

Abstract
We have studied capacitative calcium entry (CCE) under different experimental conditions in fura-2-loaded mouse pancreatic acinar cells by digital microscopic fluorimetry. CCE was investigated during [Ca2+]i decay after cell stimulation with a supramaximal concentration of ACh (10 μM) or during Ca2+ readmission in Ca2+-depleted cells (pretreated with thapsigargin or ACh).
La3+ and Zn2+ (100 μM) inhibited CCE during Ca2+ readmission but had negligible effects during ACh decay. In contrast flufenamic acid (100 μM), an inhibitor of non-selective cation channels, genistein (10 μM), a broad-range tyrosine kinase inhibitor, and piceatannol (10 μM), an inhibitor specific for non-receptor Syk tyrosine kinase, inhibited CCE during ACh decay but not during Ca2+ reintroduction.
Simultaneous detection of Mn2+ entry and [Ca2+]i measurement showed that, in the presence of extracellular calcium, application of 100 μM Mn2+ during ACh decay resulted in manganese influx without alteration of calcium influx, whilst when applied during Ca2+ readmission, Mn2+ entry was significantly smaller and induced a clear inhibition of CCE.
Application of the specific protein kinase C inhibitor GF109293X (3 μM) reduced CCE in Ca2+-depleted cells, whereas the activator phorbol 12-myristate, 13-acetate (3 μM) increased Ca2+ entry.
Based on these results we propose that cholinergic stimulation of mouse pancreatic acinar cells induces Ca2+ influx with an initial phase operated by a non-specific cation channel, sensitive to flufenamic acid and tyrosine kinase inhibitors but insensitive to lanthanum and divalent cations, followed by a moderately Ca2+-selective conductance inhibited by lanthanum and divalent cations.
Cytosolic calcium concentration ([Ca2+]i) is a key regulatory factor for a large number of cellular proccesses such as secretion, contraction, metabolism or even gene expression and apoptosis. Many hormones and neurotransmitters increase [Ca2+]i via activation of phospholipase C, which results in generation of inositol 1, 4, 5-trisphosphate (InsP3) and diacylglycerol. InsP3 releases calcium from intracellular pools by binding to specific receptors, and diacylglycerol activates several isoforms of protein kinase C (PKC), an enzyme involved in the control of numerous cellular functions.
However, to maintain Ca2+ signals and refill intracellular pools, agonists also activate entry of extracellular calcium following its electrochemical gradient through plasma membrane channels. In non-excitable cells, and also in excitable tissues, the main mechanism for calcium influx is depletion of intracellular Ca2+ stores, which opens plasma membrane Ca2+ channels using an unknown signal. This mechanism has been termed capacitative calcium entry (CCE) by Putney, and although a number of theories have been proposed, there is no convincing explanation (Clapham, 1995), in spite of its evident physiological and clinical relevance.
The identity of the plasma membrane Ca2+ channel involved in CCE is also controversial. Electrophysiological studies have described in mast cells an inward Ca2+ release-activated Ca2+ current (ICRAC) (Hoth & Penner, 1992), highly Ca2+ selective and sensitive to inhibition by La3+ and divalent cations (Hoth & Penner, 1993). ICRAC shows both fast and slow negative feedback due to inhibition by Ca2+ and PKC (Parekh & Penner, 1995; Zweifach & Lewis, 1995). However, different Ca2+ entry currents have been subsequently described, with the selectivity for calcium varying from moderate to high, depending on cell type (Clapham, 1995).
In pancreatic acinar cells there are conflicting reports regarding the possible existence of ICRAC channels. Although a moderately Ca2+-selective ICRAC has been described in rat pancreas (Bahnson et al. 1993), a recent paper describes in mouse acinar cells a La3+-insensitive non-selective cation channel as the main route for CCE (Krause et al. 1996). This current, termed INCRAC (for non-selective calcium release-activated current), can be inhibited by the tyrosine kinase inhibitor genistein, and is different from the non-selective cation channel activated by cytosolic calcium previously described in rodent exocrine pancreas (Pfeiffer et al. 1995).
The aim of our study was to characterize the route involved in Ca2+ influx in mouse pancreatic acinar cells. Our data indicate that, upon Ca2+ mobilization, there is a sequential activation of at least two different Ca2+ entry pathways or alternatively a single channel with two different states: an initial non-specific conductance, sensitive to flufenamic acid and genistein and scarcely sensitive to divalent cations and La3+, and a late conductance moderately specific for Ca2+ and inhibited by lanthanum and manganese, similar to the previously described ICRAC. In addition, we present evidence that PKC is a putative modulator of this Ca2+ influx pathway.
Some of these data were presented to the Physiological Society Meeting in Liverpool (Camello et al. 1998).
METHODS
Preparation of acinar cells
A suspension of single cells and small acini was prepared from mouse pancreas, after dislocation of the neck, by enzymatic dispersion as previously described (González et al. 1997). Briefly, the pancreas was injected with a small volume (1 ml) of collagenase solution (Worthington, 200 U ml−1) and incubated at 37°C under gentle agitation for 6-12 min. Finally, the cells were released by vigorous manual agitation. Throughout the preparation procedure, as well as during the loading and perfusion, we used a physiological solution containing (mM): 140 NaCl, 4·7 KCl, 2 CaCl2, 1·1 MgCl2, 10 glucose, 10 Hepes and 0·01 % trypsin inhibitor (soybean); pH 7·4. Ca2+-free solutions had a similar composition but Ca2+ was omitted and 1 mM EGTA was added.
Cell loading and [Ca2+]i determination
After isolation the cells were suspended in physiological solution and loaded with the fluorescent ratiometric calcium indicator fura-2 AM (1-2 μM, 30 min, room temperature, 20-25°C). Once loaded, the cells were washed and used within 2-4 h.
For experiments, a small volume of cell suspension was placed on a thin glass coverslip attached to a Perspex perfusion chamber. Perfusion (approximately 1 ml min−1) at room temperature was started after a 2 min period to allow spontaneous attachment of the cells to the coverslip. No coating treatment was necessary to immobilize the cells. The chamber was placed on the stage of an inverted fluorescence-equipped microscope (Nikon Diaphot). Cells were excited at 340 and 380 nm by a computer-controlled filter wheel, and the emitted images were captured by a cooled digital CCD camera (C-6790, Hamamatsu Photonics) and recorded using dedicated software (Argus-HisCa, Hamamatsu Photonics). After the calculation of the 340 nm/380 nm ratio pixel by pixel, the intracellular free calcium concentration ([Ca2+]i) was determined using standard methods (Grynkiewicz et al. 1985). The calibration parameters Rmax (3·2), Rmin (0·15) and Sf/Sb (4) were determined in vivo using 10 μM ionomycin in Ca2+-free and 10 mM Ca2+ solutions. We used a Kd value of 250 nM for fura-2 (Grynkiewicz et al. 1985).
