

Abstract
The actions of selective adenosine A1 and A2 receptor agonists were examined on synaptic currents in periaqueductal grey (PAG) neurons using patch-clamp recordings in brain slices.
The A1 receptor agonist 2-chloro-N-cyclopentyladenosine (CCPA), but not the A2 agonist, 2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine (CGS21680), inhibited both electrically evoked inhibitory (eIPSCs) and excitatory (eEPSCs) postsynaptic currents. The actions of CCPA were reversed by the A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX).
In the absence or presence of forskolin, DPCPX had no effect on eIPSCs, suggesting that concentrations of tonically released adenosine are not sufficient to inhibit synaptic transmission in the PAG.
CCPA decreased the frequency of spontaneous miniature action potential-independent IPSCs (mIPSCs) but had no effect on their amplitude distributions. Inhibition persisted in nominally Ca2+-free, high Mg2+ solutions and in 4-aminopyridine.
The CCPA-induced decrease in mIPSC frequency was partially blocked by the non-selective protein kinase inhibitor staurosporine, the specific protein kinase A inhibitor 8-para-chlorophenylthioadenosine-3′,5′-cyclic monophosphorothioate (Rp-8-CPT-cAMPS), and by 8-bromoadenosine cyclic 3′,5′ monophosphate (8-Br-cAMP).
These results suggest that A1 adenosine receptor agonists inhibit both GABAergic and glutamatergic synaptic transmission in the PAG. Inhibition of GABAergic transmission is mediated by presynaptic mechanisms that partly involve protein kinase A.
The midbrain periaqueductal grey (PAG) plays a pivotal role in integrating the analgesic and cardiovascular responses of an animal to threat, stress or pain (Bandler & Shipley, 1994). The PAG contains a moderate density of A1 adenosine receptors and adenosine uptake sites (Glass et al. 1996). While the functional effects of adenosine in the PAG are unknown, centrally administered adenosine receptor agonists produce analgesia (for review see Sawynok & Sweeney, 1989) and modulate cardiovascular responses (Barraco et al. 1986). The PAG is also an important site for acute opioid actions (Yaksh et al. 1976) and the expression of some signs of opioid withdrawal (for review see Christie et al. 1997). Extracellular adenosine concentrations are also increased in several brain regions during opioid withdrawal (Bonci & Williams, 1996; Chieng & Williams, 1998).
Adenosine inhibits synaptic transmission in the central nervous system (Phillis et al. 1975; Uchimura & North, 1991). We have recently reported that μ-opioid, GABAB and ORL1 receptor activation inhibits GABAergic and glutamatergic synaptic transmission in the PAG (Vaughan & Christie, 1997; Vaughan et al. 1997a, b). However, the effects of adenosine on synaptic transmission in the PAG are unknown. In the present study, the actions of adenosine on GABAergic and glutamatergic synaptic transmission were characterized in rat PAG using the thin slice patch-clamp technique.
METHODS
Sprague-Dawley rats, 12-30 days old, were anaesthetized with halothane, killed by cervical dislocation and their brains quickly removed and immersed in ice-cold extracellular solution containing (mM): NaCl, 126; KCl, 2.5; NaH2PO4, 1.4; MgCl2, 1.2; CaCl2, 2.4; glucose, 11; and NaHCO3, 25; equilibrated with 95 % O2: 5 % CO2. A vibratome was used to prepare coronal midbrain slices (250-300 μm thickness) containing the PAG, which were placed in a holding chamber containing extracellular solution maintained at 34°C.
Brain slices were placed in a recording chamber (1.5 ml volume) mounted on the stage of an upright microscope (Olympus BH-2 with a fixed-stage modification) and viewed using a water immersion objective (Zeiss, × 40). Slices were continuously superfused (2 ml min−1) with extracellular solution (34°C). Neurons located in the caudal two-thirds of the PAG were viewed using Nomarski optics and whole-cell voltage-clamp recordings were made using patch electrodes (3-5 MΩ) containing (mM): CsCl, 140; EGTA, 10; Hepes, 5; CaCl2, 2; and MgATP, 2; adjusted to pH 7.3 with CsOH, osmolarity of 280-290 mosmol l−1. Series resistance (< 16 MΩ) was compensated by 80 % and continuously monitored during experiments. Liquid junction potentials of -4 mV were corrected. At the completion of recording the location of each neuron was mapped by referring to the atlas of Paxinos & Watson (1986).
Evoked postsynaptic currents were elicited via bipolar tungsten stimulating electrodes (tip separation, 100 μm), which were placed 200-600 μm from the recorded neuron. Paired stimuli (interpulse interval, 50 ms) were applied at a frequency of 0.03 Hz (stimuli: 5-60 V, 25-125 μs) for subsequent off-line analysis (Axograph 3.5, Axon Instruments).
