ± 36 | 2.0 ± 0.1 | 72 ± 11 | 12 (8) |
| 10 | UTP, 10 μM + U-73122, 10 μM | 185 ± 37* | 1.4 ± 0.1* | 26 ± 9* | 8 (2) |
| 11 | UTP, 100 μM + L-NAME, 50 μM | 345 ± 43 | 1.4 ± 0.2* | 65 ± 21* | 4 (3) |
| 12 | UTP, 100 μM + LY-83583, 100 μM | 388 ± 41 | 1.4 ± 0.1* | 33 ± 21* | 3 (2) |
Average values of maximum [Ca2+]i (Δ[Ca2+]i,max), the maximum enhancement in CBF (F/F0)max, and the elevated plateau of [Ca2+]i are presented. The data are given as means ± S.E.M.
Significantly different from control values (Student's t test, P < 0.05).
The number of repetitions (n) is the number of culture coverslips used for each experiment (the numbers of animals from which cultures were prepared are given in parentheses).
Elevated [Ca2+]i is required for PKG-induced activation of CBF
In order to assess whether direct activation of PKG can stimulate CBF, the cells were exposed to dibutyryl-cGMP (dB-cGMP, 100 μM), the cell-permeable analogue of cGMP. dB-cGMP had no effect on [Ca2+]i or on CBF (Table 1, row 3), suggesting that PKG by itself is incapable of activating the Ca2+ influx pathway that supports the elevated [Ca2+]i plateau or of stimulating the cilia, and that an additional factor or factors are needed.
To test whether CBF activation by PKG requires Ca2+ as a co-factor, the following experimental procedure was adapted. The NO pathway was first inhibited with L-NAME, to de-couple the rise in [Ca2+]i from ciliary activation (as demonstrated in Fig. 4). PKG was then directly activated by exposing the cells to dB-cGMP (100 μM). Again, as in cells exposed earlier to dB-cGMP (Table 1, row 3), dB-cGMP had no effect on [Ca2+]i or CBF (Table 1, row 4). However, when [Ca2+]i was directly elevated by exposing an L-NAME-treated cell to ionomycin (1 μM), CBF increased 2-fold, mimicking the effects of ionomycin on untreated cells (cf. rows 1 and 5 in Table 1). A similar Ca2+-dependent activation of CBF by cGMP was observed when [Ca2+]i was elevated by extracellular ATP in cells pretreated with LY-83583 to inhibit GC (Table 1, row 6). Taken together, these results show that in order to induce a robust (≥ 2-fold) and sustained enhancement in CBF, both elevated [Ca2+]i and activated PKG are required for extracellular ATP.
Adenylyl cyclase and/or protein kinase A are not involved in ciliary activation by ATP
It has been reported that several isoenzymes of adenylyl cyclase (AC) can be directly activated by Ca2+ (Antoni, 1997), thereby stimulating production of cAMP, a well-known activator of CBF (Bonini et al. 1988; Tamaoki et al. 1989). In order to determine whether extracellular ATP enhanced CBF through a cAMP-dependent pathway, three experimental approaches were used: in the first, the cells were exposed to forskolin (25 μM), a direct activator of AC; in the second, the cells were exposed to dibutyryl-cAMP (dB-cAMP, 100-400 μM); and in the third approach, the cells were pre-treated with Rp-cAMPS (100 μM), a potent PKA inhibitor (Ki = 11 μM), and the effects of ATP were determined.
Forskolin induced a small increase (F/Fo = 1.3 ± 0.1) in CBF without changing basal [Ca2+]i (n = 10 coverslips from 8 rabbits). Similarly, dB-cAMP enhanced CBF by 20 % and did not influence [Ca2+]i (n = 6 coverslips from 5 rabbits). These results confirm previous findings that direct activation of PKA by dB-cAMP in mammalian ciliary cells produces a small (∼20-30 %) enhancement in CBF (Tamaoki et al. 1989). However, inhibiting PKA failed to attenuate the biphasic rise of [Ca2+]i or CBF enhancement induced by extracellular ATP (n = 3 coverslips from 2 rabbits; data not shown).