Determination of manganese entry
To study Mn2+ influx and the effects of this cation on Ca2+ entry, we used pulses of 100 μM MnCl2 added to the normal Ca2+-containing physiological solution. In these experiments we examined the fluorescence emitted by fura-2 under 340 and 380 nm excitation wavelengths (F340 and F380). Mn2+ entry was assessed following the procedure described by Alonso-Torre et al. (1993) and modified by Shuttleworth (1995). Briefly, F340 and F380 are added after factorization in such a way that total corrected fluorescence (Ftot= (F340+F380) ×δ) is independent of [Ca2+]i, but decreases when fura-2 is quenched by manganese. The correction factor δ was calculated separately for each individual cell by observing the effect of the initial ACh-induced large [Ca2+]i peak applied at the beginning of these experiments (see Fig. 9), in the absence of Mn2+. In our experimental conditions, δ is close to the resting fura-2 ratio divided by the peak evoked by ACh. This allows simultaneous determination of ratiometric [Ca2+]i and Mn2+ entry (Alonso-Torre et al. 1993; Suttleworth, 1995). To estimate the rate of Mn2+ entry, we calculated the decline in Ftot in response to Mn2+ quench as the fold increase of the slope with respect to the immediately preceding 60 s (Shuttleworth, 1995).
Figure 9. Effect and entry of extracellular Mn2+ during Ca2+ mobilization in response to cholinergic stimulation of mouse pancreatic acinar cells.
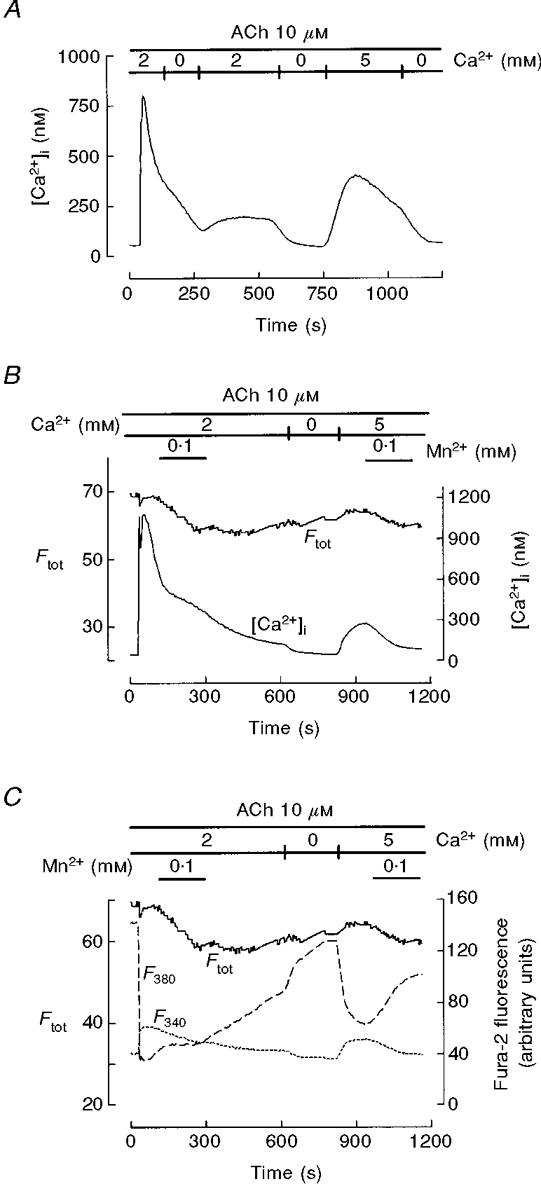
A, single cells were treated with 10 μM ACh in 2 mM Ca2+-containing solution and exposed to pulses of Ca2+-free solution during the early phase of decay and after [Ca2+]i stabilization close to resting levels. After the second exposure to Ca2+-free medium (to achieve complete Ca2+ depletion), 5 mM Ca2+ was introduced to allow calcium influx. The effect of Ca2+ removal shows the presence of Ca2+ entry during both the ACh-induced decay and the final CCE. B, cells were perifused with 100 μM Mn2+ in the presence of 2 mM extracellular Ca2+ during the decay of the ACh response and during application of 5 mM external calcium. Traces represent [Ca2+]i and the corrected total fluorescence (Ftot), which serves to detect Mn2+ entry. C, original F340 and F380 traces from the same cell recorded in B. Mn2+ entry into the cytosol during the ACh decay, as indicated by quenching of F40 and delay in F380 recovery (C) and by a decrease in Ftot (B and C), is not accompanied by inhibition of Ca2+ influx (B). In comparison, during the Ca2+ readmission-evoked plateau, Mn2+ entry is weaker but CCE inhibition is evident. This record is representative of 6 experiments.{FONT size
Materials
Chemicals were purchased from Sigma (Spain), except collagenase CLSPA, which was obtained from Worthington Biochemical Corporation (USA), fura-2, which was from Molecular Probes Europe (The Netherlands), and GF109293X and PP-1, which were from Calbiochem (USA).
RESULTS
To study CCE in fura-2-loaded mouse pancreatic acinar cells we have used commonly accepted protocols based on depletion of intracellular Ca2+ stores. Perfusion of pancreatic acinar cells with a Ca2+-free medium containing thapsigargin (1 μM), a specific inhibitor of the Ca2+ pump of internal stores (Thastrup et al. 1989), resulted in a transient [Ca2+]i increase due to release of Ca2+ from intracellular pools. Subsequent treatment with a Ca2+-containing solution induced a sustained [Ca2+]i increase indicative of CCE, as shown by its dependence on the presence of extracellular Ca2+ (Fig. 1A).
Figure 1. Capacitative calcium entry in pancreatic acinar cells is inhibited by lanthanum and zinc.
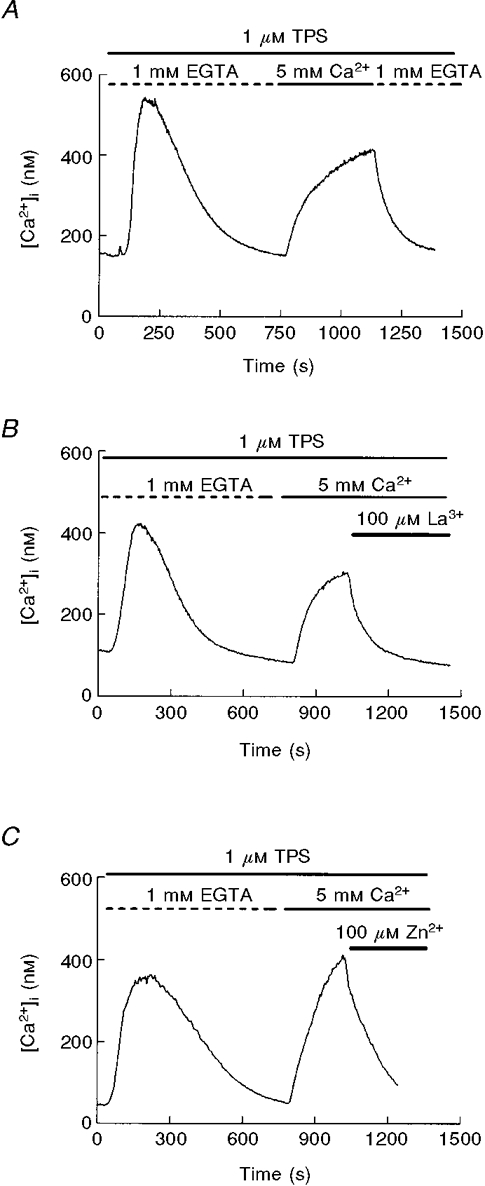
A, capacitative calcium entry in mouse pancreatic acinar cells. After depletion of intracellular Ca2+ pools with thapsigargin (TPS; 1 μM) in a Ca2+-free solution, application of 5 mM extracellular calcium results in a [Ca2+]i plateau due to Ca2+ influx. B, effect of 100 μM LaCl3 on Ca2+ influx. Lanthanum was added to the Ca2+-containing solution. C, effect of 100 μM ZnCl2 during thapsigargin-induced Ca2+ entry. All the traces are representative of at least 4 (C) or 6 (A, B) independent experiments.