Spontaneous, miniature postsynaptic currents were recorded on videotape, low-pass filtered at 1 or 2 kHz and digitized at 5 or 10 kHz (in 20-30 s epochs) for later off-line analysis (Axograph 3.5). Events were automatically analysed using a sliding variable amplitude template and were then visually examined and erroneous events were rejected before their amplitude and time of occurrence were measured. Events were ranked by amplitude and inter-event interval for preparation of cumulative probability distributions. The cumulative probability distributions were compared by the Kolmogorov-Smirnov test. All data are expressed as means ±s.e.m. and statistical comparisons were made using Student's t tests.
Stock solutions of all drugs were diluted to working concentrations in the extracellular solution immediately before use and applied by superfusion. Stock solutions of drugs were made in distilled water, except 2-chloro-N-cyclopentyladenosine (CCPA), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) and staurosporine which were made in DMSO, and phorbol 12,13-dibutyrate (PDBu) which was made in ethanol. Drugs and reagents were obtained from the following sources: tetrodotoxin (TTX) and staurosporine from Alamone Labs (Jerusalem, Israel); methionine enkephalin from Auspep (Parkville, Victoria, Australia); 8-para-chlorophenylthioadenosine-3′,5′-cyclic monophosphorothioate (Rp-8-CPT-cAMPS) from Biolog (Bremen, Germany); (-)-bicuculline methiodide, 8-Br-adenosine cyclic 3′,5′-hydrogen phosphate monosodium salt (8-Br-cAMP), CCPA, 2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine hydrochloride (CGS21680), DPCPX, forskolin, naloxone hydrochloride and PDBu from Research Biochemicals International; 4-aminopyridine (4-AP) from Sigma; and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) from Tocris (Bristol, UK).
RESULTS
Effects of adenosine agonists on evoked synaptic currents
Recordings were made from neurons located in the caudal third of the ventrolateral (n = 78 neurons) and lateral (n = 30 neurons) PAG. In the presence of a non-NMDA antagonist, CNQX (5 μM), local stimulation evoked inhibitory postsynaptic currents (eIPSCs) in PAG neurons, which had a mean amplitude of 700 ± 42 pA at -75 mV (n = 49). These eIPSCs were abolished by the GABAA receptor antagonist bicuculline (30 μM). Superfusion of the A1 adenosine receptor agonist CCPA (1 μM) reduced the amplitude of the eIPSCs in all neurons tested by 48 ± 4 % (Fig. 1A and B; n = 12). CCPA reduced the eIPSC amplitude in a concentration-dependent manner, with an IC50 of 102 nM (Fig. 1A). The CCPA-induced decrease in peak amplitude was reversed by the A1 receptor antagonist DPCPX (1 μM; Fig. 1A and B). In contrast, the adenosine A2a receptor agonist CGS21680 (1 μM) had no effect on the amplitude of eIPSCs (94 ± 6 % of control amplitude; n = 4).
Figure 1. A1 adenosine receptor agonist inhibits GABAergic IPSCs in PAG neurons.
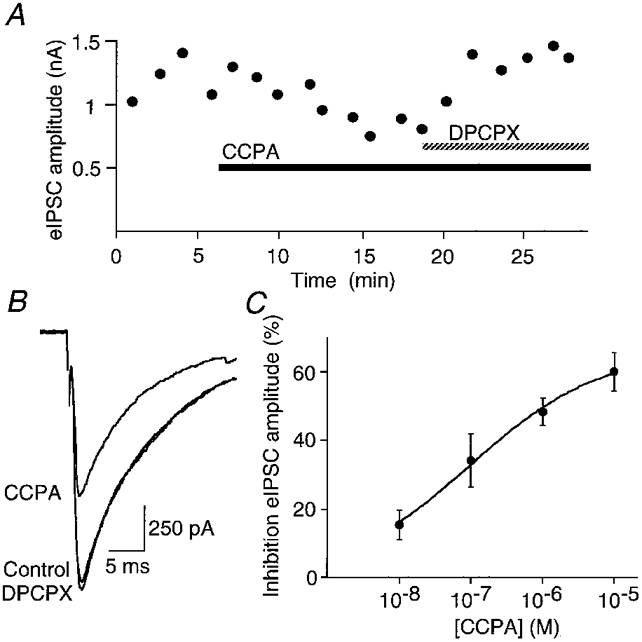
eIPSCs produced by local stimulation in the presence of CNQX (10 μM) at a holding potential of -75 mV. A, time course of the amplitude of eIPSCs during the application of CCPA (1 μM) and then CCPA plus DPCPX (1 μM). B, averaged traces (4 for each trace) of eIPSCs before and during application of CCPA (1 μM), and CCPA plus DPCPX (1 μM) from the cell shown in the time plot (A). C, concentration-response relationship for inhibition of eIPSC amplitude by CCPA. Each point shows the mean ±s.e.m. of responses of 6-12 neurons. The fitted logistic function is shown.
