

Abstract
Over recent years, it has become clear that mitochondria play a central role in many key aspects of animal physiology and pathophysiology. Their central and ubiquitous task is clearly the production of ATP. Nevertheless, they also play subtle roles in glucose homeostasis, acting as the sensor for substrate supply in the transduction pathway that promotes insulin secretion by the pancreatic β-cell and that modulates the excitability of the hypothalamic glucose-sensitive neurons involved in appetite control. Mitochondria may also act as sensors of availability of oxygen, the other major mitochondrial substrate, in the regulation of respiration. Mitochondria take up calcium, and the high capacity mitochondrial calcium uptake pathway provides a mechanism that couples energy demand to increased ATP production through the calcium-dependent upregulation of mitochondrial enzyme activity. Mitochondrial calcium accumulation may also have a substantial impact on the spatiotemporal dynamics of cellular calcium signals, with subtle differences of detail in different cell types. Recent work has also revealed the centrality of mitochondrial dysfunction as an irreversible step in the pathway to both necrotic and apoptotic cell death. This review looks at recent developments in these rapidly evolving areas of cell physiology in an attempt to draw together disparate areas of research into a common theme.
Since the award of the 1978 Nobel prize for Chemistry to Peter Mitchell for his formulation of the chemiosmotic theory of oxidative phosphorylation (Mitchell & Moyle, 1967), most physiologists have regarded mitochondria as ‘solved’. They have been perceived as divorced from those aspects of physiology that are deemed exciting and significant, and dismissed as boring structures whose sole function is to manufacture ATP unobtrusively and on demand, subservient to the energetic demands of the cell. This essay was unwittingly prompted by a colleague who sat down next to me at coffee one morning, after giving an undergraduate lecture on basic cell biology and commented ‘You work on mitochondria, don't you? Why are they interesting? What should I tell the students?’ Over the last few years, mitochondria have re-emerged into the spotlight of scientific scrutiny after many years in the wings, as unfamiliar roles are attributed to them that even Peter Mitchell would probably never have imagined (see also Miller, 1998).
‘The biggest picture?’
Oxygen is necessary for life - but only because our mitochondria use it to generate ATP. Thought to be derived from prokaryotic micro-organisms which evolved a symbiotic relationship with their eukaryotic hosts (for review, see Margulis, 1996 and see Gray, 1998), mitochondria are our primary consumers of oxygen, and it could be argued that the mitochondrial requirement for oxygen delivery has driven the evolution of the respiratory and cardiovascular systems. Mitochondria, despite all that follows here, are primarily ATP generators. This is far from trivial: ATP is the major currency in the cellular economy, required to drive by far the majority of energy-requiring reactions in all living things, from bacteria and plants to man. It is necessary for the phosphorylation reactions that modulate so many fundamental cellular processes. It may be stored and used as a (neuro)transmitter, and it controls the activity of several classes of ion channel (such as the ATP-sensitive K+ (KATP) channel, the calcium release channel of sarcoplasmic reticulum and voltage-gated calcium channels).
Mitochondria also take up calcium, and are functionally tightly integrated into mechanisms of cellular calcium signalling, and this will provide a major focal point below. It is also often forgotten that mitochondria house a number of critically important biochemical processes quite distinct and separate from energy metabolism - key enzymes involved in haemoglobin and steroid synthesis, for example, and even the carbonic anhydrase needed to make gastric acid. Mitochondrial energy production may also be harnessed in different ways, diverted from the business of ATP synthesis into the generation of heat through the expression of catecholamine-regulated uncoupling proteins in brown fat in neonates and in hibernating mammals (Palou et al. 1998). Recent developments suggest other more subtle, if speculative, actions of these proteins in cell physiology, perhaps in regulating the production of free radical species by the respiratory chain (see Boss et al. 1998 for review).
Given that mitochondria play such a major role in the life of the cell, perhaps it is not so surprising to find that mitochondrial dysfunction plays a crucial role in disease and cell death. Acute mitochondrial dysfunction causes acute energy failure and necrotic cell death, for example during ischaemia or anoxia. More subtle, insidious forms of mitochondrial dysfunction may contribute to the pathophysiology of neurodegenerative disorders such as Parkinson's disease (for review, see Schapira et al. 1998), and motoneuron disease (or amyotrophic lateral sclerosis; see Kong & Xu, 1998), while the debilitating mitochondrial encephalomyopathies result from a range of genetic disorders of mitochondrial enzyme pathways (for reviews, see Morgan-Hughes et al. 1990; Graeber & Muller, 1998). Heritable disorders of the haem synthetic pathway cause the porphyrias, and the mitochondrially generated protoporphyrin IX accounts for the photosensitivity that is so disabling in porphyria cutanea tarda, a phenomenon exploited in the design of photodynamic therapies for cancer. Most recently, it has become clear that mitochondria play a crucial role in apoptosis (for recent reviews see Kroemer et al. 1998; Green & Reed, 1998; and see below), which plays so fundamental a role both in shaping organ systems during development and as a route to delayed cell death following subacute injury or reperfusion after ischaemia (Kerr et al. 1972). Indeed, the role of mitochondria in apoptotic cell death may well underpin the pathological changes seen in the neurodegenerative disorders listed above.
A little background bioenergetics: some basic principles and definitions
A reasonable understanding of mitochondrial physiology requires a little background knowledge of basic biophysics and an appreciation of the actions of a few pharmacological agents. These are revisited in Fig. 1A, which shows a cartoon representing the key elements of oxidative phosphorylation (see figure legend, and Nicholls & Ferguson, 1992, for a comprehensive account and see also Noji et al. 1997). The fundamental principles outlined here govern mitochondrial function and inevitably dictate the approach adopted to investigate mitochondrial function in cells.
Figure 1. Imaging mitochondrial function.
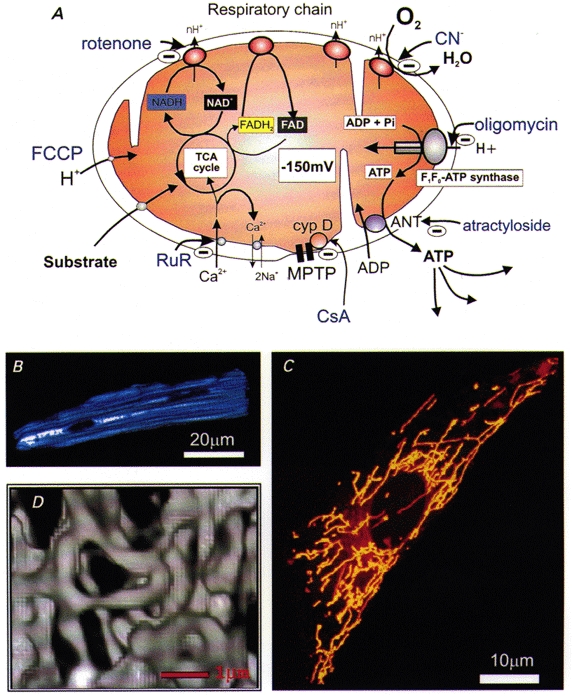
A, cartoon of a mitochondrion, illustrating the basic principles of the chemiosmotic basis for oxidative phosphorylation. The supply of substrate (only glucose is shown, but other substrates - ketones and fatty acids - are also used by some cells) to the tricarboxylic acid cycle (TCA or Krebs’ cycle) promotes the reduction of NAD+ to NADH and of FAD to FADH2. As these are re-oxidized, they supply electrons to the respiratory chain. Those electrons are transferred through the enzyme complexes of the respiratory chain to oxygen, with the production of water. In the process, protons are translocated across the inner mitochondrial membrane, generating a potential gradient of approximately -150 to -200 mV (referred to as ΔΨm). ATP synthesis takes place at a separate site, the ATP synthase, which consists of a proton channel (the F0 subunit) and an ATP synthase (the F1 subunit). The enzyme is driven by the downhill movement of protons through the F0 channel and phosphorylates ADP, producing ATP, which is transported out of the mitochondria by the adenine nucleotide translocase (ANT, which also imports ADP). The ANT forms part of the mitochondrial permeability transition pore (MPTP) but has been shown separately for clarity. The sites of action of a number of commonly used reagents are indicated in blue. B-D, images of living mitochondria within single cells. B shows a confocal image to illustrate the distribution of endogenous or ‘auto'fluorescence derived from NADH in a single rat cardiomyocyte illuminated with UV light (at 350 nm). The mitochondria within these cells run longitudinally with the axis of the cell (M. R. Duchen, unpublished observation). In C, the mitochondria of a rat cortical astrocyte have been stained with the potentiometric dye, tetramethyl rhodamine ethyl ester (TMRE), which partitions between the mitochondria and cytosol in response to ΔΨm by virtue of the positive charge on the dye. The image was obtained on a confocal system and shows a reconstruction of a series of confocal slices through the ‘z’ plane (M. R. Duchen, unpublished observations). D shows a 3-dimensional reconstruction of digital fluorescent images of mitochondria expressing targeted green fluorescent protein (GFP) in a HeLa cell, in which the mitochondria appear to form a contiguous but labile network (reproduced from Rizzuto et al. 1998, with permission).
Several points shown in this scheme may be manipulated pharmacologically (a few are indicated in blue in Fig. 1A). Agents are available that block respiration at each of the complexes of the respiratory chain, others inhibit the ATP synthase, while protonophores (such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP) and carbonyl cyanide p-trifluromethoxy-phenylhydrazone (FCCP)) short circuit the electrochemical potential for protons (primarily expressed as a mitochondrial membrane potential (ΔΨm), as the ion concentration gradient is small) and uncouple respiration from phosphorylation. As the functional consequences for each is distinct, careful use of these agents permits a systematic dissection of mitochondrial function under many conditions. The scheme also introduces several variables which are accessible for measurement and which can be used to inform us about the behaviour of mitochondria in cells, most particularly mitochondrial potential and redox state (Fig. 1A and D).