Since inhibition by divalent cations and La3+ is a key figure in the original description of the store-operated Ca2+ channel ICRAC (Hoth & Penner, 1993) we tested the effects of La3+ and Zn2+. As shown in Fig. 1B, CCE evoked by thapsigargin was clearly inhibited by La3+ in all the cells tested. This inhibition was also present when CCE was achieved using a physiological extracellular Ca2+ level (2 mM; data not shown) instead of 5 mM. The inhibition was fast and dose dependent (not shown), and was reproduced by Zn2+ (Fig. 1C).
Another tool to inhibit CCE is flufenamic acid, an inhibitor of non-selective cation channels (Schumann et al. 1994; Weiser & Wienrich, 1996), which is a potential secondary route for Ca2+ in rodent pancreatic acinar cells given the existence of non-selective cation channels in these cells (Thorn & Petersen, 1992). As can be observed in Fig. 2, flufenamic acid did not reduce entry of extracellular calcium after depletion of intracellular Ca2+ pools, indicating that the non-selective cation channel of rodent pancreas is not the main entry pathway involved in CCE in Ca2+-depleted cells. Moreover, the sustained [Ca2+]i during calcium entry was enhanced in the presence of flufenamic acid, suggesting that a non-specific cation channel reduces the electrochemical gradient for calcium entry.
Figure 2. Effect of flufenamic acid on thapsigargin-evoked CCE.
Once Ca2+ influx was established in Ca2+-depleted cells, application of flufenamic acid (100 μM) did not reduce the entry of extracellular calcium. The trace is representative of 4 experiments.
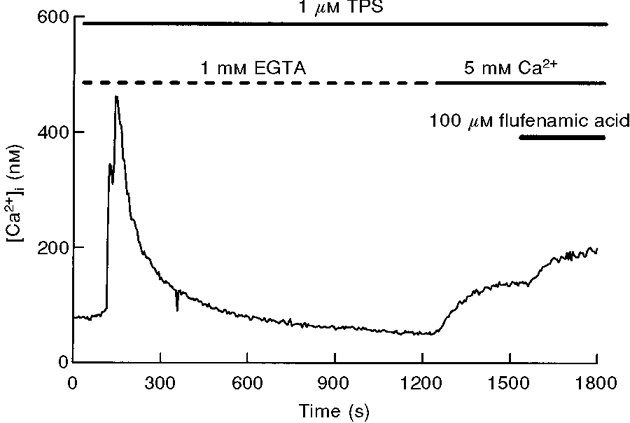
It has previously been reported that, in pancreatic acinar cells, tyrosine kinase inhibitors such as genistein and tyrphostins acutely block CCE (Yule et al. 1994; Pfeiffer et al. 1995). To test this possibility in thapsigargin-treated cells we used 10 μM genistein (Fig. 3A), tyrphostin A25 (Fig. 3B), another broad-range tyrosine kinase inhibitor, and PP-1 and piceatannol, inhibitors specific for several non-receptor tyrosine kinases (Oliver et al. 1994; Hanke et al. 1996), during the Ca2+ entry-induced plateau (Fig. 3) or several minutes before readmission of extracellular Ca2+ (Fig. 4). As shown in Figs 3 and 4, in the two experimental conditions the effect was residual or not significant. The only exception was the acute application of genistein, which induced a 30 % inhibition of CCE; however, this result is not consistent with the lack of effect in the case of pretreatment, a condition in which the effect was expected to be larger (Figs 3C and 4B).
Figure 3. Effect of application of genistein or tyrphostin A25 during CCE in mouse pancreatic acinar cells.
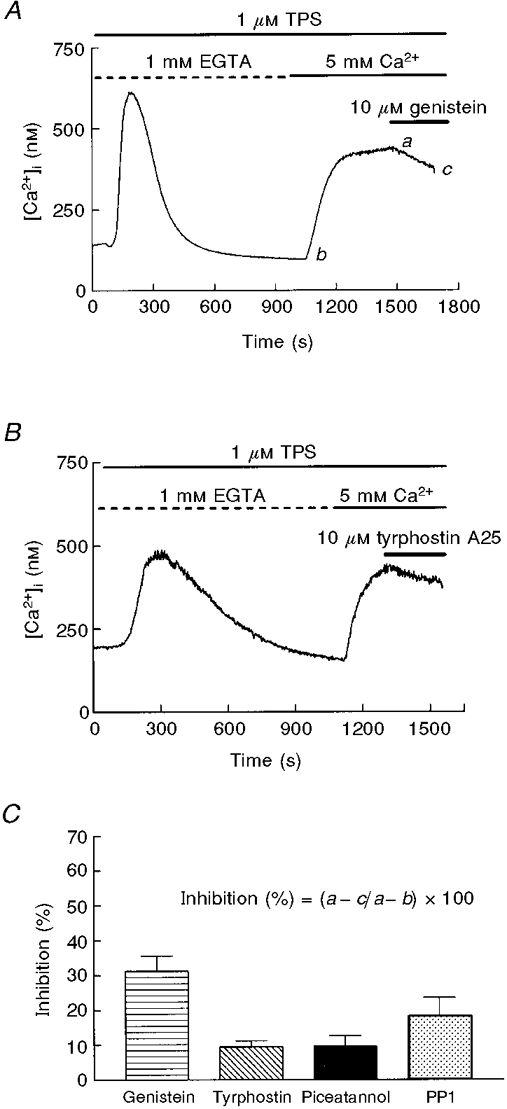
Genistein (A; 10 μM) and tyrphostin A25 (B; 10 μM), were applied during the plateau phase of Ca2+ entry. Data are typical of 5 (A) or 3 (B) separate experiments. C, the effects of different tyrosine kinase inhibitors following the protocol shown in A and B. Percentage inhibition is calculated by substracting the [Ca2+]i decrease evoked by the inhibitor (a - c in A) from the previous [Ca2+]i increase induced by Ca2+ readmission (a - b in A). Data (means ±s.e.m.) are from 4 separate experiments, except for genistein (5 experiments).
Figure 4. Effect of pretreatment with tyrosine kinase inhibitors on thapsigargin-evoked CCE.
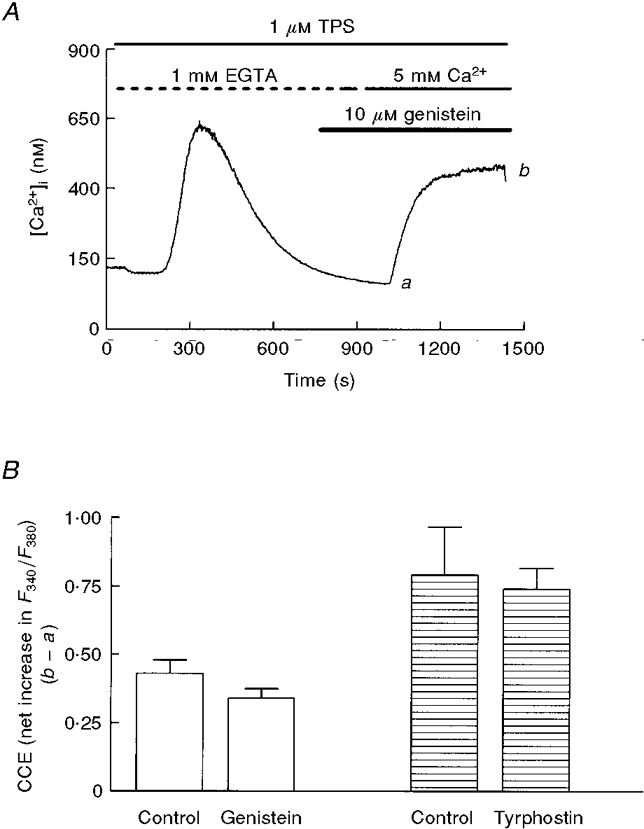
A, following a protocol similar to that in Fig. 3, mouse pancreatic acinar cells were Ca2+ depleted before activation of CCE by admission of extracellular Ca2+. Genistein (10 μM) was applied from the beginning of the experiment. B, effects of genistein and tyrphostin A25 following the protocol described in A. Bars represent means ±s.e.m. of the net increment of F340/F380 (taken as b - a in A) during the CCE-evoked plateau in control and pretreated cells from the same preparations (5 independent experiments).