Extracellular concentrations of adenosine in the PAG are insufficient to modulate eIPSCs as superfusion of the A1 receptor antagonist DPCPX (1 μM) alone had no effect on the eIPSC amplitude (98 ± 2 % of control amplitude; n = 7). In the ventral tegmental area and nucleus accumbens, activation of adenylyl cyclase by forskolin, and thus enhancement of cAMP formation, leads to increased extracellular accumulation of adenosine, which inhibits GABAergic synaptic transmission (Bonci & Williams, 1996; Chieng & Williams, 1998). However, in the PAG after adenylyl cyclase activation with forskolin (10 μM), superfusion of DPCPX (1 μM) did not alter eIPSC amplitude (109 ± 16 % of control amplitude; n = 6).
In the presence of the GABAA antagonist bicuculline (30 μM), local stimulation evoked excitatory postsynaptic currents (eEPSCs) in PAG neurons with a mean amplitude of 778 ± 153 pA at -75 mV (n = 8). These eEPSCs were largely abolished by the non-NMDA receptor antagonist CNQX (5 μM). Superfusion of CCPA (1 μM) reduced the amplitude of the eEPSC by 52 ± 9 % (P < 0.02, Student's paired t test; n = 8) and this decrease was reversed by DPCPX (1 μM, Fig. 2A and B).
Figure 2. A1 adenosine receptor agonist inhibits glutamatergic EPSCs in PAG neurons.
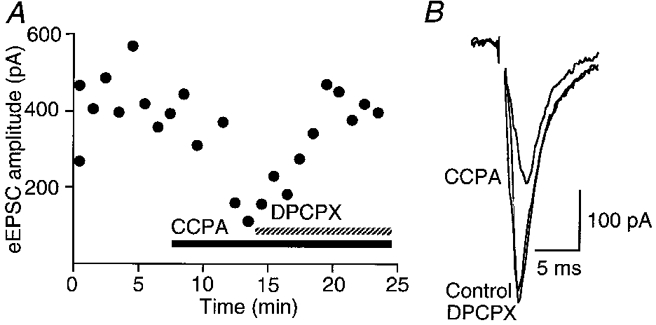
eEPSCs produced by local stimulation in the presence of bicuculline (30 μM) at a holding potential of -75 mV. A, time course of the amplitude of eEPSCs during the application of CCPA (1 μM) and then CCPA plus DPCPX (1 μM). B, averaged traces (4 for each trace) of eEPSCs before and during application of CCPA (1 μM), and CCPA plus DPCPX (1 μM) for the cell shown in the time plot (A).
Effects of an A1 receptor agonist on miniature synaptic currents
Spontaneous miniature inhibitory postsynaptic currents (mIPSCs) were readily observed in the presence of CNQX (3 μM) and TTX (0.3 μM, a concentration that blocked sodium channel-dependent action potentials and evoked postsynaptic currents). mIPSCs had a mean frequency of 2.8 ± 0.5 Hz (range, 0.19-14.56 Hz; n = 40) and a mean amplitude of 72 ± 3 pA (range, 23-123 pA) at -75 mV.
Superfusion of CCPA (1 μM) decreased the frequency of mIPSCs (Fig. 3A and B) but had no effect on the cumulative distribution of mIPSC amplitudes (Fig. 3A). CCPA reduced the mean mIPSC frequency by 41 ± 3 % (P < 0.01, Student's paired t test; n = 15), whereas the mean mIPSC amplitude was 97 ± 4 % of the control amplitude. The CCPA-induced reduction in mIPSC frequency was reversed by DPCPX (1 μM; n = 15).
Figure 3. A1 adenosine receptor agonist decreases the frequency of mIPSCs in PAG neurons.