Apart from ATP synthesis, mitochondrial Ca2+ uptake represents a major function of mitochondria which is also driven by chemiosmotic principles. The pathway is provided by a uniporter, an electrogenic mechanism, quite possibly a channel opened when [Ca2+]c is high, which is blocked by Ruthenium Red (RuR) and various RuR derivatives and analogues (e.g. see Crompton & Andreeva, 1994; Matlib et al. 1998). Calcium uptake is driven by the electrochemical potential gradient generated by the combination of the mitochondrial inner membrane potential and the low concentration of Ca2+ in the matrix. The latter is maintained by efflux pathways - carriers which exchange Ca2+ for protons or Na+ ions. More recently, evidence has emerged suggesting that a rapid higher-affinity uptake pathway may also be expressed in heart and, with some subtle differences, in liver, which may contribute specifically to the mitochondrial responses to brief physiological stimuli (see Gunter et al. 1998).
The mitochondrial permeability transition (MPT) or megachannel
This has has recently been recognized as a central participant in several important pathological and possibly physiological processes, and needs to be introduced. This is a pore with a large maximal conductance (∼1200 pS), but with multiple subconductance states, and an estimated pore diameter of ∼2 nm (Crompton & Costi, 1990), apparently concentrated at contact sites between the mitochondrial inner and outer membranes (Fig. 1A). First discovered by the observation of Ca2+-induced mitochondrial swelling (Haworth & Hunter, 1979a, b, c), this phenomenon was intensively studied for many years in a small number of laboratories before leaping into prominence recently through the proposal that it may play a role in apoptosis (reviewed by Kroemer et al. 1998 and see below). The MPT is triggered by a host of different conditions (see the extensive review by Zoratti & Szabo, 1995), but most notably by a rise in intramitochondrial [Ca2+] ([Ca2+]m), accompanied by a fall in ATP/ADP ratio, a rise in inorganic Pi, a state of oxidative stress (excessive generation of reactive oxygen species coupled with depletion of cellular antioxidant defences) and resulting oxidation of critical thiol groups that appear to govern the channel properties (Petronilli et al. 1994). The channel is thought to arise from an alternate function of the adenine nucleotide translocase, which in isolation, can show channel-like properties in the presence of high [Ca2+] (Brustovetsky & Klingenberg, 1996). Perhaps most significantly, the channel is closed by cyclosporin A (CsA; Crompton et al. 1988), which binds to cyclophilin D (cyp D), a mitochondrial protein that mediates channel opening (Fig. 1A). The pharmacology is unsatisfactory as CsA has many sites of action, interacting with a whole family of cyclophilins whose properties remain poorly characterized. Recently, methyl-valine CsA was made available with a more selective action on the channel. A detailed review of the channel is out of place here, and the reader is referred to a number of recent reviews (Zoratti & Szabo, 1995; Ichas & Mazat, 1998), while its possible physiological and pathophysiological roles will be discussed below.
Causes and consequences of mitochondrial depolarization; mitochondria as ATP consumers
It is perhaps important here to emphasise that mitochondrial depolarization is not specific to any single process. Influx of cations such as Ca2+ or K+ will carry a current and depolarize the membrane, just as an inward current depolarizes the plasma membrane. Opening of a channel such as the large conductance pathway provided by the MPT, or the action of the protonophoric uncouplers, such as FCCP or CCCP, which shuttle protons across the membrane, short circuits ΔΨm. The intact respiratory chain responds to the depolarization by consuming oxygen at a maximal rate. Under these conditions, there is no driving force for ATP synthesis, and ATP may be consumed as the F1F0-ATP synthase runs in reverse mode: the equilibrium of the ATP synthase is determined both by ΔΨm and by the ATP/ADP.Pi ratio. Indeed, the enzyme may be regarded simply as an ATPase which is driven as an ATP synthase by virtue of the mitochondrial potential. In response to mitochondrial depolarization, the enzyme activity reverts to its ATPase activity, i.e. it now consumes ATP and pumps protons outwards, in a futile, energy-consuming cycle. Indeed, ATP may be consumed in minutes in response to an uncoupler (e.g. see Leyssens et al. 1996). Removal of substrate - either of oxygen or of carbon sources - or exposure to inhibitors of the respiratory chain stops respiration, the process that sustains the potential. The consequence then depends on the leakiness of the inner mitochondrial membrane and on the activity of the ATPase in reverse mode, which, by acting as a proton pump, can sustain the potential in some cells until ATP is depleted (e.g. see Duchen et al. 1993a). Is it not remarkable that the ‘powerhouse of the cell’ may provide a primary mechanism of ATP depletion and may precipitate cell death when function is compromised? This activity can be ascribed to the relative independence of mitochondria: it is useful for the mitochondrion to consume ATP and conserve ΔΨm as this prevents mitochondrial swelling. However, it is not in the best interests of the cell, and many cells have evolved a process to impose their own interests over and above that of the mitochondrial pool, and express a protein known as IF1 (the inhibitory protein - see Walker, 1994), a component of the ATP synthase complex, which inhibits mitochondrial ATP consumption. Under these conditions, the power play between the interests of mitochondrial and cell requirements is revealed, perhaps giving a reminder of their evolutionary origins.
Studies of mitochondrial function in single cells
Until recently, almost all the literature concerning mitochondrial function was derived from studies of isolated mitochondria in suspension. Typically, this means brain, heart or liver. It was very difficult to gather data about the specific properties of mitochondria in discrete cell types - in cerebellar granule cells as opposed to Purkinje cells or hippocampal pyramidal neurons for example. The growth of our knowledge about the behaviour of mitochondria in living cells owes much to the development of fluorescence measurement and imaging technologies (see Fig. 1B-D). Measurements of mitochondrial potential, of intramitochondrial [Ca2+] ([Ca2+]m) and of mitochondrial redox state in single cells has made it possible to explore the properties of mitochondria within identifiable cell types and their responses to physiological and pathophysiological stimuli in great detail, although it has to be said that these measurements are not without pitfalls, as interpretation of the fluorescence signals from some of the indicators used remains somewhat ambiguous. Presumably there is still much to be learned about the specializations of mitochondrial function in different cell types.
Mitochondrial contributions to ‘macrophysiology’: mitochondria play a central role in sensing glucose and oxygen
The primary energy supply for mitochondrial metabolism is derived from glucose and oxygen. It might, therefore, seem appropriate that mitochondria are used as substrate sensors in the major biological systems involved in the homeostatic regulation of these fundamental variables - in the pancreatic β-cell (Ashcroft & Gribble, 1998), in the glucose-sensing neurons of the hypothalamus (Ashford et al. 1990) and in the oxygen-sensing carotid body (Duchen & Biscoe, 1992a, b).
Role of mitochondria in glucose homeostasis
In both the pancreatic β-cell and the glucose-sensing neurons of the hypothalamus, delivery of substrate stimulates mitochondrial respiration, and the resulting increase in ATP/ADP.Pi ratio closes a class of K+ channels in the plasmalemma (KATP channels). The cell membrane potential depolarizes, opening voltage-gated Ca2+ channels, raising [Ca2+]i. In the β-cell, this promotes insulin secretion, the insulin lowers plasma [glucose] and closes the feedback loop. In the hypothalamus, regulation of neuronal excitability by ATP-dependent K+ channels also appears to play a role in the control of total body glucose through the regulation of appetite. Indeed, the situation is even more complex: two cell types have been described, some expressing a KATP channel and depolarizing in response to an increased supply of glucose, while another class of cells express an ATP-activated K+ channel, and therefore hyperpolarize on the supply of glucose (Rowe et al. 1996). The modulation of these conductances by leptin, the modulator of appetite, (Spanswick et al. 1997), and abnormalities in the genetically obese mouse seem to place their role firmly in the realm of appetite control (and see Friedman & Halaas, 1998).
In the β-cell, the supply of glucose to the cell increases substrate supply to the TCA cycle, increases the provision of NADH and FADH2 to the respiratory chain, and increases respiratory rate. This drives the hyperpolarization of the mitochondrial inner membrane which is the basis for the increased rate of ATP production (Duchen et al. 1993b; for review, see Ashcroft & Rorsman, 1989). This sequence results in a high [Ca2+]c in the presence of hyperpolarized mitochondria, encouraging mitochondrial Ca2+ accumulation. A most surprising story to emerge recently in the β-cell literature suggests that, following mitochondrial Ca2+ uptake, and given an appropriate carbon substrate for the TCA cycle, mitochondria release a (still unidentified) factor which facilitates insulin secretion at a permissive [Ca2+]c (Maechler et al. 1997). These authors have also suggested that the modulation of mitochondrial Ca2+ uptake may be involved in the desensitization of insulin secretion in response to repeated exposures to metabolic substrate: a dissection of the metabolic pathway (Maechler et al. 1998) revealed that ATP production was equivalent for each exposure, but the key difference between responses was a decrease in associated mitochondrial Ca2+ accumulation. These authors suggested that a reduction in the mitochondrial [Ca2+] load could account for the reduction in insulin secretion, although the mechanism for this process remains unknown. While this factor remains mysterious, the observations emphasize the pivotal role of mitochondria in the regulation of insulin secretion (see also, Kennedy & Wollheim, 1998). Adding a little flavour of integrative physiology to the story, the β-cell KATP channels (at least, in an insulin-secreting cell line, Harvey et al. 1997) also are regulated by leptin, and so appetite control and glucose homeostasis appear closely linked. The central role of mitochondria in the transduction pathway has led to suggestions that at least some forms of late onset diabetes mellitus, a disorder responsible for such widespread morbidity and mortality, may involve mitochondrial dysfunction.