To check a possible involvement of PKC in CCE we used the exogenous activator of PKC phorbol 12-myristate, 13-acetate (PMA) and the specific inhibitor, GF109293X (Toullec et al. 1991). After depletion of internal pools with thapsigargin, pancreatic acinar cells were perifused with either PMA or GF109293X (3 μM) 5 min before initiation of CCE (Fig. 5). Calcium influx was significantly enhanced in PMA-treated cells, while GF109293X induced a significant impairment of calcium entry. Moreover, a similar finding was attained when PMA or GF109293X was applied during the sustained plateau of CCE (data not shown), showing that PKC can promote Ca2+ influx.
Figure 5. PKC modulates CCE in pancreatic acinar cells.
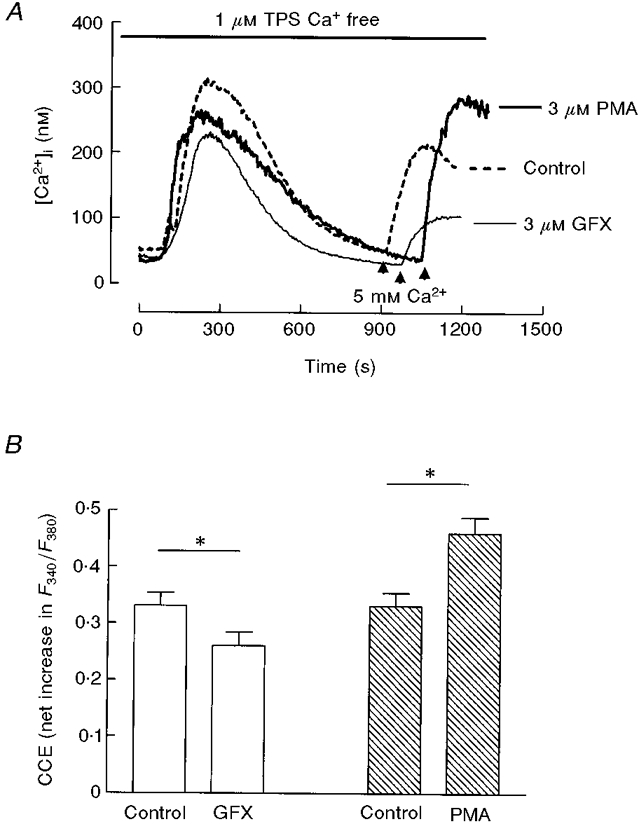
A, the PKC inhibitor GF109293X (bisindolylmaleimide I; 3 μM) and the exogenous activator of PKC PMA (3 μM) were applied 5 min before readmission of extracellular calcium, indicated by the arrows. B, histogram depicting means ±s.e.m. of CCE (net increase in F340/F380) from 5 independent experiments following the protocol shown in A. *P < 0·05, t test.
Another index of capacitative calcium entry is the plateau phase of the Ca2+ response to calcium-mediated agonists: during Ca2+ mobilization by physiological agonists of pancreatic acinar cells, such as acetylcholine (ACh) and cholecystokinin (CCK), activation of CCE contributes to the maintenance of the Ca2+ signal (Yule & Gallacher, 1988; Hurley & Brinck, 1990). As shown in Fig. 6A, impairment of Ca2+ influx during the sustained phase of the ACh-evoked response by removal of extracellular calcium induced an accelerated decay of [Ca2+]i. Upon application of Ca2+-free solution, the rate of decay was enhanced about 2-fold (slopes of decay 0·0032 ± 0·0008 before calcium removal vs. 0·0061 ± 0·001 after removal, means ±s.e.m., P < 0·01, t test, 7 experiments). Also, the whole time course of the response to ACh was partially dependent on calcium entry, as shown in the inset of Fig. 6A. Therefore, in agreement with previous studies, we used maintenance of this decay as an index of calcium influx (Muallem et al. 1989, 1990). When La3+ was applied in the presence of ACh we obtained a small inhibition in [Ca2+]i (Fig. 6B). This pattern was observed in 72 % of 67 studied cells, without appreciable effect in the remaining 28 %. A similar inhibition was obtained when cells were perfused with Zn2+ during the response to ACh (Fig. 6C) (64 % of 50 cells, 36 % without effect). This is in contrast with the clear effect of the two cations in Ca2+-depleted cells shown in Fig. 1.
Figure 6. Effect of extracellular calcium, La3+ and Zn2+ on the ACh-evoked [Ca2+]i decay.
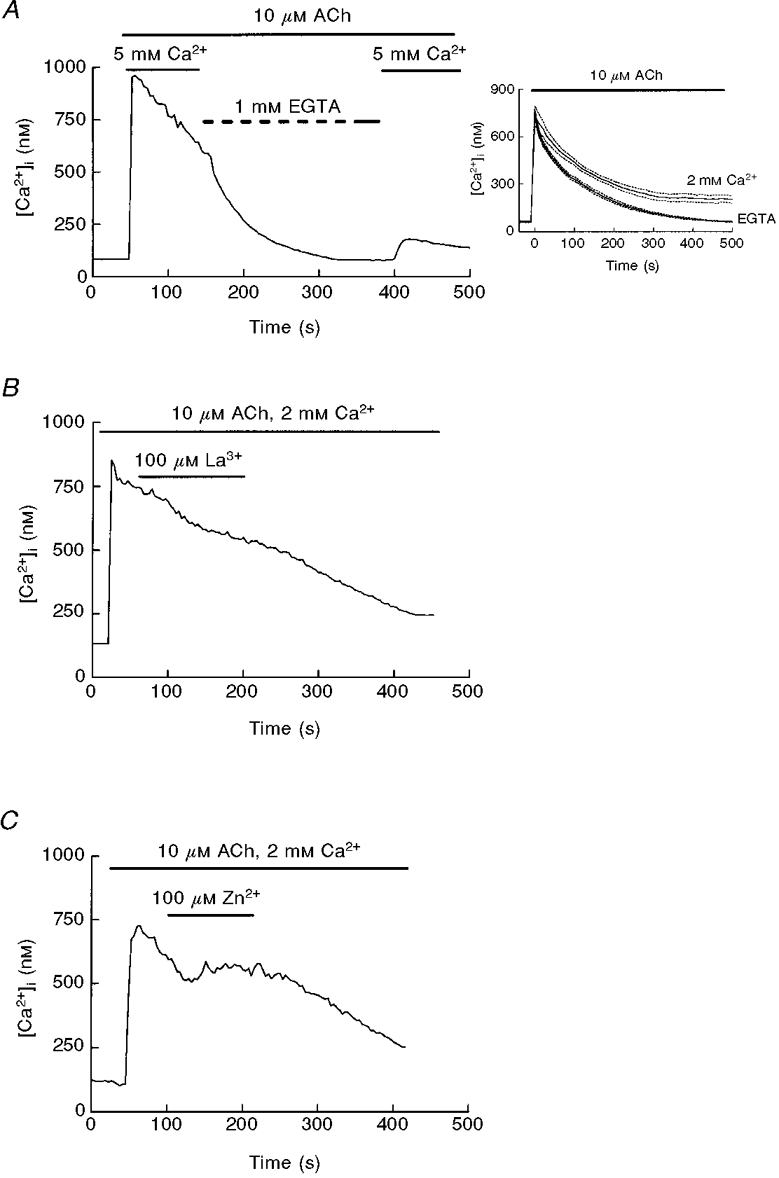
A, treatment of mouse pancreatic acinar cells with 10 μM ACh in 2 mM Ca2+-containing medium results in a [Ca2+]i transient with a sustained plateau phase dependent on influx of extracellular Ca2+, as shown by the effect of a Ca2+-free solution. The inset shows the averaged responses (means ±s.e.m.) to ACh in the presence (2 mM Ca2+) and the absence (1 mM EGTA) of extracellular calcium. All the responses were previously aligned at the onset of the response. B and C, addition of 100 μM La3+ (B) or Zn2+ (C) during the plateau phase of the Ca2+ response to ACh caused only a slight reduction in Ca2+ entry. Traces are typical of 7 (A), 8 (B) or 4 experiments (C).