It is possible that PKA requires Ca2+ as a co-factor, as shown above for PKG. In order to test this possibility, the NO pathway was blocked with L-NAME (50 μM), while [Ca2+]i was elevated with extracellular ATP (100 μM) in the presence of forskolin (25 μM) or dB-cAMP (200 μM). In these experiments, no additional enhancement in CBF (above the 30 % enhancement observed in the absence of elevated [Ca2+]i) was observed either for forskolin (n = 3 coverslips from 2 rabbits) or for dB-cAMP (n = 5 coverslips from 2 rabbits).
These results suggest that PKA is not involved in purinergically induced CBF enhancement. Moreover, the inability of either forskolin or dB-cAMP to produce a robust enhancement in CBF in the presence and absence of elevated [Ca2+]i strongly indicates that cAMP, adenylyl cyclase and PKA do not play an important role in CBF stimulation in rabbit airway epithelium.
UTP and ATP activate CBF via the same pathways
Cell surface receptors for purines and pyridines are classified into two major types (Abbracchio & Burnstock, 1994): G protein-coupled receptors (P2Y receptors) and ligand-gated ion channels (P2X receptors). One of the cloned P2Y receptors (P2Y2), which is equally sensitive to UTP and ATP, is expressed in lung (Erb et al. 1993) and may therefore underlie some of the effects presented above. In addition, a UTP-sensitive purinoceptor was found in ciliary cells from human nasal polyps (Parr et al. 1994). Accordingly, the effects of UTP on CBF and [Ca2+]i were examined. Figure 5 shows that the effects of ATP (100 μM) (A and B) and UTP (100 μM) (C and D) on the time course and magnitude of [Ca2+]i and CBF were almost identical. Moreover, within the accuracy of our measurement (P < 0.05), the average maximal rise in [Ca2+]i (Fig. 5A) and average maximal rise in CBF (Fig. 5A) induced by UTP and ATP were similar over a range of concentrations. In addition, suramin (100 μM), a competitive antagonist to most of the P2X and P2Y purinoceptors, completely abolished the increase in CBF induced by ATP (10 μM) or UTP (10 μM) (n = 6 coverslips from 2 rabbits for ATP and n = 3 coverslips from 2 rabbits for UTP).
Figure 5. ATP and UTP have similar effects on [Ca2+]i and CBF.
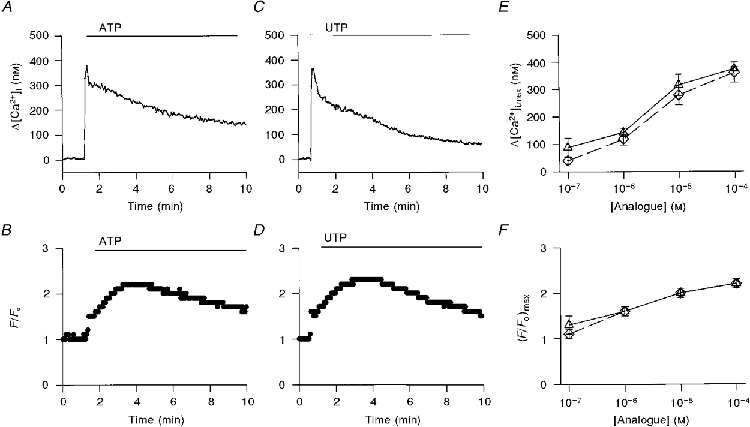
The time course of the effect of extracellular ATP (A and B) or UTP (C and D) on [Ca2+]i and CBF, respectively, is shown. [Ca2+]i and CBF were measured simultaneously from the same ciliary area in each experiment. The similarity between the initial rise of [Ca2+]i (A and C) or CBF enhancement (B and D) and their time course induced by ATP (100 μM) or UTP (100 μM) is evident. In addition, the dose-response relationships of the average maximal rise in [Ca2+]i (E) and the average maximal rise in CBF (F) to a given concentration of ATP (▵) or UTP (⋄) are presented. Each concentration of ATP or UTP represents the mean ±s.e.m. averaged over 3-40 experiments from 2-33 animals. The dose responses in the rise of [Ca2+]i (E) or CBF (F) enhancement induced by extracellular ATP or UTP were found to be similar within P < 0.05 (Student's t test).