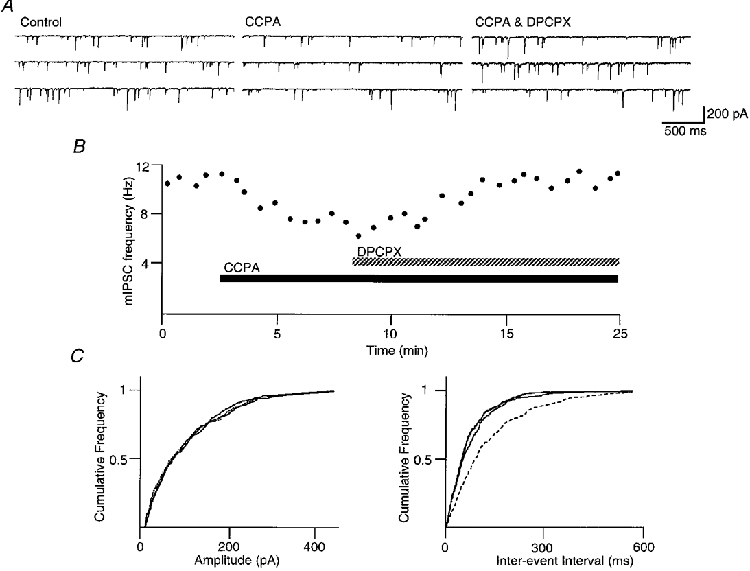
mIPSCs were recorded in the presence of TTX (0.3 μM) and CNQX (3 μM). All data shown are from the same neuron, which was clamped at -75 mV. A, 3 consecutive sweeps before and during application of CCPA (1 μM), and CCPA plus DPCPX (1 μM). B, time course of the frequency of mIPSCs before and during application of CCPA (1 μM), and CCPA plus DPCPX (1 μM). C, cumulative distribution plots of mIPSC amplitude and inter-event interval before and during application of CCPA (dashed line), and CCPA plus DPCPX. CCPA had no effect on the amplitude distribution (P > 0.3, Kolmogorov-Smirnov statistics for control - CCPA), but reversibly shifted the frequency distribution to longer inter-event intervals (P < 0.01, Kolmogorov-Smirnov statistics for control - CCPA).
Mechanisms of inhibition of IPSCs
The involvement of the adenylyl cyclase-protein kinase A cascade in the CCPA-induced inhibition of GABAergic synaptic transmission was examined because agonist stimulation of A1 adenosine receptors inhibits adenylyl cyclase (van Calker et al. 1978) and stimulation of the cAMP-protein kinase A cascade enhances synaptic transmission (Llano & Gerschenfeld, 1993; Vaughan et al. 1997B). Superfusion of the selective protein kinase A inhibitor Rp-8-CPT-cAMPS (100 μM) alone decreased mIPSC frequency (50 ± 15 % of control; P < 0.05, Student's paired t test; n = 4) but produced a small inhibition of mIPSC amplitude (68 ± 9 % of control; P = 0.052, Student's paired t test). The basal effects of the non-specific protein kinase inhibitor staurosporine (1 μM) could not be examined because it was superfused for at least 30 min prior to establishing recordings. Superfusion of the metabolically stable cAMP analogue 8-Br-cAMP (1 mM) alone did not alter mIPSC frequency (118 ± 16 % of control; P > 0.05, Student's paired t test; n = 4) or amplitude (86 ± 8 % of control; P > 0.05, Student's paired t test).
Staurosporine (1 μM) partly prevented the CCPA-induced inhibition in mIPSC frequency (P < 0.01, Student's unpaired t test; n = 5; Fig. 4). Furthermore, Rp-8-CPT-cAMPS (100 μM) also reduced the CCPA-induced inhibition of mIPSC frequency (P < 0.01, Student's unpaired t test; n = 4; Fig. 4). The CCPA-induced decrease in mIPSC frequency was also reduced in the presence of 8-Br-cAMP (1 mM, P < 0.01, Student's unpaired t test; n = 4; Fig. 4). CCPA had no effect on mIPSC amplitude in the presence of 8-Br-cAMP, staurosporine or Rp-8-CPT-cAMPS (96 ± 3 % of control; pooled n = 13).
Figure 4. Inhibition of mIPSC frequency is partially mediated by adenylyl cyclase and protein kinase A.
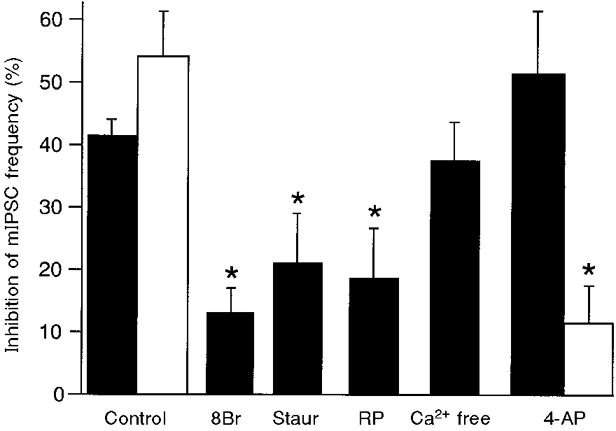
Inhibition of mIPSC frequency by CCPA (1 μM; ▪) and methionine enkephalin (10 μM; □) during superfusion of the indicated drugs is presented. 8Br, 8-Br-cAMP (1 mM); Staur, staurosporine (1 μM); RP, Rp-8-CPT-cAMPS (100 μM); Ca2+ free, 0 mM Ca2+ and 10 mM Mg2+; 4-AP, 4-aminopyridine (0.1-1 mM). Asterisks signify statistical significance for comparisons between inhibition of mIPSC frequency in control and in the specified treatment (P < 0.01, Student's unpaired t tests).