When flufenamic acid was applied during the decay of the sustained response to ACh there was a clear reduction in [Ca2+]i (Fig. 7) (the rate of decay expressed as slope of the signal before and after addition of flufenamic acid was 0·0044 ± 0·001 vs. 0·0099 ± 0·002, P < 0·05, 5 experiments), while this drug was without effect when applied during Ca2+ entry in Ca2+-depleted cells (see Fig. 2). Thus the pattern of inhibition of CCE by flufenamic acid (i.e. inhibition during ACh decay but not during the Ca2+ readmission plateau) is opposite to that of La3+ and Zn2+ (weak reduction during decay and strong inhibition of late entry).
Figure 7. Inhibition of ACh-evoked [Ca2+]i decay by flufenamic acid.
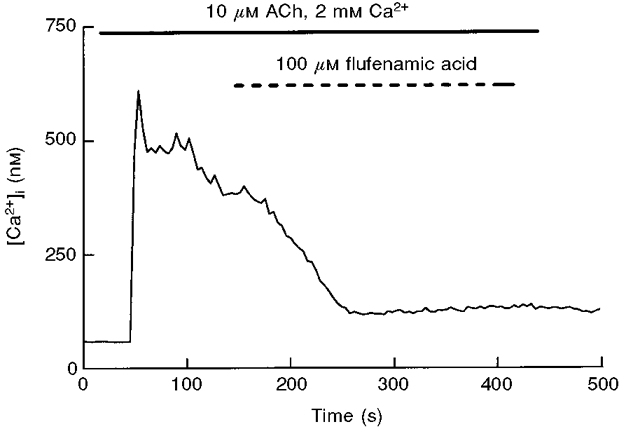
Pancreatic acinar cells were perifused with 100 μM flufenamic acid during the plateau phase of the [Ca2+]i response induced by 10 μM ACh. Data are representative of 5 experiments.
We also investigated the effects of tyrosine kinase inhibitors on the ACh-evoked [Ca2+]i decay. Unlike their scarce or non-existent effects during calcium influx in Ca2+-depleted cells (Figs 3 and 4), when genistein and piceatannol were applied during the sustained response to ACh we observed a clear reduction in [Ca2+]i, indicative of a reduction of Ca2+ influx (Fig. 8) (rate of decay before genistein 0·0033 ± 0·001 vs. 0·0080 ± 0·0007 after genistein, P < 0·05, 5 experiments; decay before piceatannol, 0·0023 ± 0·0006 vs. 0·0047 ± 0·0004, P < 0·05, 5 experiments). Therefore their pattern of effect on CCE was similar to that of flufenamic acid.
Figure 8. Effects of tyrosine kinase inhibitors on ACh-induced [Ca2+]i decay.
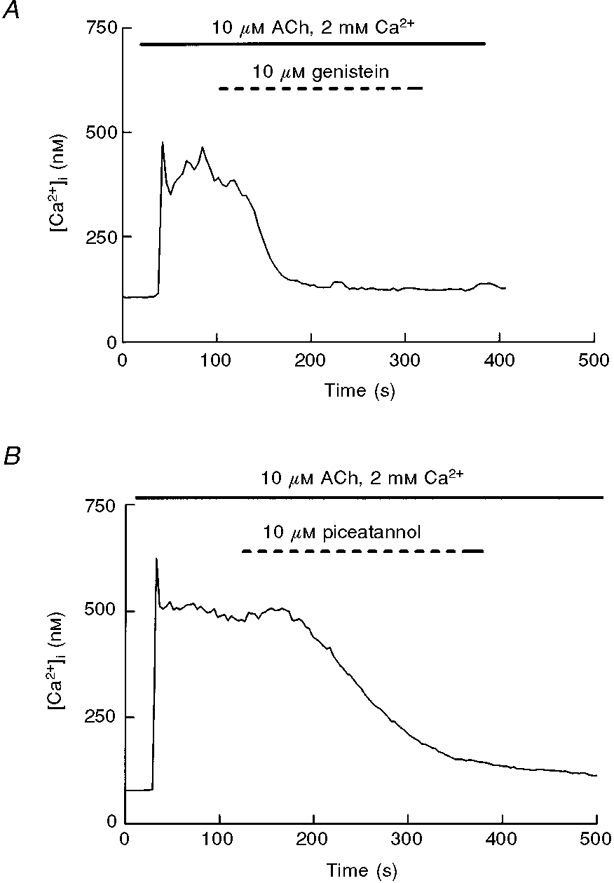
ACh (10 μM)-stimulated pancreatic acinar cells were perfused with either genistein (A, 10 μM) or piceatannol (B, 10 μM) during the decay phase of the ACh response. Traces are typical of 6 (A) and 5 (B) experiments.
The differences in the sensitivity of the ACh-evoked decay and the Ca2+ readmission-induced plateau to several experimental conditions suggested the existence of two different Ca2+ entry pathways in mouse pancreatic acinar cells: while the initial Ca2+ entry could operate through a non-specific channel, in Ca2+-depleted cells influx could be explained by the presence of ICRAC-like conductance. Therefore, we wanted to explore this possibility further in experiments designed to evaluate in the same individual cell the two phases of calcium entry, i.e. ACh-induced decay and the Ca2+ readmission-evoked plateau. As shown in Fig. 9A, in the presence of extracellular Ca2+, 10 μM ACh induced a [Ca2+]i peak followed by a decay that is sensitive to removal of external calcium and eventually reaches a steady level slightly higher than resting values. If extracellular calcium is removed after stabilization (to complete Ca2+ depletion), perfusion with a Ca2+-containing solution induces a plateau in [Ca2+]i due to CCE. As can be seen in Fig. 9A, the two stages of calcium influx (i.e. early phase of ACh decay and the Ca2+ readmission-induced plateau) are sensitive to removal of external calcium.