In order to assess whether the molecular events that underlie the effects of UTP are similar to those detected for ATP, most of the experimental manipulations described above were repeated for UTP. These included inhibition of PLC (Table 1, row 10), inhibition of NOS (Table 1, row 11) and inhibition of GC (Table 1, row 12). Results similar to those observed with extracellular ATP were obtained, indicating that UTP stimulates CBF via the same pathway as extracellular ATP, and suggesting that the same receptor underlies the effects of ATP and UTP.
DISCUSSION
Recently, it was shown that extracellular ATP induced a rapid and strong (4- to 5-fold) increase in [Ca2+]i in the airway epithelia of rabbit. This effect was transient, decaying after 3-4 min, while CBF enhancement induced by extracellular ATP was rapid and prolonged, continuing for 20-30 min. Careful examination of these phenomena using a technique that allows simultaneous measurement of [Ca2+]i and CBF from the same cell (Korngreen & Priel, 1994) revealed that [Ca2+]i decayed to a sustained, higher than basal, plateau. Manipulations that lowered this sustained plateau of [Ca2+]i caused the CBF to decay rapidly to its basal value (Korngreen & Priel, 1996). The main goal of the present study was to elucidate the underlying molecular events that lead to [Ca2+]i and CBF enhancement, and the interplay between them.
Pharmacological tools were used to achieve this goal. The main strength of this approach is that it enables the examination of the possible interdependence between physiological response and intracellular molecular events in intact epithelium by non-destructive methods. On the other hand, introduction of inhibitors into the cell could produce non-specific effects. In addition, it should be noted that the actual cytosolic concentrations of the applied inhibitors are unknown and could be lower, to various extents depending on the material, than their concentration in the extracellular medium. In order to minimize possible artifacts, we used (whenever possible) two well-known, structurally different inhibitors at their widely accepted extracellular concentrations (as close as possible to the IC50 for pure enzyme). Moreover, different enzymes along the signal transduction pathway were inhibited using different blockers. In addition, persistent powerful activators were used to test the possible role of suggested pathways in the physiological responses. The consistency of the results obtained strengthens our premise regarding the action of each individual inhibitor, while collectively the results support the proposed mechanism for ciliary activation by extracellular ATP.
The molecular events induced by extracellular ATP
It was shown that strong and sustained enhancement of CBF and elevation of [Ca2+]i were mainly mediated by a suramin-sensitive P2Y receptor equally sensitive to UTP and ATP (Fig. 5). It is well established that P2Y receptors activate PLC, which produces IP3 and DAG. The former interacts with the Ca2+ stores to initiate the release of Ca2+, and the latter activates PKC. Indeed, inhibition of PLC by U-73122, a well-known PLC inhibitor, drastically reduced the [Ca2+]i rise, abolished the elevated plateau, and attenuated the enhancement in CBF induced by ATP (Fig. 1). Interestingly, even at a high concentration (15 μM), the PLC blocker did not completely abolish the effect of extracellular ATP. In order to examine whether the residual rise in [Ca2+]i and concomitant small enhancement of CBF resulted from mobilization of Ca2+ from intracellular stores, the extracellular Ca2+ concentration was reduced to 0.5 μM. Preventing Ca2+ influx in the presence of a PLC blocker did not abolish the observed effect (data not shown), strongly indicating that the residual Ca2+ was mobilized from intracellular stores. At this stage it is not possible to determine whether the inhibitor failed to completely block PLC activity, or whether an additional pathway was activated by ATP. Nevertheless, based on these findings it is possible to conclude that the initial [Ca2+]i surge induced by extracellular ATP is an outcome of Ca2+ mobilization from intracellular stores via the PLC pathway. In addition, the robust and sustained enhancement in CBF is also mainly mediated by the PLC pathway.