Role of the mitochondria in oxygen sensing
The role of the mitochondria in oxygen sensingis far more contentious. As I have a personal interest in this story, I will rehearse it, if only briefly, but stress that this mechanism is by no means widely accepted (for counter views, see Acker, 1994; Lopez Barneo, 1996; Buckler, 1997). The carotid body senses a fall in arterial oxygen tension. Excitation increases the frequency of action potentials conveyed by the sinus nerve to the respiratory centres. It is the excitation of the carotid body that makes you feel breathless in the rarefied atmosphere at high altitude and that contributes to the sense of breathlessness of the chronic bronchitic. In response to a fall in oxygen tension, the type I (or ‘glomus’) cells of the structure depolarize, [Ca2+]i rises and catecholamines are secreted. The basis for the transduction mechanism is still troublesome - how do the cells sense subtle changes in oxygen tension that would be insignificant for most other cells? There is evidence for a specialized NADPH oxidase in the plasma membrane (Acker, 1994), for K+channels that directly sense oxygen (Lopez-Barneo, 1996) and for modulation of Ca2+-dependent K+ channels (Peers, 1997; but see also Lahiri et al. 1998 and Donnelly, 1997). Nevertheless, one of the remarkable features of the carotid body is that it is excited by all agents known to impair mitochondrial oxidative phosphorylation, agents that have no structural pharmacokinetic features in common (Anichkov & Belen'kii, 1963; Biscoe & Duchen, 1990; Duchen & Biscoe, 1992a, b; see also Buckler & Vaughan-Jones, 1998). T. Biscoe and I found that mitochondrial membrane potential and redox state of the Type I cells show a unique sensitivity to moderately low oxygen tensions, changing at PO2 values of 20-30 mmHg, which have no effect on mitochondrial function in any other cell that we have ever studied. The jury is still out, but there is a case to be addressed for a specialized role of mitochondria in these cells coupled with evidence for expression of a specialized cytochrome aa3 (Mills & Jöbsis, 1970, 1972; see also Lahiri et al. 1996), and it is tempting to speculate that this is another example in which mitochondrial substrate supply plays a central role in a cellular transduction process. There is an attractive symmetry in a scheme whereby the organelles that require oxygen and glucose are also responsible for ensuring the homeostasis of those very substrates. Even local oxygen delivery to tissues is regulated by the energy-transducing KATP channels expressed in vascular smooth muscle cells (see Quayle et al. 1997), although these are probably mainly regulated by transmitter (rather than metabolic) activity.
Mitochondrial calcium uptake and physiological calcium signalling
Apart from the production of ATP, the major contribution of mitochondria to cell physiology must lie in their impact on Ca2+ signalling. Isolated mitochondria will accumulate huge amounts of Ca2+ in the presence of inorganic phosphate. Ca2+ influx is driven primarily by the mitochondrial inner membrane potential. Thus, the driving force for Ca2+ movement will favour uptake into the matrix once the concentration of Ca2+ close to the mitochondria rises into the range of the affinity of the uniporter, typically described as a low affinity (Kd≡ 1-5 μM), high capacity pathway (see Rizzuto et al. 1994; Gunter et al. 1998), although it must be said that the concept of a Kd may be misleading, and net Ca2+ uptake is rather a kinetic balance between uniporter activity and the efflux pathways (Fig. 1A). Until recently, two major questions remained: firstly, is mitochondrial Ca2+ uptake significant during physiological Ca2+ signalling? Secondly, a more teleological question - what is that uptake for, or more objectively, what is its functional significance? More recently, these questions have been extended to ask about the impact of mitochondrial Ca2+ uptake on the quantitative and spatiotemporal characteristics of Ca2+ signals (see also recent reviews by Miller, 1998; Babcock & Hille, 1998; Simpson & Russell, 1998).
A substantial impetus was given to attempts to address these issues by the discovery that the key rate-limiting enzymes of the citric acid cycle are upregulated by Ca2+, either directly or through the regulation of phosphorylation reactions (for review, see McCormack et al. 1990). There is now clear evidence that this process is significant during physiological [Ca2+]c signalling. The first direct study in intact cells was probably that of Fein & Tsacopoulos (1988), who showed that a rise of [Ca2+]c following a flash of light increased oxygen consumption in Limulus photoreceptors. In sensory neurons, a rise in [Ca2+]c in response to voltage-gated Ca2+ influx was associated with a modest transient mitochondrial depolarization and a prolonged increase in NADH autofluorescence (an increase in NADH/NAD+ ratio, consistent with upregulation of the TCA cycle) which was blocked by inhibition of the mitochondrial uniporter by microinjection of RuR (Duchen, 1992). More recent studies have extended these observations to explore the metabolic consequences of Ca2+ mobilization from IP3-dependent internal stores (Pralong et al. 1994; Hajnoczky et al. 1995; Robb-Gaspers et al. 1998). Most intriguingly, there appear to be subtle differences in the responses of different cell types. Thus, IP3-dependent oscillations of [Ca2+]c in hepatocytes, although communicated to mitochondria as oscillatory changes in [Ca2+]m (Hajnoczky et al. 1995), were translated as graded but sustained changes in the reduction of mitochondrial NAD+ to NADH (Pralong et al. 1994; Hajnoczky et al. 1995). In the hepatocytes, the Ca2+-dependent metabolic activation clearly outlasted the [Ca2+]m signal, thereby acting as an integrator of oscillatory [Ca2+]i-signalling activity. The rise in [Ca2+]m was shown to upregulate pyruvate dehydrogenase, increase NADH autofluorescence and then to initiate a slower increase in mitochondrial potential (Robb-Gaspers et al. 1998), which would then drive an increase in ATP production. In contrast, [Ca2+]c oscillations in pancreatic β-cells and adrenal glomerulosa cells were associated with oscillatory changes in NADH (Pralong et al. 1994). These apparent differences in signal need to be explored further to establish the degree and origin of variability in mitochondrial function between different cell types.
It now seems likely that the major consequence of the Ca2+ uptake pathway in terms of mitochondrial function is the upregulation of energy metabolism. This makes good sense - oxidative phosphorylation is modulated in relation to energy demand by the relayed Ca2+ signal, as increased energy demand is almost always accompanied by a rise in [Ca2+]c - for secretion or contraction for example. The functional importance of this mechanism is illustrated nicely by studies of the cardiomyopathic hamster (Di Lisa et al. 1993), in which an impaired [Ca2+]c signal in cardiac muscle is associated with impaired energy metabolism.
Not only has this body of work demonstrated the functional significance of mitochondrial Ca2+ uptake, but it also showed that physiological Ca2+ signalling is associated with significant mitochondrial Ca2+ uptake. Indeed, it is now clear that mitochondria do indeed take up Ca2+ during physiological signalling in most cells that have been studied. The technology required to answer these questions has developed largely through improvements in fluorescence microscopy and imaging techniques, especially through improvements in confocal microscopy, and, perhaps most dramatically, through the introduction of genetically engineered, site-directed expression of the Ca2+-sensitive photoprotein, aequorin, that permits specific measurement of [Ca2+] within unambiguously identifiable compartments (see Brini et al. 1995 and Fig. 2) or using mitochondrially localized fluorescent indicators, such as rhod-2 (e.g. see Hajnoczky et al. 1995; Babcock et al. 1997; Robb-Gaspers et al. 1998; Fig. 2).
Figure 2. Direct measurements of mitochondrial Ca2+ uptake.
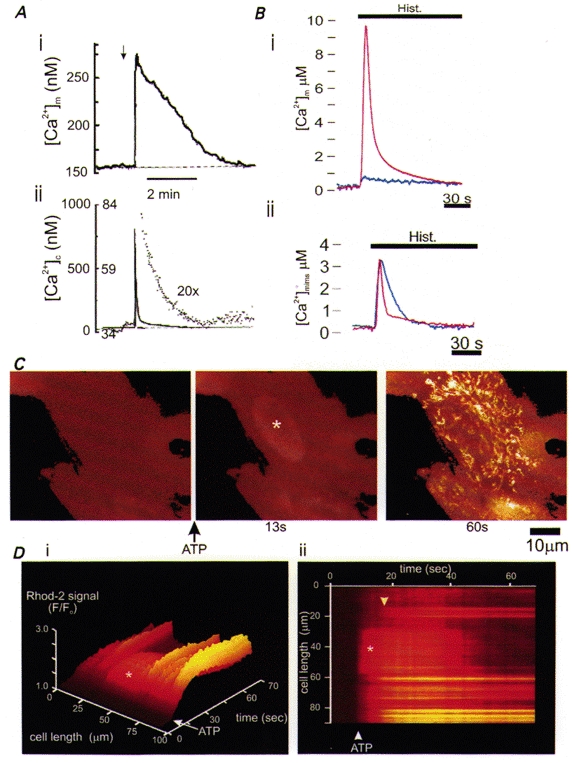
A, measurements of mitochondrial [Ca2+] (i) and cytosolic [Ca2+] (ii) in a rat adrenal chromaffin cell following depolarization of the plasma membrane and Ca2+ influx through voltage-gated channels. The brisk [Ca2+]c increase and early recovery (ii) were followed by a prolonged slower decay phase (seen as the dotted and enlarged trace) which coincided with the time course of the slow decline in [Ca2+]m revealed with rhod-2 fluorescence (i) (modified from Babcock et al. 1997, with permission). B: (i) stimulation of a HeLa cell with histamine mobilizes Ca2+ from IP3-sensitive ER stores. The red trace shows the rise in [Ca2+]m revealed with aequorin targeted to the mitochondrial matrix. Note the very large increase in [Ca2+]m which was abolished by pretreatment with FCCP (blue trace), preventing mitochondrial Ca2+ uptake. (ii), an aequorin construct targeted to the mitochondrial intermembrane space (mims) to sense local changes in [Ca2+]c showed a brisk but smaller rise in [Ca2+] in response to the same stimulus. Pretreatment with FCCP had no significant effect on the amplitude of the response but significantly slowed the decay phase, consistent with a role of mitochondrial uptake in removing [Ca2+]c. (These data were obtained from populations of cells; from Rizzuto et al. 1998, with permission.) C shows digital fluorescence images of rat cortical astrocytes loaded with rhod-2, normalized with respect to the first image to reveal relative changes in signal. Stimulation with ATP (100 μM) to mobilize Ca2+ from ER stores raised [Ca2+]c first, best seen over the nucleus (*, at 13 s into the sequence), followed later by a large and sustained increase over the mitochondria. This is illustrated further by the images in D, which show the evolution of the signal with time, measured as an intensity profile along a line selected along the axis of the cell and displayed as a surface plot in (i) and as a ‘line image’, seen as if from above in (ii). The relatively fast and transient increase over the nucleus (*) contrasts with the slower but sustained increase in signal over the mitochondria (downward-pointing arrowhead) (M. R. Duchen & E. Boitier, unpublished observations).