The CCPA-induced inhibition of GABAergic synaptic transmission might have been due to modulation of presynaptic K+ or Ca2+ conductances. In the PAG, μ-opioids inhibit GABAergic synaptic transmission via a presynaptic 4-AP-/dendrotoxin-sensitive K+ conductance (Vaughan et al. 1997B). Thus, the methionine enkephalin (10 μM)-induced inhibition of mIPSC frequency was largely abolished by 4-AP (0.1-1 mM; P < 0.01, Student's unpaired t test; n = 4; Fig. 4), as previously reported (Vaughan et al. 1997B). In contrast, CCPA (1-3 μM) reduced mIPSC frequency to the same extent in the presence (n = 4) or absence (n = 15) of 4-AP (P > 0.1, Student's unpaired t test; Fig. 4). The effect of Ca2+ entry blockade was then examined by superfusing with nominally Ca2+-free extracellular solution (0 mM Ca2+, 10 mM Mg2+). The CCPA-induced reduction of mIPSC frequency was similar in control and Ca2+-free solutions (P > 0.5, Student's t test; n = 3; Fig. 4). CCPA and methionine enkephalin had no effect on mIPSC amplitude in Ca2+-free or 4-AP-containing solutions (105 ± 9 % of control amplitude; pooled n = 7).
DISCUSSION
In this study it has been demonstrated that CCPA acts via A1 adenosine receptors to inhibit GABAergic and glutamatergic synaptic transmission in the PAG. The inhibition of GABAergic synaptic transmission is mediated by a direct action on the presynaptic terminal. In addition, this presynaptic inhibition is at least partly mediated by modulation of the adenylyl cyclase-protein kinase A cascade.
CCPA inhibited both GABAergic and glutamatergic (non-NMDA) synaptic transmission in the present study. Similarly, A1 adenosine receptor activation inhibited both GABAergic and glutamatergic synaptic transmission in the nucleus accumbens (Uchimura & North, 1991), substantia nigra and ventral tegmental area (Wu et al. 1995). In contrast, adenosine selectively inhibits glutamatergic synaptic transmission in other brain regions such as the hippocampus (Lambert & Teyler, 1991).
Inhibition of GABAergic synaptic transmission was mediated by A1 adenosine receptors because CCPA is a specific A1 adenosine receptor agonist (Lohse et al. 1988) and the inhibition was reversed by the specific A1 adenosine receptor antagonist DPCPX (Lohse et al. 1987). The potency of CCPA for inhibition of GABAergic synaptic transmission in the present study (IC50 = 102 nM) is comparable to that previously found for inhibition of adenylyl cyclase in the cerebral cortex (IC50 = 18 nM; Alexander et al. 1994) and rat adipocytes (IC50 = 33 nM; Lohse et al. 1988). Activation of A2a adenosine receptors did not affect GABAergic synaptic transmission in the present study, suggesting that A2a receptor activation is unlikely to contribute to the actions of adenosine on synaptic transmission in the PAG. The latter finding is in contrast to studies showing that A2a receptor activation inhibits synaptic transmission in hypothalamic culture (Chen & van den Pol, 1997) and facilitates synaptic transmission in hypoglossal neurons (Umemiya & Berger, 1994).
The CCPA-mediated inhibition of GABAergic synaptic transmission was due to a reduced probability of presynaptic transmitter release rather than a reduction in postsynaptic receptor sensitivity, as evidenced by inhibition of the frequency of mIPSCs without affecting their amplitude distributions. Inhibition of the frequency but not the amplitude of miniature synaptic potentials by adenosine has previously been observed in the rat phrenic nerve diaphragm (Ginsborg & Hirst, 1972). The site of inhibition of glutamatergic synaptic transmission was not examined in the present study. Inhibition of glutamatergic synaptic transmission in the hippocampus is also thought to be mediated by a presynaptic mechanism (Scholz & Miller, 1992).
The A1 adenosine receptor-mediated inhibition of GABAergic synaptic transmission in the PAG is at least partly mediated by modulation of the adenylyl cyclase-protein kinase A cascade. The specific protein kinase A inhibitor Rp-8-CPT-AMPS and the non-specific protein kinase inhibitor staurosporine both reduced the CCPA inhibition of mIPSC frequency. In addition, the stable cAMP analogue 8-Br-cAMP partially occluded the effect of CCPA on mIPSC frequency. In contrast, inhibition of synaptic transmission by adenosine is not decreased by cAMP analogues in the hippocampus (Dunwiddie & Fredholm, 1985), or by protein kinase A inhibitors in dorsal root ganglion cells (Hu & Li, 1997).