Stimulation of PLC inevitably results in the production of DAG, the natural activator of PKC. Inhibition of PKC by chelerythrine chloride or by GF 109203X failed to attenuate the rise in [Ca2+]i and CBF induced by extracellular ATP. These results suggest that PKC is not involved in ciliary activation induced by extracellular ATP in rabbit airway.
It is well established that a rise in [Ca2+]i can activate several types of AC (Antoni, 1997) and/or isoenzymes of NOS (Motte et al. 1995), leading to the stimulation of PKA and PKG, respectively. Inhibition of PKA by Rp-cAMPS failed to attenuate the rise in [Ca2+]i or the CBF enhancement caused by extracellular ATP, indicating that PKA is not involved in this process. On the other hand, inhibition of PKG by KT-5823 drastically reduced the ATP-induced enhancement in CBF (from 2.2- to 1.2-fold) and lowered the elevated plateau of [Ca2+]i (by 67 %), but did not attenuate the initial rise in [Ca2+]i to its maximum value. Moreover, inhibition of GC by LY-83583, or inhibition of NOS by L-NAME, produced effects similar to those induced by inhibition of PKG (Fig. 3). These results strongly suggest that PKG, stimulated via the NO pathway, plays a key role in CBF enhancement induced by extracellular ATP, and that the Ca2+ influx which supports the elevated plateau of [Ca2+]i induced by extracellular ATP requires activation of the NO pathway.
The role of PKC, PKG or PKA in CBF enhancement
Membrane-permeable activators of PKC, PKG or PKA were used in order to determine whether these kinases stimulated CBF. Direct activation of PKC by TPA had no apparent effect on CBF or on [Ca2+]i. In cultured cells from rabbit trachea, Kobayashi et al. (1988) observed that TPA induced a small decrease in CBF. The discrepancy between our findings and those of Kobayashi et al. may be due to the relatively high concentration of TPA used by the latter.
Direct activation of PKA by dB-cAMP induced a relatively small enhancement of CBF (∼20 %), in agreement with previously reported results (Tamaoki et al. 1989). A similar response was obtained after the addition of forskolin (Fmax/Fo = 1.3). Forskolin is known to induce powerful and persistent activation of adenylyl cyclase, leading to accumulation of a large amount of cAMP that may far exceed levels achieved by addition of dB-cAMP or by an agonist. However, activation of PKA by either dB-cAMP or forskolin did not induce measurable changes in [Ca2+]i.
Surprisingly, direct activation of PKG with dB-cGMP failed to yield any measurable influence on CBF or a rise in [Ca2+]i. Tamaoki et al. (1991) reported that cGMP inhibited ciliary motility in rabbit airway epithelium, while Geary et al. (1995) found that the application of 8-Br-cGMP to nasal airway epithelium enhanced CBF. At present we can offer no explanation for the discrepancy between these earlier observations and the findings reported in this study.
In conclusion, while activated PKA alone induced a small enhancement in CBF, activated PKC or PKG did not.
The dual role of elevated [Ca2+]i and activated PKG in CBF enhancement
Preventing a rise in [Ca2+]i with BAPTA completely abolished ciliary activation by extracellular ATP (data not shown). Likewise, discharging the intracellular Ca2+ stores with ionomycin in the presence of low levels of extracellular Ca2+ cancelled the enhancement in CBF induced by extracellular ATP (Korngreen & Priel, 1996). From these results it may be inferred that a rise in [Ca2+]i is essential for CBF enhancement by extracellular ATP. However, that is a necessary but not a sufficient condition, and activation of PKG is also required (Figs 2 and 3). Similarly, activated PKG by itself failed to enhance CBF (Table 1, row 3), while in the presence of elevated [Ca2+]i a strong and sustained CBF enhancement was observed (Table 1, rows 5 and 6), cf. the small enhancement in CBF induced by activated PKA independently of [Ca2+]i level.