If our hypothesis was correct the two phases should display a different behaviour in the presence of Mn2+, since this cation, provided it penetrates the cells, can be used as a surrogate for calcium entry given its quenching effect on fura-2 (e.g. Muallem et al. 1990; Jacob, 1992), and at the same time is an inhibitor of ICRAC (Hoth & Penner, 1993). To assess Mn2+ entry we examined its effect on the recorded fura-2 fluorescence under 340 and 380 nm excitation wavelengths with simultaneous ratiometric [Ca2+]i determination (see Methods).
We expected Mn2+ to enter the cell during the ACh decay, while the final Ca2+ reintroduction-induced plateau should display inhibition by manganese and a small permeation of this cation. As seen in Fig. 9B, this was the case. Mn2+ influx was much more patent during the initial [Ca2+]i decay than during the Ca2+ readmission phase: the rate of Mn2+ quench, expressed as the fold increase in the slope of Ftot evoked by Mn2+ application with respect to the immediately preceding 60 s, was 22·58 ± 2·9 during the ACh decay compared with 1·76 ± 0·36 during the plateau phase (6 experiments, P < 0·05). On the other hand, manganese only inhibited CCE when applied during Ca2+ readmission (note the lack of effect of Mn2+ on the ACh decay, Fig. 9B). This behaviour was present in most of the cells examined (78 % of 61 studied cells, 6 independent experiments), and indicates different selectivity and sensitivity during the two phases of calcium entry. This result supports the hypothesis that at least two Ca2+ conductances (or one conductance with two different stages) are present in mouse pancreatic acinar cells.
The presented data cannot rule out the possibility that the initial phase of influx is not CCE, but is elicited by cholinergic activation of a non-capacitative mechanism, independent of Ca2+ depletion. If this is true one would expect a [Ca2+]i increase when ACh is applied in the presence of extracellular calcium after depletion of the stores using thapsigargin, since ACh would open the non-capacitative conductance. However, as shown in Fig. 10A, ACh reduces[Ca2+]i, probably by enhancement of Ca2+ extrusion. Another indication that store depletion can activate the two phases of calcium entry is presented in Fig. 10B. The response to thapsigargin in the presence of extracellular Ca2+ has a low sensitivity to La3+ during the initial phase, which is still inhibited by a Ca2+-free solution, while the late phase is clearly inhibited by La3+. This pattern was present in 85 % of 34 studied cells (4 independent experiments).
Figure 10. Effects of lanthanum and ACh on capacitative calcium entry evoked by thapsigargin.
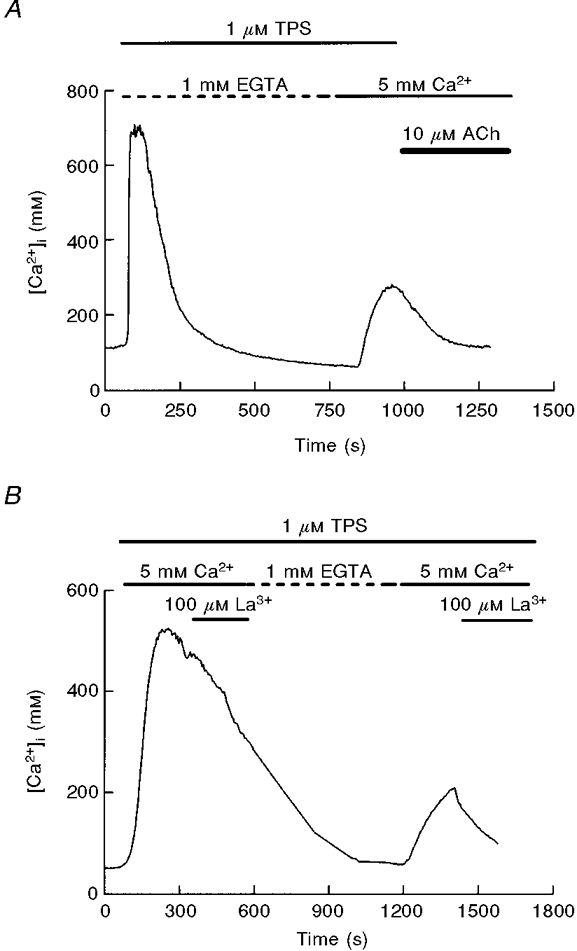
A, effect of 10 μM ACh application during the sustained plateau evoked by Ca2+ influx in cells pretreated with thapsigargin (1 μM). The decrease in [Ca2+]i was observed in 4 experiments. B, CCE evoked by depletion of the stores shows temporal change in sensitivity to lanthanum. Cells were treated initially with 1 μM thapsigargin in the presence of external calcium, and subsequentially perifused with 100 μM La3+- and EGTA-containing solutions. After posterior readmission of extracellular calcium a second application of lanthanum was given. The trace, showing a differential effect of lanthanum on the initial and late phases of CCE, is representative of 4 separate experiments.
DISCUSSION
Similar to other non-excitable tissues, influx of extracellular calcium in pancreatic acinar cells is mediated by a voltage-independent channel, but other features are poorly understood. Our data show that, upon cholinergic stimulation, two different conductances are sequentially activated: an initial non-specific cation channel, inhibited by genistein and flufenamic acid and scarcely sensitive to La3+ and Zn2+, followed by a more specific Ca2+ entry pathway, inhibited by Mn2+, La3+ and Zn2+. Other authors have previously reported the coexistence of different Ca2+ entry pathways in other cell types (Estacion & Mordan, 1993; Ross & Cahalan, 1995; Hug et al. 1996), although the temporal evolution of the agonist-evoked Ca2+ entry is, to our knowledge, a novel feature.
The inhibitory effect of flufenamic acid and genistein, as well as the permeation of Mn2+, indicates early activation of the non-selective cation channel in response to cholinergic stimulation. This is in agreement with a recent paper describing INCRAC, a non-selective cation current inhibited by genistein, as the main route for CCE in mouse pancreas (Krause et al. 1996). In addition, other authors have also proposed that non-specific cation channels can support Ca2+ influx in several cell types (Schumann et al. 1994; Weiser & Wienrich, 1996). However, Krause et al. (1996) concluded that Ca2+ influx was insensitive to La3+, which differs from our results. In our opinion this can be explained by differences in the sensitivity of the methods (we used fura-2, which is more sensitive than fluo-3 used by Krause et al.) and/or by the fact that at that stage the second Ca2+ entry phase is already slightly active in our experiments (see below).
The temporal change in the sensitivity of CCE to different experimental manipulations clearly reveals that, after the initial non-specific conductance discussed above, the Ca2+ entry pathway behaves as a relatively specific calcium channel, in keeping with the report of the presence of ICRAC in exocrine pancreas by Bahnson et al. (1993), who showed a low permeation for Mn2+. In this particular aspect our data are clearly different from Krause et al. (1996), the discrepancies being probably due to differences in the timing of the experimental protocol, since we have used a period of Ca2+ depletion greater than 10 min, while Krause et al. studied the behaviour of the Ca2+ entry pathway after shorter periods.