The kinetics of mitochondrial Ca2+ uptake and release appear to differ between cell types (Fig. 2), perhaps reflecting differences in [Na+]i availability for the mitochondrial Na+-Ca2+ exchanger. Almost all reports to date describe a rapid uptake followed by much slower re-equilibration, with the exception of the measurements by Rizzuto et al. (1992, 1994) in HeLa cells, using site-directed aequorin and by Robb-Gaspers et al. (1998) in hepatocytes, using rhod-2. In both these studies, recovery of [Ca2+]m appeared relatively fast despite a sustained rise in [Ca2+]c. Our own imaging study of mitochondrial Ca2+ uptake in adult rat cortical astrocytes using the mitochondrially localized Ca2+ indicator rhod-2 suggests that a brief transient rise in [Ca2+]c was followed by a sustained increase in [Ca2+]m that could last for many minutes (Boitier et al. 1999; see Fig. 2A and D), although Jou et al. (1996) described oscillations of [Ca2+]m associated with [Ca2+]c oscillations in a neonatal astrocytic cell line. It is not yet clear whether these are real differences in [Ca2+]m homeostasis between cell types, or technical issues related to the properties (consumption, affinity, loading conditions etc.) of the indicators used. However, the kinetics are important and have a number of fascinating functional consequences. At nerve terminals, for example, the slow release of the Ca2+ that accumulates within mitochondria during periods of rapid repeated stimulation maintains an elevated [Ca2+]c in the terminal for a substantial period of time. Further stimulation during that period therefore raises [Ca2+]c more than would otherwise be expected and potentiates transmitter release - in other words, the temporal buffering of [Ca2+]c by mitochondrial Ca2+ uptake plays a central role in post-tetanic potentiation (Tang & Zucker, 1997; David et al. 1998; Melamed-Book & Rahamimoff, 1998).
Most obviously, if mitochondria take up Ca2+, they may act as a fixed buffering system. The impact that this has on cellular [Ca2+]c signalling will depend on a number of factors, not least the balance of Ca2+-regulating systems expressed by the cell, the relative volume of mitochondria in the cell and the mitochondrial membrane potential. The consequences of mitochondrial Ca2+ uptake for [Ca2+]c signalling has now been widely studied, revealing profound alterations in shaping global [Ca2+]c responses to depolarization in adrenal chromaffin cells, sensory neurons and central neurons when mitochondrial Ca2+ uptake was disabled, achieved most simply by dissipating ΔΨm using an uncoupler (Thayer & Miller, 1990; Werth & Thayer, 1994; Friel & Tsien, 1994; White & Reynolds, 1995; Herrington et al. 1996; Khodorov et al. 1996; Stout et al. 1998). On this point it is crucial to differentiate between changes in [Ca2+]c signal due specifically to reduced mitochondrial Ca2+ uptake, and changes due to ATP depletion that may follow a loss of ΔΨm (see also Landolfi et al. 1998), as depletion of ATP will clearly alter the ability of cells to extrude or sequester a Ca2+ load quite independently of mitochondrial Ca2+ uptake. In many excitable cells, depolarization causes Ca2+ influx through voltage gated channels and a fast [Ca2+]c transient which shows a biphasic recovery; a rapid early phase followed by a long slow phase that may last for minutes (Fig. 3A and B). Inhibition of mitochondrial Ca2+ uptake generally increases the amplitude of the transient response and removes the slow recovery phase (Fig. 3A and B; Werth & Thayer, 1994; Herrington et al. 1996). Mitochondrial depolarization during the slow recovery phase causes a large [Ca2+]c transient (Fig. 3B), suggesting Ca2+ release from Ca2+-loaded mitochondria. The slow phase is also impaired by inhibition of the mitochondrial Na+-Ca2+ exchange by agents such as diltiazem or CGP-37157 (White & Reynolds, 1997). This mechanism thus shapes the global [Ca2+]c transient following periods of depolarization, promoting rapid recovery from a peak of high [Ca2+]c followed by a sustained plateau of raised [Ca2+]c as the mitochondrial Ca2+ homeostatic systems re-equilibrate.
Figure 3. Impact of mitochondrial Ca2+ uptake on global [Ca2+]c signalling in a variety of cell types.
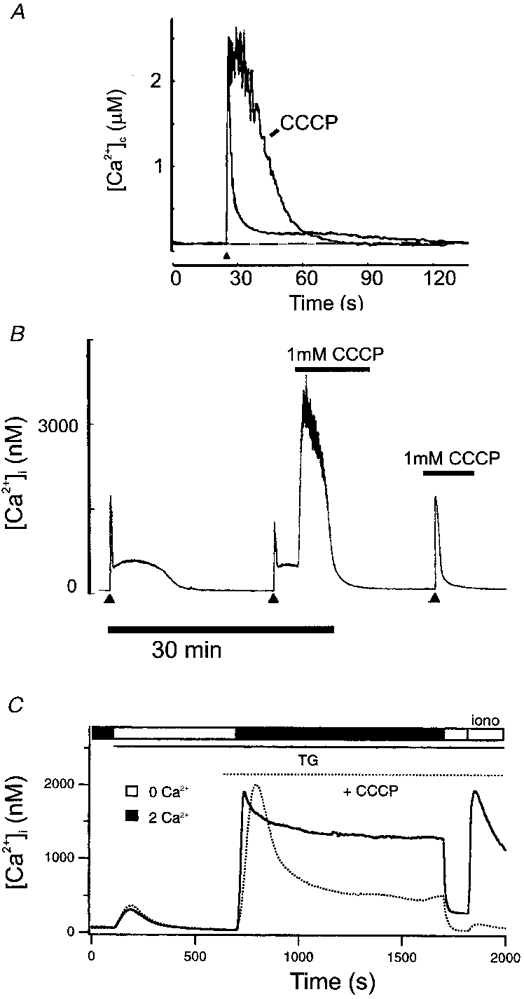
A, in a bovine adrenal chromaffin cell, a brief depolarization caused a transient rise and rapid recovery of [Ca2+]c which was followed by a prolonged ‘tail’ phase of slow recovery (cf. Fig. 2A). Preventing mitochondrial Ca2+ uptake by prior depolarization of ΔΨm with CCCP: (i) increased the amplitude of the response; (ii) greatly slowed recovery; and (iii) abolished the slow recovery phase (modified from Herrington et al. 1996, with permission). B, a similar pattern of [Ca2+]c signal was seen in sensory neurons: a large [Ca2+]c load following prolonged depolarization showed a biphasic recovery - a rapid phase followed by a prolonged secondary plateau. Depolarization of ΔΨm by the application of CCCP during the plateau caused a massive increase in [Ca2+]c, suggesting Ca2+ release from Ca2+-loaded mitochondria. Prior application of CCCP abolished the plateau phase completely (modified from Werth & Thayer, 1994, with permission). C, in a population of T lymphocytes, capacitative Ca2+ influx was studied thus: internal stores were emptied by inhibition of the ER Ca2+ ATPase with thapsigargin (TG) in the absence of external Ca2+. Reintroduction of [Ca2+]o caused a large sustained [Ca2+]c signal. If Ca2+ was then again removed, a challenge with ionomycin revealed that an internal store, probably mitochondrial, had now been filled. With mitochondrial uptake disabled with CCCP (dotted line), the sustained component of the capacitative influx signal was much reduced, and the final ionomycin challenge confirmed the lack of mitochondrial Ca2+ accumulation. This, along with other data from the study, showed that mitochondrial uptake helps to define the characteristics of capacitative Ca2+ influx (modified from Hoth et al. 1997, with permission).
In some other models, the functional consequences of mitochondrial Ca2+ uptake are rather more complex and subtle. For example, during measurements of capacitative Ca2+ signalling in populations of T lymphocytes (Hoth et al. 1997), the plateau phase of sustained Ca2+ influx seen in measurements from cell populations was impaired by the uncoupler CCCP (Fig. 3A). High [Ca2+] reduces the open probability of the channel that carries the Ca2+ flux in response to emptying of ER stores (ICRAC - the calcium release-activated calcium current). These authors therefore proposed that the spatial buffering of Ca2+ by mitochondrial Ca2+ uptake limits the rise in [Ca2+]c close to the channel mouth, the ICRAC channels stay open, the total Ca2+ flux is greater and ER store refilling is more effective. Remove mitochondrial Ca2+ uptake, and the local accumulation of [Ca2+]c close to the channel mouth will close the channels, and therefore limits store refilling. Mitochondrial Ca2+ export through a Na+-Ca2+ exchange mechanism in turn apparently maintains the high plateau global[Ca2+]c signal during the activation of the Ca2+ influx pathway. This whole process is perhaps more complex than it initially seems - it must of necessity require some spatial separation of the mitochondrial efflux pathway from the focal high [Ca2+]c near the channel mouth responsible for channel inactivation, or the model could not work - mitochondrial efflux would simply sustain the high local [Ca2+]c and cause channel inactivation. These considerations might prompt speculation about the spatial organization of mitochondrial structures: the observations of Rizzuto et al. (1998) in HeLa cells transfected with a mitochondrially localized green fluorescent protein (GFP) suggested that, at least in these cells, mitochondria may form a contiguous network (Fig. 1A) and even the more conventional confocal reconstruction shown in Fig. 1A shows a substantial network structure. Does this mean that Ca2+ could be taken up in one part of the cell and released at some distant site from another part of the same mitochondrial network - a notion of Ca2+ tunnelling through an organelle structure akin to that described for the ER network of pancreatic acinar cells (Mogami et al. 1997)?