A second role of elevated [Ca2+]i is to stimulate NOS, which activates PKG via the NO pathway. Activated PKG plays a crucial role by inducing the elevated [Ca2+]i levels required for sustained activation of NOS and CBF enhancement (positive feedback). The failure of dB-cGMP to induce Ca2+ influx indicates that an additional factor other than activated PKG is at work. Elucidating the mechanism of the ATP-induced influx of Ca2+ lies beyond the scope of this study. However, the ability of extracellular ATP to completely restore elevated [Ca2+]i in the presence of dB-cGMP and inhibited GC (Table 1, row 6) suggests that the additional factor involved could be associated with the PLC pathway.
It appears therefore that the main role of the NO pathway is to stimulate PKG, which in the presence of elevated [Ca2+]i leads to strong and prolonged enhancement of CBF. In support of this conclusion, when the NO pathway was bypassed by inhibiting GC and [Ca2+]i was subsequently elevated by adding extracellular ATP in the presence of dB-cGMP, the consequent increase in CBF and the height of the elevated [Ca2+]i plateau were similar to those induced by extracellular ATP under control conditions (Table 1, row 6).
Suggested sequence of molecular events leading to CBF enhancement
Activation of a P2Y receptor by extracellular ATP or UTP stimulates PLC, producing a massive release of Ca2+ from internal stores by IP3. Elevated [Ca2+]i activates NOS, leading to the production of NO, which activates GC and thus raises the concentration of cGMP. PKG, activated by the cGMP, in the presence of elevated [Ca2+]i, induces a rapid and robust enhancement in CBF. In parallel, the activated PKG, together with additional as yet unidentified factor(s), induce a Ca2+ influx that supports the elevated sustained enhancement in [Ca2+]i and the prolonged robust enhancement in CBF in the continuous presence of ATP (Fig. 6). According to this model [Ca2+]i and PKG are coupled via the NO pathway in an intricate relationship which culminates synergistically in robust ciliary activation.
Figure 6. Flow chart of the molecular events induced by extracellular ATP.
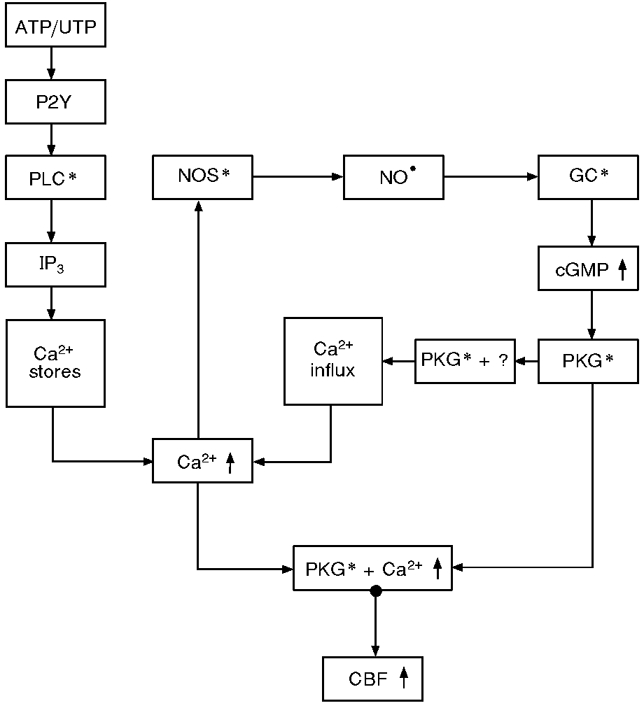
A P2Y receptor equally sensitive to ATP and UTP activates the PLC pathway, leading to discharge of intracellular Ca2+ stores, which results in strong rapid [Ca2+]i elevation that activates the NO pathway. Activated PKG in the presence of elevated [Ca2+]i induces strong CBF enhancement. Both effects produced by Ca2+ store discharge are transient and cease within 3-4 min of onset. Concurrently, activated PKG co-operatively with additional, as yet unidentified, factor (s) induce Ca2+ influx from the extracellular space, leading to prolonged elevation of [Ca2+]i (elevated plateau) and to positive feedback, which in turn results in robust sustained CBF enhancement. * indicates activated condition of enzyme.