The inhibition of store depletion-evoked CCE by lanthanum and divalent cations is in keeping with numerous reports describing this cation as an efficient tool for blocking Ca2+ influx (Pandol et al. 1987; Muallem et al. 1989; Estacion & Mordan, 1993). This feature is characteristic of the Ca2+-selective ICRAC channels (Hoth & Penner, 1993). Although previous authors have used permeation of Mn2+ to study CCE in exocrine pancreas (e.g. Muallem et al. 1990), our data clearly demonstrate that Mn2+ can inhibit influx (see Fig. 9) in Ca2+-depleted cells. Experimental conditions could account, at least in part, for this difference. Extracellular calcium concentration is usually reduced while using Mn2+ as a surrogate for Ca2+ influx (Jacob, 1990; Muallem et al. 1990; Yule et al. 1994; Shuttleworth, 1995), which could lead to reduction of selectivity of Ca2+ channels (Hess & Tsien, 1984). In fact we have found that activation of Mn2+ entry upon cholinergic stimulation is higher when extracellular calcium is removed (data not shown). In any case, a previous report also describes inhibition of Ca2+ fluxes by Mn2+ in pancreas (Rorsman & Hellman, 1983).
Our results allow us to propose the hypothesis that, once a Ca2+-mobilizing agonist binds to its receptors in pancreatic acinar cells, it activates sequentially two conductances for Ca2+ influx. In an early phase a non-selective cation channel is activated, and after some time it is replaced by a more selective Ca2+ channel, similar to the ICRAC channel. At any given moment, the detectable Ca2+ entry is the result of the overall opening of the two channels, and the experimental behaviour will fit to the predominant conductance. This would explain the increase in sensitivity to La3+ and divalent cations with time, as well as the apparent decrease of Mn2+ permeability and the loss of effect of flufenamic acid and tyrosine kinase inhibitors. Of course it is not possible to rule out the possibility that a single channel with two stages of different permeability is the cause of our observations. In any case, this explanation introduces temporal evolution as a new aspect in the physiology of calcium entry pathways.
Whether the two calcium entry mechanisms are strictly capacitative (i.e. activated by depletion of stores) or not, cannot be deduced from our results, but the results in Fig. 10 strongly support this concept.
Although speculative, there is a potential relationship between our results and the presence of trp- and trp-l-encoded Ca2+ channels in mammals. TRP forms a Ca2+-selective channel, sensitive to La3+ and activated by store depletion, while TRP-L is poorly selective for Ca2+ and relatively insensitive to inhibition by La3+ (for a review, see Montell, 1997). This pattern closely resembles our finding of two different conductances. Since coexpression of TRP and TRP-L produces a functional CCE pathway forming heteromultimers (Niemeyer et al. 1996), it is possible that in pancreatic acinar cells CCE is driven by these proteins, and one single complex with two different states of permeability could account for the apparent diversity of Ca2+ influx channels.
The putative mechanism allowing temporal co-ordination of these two pathways is not known, but current data suggest that protein kinases are involved in the process. The effect of PKC on CCE depends on the experimental conditions and the cell type: PKC inactivates ICRAC in RBL-2H3 cells (Parekh & Penner, 1995) as well as Ca2+ influx in other cell types (Montero et al. 1993; Törnquist, 1993), whilst activating influx in NIH 3T3 (Pedrosa Ribeiro & Putney, 1996) and RINm5F cells (Bode & Goke, 1994), in line with our results. Interestingly, in Drosophila PKC forms a supramolecular complex with TRP, TRP-L and other proteins involved in Ca2+ mobilization (Huber et al. 1996). This complex has been postulated as a regulatory unit for Ca2+ influx, and could convey complex inhibitory and/or activating signals to modulate the status of the channels (Huber et al. 1998). In mouse pancreatic acinar cells, PKC could serve as a signal to switch from the initial Ca2+ conductance to a more specific Ca2+ channel.
Regarding the role of tyrosine kinases in the control of CCE, the situation is controversial. Although these enzymes were proposed by Vostal as the signal activating CCE (Vostal et al. 1991), and despite the observation that CCE is blocked by tyrosine kinase inhibitors in rat pancreatic acinar cells (Yule et al. 1994), our results show a residual role in the control of the late CCE, while the initial Ca2+ influx is more sensitive to tyrosine kinase inhibitors in agreement with Krause et al. (1996). Vostal (Vostal & Shafer, 1996) recently corrected his original hypothesis and report that genistein inhibition of calcium influx is not associated with a decrease in tyrosine kinase activity. The discrepancies in the effect of genistein depending on the experimental conditions (Figs 3 and 4) reinforces the conclusion that the effects of tyrosine kinase inhibitors on CCE must be evaluated with caution. However, the putative link between tyrosine kinase and CCE cannot be ruled out. In fact, our finding that piceatannol, an inhibitor structurally different from genistein, inhibits Ca2+ entry during ACh decay supports the hypothesis CCE channels have some relationship with its target, the non-receptor tyrosine kinase p72Syk, a protein apparently involved in Ca2+ transport mechanisms (Jenner et al. 1997).
In conclusion, our results show that cholinergic stimulation of mouse pancreatic acinar cells activates an initial phase of CCE relatively insensitive to La3+, inhibited by tyrosine kinase inhibitors and carried by a non-selective Ca2+ channel, followed by a more selective Ca2+ conductance inhibited by La3+, Mn2+ and Zn2+. In addition, PKC seems to be involved in the control of CCE in pancreatic acinar cells. The identity of the channels underlying this pattern and its regulatory mechanisms deserve further investigation.
Acknowledgments
We thank Mrs Mercedes Gómez Blázquez for technical assistance. Cristina Camello is the recipent of a research fellowship from Junta de Extremadura (Spain).
References
- Alonso-Torre SR, Alvarez J, Montero M, Sanchez A, Garcia-Sancho J. Control of Ca2+ entry into HL60 and U937 human leukaemia cells by the filling state of the intracellular Ca2+ stores. Biochemical Journal. 1993;289:761–76. doi: 10.1042/bj2890761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahnson TD, Pandol SJ, Dionne VE. Cyclic GMP modulates depletion-activated Ca2+ entry in pancreatic acinar cells. Journal of Biological Chemistry. 1993;268:10808–10812. [PubMed] [Google Scholar]
- Bode HP, Goke B. Protein kinase C activates capacitative calcium entry in the insulin secreting cell line RINm5F. FEBS Letters. 1994;339:307–311. doi: 10.1016/0014-5793(94)80436-2. [DOI] [PubMed] [Google Scholar]
- Camello C, Salido GM, Camello PJ. Lanthanum and PK-C modulate capacitative Ca2+ entry in mouse pancreatic acinar cells. The Journal of Physiology. 1998;509.P:18. doi: 10.1111/j.1469-7793.1999.0399v.x. P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Estacion M, Mordan LJ. Competence induction by PDGF requires sustained calcium influx by a mechanism distinct from storage-dependent calcium influx. Cell Calcium. 1993;14:439–454. doi: 10.1016/0143-4160(93)90003-o. 10.1016/0143-4160(93)90003-O. [DOI] [PubMed] [Google Scholar]