Modulation of spatiotemporal [Ca2+]c signals by mitochondria
The elegant experiments of Rizzuto et al. (1998) using site-directed transfection of the Ca2+-sensitive photoprotein aequorin, more recently combined with GFP targeted to ER membranes and a separate aequorin construct targeted to the outer leaflet of the inner mitochondrial membrane (Rizzuto et al. 1998), have revealed a tight apposition of mitochondria with ER membranes and a functional exchange of ER Ca2+ release and mitochondrial Ca2+ uptake. This means that in cells that depend primarily on IP3-mediated Ca2+ signalling from ER (or perhaps, ryanodine-sensitive Ca2+-induced Ca2+ release from SR), the anatomical organization of mitochondria results in the exposure of mitochondria to microdomains of high [Ca2+]c close to ER Ca2+ release sites (Fig. 2A). This is important, as it implies that mitochondria within the cell may be exposed in some sites to a local [Ca2+]c far higher than the averaged [Ca2+]c measured across the cell. Thus attempts to extrapolate from studies of Ca2+ handling by isolated mitochondria in order to predict the functional consequences of mitochondrial Ca2+ uptake must consider the local not the global Ca2+ concentrations. Observations from our own laboratory also point to a tight local coupling of ER/SR Ca2+ release and Ca2+ uptake by nearby mitochondria. We have measured local changes in mitochondrial potential in cardiomyocytes and, more recently, in astrocytes in culture, and noted highly localized transient mitochondrial depolarizations which were dependent on local Ca2+ release from internal stores and subsequent mitochondrial Ca2+ uptake (Fig. 4A; Duchen et al. 1998; Jacobson & Duchen, 1998). Ultrastructural studies also suggest that the cellular organization of mitochondria may be arranged in relation to cell function: EM studies described by Lawrie et al. (1996) suggest that while mitochondria may be juxtaposed to ER in inexcitable cells, they are more likely to lie close to the plasmalemma in excitable cells. As far as I am aware, there are no clues to tell us how this organization is controlled.
Figure 4. Impact of mitochondrial Ca2+ uptake on propagated [Ca2+]c waves.
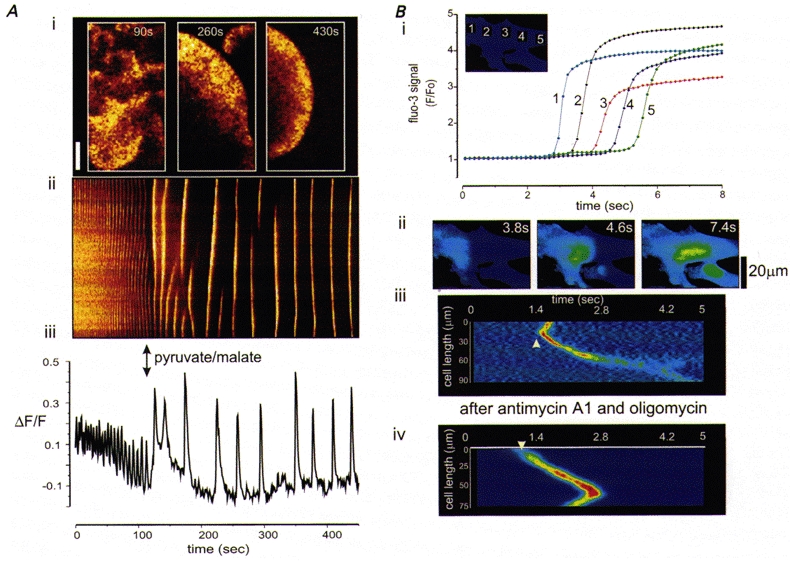
A shows the effect of mitochondrial energization on the properties of [Ca2+]c waves in Xenopus oocytes. [Ca2+]c waves in a Xenopus oocyte triggered by flash photolytic release of a caged, non-hydrolysable analogue of IP3 were initially chaotic and small in amplitude. Following microinjection of the mitochondrial substrates pyruvate and malate (at the time indicated by the arrows), mitochondrial potential increased (measured separately) and the waves became increasingly co-ordinated, larger in amplitude, and the rate of propagation increased. A(i) shows examples of the waves before (90 s) and after (at 240 and 480 s) injection of the substrates. A(ii) shows a line image to illustrate the characteristics of the waves with time, and a plot of amplitude with time for a single point in the cell is shown in (iii) (modified from Jouaville et al. 1995, with permission). B, mitochondrial Ca2+ uptake has opposite effects on the propagation of [Ca2+]c waves in rat cortical astrocytes in culture. B (i) shows a plot of intensity with time at a series of equidistant points through a single cell. The images in B (ii) illustrate the spread of the [Ca2+]c signal through the cell with time. To obtain the images shown in (iii) and (iv), which illustrate the propagating wave front, the image series was differentiated (a process of sequential subtractions that reveals a signal only in pixels in which the signal changed). A line was then selected along the axis of the cell and the colour-coded intensity profile along that line was plotted as a function of time, revealing the wave front as a diagonal band as it progressed across the cell with time. The rate of propagation of the wave clearly increased in cells in which mitochondrial Ca2+ accumulation was abolished by dissipating ΔΨm (iv), using antimycin A1 combined with oligomycin, to limit mitochondrial ATP consumption (M. R. Duchen, R. Rea & E. Boitier, unpublished observations).
The functional consequences of the local juxtaposition of mitochondria and ER/SR for [Ca2+]c signalling are not so easy to predict. Intuitively, two quite opposing consequences suggest themselves: (i) mitochondria take up Ca2+ as it is released and therefore limit the spatial spread of the [Ca2+]c signal, i.e. acting as a spatial buffer; (ii) the regulation of local [Ca2+]c by strategically placed mitochondria may regulate local Ca2+ flux through Ca2+-sensitive Ca2+ release channels and thus modulate [Ca2+]c signalling, much as described above for ICRAC. For example, the IP3 (type 1) Ca2+ release channel is sensitive to [Ca2+], enhanced at low [Ca2+] (< ∼500 nM), but inhibited as the Ca2+ concentration rises into the micromolar range, giving rise to a bell-shaped [Ca2+]c/Ca2+ release curve (Iino, 1990; Bezprozvanny et al. 1991). Thus the local buffering of [Ca2+]c by mitochondria may have a major impact on ER Ca2+ release. The peak of the Ca2+ release/[Ca2+]c curve lies around 400-500 nM, the value at which mitochondrial Ca2+ uptake becomes significant. As [Ca2+]c rises to levels closer to 1 μM, channel opening is inhibited. Removal of local Ca2+ by mitochondria may thus serve to keep the channels open and therefore help sustain ER Ca2+ efflux, while the spatial orientation of mitochondria may support the dispersal of the Ca2+ in the cytosol away from the mouth of open Ca2+-sensitive IP3-sensitive channels. This is the mechanism proposed by Jouaville et al. (1995) to account for observations in Xenopus oocytes, in which the rate of propagation of [Ca2+]c waves was modulated by varying mitochondrial potential - the larger the mitochondrial potential, the faster and more co-ordinated the wave (Fig. 4A). This might seem counter-intuitive - with a larger potential, mitochondrial Ca2+ uptake is more efficient and should serve as a more powerful buffer. However, by buffering microdomains of [Ca2+]c, mitochondria could regulate the rate of efflux from the ER Ca2+ release channels, pulling the [Ca2+]c into a region in which the [Ca2+]c dependence of Ca2+ release lay at the rising phase of the bell-shaped curve.
We have recently examined the role of mitochondrial Ca2+ uptake as a determinant of the rate of Ca2+ wave propagation in mammalian astrocytes, and have found the opposite - [Ca2+]c wave propagation was clearly significantly faster when mitochondrial Ca2+ uptake was impaired (Boitier et al. 1999; Fig. 4A). Why should these similar experimental models yield different answers? One possibility is simply that, in the astrocytes, local [Ca2+]c may not have reached levels at which ER Ca2+ release is impaired. Alternatively, the answer may lie in the properties of different classes of IP3 receptors. Astrocytes predominantly express type 2 IP3-dependent ER Ca2+ release channels (Shepherd et al. 1997; E. Boitier, S. Brind & M. R. Duchen, unpublished observations) which do not show the same bell-shaped [Ca2+]c sensitivity as the type 1 receptors (Ramos-Franco et al. 1998). Indeed, type 3 receptors also show a monotonic [Ca2+]c sensitivity (Hagar et al. 1998). Thus, the consequence of this focal interaction between organelles may vary between cells, depending on the relative expression of these proteins. In the astrocyte, with type 2 receptors dominant, mitochondria act effectively as a spatial buffer, limiting the rate and extent of spread of a [Ca2+]c signal. This may have significant implications for the propagation of Ca2+ signals during periods of anoxia/ischaemia in the CNS, during which mitochondrial potential will dissipate, and becomes particularly interesting in the light of recent demonstration that ‘death’ signals may also propagate through cellular networks (Lin et al. 1998).