Acknowledgments
This work was supported by a grant from the Israeli Science Foundation.
References
- Abbracchio MP, Burnstock G. Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacology and Therapeutics. 1994;64:445–475. doi: 10.1016/0163-7258(94)00048-4. 10.1016/0163-7258(94)00048-4. [DOI] [PubMed] [Google Scholar]
- Antoni FA. Calcium regulation of adenylyl cyclase. Trends in Endocrinology and Metabolism. 1997;8:7–14. doi: 10.1016/s1043-2760(96)00206-8. 10.1016/S1043-2760(96)00206-8. [DOI] [PubMed] [Google Scholar]
- Bleasdale JE, Bundy GL, Bunting S, Fitzpatrick FA, Huff RM, Sun FF, Pike JE. Inhibition of phospholipase C dependent processes by U-73122. Advances in Prostaglandin, Thromboxane and Leukotriene Research. 1989;19:590–593. [PubMed] [Google Scholar]
- Bonini NM, Gustin MC, Nelson DL. Regulation of ciliary motility by membrane potential in Paramecium: a role for cyclic AMP. Cell Motility and the Cytoskeleton. 1988;6:256–272. doi: 10.1002/cm.970060303. [DOI] [PubMed] [Google Scholar]
- Braiman A, Zagoory O, Priel Z. Protein kinase A induces calcium release and enhances ciliary beat frequency in a calcium dependent and independent manner. American Journal of Physiology. 1998;275:C790–797. doi: 10.1152/ajpcell.1998.275.3.C790. [DOI] [PubMed] [Google Scholar]
- Di Benedetto G, Magnus CJ, Gray PT, Mehta A. Calcium regulation of ciliary beat frequency in human respiratory epithelium in vitro. The Journal of Physiology. 1991;439:103–113. doi: 10.1113/jphysiol.1991.sp018659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb L, Lustig K, Sullivan D, Turner J, Weisman GA. Functional expression and photoaffinity labeling of a cloned P2U purinergic receptor. Proceedings of the National Academy of Sciences of the USA. 1993;90:10449–10453. doi: 10.1073/pnas.90.22.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary CA, Davis CW, Paradiso AM, Boucher RC. Role of CNP in human airways: cGMP-mediated stimulation of ciliary beat frequency. American Journal of Physiology. 1995;268:L1021–1028. doi: 10.1152/ajplung.1995.268.6.L1021. [DOI] [PubMed] [Google Scholar]
- Geary CA, Goy MF, Boucher RC. Synthesis and vectorial export of cGMP in airway epithelium: expression of soluble and CNP-specific guanylate cyclases. American Journal of Physiology. 1993;265:L598–605. doi: 10.1152/ajplung.1993.265.6.L598. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guo FH, Uetani K, Haque SJ, Williams BRG, Dweik RA, Thunnissen FBJM, Calhoun W, Erzurum SC. Interferon γ and interleukin 4 stimulate prolonged expression of inducible nitric oxide synthase in human airway epithelium through synthesis of soluble mediators. Journal of Clinical Investigation. 1997;100:829–838. doi: 10.1172/JCI119598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki T, Barkalow J, Richmond J, Satir P. A cAMP-stimulated phosphorylation of an axonemal polypeptide that copurifies with the 22S dynein arm regulates microtubule translocation velocity and swimming speed in Paramecium. Proceedings of the National Academy of Sciences of the USA. 1991;88:7918–7922. doi: 10.1073/pnas.88.18.7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochemical and Biophysical Research Communications. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- Jain B, Rubinstein I, Robbins R, Leise KL, Sisson JH. Modulation of airway epithelial cell ciliary beat frequency by nitric oxide. Biochemical and Biophysical Research Communications. 1993;191:83–88. doi: 10.1006/bbrc.1993.1187. 10.1006/bbrc.1993.1187. [DOI] [PubMed] [Google Scholar]