- González A, Camello PJ, Pariente JA, Salido GM. Free cytosolic calcium levels modify intracellular pH in rat pancreatic acinar cells. Biochemical and Biophysical Research Communications. 1997;230:652–656. doi: 10.1006/bbrc.1996.6026. 10.1006/bbrc.1996.6026. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz C, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. Journal of Biological Chemistry. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Hess P, Tsien RW. Mechanism of ion permeation through calcium channels. Nature. 1984;309:453–456. doi: 10.1038/309453a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. The Journal of Physiology. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber A, Sander P, Bahner M, Paulsen R. The TRP Ca2+ channel assembled in a signaling complex by the PDZ domain protein INAD is phosphorylated through the interaction with protein kinase C (ePKC) FEBS Letters. 1998;425:317–322. doi: 10.1016/s0014-5793(98)00248-8. 10.1016/S0014-5793(98)00248-8. [DOI] [PubMed] [Google Scholar]
- Huber A, Sander P, Gobert A, Bahner M, Hermann R, Paulsen R. The transient receptor potential protein (Trp), a putative store-operated Ca2+ channel essential for phophoinositide-mediated photoreception, forms a signaling complex with NorpA, InaC and InaD. EMBO Journal. 1996;15:7036–7045. [PMC free article] [PubMed] [Google Scholar]
- Hug MJ, Pahl C, Novak I. Calcium influx pathways in rat pancreatic ducts. Pflügers Archiv. 1996;432:278–285. doi: 10.1007/s004240050134. [DOI] [PubMed] [Google Scholar]
- Hurley T, Brinck RW. Regulating trasient and sustained changes of cytosolic Ca2+ in rat pancreatic acini. American Journal of Physiology. 1990;258:C54–61. doi: 10.1152/ajpcell.1990.258.1.C54. [DOI] [PubMed] [Google Scholar]
- Jacob R. Agonist-stimulated divalent cation entry into single cultured human umbilical vein endothelial cells. The Journal of Physiology. 1992;421:55–77. doi: 10.1113/jphysiol.1990.sp017933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner S, Sage S, Farndale R. The effect of calcium store depletion on the activity of the non-receptor protein tyrosine kinase p72syk in human platelets. The Journal of Physiology. 1997;499.P:36–37. P. [Google Scholar]
- Krause E, Pfeiffer F, Schmid A, Schulz I. Depletion of intracellular calcium stores activates a calcium conducting non-selective cation current in mouse pancreatic acinar cells. Journal of Biological Chemistry. 1996;271:32523–32528. doi: 10.1074/jbc.271.51.32523. [DOI] [PubMed] [Google Scholar]
- Montell C. New light on TRP and TRPL. Molecular Pharmacology. 1997;52:755–763. doi: 10.1124/mol.52.5.755. [DOI] [PubMed] [Google Scholar]
- Montero M, Garcia-Sancho J, Alvarez J. Transient inhibition by chemotactic peptide of a store-operated Ca2+ entry pathway in human neutrophils. Journal of Biological Chemistry. 1993;268:13055–13061. [PubMed] [Google Scholar]
- Muallem S, Khademazad M, Sachs G. The route of Ca2+ entry during reloading of the intracellular Ca2+ pool in pancreatic acini. Journal of Biological Chemistry. 1990;265:2011–2016. [PubMed] [Google Scholar]
- Muallem S, Pandol SJ, Beeker TG. Modulation of agonist-activated calcium influx by extracellular pH in rat pancreatic acini. American Journal of Physiology. 1989;257:G917–924. doi: 10.1152/ajpgi.1989.257.6.G917. [DOI] [PubMed] [Google Scholar]
- Niemeyer BA, Suzuki E, Scott K, Jalink K, Zuker CS. The Drosophila light-activated conductance is composed of the two channels TRP and TRPL. Cell. 1996;85:651–659. doi: 10.1016/s0092-8674(00)81232-5. [DOI] [PubMed] [Google Scholar]
- Oliver JM, Burg DL, Wilson BS, Mclaughlin JL, Geahlen RL. Inhibition of mast cell Fc epsilon R1-mediated signaling and effector function by the Syk-selective inhibitor, piceatannol. Journal of Biological Chemistry. 1994;269:29697–29703. [PubMed] [Google Scholar]
- Pandol SJ, Schoeffield MS, Fimmel CJ, Muallem S. The agonist-sensitive calcium pool in the pancreatic acinar cell. Journal of Biological Chemistry. 1987;262:16963–16968. [PubMed] [Google Scholar]
- Parekh AB, Penner R. Depletion-activated calcium current is inhibited by protein kinase in RBL-2H3 cells. Proceedings of the National Academy of Sciences of the USA. 1995;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrosa Ribeiro CM, Putney JW., Jr Differential effects of protein kinase C activation on calcium storage and capacitative calcium entry in NIH 3T3 cells. Journal of Biological Chemistry. 1996;271:21522–21528. doi: 10.1074/jbc.271.35.21522. [DOI] [PubMed] [Google Scholar]
- Pfeiffer F, Schmid A, Schulz I. Capacitative Ca2+ influx and a Ca2+-dependent non-selective cation pathway are discriminated by genistein in mouse pancreatic acinar cells. Pflügers Archiv. 1995;430:916–922. doi: 10.1007/BF01837405. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Hellman B. The interaction between manganese and calcium fluxes in pancreatic β cells. Biochemical Journal. 1983;210:307–314. doi: 10.1042/bj2100307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PE, Cahalan MD. Ca2+ influx pathways mediated by swelling or stores depletion in mouse thymocytes. Journal of General Physiology. 1995;106:415–444. doi: 10.1085/jgp.106.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann F, Greger R, Leipziger J. Flufenamate and Gd3+ inhibit stimulated Ca2+ influx in the epithelial cell line CFPAC-1. Pflügers Archiv. 1994;428:583–589. doi: 10.1007/BF00374581. [DOI] [PubMed] [Google Scholar]
- Shuttleworth TJ. Temporal relationships between Ca2+ store mobilization and Ca2+ entry in an exocrine cell. Cell Calcium. 1995;15:457–466. doi: 10.1016/0143-4160(94)90110-4. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Dawson AP, Scharff O, Foder B, Cullen PJ, Drobak BK, Bjerrum PJ, Christensen SB, Sugimura T. Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents and Actions. 1989;27:17–23. doi: 10.1007/BF02222186. [DOI] [PubMed] [Google Scholar]
- Thorn P, Petersen OH. Activation of non-selective cation channels by physiological cholecystokinin concentrations in mouse pancreatic acinar cells. Journal of General Physiology. 1992;100:11–25. doi: 10.1085/jgp.100.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törnquist K. Modulatory effect of protein kinase C on thapsigargin-induced calcium entry in thyroid FRTL-5 cells. Biochemical Journal. 1993;290:443–447. doi: 10.1042/bj2900443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. Journal of Biological Chemistry. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Vostal JG, Jackson WL, Shilman NR. Cytosolic and stored calcium antagonistically control tyrosine phosphorylation of specific platelet proteins. Journal of Biological Chemistry. 1991;266:16911–16916. [PubMed] [Google Scholar]
- Vostal JG, Shafer B. Thapsigargin-induced calcium influx in the absence of detectable tyrosine phosphorylation in human platelets. Journal of Biological Chemistry. 1996;271:19524–19529. doi: 10.1074/jbc.271.32.19524. [DOI] [PubMed] [Google Scholar]
- Weiser T, Wienrich M. Investigations on the mechanism of action of the antiproliferant and ion channel antagonist flufenamic acid. Naunyn-Schmiedeberg's Archives of Pharmacology. 1996;353:452–460. doi: 10.1007/BF00261443. [DOI] [PubMed] [Google Scholar]
- Yule DI, Gallacher DV. Oscillations of cytosolic calcium in single pancreatic acinar cells stimulated by acetylcholine. FEBS Letters. 1988;239:358–362. doi: 10.1016/0014-5793(88)80951-7. [DOI] [PubMed] [Google Scholar]
- Yule DI, Kim ET, Williams JA. Tyrosine kinase inhibitors attenuate ‘capacitative’ Ca2+ influx in rat pancreatic acinar cells. Biochemical and Biophysical Research Communications. 1994;202:1697–1704. doi: 10.1006/bbrc.1994.2130. [DOI] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. Journal of General Physiology. 1995;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]