A most intriguing contribution to this story has evolved recently, growing out of a number of related observations. It has been suggested that mitochondria may participate in the active propagation of a [Ca2+]c signal across the cell through a process of Ca2+-induced Ca2+ release (CICR; see Ichas & Mazat, 1998, for review). The mechanism that underpins this story is the mitochondrial permeability transition, or MPT (see above). While most of the relevant literature deals with pathological consequences of the MPT - with proposed roles for initiation of apoptosis, ATP depletion and reperfusion injury - Ichas et al. (1997) suggested that the MPT may open transiently at a low conductance state during normal [Ca2+]c signalling. It is proposed that a rise in [Ca2+]m and a resultant small fall in intramitochondrial pH might suffice to trigger transient flickering openings of the channel in a low conductance state; once open, the accumulated Ca2+ is released, causing a local microdomain of high [Ca2+]c. This may trigger local CICR from ER or may be taken up by the next bank of quiescent mitochondria which will in turn release the Ca2+. The authors were able to create a propagating, CsA-sensitive [Ca2+] wave through a ‘lawn’ of isolated mitochondria seeded onto a coverslip, simply in response to a microdomain of [Ca2+]c generated at one corner of the lawn, i.e. a wave of Ca2+ through the mitochondrial lawn was generated in the absence of any other components of the cytosol. This was, of course, a wave of [Ca2+]m, not the propagation of a cytosolic wave. Evidence for the role of this mechanism in cellular [Ca2+]c signalling was based on the observations that the amplitude of cytosolic [Ca2+]c signals were modulated by CsA, which inhibits the MPT. It would be nice to be completely confident that the CsA was doing nothing else in the cell, as CsA is also a potent inhibitor of the Ca2+-dependent phosphatase, calcineurin, but the possibility that mitochondria may play an active role in [Ca2+]c signalling, at least in some cells, remains highly intriguing.
At the very least, it should be clear from the arguments presented that mitochondria play a significant, although varied, role in shaping cellular [Ca2+]i signals. [Ca2+]i signalling is so central to all normal physiological functions in all cell types that any disturbances in the subtleties of [Ca2+]i signalling that result from mitochondrial dysfuction will inevitably have a substantial impact on cell function, and more broadly, on the integrative function of more complex organ systems.
Consequences of mitochondrial Ca2+ uptake for mitochondrial function: from cell signalling to cell death
The most immediate consequence of mitochondrial Ca2+ uptake is a mitochondrial depolarization. This is most probably simply the consequence of the flow of a Ca2+ current, as the uniporter is electrogenic. It is seen as a response to the addition of Ca2+ to preparations of isolated mitochondria, and is seen in intact cells as a reponse to physiological [Ca2+]c signals. Transient mitochondrial depolarizations have been described in excitable cells in response to depolarization in sensory neurons (Duchen, 1992), hippocampal neurons in slices (Bindokas et al. 1998) or in culture (Keelan et al. 1998 and see Fig. 5A) and in response to a propagating [Ca2+]c wave in cardiomyocytes (Fig. 5B; Duchen et al. 1998), and have also been seen in inexcitable cells in response to mobilization of ER Ca2+ (Peuchen et al. 1996; Boitier, Rae & M. R. Duchen, submitted). Recently we have also described focal mitochondrial depolarizations in relation to focal SR/ER Ca2+ release (Duchen et al. 1998; Fig. 5A), a functional consequence of the close apposition of ER Ca2+ release sites and mitochondria.
Figure 5. Impact of mitochondrial Ca2+ uptake on mitochondrial potential.
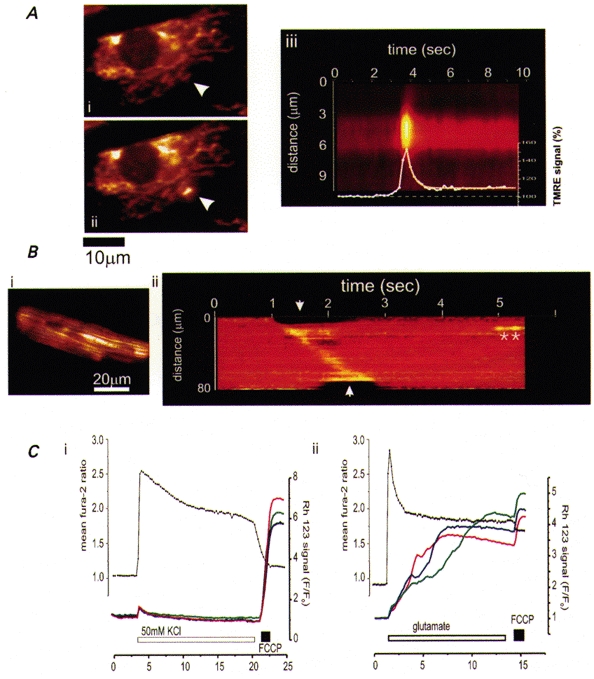
A, microdomains of high [Ca2+]c due to release from SR or ER cause brief transient depolarizations of mitochondria. These events were seen in single cells loaded with the potentiometric probe, tetramethyl rhodamine ethyl ester (TMRE), involving either clusters of mitochondria or even a single mitochondrion, as shown in (ii) in a neonatal cardiomyocyte in culture. The time course of the event is illustrated in (iii) in which the intensity profile along a line selected through the mitochondrion is shown with time. The mean intensity plotted with time is superimposed. These events were abolished by ryanodine or intracellular calcium chelation. B, a similar process operates during co-ordinated [Ca2+]c signalling, illustrated during a [Ca2+]i wave in a cardiomyocyte (also loaded with TMRE). A [Ca2+]i wave in these cells causes a contractile wave which was associated here with a wave of mitochondrial depolarization, seen as an increase in fluorescence signal. The line image (ii) illustrates the evolution of the signal with time along a line selected along the axis of the cell (shown in (i), white line). The contraction started first at the upper margin (the top left in (i) and propagated along the length of the cell. The contraction can be seen at the edges of the line image (arrows) as the line extended beyond the edges of the cell (modified from Duchen et al. 1998, with permission). Two spontaneous transient events of similar amplitude as the wave were seen later (*). C shows simultaneous measurements of [Ca2+]c and ΔΨm made in rat hippocampal neurons in culture using fura-2 (•) and rhodamine 123 (continuous lines). (i), depolarization of the plasma membrane with 50 mM KCl for 10 min raised [Ca2+]c but caused only a small transient mitochondrial depolarization. (ii), depolarization for 10 min with glutamate under conditions that caused 75 % cell death 24 h later, also raised [Ca2+]c but caused a progressive and very nearly complete dissipation of ΔΨm (note that the response could barely be increased further by application of FCCP; J. Keelan, O. Vergun & M. R. Duchen, unpublished observations, but see Keelan et al. 1998).
The depolarization of ΔΨm in response to a rise in [Ca2+]c lies at the borderlines between mitochondrial physiology and mitochondrial pathology. It has been clear for many years that mitochondrial Ca2+ accumulation during a state of oxidative stress may precipitate opening of the megachannel or mitochondrial permeability transition pore MPT (see above, and Lemasters et al. 1998). While flickering subconductance states may play roles in [Ca2+]c signalling (Ichas et al. 1997), the MPT generally represents a catastrophe for the cell and will lead inexorably to cell death, either through ATP consumption, acute energy failure and necrosis or through the initiation of apoptosis. The latter may be driven by the release of apoptosis-inducing factor (AIF; Kroemer et al. 1998) or of cytochrome c (see Liu et al. 1996) or both, leading to caspase (cysteine protease) activation and a suicidal (apoptotic) cascade. It seems increasingly likely that a key step in several routes to programmed cell death involves the release of mitochondrial cytochrome c from the intermembrane space and activation of caspases, initiating a positive feedback cycle in which caspase activation releases more cytochrome c or AIF, which in turn triggers the permeability transition (i.e. pore opening), and drives a relentless progression to cell death. There is some controversy at present about the exact sequence - perhaps even by the time this essay has appeared on paper the answer will be known, so fast is this field evolving (for recent reviews, see Kroemer et al. 1998; Green & Reed, 1998). It is also perhaps worth remarking on the paradox embedded in this story: apoptosis is an ATP-dependent process and yet opening of the megachannel is likely to lead rapidly to ATP depletion, much as seen in response to an uncoupler (see above). How then is the MPT compatible with a role in apoptosis? Perhaps initiation of apoptosis requires only flickering transient openings of the megachannel sufficient to release some cytochrome c. Perhaps the full opening of the megachannel would lead rather to ATP depletion, energetic collapse and necrotic cell death. Perhaps apoptosis will only result from MPT if mitochondrial ATP consumption is somehow limited. Clearly, the details of this process remain to be clarified. It is also important to emphasise that the interpretation of experimental evidence in favour of a role of the MPT in apoptotic cell death relies heavily on the use of pharmacological agents, such as cyclosporin A, which itself is not specific, and to measurements of ΔΨm which are fraught with inaccuracies and misinterpretations, and so this remains an area in which it is necessary to tread carefully.
Most of the models of apoptotic cell death involve the application of toxic agents to or the withdrawal of growth factors from cell lines in culture. Perhaps the most actively studied model that bears an obvious relationship to cell death in a common human pathology involves glutamate toxicity. During periods of anoxia and ischaemia in the CNS, during the evolution of a stroke or following neonatal asphyxia, for example, glutamate accumulates in the extracellular space. The toxicity of glutamate under these conditions has been recognized and attributed to a cellular Ca2+ overload for many years (see Choi, 1994). However, recent evidence suggests that a key process that links the rise in [Ca2+]c to toxicity is mitochondrial Ca2+ accumulation and resultant mitochondrial depolarization. The loss of mitochondrial potential in this model is clearly linked to cell death, and may result from MPT, but the evidence for a role of the MPT in this model is still weak, and other processes - notably inhibition of the respiratory chain - could produce similar consequences (Fig. 5C; Schinder et al. 1996; Nieminen et al. 1996; Khodorov et al 1996; Castilho et al. 1998; Keelan et al. 1998). A similar process may take place in the heart following reperfusion after an ischaemic episode - again conditions in which mitochondrial Ca2+ loading will be combined with oxidative stress, leading to MPT and cell death (Duchen et al. 1993a;Griffiths & Halestrap, 1995).