- Jain B, Rubinstein I, Robbins R, Sisson JH. TNF-α and IL-1β upregulate nitric oxide-dependent ciliary motility in bovine airway epithelium. American Journal of Physiology. 1995;268:L911–917. doi: 10.1152/ajplung.1995.268.6.L911. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Tamaoki J, Sakai N, Chiyotani A, Takizawa T. Inhibition of ciliary activity by phorbol esters in rabbit tracheal epithelial cells. Lung. 1988;167:277–284. doi: 10.1007/BF02714957. [DOI] [PubMed] [Google Scholar]
- Korngreen A, Priel Z. Simultaneous measurement of ciliary beating and intracellular calcium. Biophysical Journal. 1994;67:377–380. doi: 10.1016/S0006-3495(94)80492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korngreen A, Priel Z. Purinergic stimulation of rabbit ciliated airway epithelia: control by multiple calcium sources. The Journal of Physiology. 1996;497:53–66. doi: 10.1113/jphysiol.1996.sp021749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansley AB, Sanderson MJ, Dirksen ER. Control of the beat cycle of respiratory tract cilia by Ca2+ and cAMP. American Journal of Physiology. 1992;263:L232–242. doi: 10.1152/ajplung.1992.263.2.L232. [DOI] [PubMed] [Google Scholar]
- Levin R, Braiman A, Priel Z. Protein kinase C induced calcium influx and sustained enhancement of ciliary beating by extracellular ATP. Cell Calcium. 1997;21:103–113. doi: 10.1016/s0143-4160(97)90034-8. 10.1016/S0143-4160(97)90034-8. [DOI] [PubMed] [Google Scholar]
- Mason SJ, Paradiso AM, Boucher RC. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. British Journal of Pharmacology. 1991;103:1649–1656. doi: 10.1111/j.1476-5381.1991.tb09842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motte S, Communi D, Pirotton S, Boeynaems JM. Involvement of multiple receptors in the actions of extracellular ATP: the example of vascular endothelial cells. International Journal of Biochemistry and Cell Biology. 1995;27:1–7. doi: 10.1016/1357-2725(94)00059-x. 10.1016/1357-2725(94)00059-X. [DOI] [PubMed] [Google Scholar]
- Ovadyahu D, Eshel D, Priel Z. Investigation of ciliary motility by extracellular ATP. Biorheology. 1988;25:489–501. doi: 10.3233/bir-1988-25309. [DOI] [PubMed] [Google Scholar]
- Parr C, Sullivan D, Paradiso A, Lazarowski E, Burch L, Olsen J, Erb L, Weisman G, Boucher R, Turner J. Cloning and expression of a human P2u nucleotide receptor, a target for cystic fibrosis pharmacology. Proceedings of the National Academy of Sciences of the USA. 1994;91:3275–3279. doi: 10.1073/pnas.91.8.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins RA, Sisson JH, Springall DR, Nelson KJ, Taylor JA, Mason NA, Polak JM, Townley RG. Human lung mononuclear cells induce nitric oxide synthase in murine airway epithelial cells in vitro: role of TNFα and IL-1β. American Journal of Respiratory and Critical Medicine. 1997;155:268–273. doi: 10.1164/ajrccm.155.1.9001323. [DOI] [PubMed] [Google Scholar]
- Salathe M, Bookman RJ. Coupling of [Ca2+]i and ciliary beating in cultured trachea epithelial cells. Journal of Cell Science. 1995;108:431–440. doi: 10.1242/jcs.108.2.431. [DOI] [PubMed] [Google Scholar]
- Salathe M, Pratt MM, Wanner A. Cyclic AMP-dependent phosphorylation of a 26 kD axonemal protein in ovine cilia isolated from small tissue pieces. American Journal of Respiratory Cell and Molecular Biology. 1993;9:306–314. doi: 10.1165/ajrcmb/9.3.306. [DOI] [PubMed] [Google Scholar]