It has previously been demonstrated that A1 adenosine receptor activation inhibits adenylyl cyclase activity in the central nervous system (van Calker et al. 1978). Activators (Llano & Gerschenfeld, 1993; Vaughan et al. 1997B) and inhibitors (Trudeau et al. 1996) of the adenylyl cyclase- protein kinase A cascade increase and decrease the probability of transmitter release, respectively. In the present study, the protein kinase A inhibitor Rp-8-CPT-cAMPS alone decreased mIPSC frequency. Although superfusion of 8-Br-cAMP alone did not increase mIPSC frequency, it did partly occlude the presynaptic inhibitory effects of adenosine. This cAMP analogue has previously been reported to produce variable effects on mIPSC frequency in PAG although forskolin consistently increases release probability (Vaughan et al. 1997B), suggesting that 8-Br-cAMP might have actions in addition to stimulation of adenylyl cyclase. Taken together these findings suggest that protein kinase A activity modulates GABAergic synaptic transmission in the PAG, and A1 adenosine receptor inhibition is mediated partly by inhibition of adenylyl cyclase, reduced cAMP production and reduced activation of protein kinase A. The critical substrate/s of protein kinase A remain unclear. One possibility is that protein kinase A directly modulates neurotransmitter release through phosphorylation of proteins important in neurotransmitter exocytosis. Protein kinase A has been shown to phosphorylate several proteins involved in neurotransmitter exocytosis such as rabphilin-3A (Fykse et al. 1995) and αSNAP (Hirling & Scheller, 1996).
Modulation of presynaptic K+ conductances is unlikely to be responsible for the presynaptic A1 adenosine receptor-mediated inhibition of GABAergic synaptic transmission. μ-Opioid inhibition of GABA release from terminals in the PAG is mediated by activation of a presynaptic 4-AP- and dendrotoxin-sensitive K+ conductance via formation of 12-lipoxygenase metabolites of arachidonic acid (Vaughan et al. 1997B). However, the inhibitory action of methionine enkephalin, but not of CCPA, on mIPSC frequency was abolished by 4-AP. This indicates that A1 adenosine receptor agonists act through a distinct presynaptic mechanism to μ-opioids. These results are consistent with findings that metabolites of arachidonic acid do not mediate adenosine inhibition of synaptic transmission in the hippocampus or the striatum (Dunwiddie et al. 1990). However, the involvement of a barium-sensitive K+ conductance (Gerber et al. 1989) or another postsynaptic K+ conductance cannot be excluded.
CCPA reduced the frequency of mIPSCs to a similar extent in normal and Ca2+-free solutions. A similar lack of effect of both nominally Ca2+-free and Cd2+-containing solutions has previously been reported for μ-opioid- and GABAB receptor-mediated presynaptic inhibition in the PAG (Vaughan & Christie, 1997; Vaughan et al. 1997B). Inhibition of mIPSC frequency by A1 adenosine receptor activation in the hippocampus (Scholz & Miller, 1992) is also independent of presynaptic Ca2+ entry. However, the possible involvement of inhibition of voltage-dependent Ca2+ conductances in the actions of A1 adenosine receptor agonists on eIPSCs cannot be ruled out. A1 adenosine receptor activation inhibits postsynaptic voltage-dependent Ca2+ conductances (Scholz & Miller, 1991) and inhibition of Ca2+ conductances in nerve terminals results in decreased transmitter release in the hippocampus (Wu & Saggau, 1994).
Tonic extracellular concentrations of adenosine in the PAG were insufficient to modulate GABAergic synaptic transmission. Application of the A1 adenosine receptor antagonist DPCPX did not alter GABAergic synaptic transmission, as has been demonstrated in the thalamus (Ulrich & Huguenard, 1995) and hypoglossal neurons (Umemiya & Berger, 1994). In contrast, tonic adenosine inhibition of synaptic transmission has been observed in the hippocampus (Dunwiddie & Diao, 1994) and ventral tegmental area (Bonci & Williams, 1996). In the ventral tegmental area opioid withdrawal activates adenylyl cyclase and thus enhances cAMP formation. Breakdown of this extra cAMP leads to increased extracellular accumulation of adenosine, which inhibits GABAergic synaptic transmission (Bonci & Williams, 1996). Like the ventral tegmental area, the PAG plays an important role in acute opioid actions and opioid dependence. However, forskolin activation of adenylyl cyclase did not increase the extracellular adenosine concentrations sufficiently to inhibit GABAergic synaptic transmission in the present study.