Why should the cell place such fundamental decisions about life and death in the hands of the mitochondrial interlopers? Perhaps the key lies in the heart of the evolutionary history of the organelles. Although mitochondria have become obligate symbionts, dependent on the cellular machinery for production of many proteins, they still retain many features of their bacterial origins. The pore that underlies the ‘permeability transition’ consists of a protein complex that includes at least VDAC - the voltage-dependent anion channel that has a strong homology to bacterial porins, situated in the outer mitochondrial membrane - and the adenine nucleotide translocase (ANT), which sits in the inner mitochondrial membrane but especially at contact sites between inner and outer membranes. Cyclophilin D and the benzodiazepine receptor are also attached to this complex and appear to regulate pore opening (Crompton et al. 1998). Apparently in close association with, or even a constituent of the complex, is the proapoptic protein bax (Marzo et al. 1998), a channel-forming protein reminiscent of the bacterial pore-forming colicins, which are used by bacteria for self defence. It seems likely that the antiapoptotic proteins bcl-2 and bcl-xL also act as mitochondrial pores or regulators (Vander Heiden et al. 1997). Is it not remarkable that if you wish to express bax in bacteria you must also express bcl-2 or the bacteria die? Why cytochrome c should have become a major trigger to cell death is harder (for me) to understand but perhaps its presence in the cytosol informs the cell that its mitochondria are damaged, that energetic function will become compromised and the mitochondria may become a source of free radicals and oxidative injury (see also Skulachev, 1996). As oxidative stress may promote DNA damage and possible neoplastic transformation, it is perhaps safer for the cell to commit suicide than to put the host organism at risk.
CODA
Mitochondria have become recognized as fascinating structures that have a major impact on the physiology and pathophysiology of the organism that they inhabit at many levels. Their relative inaccessibility and the difficulties involved in trying to study them in their native habitat - the cytosol - has delayed much of this understanding and they still have many secrets to yield. What is perhaps most fascinating is the combination of a curiously wild and independent streak, a residue of their independent bacterial history, with an extraordinary integration with cellular specializations. However, why do mitochondrial structures and shape vary between one cell type and another? To what extent are mitochondrial functions different in different cell types? How do cells regulate mitochondrial numbers, morphology, function? And to what extent are changes in mitochondrial function at the heart of many diseases?
Acknowledgments
Work in my laboratory is supported by The Wellcome Trust, the Royal Society, British Heart Foundation and the BBSRC. I would like to thank my friends and colleagues Eric Boitier, D. ‘Jake’ Jacobson, Julie Keelan, Mart Mojet and Martin Crompton for their contributions to work discussed and for helpful comments on this manuscript for which I also thank Boris Khodorov, Frances Ashcroft and John B. Clark. I also thank Rosario Rizzuto, Markus Hoth, Bertil Hille, Jim Lechleiter and Stanley Thayer for permission to use their work to illustrate this essay. Obviously, in a relatively short piece such as this, references to important areas of work must be omitted, and I apologize to those whose work has not been cited.
References
- Acker H. Oxygen sensing in the carotid body: ideas and models. Advances in Experimental Medicine and Biology. 1994;360:21–27. doi: 10.1007/978-1-4615-2572-1_3. [DOI] [PubMed] [Google Scholar]
- Anichkov SV, Belen'kii ML. Pharmacology of the Carotid Body Chemo-receptors. New York: MacMillan; 1963. [Google Scholar]
- Ashcroft FM, Gribble FM. Correlating structure and function in ATP-sensitive K+ channels. Trends in Neurosciences. 1998;21:288–294. doi: 10.1016/s0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. ATP-sensitive K+ channels: a link between B-cell metabolism and insulin secretion. Biochemical Society Transactions. 1989;18:109–111. doi: 10.1042/bst0180109. [DOI] [PubMed] [Google Scholar]
- Ashford ML, Boden PR, Treherne JM. Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pflügers Archiv. 1990;415:479–483. doi: 10.1007/BF00373626. [DOI] [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. Journal of Cell Biology. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DF, Hille B. Mitochondrial oversight of cellular Ca2+ signaling. Current Opinion in Neurobiology. 1998;8:398–404. doi: 10.1016/s0959-4388(98)80067-6. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Chong C, Lee CC, Colmers WF, Miller RJ. Changes in mitochondrial function resulting from synaptic activity in the rat hippocampal slice. Journal of Neuroscience. 1998;18:4570–4587. doi: 10.1523/JNEUROSCI.18-12-04570.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscoe TJ, Duchen MR. Responses of type I cells dissociated from the rabbit carotid body to hypoxia. The Journal of Physiology. 1990;428:39–59. doi: 10.1113/jphysiol.1990.sp018199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitier E, Rea R, Duchen MR. Modulation of the propagation of intracellular calcium waves by mitochondria in rat cortical astrocytes. Biophysical Journal. 1999;76:A223. doi: 10.1083/jcb.145.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boss O, Muzzin P, Giacobino JP. The uncoupling proteins, a review. European Journal of Endocrinology. 1998;139:1–9. doi: 10.1530/eje.0.1390001. [DOI] [PubMed] [Google Scholar]
- Brini M, Marsault R, Bastianutto C, Alvarez J, Pozzan T, Rizzuto R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c). A critical evaluation. Journal of Biological Chemistry. 1995;270:9896–9903. doi: 10.1074/jbc.270.17.9896. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ Biochemistry. 1996;35:8483–8488. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. A novel oxygen-sensitive potassium current in rat carotid body type I cells. The Journal of Physiology. 1997;498:649–662. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones R. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. The Journal of Physiology. 1998;513:819–833. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho RF, Hansson O, Ward MW, Budd SL, Nicholls DG. Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. Journal of Neuroscience. 1998;18:10277–10288. doi: 10.1523/JNEUROSCI.18-24-10277.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Calcium and excitotoxic neuronal injury. Annals of the New York Academy of Sciences. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- Crompton M, Andreeva L. On the interactions of Ca2+ and cyclosporin A with a mitochondrial inner membrane pore: a study using cobaltammine complex inhibitors of the Ca2+ uniporter. Biochemical Journal. 1994;302:181–185. doi: 10.1042/bj3020181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Costi A. A heart mitochondrial pore with possible relevance to reperfusion injury. Biochemical Journal. 1990;266:33–39. doi: 10.1042/bj2660033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochemical Journal. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Virji S, Ward JM. Cyclophilin D binds strongly to complexes of the voltage dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. European Journal of Biochemistry. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- David G, Barrett DG, Barrett EF. Evidence that mitochondria bufffer physiological Ca2+ loads in lizard motor nerve terminals. The Journal of Physiology. 1998;509:59–97. doi: 10.1111/j.1469-7793.1998.059bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Fan CZ, Gambassi G, Hogue BA, Kudryashova I, Hansford RG. Altered pyruvate dehydrogenase control and mitochondrial free Ca2+ in hearts of cardiomyopathic hamsters. American Journal of Physiology. 1993;264:H2188–2197. doi: 10.1152/ajpheart.1993.264.6.H2188. [DOI] [PubMed] [Google Scholar]
- Donnelly DF. Are oxygen dependent K+ channels essential for carotid body chemo-transduction? Respiratory Physiology. 1997;110:211–218. doi: 10.1016/s0034-5687(97)00085-6. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Ca2+-dependent changes in the mitochondrial energetics of single mouse sensory neurones. Biochemical Journal. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Mitochondrial function in type I cells isolated from the rabbit carotid body. The Journal of Physiology. 1992a;450:13–31. doi: 10.1113/jphysiol.1992.sp019114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. The Journal of Physiology. 1992b;450:31–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Leyssens A, Crompton M. Transient mitochondrial depolarisations in response to focal SR calcium release in single rat cardiomyocytes. Journal of Cell Biology. 1998;142:975–988. doi: 10.1083/jcb.142.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, McGuinness O, Brown L, Crompton M. The role of the cyclosporin-A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovascular Research. 1993a;27:1790–1794. doi: 10.1093/cvr/27.10.1790. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Smith PA, Ashcroft FM. Substrate-dependent changes in mitochondrial function, [Ca2+]i and membrane channels in pancreatic beta cells in culture. Biochemical Journal. 1993b;294:35–42. doi: 10.1042/bj2940035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fein A, Tsacopoulos M. Activation of mitochondrial oxidative metabolism by calcium ions in Limulus ventral photoreceptor. Nature. 1988;331:437–440. doi: 10.1038/331437a0. [DOI] [PubMed] [Google Scholar]
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. Journal of Neuroscience. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Muller U. Recent developments in the molecular genetics of mitochondrial disorders. Journal of the Neurological Sciences. 1998;153:251–263. doi: 10.1016/s0022-510x(97)00295-5. [DOI] [PubMed] [Google Scholar]
- Gray MW. Rickettsia, typhus and the mitochondrial connection. Nature. 1998;396:109–110. doi: 10.1038/24030. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochemical Journal. 1995;307:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Buntinas L, Sparagna GC, Gunter KK. The Ca2+ transport mechanisms of mitochondria and Ca2+ uptake from physiological-type Ca2+ transients. Biochimica et Biophysica Acta. 1998;1366:5–15. doi: 10.1016/s0005-2728(98)00117-0. [DOI] [PubMed] [Google Scholar]