- Satir P, Sleigh MA. The physiology of cilia and mucociliary interactions. Annual Review of Physiology. 1990;52:137–155. doi: 10.1146/annurev.ph.52.030190.001033. 10.1146/annurev.ph.52.030190.001033. [DOI] [PubMed] [Google Scholar]
- Tamaoki J, Chiyotani A, Kondo M, Takemura H, Konno K. Role of NO generation in β-adrenoceptor-mediated stimulation of rabbit airway ciliary motility. American Journal of Physiology. 1995a;268:C1342–1347. doi: 10.1152/ajpcell.1995.268.6.C1342. [DOI] [PubMed] [Google Scholar]
- Tamaoki J, Kobayashi K, Sakai N, Kanemura T, Horii S, Isono K, Takeuchi S, Chiyotani A, Yamawaki I, Takizawa T. Atrial natriuretic factor inhibits ciliary motility in cultured rabbit tracheal epithelium. American Journal of Physiology. 1991;260:C201–205. doi: 10.1152/ajpcell.1991.260.2.C201. [DOI] [PubMed] [Google Scholar]
- Tamaoki J, Kondo M, Takizawa T. Effect of cAMP on ciliary function in rabbit tracheal epithelial cells. Journal of Applied Physiology. 1989;66:1035–1039. doi: 10.1152/jappl.1989.66.3.1035. [DOI] [PubMed] [Google Scholar]
- Tamaoki J, Sakai A, Kondo M, Takemura H, Konno K. Role of nitric oxide in tachykinin-induced increase in potential difference of rabbit tracheal mucosa. The Journal of Physiology. 1995b;488:115–122. doi: 10.1113/jphysiol.1995.sp020950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasiuk A, Bar-Shimon M, Gheber L, Korngreen A, Grossman Y, Priel Z. Extracellular ATP induces hyperpolarization and motility stimulation of ciliary cells. Biophysical Journal. 1995;68:1163–1169. doi: 10.1016/S0006-3495(95)80292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. Journal of Biological Chemistry. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Uzlaner N, Priel Z. 6th International Symposium on Adenosine and Adenine Nucleotides. Ferrara, Italy: 1998. Mechanism of ciliary beat frequency enhancement in rabbit trachea epithelia by extracellular ATP. May 19-24. [Google Scholar]
- Verdugo P. Ca2+-dependent hormonal stimulation of ciliary activity. Nature. 1980;283:764–765. doi: 10.1038/283764a0. [DOI] [PubMed] [Google Scholar]
- Verdugo P, Johnson NT, Tam PY. β-Adrenergic stimulation of respiratory ciliary activity. Journal of Applied Physiology. 1980;48:868–871. doi: 10.1152/jappl.1980.48.5.868. [DOI] [PubMed] [Google Scholar]
- Villalon M, Hinds TR, Verdugo P. Stimulus-response coupling in mammalian ciliated cells. Demonstration of two mechanisms of control for cytosolic [Ca2+] Biophysical Journal. 1989;56:1255–1258. doi: 10.1016/S0006-3495(89)82772-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss T, Gheber L, Shoshan-Barmatz V, Priel Z. Possible mechanism of ciliary stimulation by extracellular ATP: involvement of calcium-dependent potassium channels and exogenous Ca2+ Journal of Membrane Biology. 1992;127:185–193. doi: 10.1007/BF00231506. [DOI] [PubMed] [Google Scholar]
- Wong LB, Yeates DB. Luminal purinergic regulatory mechanisms of tracheal ciliary beat frequency. American Journal of Respiratory Cell and Molecular Biology. 1992;7:447–454. doi: 10.1165/ajrcmb/7.4.447. [DOI] [PubMed] [Google Scholar]
- Yang B, Schlosser RJ, McCaffrey TV. Dual signal transduction mechanisms modulate ciliary beat frequency in upper airway epithelium. American Journal of Physiology. 1996;270:L745–751. doi: 10.1152/ajplung.1996.270.5.L745. [DOI] [PubMed] [Google Scholar]