The present results demonstrate that A1 adenosine receptor activation inhibits synaptic transmission by acting presynaptically, partly via inhibition of the adenylyl cyclase- cAMP-protein kinase A cascade, to decrease release of GABA. It remains unclear what the overall functional effects of adenosine receptor activation in the PAG would be, as both GABAergic and glutamatergic synaptic transmission are inhibited via A1 adenosine receptors. Furthermore, the postsynaptic actions of adenosine in the PAG have not been investigated. It has been demonstrated that μ-opioids (Yaksh et al. 1976) and GABAB receptor agonists (Levy & Proudfit, 1979), which also inhibit GABAergic and glutamatergic synaptic transmission in the PAG, produce analgesia when directly microinjected into this region. Thus, it is possible that adenosine receptor activation within the PAG might also modulate nociception and perhaps the other behavioural and cardio-respiratory functions of the PAG.
Acknowledgments
Dr Mark Connor is acknowledged for helpful suggestions. This work was supported by the National Health and Medical Research Council of Australia, and The Medical Foundation of The University of Sydney.
References
- Alexander SP, Curtis AR, Kendall DA, Hill SJ. A1 adenosine receptor inhibition of cyclic AMP formation and radioligand binding in the guinea-pig cerebral cortex. British Journal of Pharmacology. 1994;113:1501–1507. doi: 10.1111/j.1476-5381.1994.tb17166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandler R, Shipley MT. Columnar organization in the midbrain periaqueductal gray: modules for emotional expression? Trends in Neurosciences. 1994;17:379–389. doi: 10.1016/0166-2236(94)90047-7. [DOI] [PubMed] [Google Scholar]
- Barraco RA, Phillis JW, Campbell WR, Marcantonio DR, Salah RS. The effects of central injections of adenosine analogs on blood pressure and heart rate in the rat. Neuropharmacology. 1986;25:675–680. doi: 10.1016/0028-3908(86)90081-x. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;16:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- Chen G, van den Pol AN. Adenosine modulation of calcium currents and presynaptic inhibition of GABA release in suprachiasmatic and arcuate nucleus neurons. Journal of Neurophysiology. 1997;77:3035–3047. doi: 10.1152/jn.1997.77.6.3035. [DOI] [PubMed] [Google Scholar]
- Chieng B, Williams JT. Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. Journal of Neuroscience. 1998;18:7033–7039. doi: 10.1523/JNEUROSCI.18-17-07033.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, Osborne PB, Bellchambers CE. Where is the locus in opioid withdrawal? Trends in Pharmacological Sciences. 1997;18:134–140. doi: 10.1016/s0165-6147(97)01045-6. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. Journal of Pharmacology and Experimental Therapeutics. 1994;268:537–545. [PubMed] [Google Scholar]
- Dunwiddie TV, Fredholm BB. Adenosine modulation of synaptic responses in rat hippocampus: possible role of inhibition or activation of adenylate cyclase. Advances in Cyclic Nucleotide and Protein Phosphorylation Research. 1985;19:259–272. [PubMed] [Google Scholar]
- Dunwiddie TV, Taylor M, Cass WA, Fitzpatrick FA, Zahniser NR. Arachidonic acid metabolites do not mediate modulation of neurotransmitter release by adenosine in rat hippocampus or striatum. Brain Research. 1990;527:76–80. doi: 10.1016/0006-8993(90)91062-l. 10.1016/0006-8993(90)91062-L. [DOI] [PubMed] [Google Scholar]
- Fykse EM, Li C, Sudhof TC. Phosphorylation of rabphilin-3A by Ca2+/calmodulin- and cAMP-dependent protein kinases in vitro. Journal of Neuroscience. 1995;15:2385–2395. doi: 10.1523/JNEUROSCI.15-03-02385.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber U, Greene RW, Haas H, Stevens DR. Characterization of inhibition mediated by adenosine in the hippocampus of the rat in vitro. The Journal of Physiology. 1989;417:567–578. doi: 10.1113/jphysiol.1989.sp017819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsborg BL, Hirst GD. The effect of adenosine on the release of the transmitter from the phrenic nerve of the rat. The Journal of Physiology. 1972;224:629–645. doi: 10.1113/jphysiol.1972.sp009916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Faull RL, Dragunow M. Localisation of the adenosine uptake site in the human brain: a comparison with the distribution of adenosine A1 receptors. Brain Research. 1996;710:79–91. doi: 10.1016/0006-8993(95)01318-0. 10.1016/0006-8993(95)01318-0. [DOI] [PubMed] [Google Scholar]