- Hagar RE, Burgstahler AD, Nathanson MH, Ehrlich BE. Type III InsP3 receptor channel stays open in the presence of increased calcium. Nature. 1998;396:81–84. doi: 10.1038/23954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Harvey J, McKenna F, Herson PS, Spanswick D, Ashford ML. Leptin activates ATP-sensitive potassium channels in the rat insulin-secreting cell line, CRI-G1. The Journal of Physiology. 1997;504:527–535. doi: 10.1111/j.1469-7793.1997.527bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Archives of Biochemistry and Biophysics. 1979a;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Archives of Biochemistry and Biophysics. 1979b;195:460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Archives of Biochemistry and Biophysics. 1979c;195:468–477. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock D, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. Journal of Cell Biology. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Ichas F, Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochimica et Biophysica Acta. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca2+ release in smooth muscle cells of the guinea pig taenia caeci. Journal of General Physiology. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson D, Duchen MR. Fluorescence imaging of the mitochondrial permeability transition in rat cortical astrocytes in culture. The Journal of Physiology. 1998;506.P:75P. [Google Scholar]
- Jou MJ, Peng TI, Sheu SS. Histamine induces oscillations of mitochondrial free Ca2+ concentration in single cultured rat brain astrocytes. The Journal of Physiology. 1996;497:299–308. doi: 10.1113/jphysiol.1996.sp021769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature. 1995;377:438–441. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Keelan J, Vergun O, Khodorov B, Duchen MR. Calcium-dependent mitochondrial depolarization specific to toxic glutamate stimulation in cultured rat hippocampal neurones. The Journal of Physiology. 1998;506.P:75P. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy ED, Wollheim CB. Role of mitochondrial calcium in metabolism-secretion coupling in nutrient-stimulated insulin release. Diabetes and Metabolism. 1998;24:15–24. [PubMed] [Google Scholar]
- Kerr JFR, Wyllie AM, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British Journal of Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Storozhevykh T, Vergun O, Vinskaya N. Dominant role of mitochondria in protection against a delayed neuronal Ca2+ overload induced by endogenous excitatory amino acids following a glutamate pulse. FEBS Letters. 1996;393:135–138. doi: 10.1016/0014-5793(96)00873-3. [DOI] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Vergun O, Storozhevykh T, Vinskaya N. Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Letters. 1996;397:230–234. doi: 10.1016/s0014-5793(96)01139-8. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. Journal of Neuroscience. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annual Review of Physiology. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Chugh DK, Mokashi A, Vinogradov S, Osanai S, Wilson DF. Cytochrome oxidase is the primary oxygen sensor in the cat carotid body. Advances in Experimental Medicine and Biology. 1996;388:213–217. doi: 10.1007/978-1-4613-0333-6_26. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Roy A, Rozanov C, Mokashi A. K+-current modulated by PO2 in type I cells in rat carotid body is not a chemosensor. Brain Research. 1998;794:162–165. doi: 10.1016/s0006-8993(98)00276-5. [DOI] [PubMed] [Google Scholar]
- Landolfi B, Curci S, Debellis L, Pozzan T, Hofer AM. Ca2+ homeostasis in the agonist-sensitive internal store: functional interactions between mitochondria and the ER measured in situ in intact cells. Journal of Cell Biology. 1998;142:1235–1243. doi: 10.1083/jcb.142.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrie AM, Rizzuto R, Pozzan T, Simpson AW. A role for calcium influx in the regulation of mitochondrial calcium in endothelial cells. Journal of Biological Chemistry. 1996;271:10753–10759. doi: 10.1074/jbc.271.18.10753. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochimica et Biophysica Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- Leyssens A, Nowicky AV, Patterson DL, Crompton M, Duchen MR. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. The Journal of Physiology. 1996;496:111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J H-C, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, Hansen TW, Goldman S, Nedergaard M. Gap-junction mediated propagation and amplification of cell injury. Nature Neuroscience. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J. Oxygen-sensing by ion channels and the regulation of cellular functions. Trends in Neurosciences. 1996;19:435–440. doi: 10.1016/0166-2236(96)10050-3. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiological Reviews. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Maechler P, Kennedy ED, Pozzan T, Wollheim CB. Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic beta-cells. EMBO Journal. 1997;16:3833–3841. doi: 10.1093/emboj/16.13.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler P, Kennedy ED, Wang H, Wollheim CB. Desensitization of mitochondrial Ca2+ and insulin secretion responses in the beta cell. Journal of Biological Chemistry. 1998;273:20770–20778. doi: 10.1074/jbc.273.33.20770. [DOI] [PubMed] [Google Scholar]
- Margulis L. Archaeal-eubacterial mergers in the origin of Eukarya: phylogenetic classification of life. Proceedings of the National Academy of Sciences of the USA. 1996;93:1071–1076. doi: 10.1073/pnas.93.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–203. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, Bers DM. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. Journal of Biological Chemistry. 1998;273:10223–10231. doi: 10.1074/jbc.273.17.10223. [DOI] [PubMed] [Google Scholar]
- Melamed-Book N, Rahamimoff R. The revival of the role of the mitochondrion in regulation of transmitter release. The Journal of Physiology. 1998;509:2. doi: 10.1111/j.1469-7793.1998.002bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RJ. Mitochondria - the Kraken wakes. Trends in Neurosciences. 1998;21:95–97. doi: 10.1016/s0166-2236(97)01206-x. [DOI] [PubMed] [Google Scholar]
- Mills E, Jöbsis FF. Simultaneous measurement of cytochrome a3 reduction and chemoreceptor afferent activity in the carotid body. Nature. 1970;225:1147–1149. doi: 10.1038/2251147a0. [DOI] [PubMed] [Google Scholar]
- Mills E, Jöbsis FF. Mitochondrial respiratory chain of carotid body and chemoreceptor response to changes in oxygen tension. Journal of Neurophysiology. 1972;35:405–428. doi: 10.1152/jn.1972.35.4.405. [DOI] [PubMed] [Google Scholar]
- Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. 1967;213:137–139. doi: 10.1038/213137a0. [DOI] [PubMed] [Google Scholar]
- Mogami H, Nakano K, Tepikin AV, Petersen OH. Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell. 1997;88:49–55. doi: 10.1016/s0092-8674(00)81857-7. [DOI] [PubMed] [Google Scholar]
- Morgan-Hughes JA, Schapira AH, Cooper JM, Holt IJ, Harding AE, Clark JB. The molecular pathology of respiratory-chain dysfunction in human mitochondrial myopathies. Biochimica et Biophysica Acta. 1990;1018:217–222. doi: 10.1016/0005-2728(90)90252-y. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ferguson SJ. Bioenergetics 2. Academic Press; 1992. [Google Scholar]
- Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-methyl-D-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- Noji H, Yasuda R, Yoshida M, Kinoshita K., Jr Direct observation of the rotation of F1-ATPase. Nature. 1997;386:299–302. doi: 10.1038/386299a0. [DOI] [PubMed] [Google Scholar]
- Palou A, Pico C, Bonet ML, Oliver P. The uncoupling protein, thermogenin. International Journal of Biochemistry and Cell Biology. 1998;30:7–11. doi: 10.1016/s1357-2725(97)00065-4. [DOI] [PubMed] [Google Scholar]
- Peers C. Oxygen-sensitive ion channels. Trends in Pharmacological Sciences. 1997;18:405–408. doi: 10.1016/s0165-6147(97)01120-6. [DOI] [PubMed] [Google Scholar]
- Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. Journal of Biological Chemistry. 1994;269:16638–16642. [PubMed] [Google Scholar]
- Peuchen S, Duchen MR, Clark JB. Energy metabolism of adult astrocytes in vitro. Neuroscience. 1996;71:855–870. doi: 10.1016/0306-4522(95)00480-7. [DOI] [PubMed] [Google Scholar]
- Pralong WF, Spat A, Wollheim CB. Dynamic pacing of cell metabolism by intracellular Ca2+ transients. Journal of Biological Chemistry. 1994;269:27310–27314. [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiological Reviews. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Ramos-Franco J, Fill M, Mignery GA. Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophysical Journal. 1998;75:834–839. doi: 10.1016/S0006-3495(98)77572-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. Journal of Cell Biology. 1994;126:1183–1194. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–327. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO Journal. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe IC, Treherne JM, Ashford ML. Activation by intracellular ATP of a potassium channel in neurones from rat basomedial hypothalamus. The Journal of Physiology. 1996;490:97–113. doi: 10.1113/jphysiol.1996.sp021129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Gu M, Taanman JW, Tabrizi SJ, Seaton T, Cleeter M, Cooper JM. Mitochondria in the etiology and pathogenesis of Parkinson's disease. Annals of Neurology. 1998;44(suppl. 1):S89–98. doi: 10.1002/ana.410440714. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. Journal of Neuroscience. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd CA, Simpson PB, Sharp AH, Nucifora FC, Ross CA, Lange GD, Russell JT. Comparison of Type 2 inositol 1,4,5,-trisphosphate receptor distribution and subcellular Ca2+ release sites that support Ca2+ waves in cultured astrocytes. Journal of Neurochemistry. 1997;68:2317–2327. doi: 10.1046/j.1471-4159.1997.68062317.x. [DOI] [PubMed] [Google Scholar]
- Simpson PB, Russell JT. Role of mitochondrial Ca2+ regulation in neuronal and glial cell signalling. Brain Research Reviews. 1998;26:72–81. doi: 10.1016/s0165-0173(97)00056-8. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Letters. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390:521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nature Neuroscience. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Tang YG, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. The Journal of Physiology. 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Walker JE. The regulation of catalysis in ATP synthase. Current Opinion in Structural Biology. 1994;4:912–918. doi: 10.1016/0959-440x(94)90274-7. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. Journal of Neuroscience. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. Journal of Neuroscience. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurones. The Journal of Physiology. 1997;498:31–47. doi: 10.1113/jphysiol.1997.sp021839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochimica et Biophysica Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]