- Hirling H, Scheller RH. Phosphorylation of synaptic vesicle proteins: modulation of the alpha SNAP interaction with the core complex. Proceedings of the National Academy of Sciences of the USA. 1996;93:11945–11949. doi: 10.1073/pnas.93.21.11945. 10.1073/pnas.93.21.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HZ, Li ZW. Modulation by adenosine of GABA-activated current in rat dorsal root ganglion neurons. The Journal of Physiology. 1997;501:67–75. doi: 10.1111/j.1469-7793.1997.067bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert NA, Teyler TJ. Adenosine depresses excitatory but not fast inhibitory synaptic transmission in area CA1 of the rat hippocampus. Neuroscience Letters. 1991;122:50–52. doi: 10.1016/0304-3940(91)90190-5. 10.1016/0304-3940(91)90190-5. [DOI] [PubMed] [Google Scholar]
- Levy RA, Proudfit HK. Analgesia produced by microinjection of baclofen and morphine at brainstem sites. European Journal of Pharmacology. 1979;57:43–55. doi: 10.1016/0014-2999(79)90102-x. 10.1016/0014-2999(79)90102-X. [DOI] [PubMed] [Google Scholar]
- Llano I, Gerschenfeld HM. Beta-adrenergic enhancement of inhibitory synaptic activity in rat cerebellar stellate and Purkinje cells. The Journal of Physiology. 1993;468:201–224. doi: 10.1113/jphysiol.1993.sp019767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Klotz KN, Lindenborn-Fotinos J, Reddington M, Schwabe U, Olsson RA. 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX) - a selective high affinity antagonist radioligand for A1 adenosine receptors. Naunyn-Schmiedeberg's Archives of Pharmacology. 1987;336:204–210. doi: 10.1007/BF00165806. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Klotz KN, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-N6-cyclopentyladenosine: a highly selective agonist at A1 adenosine receptors. Naunyn- Schmiedeberg's Archives of Pharmacology. 1988;337:687–689. doi: 10.1007/BF00175797. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 2. Sydney: Academic Press Australia; 1986. [Google Scholar]
- Phillis JW, Kostopoulos GK, Limacher JJ. A potent depressant action of adenine derivatives on cerebral cortical neurones. European Journal of Pharmacology. 1975;30:125–129. doi: 10.1016/0014-2999(75)90214-9. 10.1016/0014-2999(75)90214-9. [DOI] [PubMed] [Google Scholar]
- Sawynok J, Sweeney MI. The role of purines in nociception. Neuroscience. 1989;32:557–569. doi: 10.1016/0306-4522(89)90278-9. 10.1016/0306-4522(89)90278-9. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Analysis of adenosine actions on Ca2+ currents and synaptic transmission in cultured rat hippocampal pyramidal neurones. The Journal of Physiology. 1991;435:373–393. doi: 10.1113/jphysiol.1991.sp018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Inhibition of quantal transmitter release in the absence of calcium influx by a G protein-linked adenosine receptor at hippocampal synapses. Neuron. 1992;8:1139–1150. doi: 10.1016/0896-6273(92)90134-y. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. 10.1016/S0896-6273(00)80210-X. [DOI] [PubMed] [Google Scholar]
- Uchimura N, North RA. Baclofen and adenosine inhibit synaptic potentials mediated by gamma-aminobutyric acid and glutamate release in rat nucleus accumbens. Journal of Pharmacology and Experimental Therapeutics. 1991;258:663–668. [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR. Purinergic inhibition of GABA and glutamate release in the thalamus: implications for thalamic network activity. Neuron. 1995;15:909–918. doi: 10.1016/0896-6273(95)90181-7. 10.1016/0896-6273(95)90181-7. [DOI] [PubMed] [Google Scholar]
- Umemiya M, Berger AJ. Activation of adenosine A1 and A2 receptors differentially modulates calcium channels and glycinergic synaptic transmission in rat brainstem. Neuron. 1994;13:1439–1446. doi: 10.1016/0896-6273(94)90429-4. 10.1016/0896-6273(94)90429-4. [DOI] [PubMed] [Google Scholar]
- van Calker D, Muller M, Hamprecht B. Adenosine inhibits the accumulation of cyclic AMP in cultured brain cells. Nature. 1978;276:839–841. doi: 10.1038/276839a0. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Christie MJ. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. The Journal of Physiology. 1997;498:463–472. doi: 10.1113/jphysiol.1997.sp021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Christie MJ. Actions of the ORL1 receptor ligand nociceptin on membrane properties of rat periaqueductal gray neurons in vitro. Journal of Neuroscience. 1997a;17:996–1003. doi: 10.1523/JNEUROSCI.17-03-00996.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997b;390:611–614. doi: 10.1038/37610. 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Wu YN, Mercuri NB, Johnson SW. Presynaptic inhibition of gamma-aminobutyric acidB-mediated synaptic current by adenosine recorded in vitro in midbrain dopamine neurons. Journal of Pharmacology and Experimental Therapeutics. 1995;273:576–581. [PubMed] [Google Scholar]
- Yaksh TL, Yeung JC, Rudy TA. Systematic examination in the rat of brain stem sites sensitive to the direct application of morphine: observation of differential effects within the periaqueductal gray. Brain Research. 1976;114:83–103. doi: 10.1016/0006-8993(76)91009-x. 10.1016/0006-8993(76)91009-X. [DOI] [PubMed] [Google Scholar]