

Abstract
The effects of adrenergic stimulation on the ultrarapid delayed rectifier K+ current (IKur,d) of dog atrial myocytes was studied with patch-clamp methods.
Isoproterenol (isoprenaline) increased IKur,d in a concentration-dependent fashion with an EC50 of 7·3 ± 0·8 nM. The effect of isoproterenol was blocked by propranolol, mimicked by forskolin and 8-bromo-cAMP, and prevented by inhibition of protein kinase A.
Phenylephrine (in the presence of propranolol) increased IKur,d with an EC50 of 0·49 ± 0·06 μM. The effect of phenylephrine was blocked by prazosin, prevented by inhibition of protein kinase C, and mimicked by activation of protein kinase C with phorbol ester.
Phenylephrine significantly abbreviated canine atrial action potential duration in the absence of tetraethylammonium (TEA). When TEA was present under both control conditions and in the presence of phenylephrine, phenylephrine failed to alter canine atrial repolarization.
We conclude that β- and α-adrenergic stimulation increase IKur,d via protein kinase A and C, respectively, and that the induced changes in IKur,d may play a role in adrenergic control of canine atrial repolarization.
The adrenergic nervous system is well known to be an important physiological control mechanism for cardiac electrophysiological function. Adrenergic stimulation causes changes in cardiac contractility and/or excitability (Bruckner et al. 1985), changes in cardiac metabolism and gene expression (Iwaki et al. 1990), and alters the occurrence of cardiac arrhythmias in animal models and man (Zipes, 1991). Adrenergic stimulation can affect a wide variety of cardiac ion channels, including K+ (Walsh et al. 1989; Fedida et al. 1990; Braun et al. 1992; Koumi et al. 1995; Li et al. 1996), Na+ (Gintant & Liu, 1992), Ca2+ (Walsh et al. 1989) and Cl− channels (Harvey & Hume, 1989; Duan et al. 1995).
Recent work has shown that cardiac tissues of a variety of species possess cardiac delayed rectifier currents that activate much more rapidly than the rapid (IKr) and slow (IKs) components of the classical delayed rectifier current (Noble & Tsien, 1969; Sanguinetti & Jurkiewicz, 1990). Examples of such ‘ultrarapid’ delayed rectifier currents include the ‘plateau’ current of guinea-pig ventricular myocytes (Backx & Marban, 1993), a current found in a minority of neonatal puppy dogs (Jeck & Boyden, 1992), rat atrial (Boyle & Nerbonne, 1991) and ventricular (Apkon & Nerbonne, 1991) currents, a current in mouse ventricle (Fiset et al. 1997) and the human atrial ultrarapid delayed rectifier current IKur (Wang et al. 1993). IKur plays a significant role in human atrial repolarization (Wang et al. 1993) and appears to be encoded by the Kv1.5 channel gene (Wang et al. 1993; Feng et al. 1997). IKur is under dual control by adrenergic neurotransmitters, being enhanced by β-adrenergic agonists and inhibited by α-adrenergic stimulation (Li et al. 1996).
We have recently characterized a novel ultrarapid delayed rectifier current in canine atrial myocytes (Yue et al. 1996), and referred to it as IKur,d (for ‘ultrarapid delayed rectifier, dog’). Like human atrial IKur, IKur,d activates very rapidly, shows slow time-dependent inactivation and is very sensitive to 4-aminopyridine (4-AP). Unlike the human counterpart, IKur,d is also sensitive to tetraethylammonium (TEA) and shows inward rectification at voltages positive to +30 mV (Yue et al. 1996). Canine atrial refractoriness is abbreviated by sympathetic nerve stimulation (Liu & Nattel, 1997). In the light of the adrenergic regulation of human IKur (Li et al. 1996), it is possible that IKur,d is also subject to modulation by adrenergic agonists. The present study was therefore designed to determine (1) whether IKur,d is affected by exposure to α- or β-adrenergic receptor agonists, (2) what signal transduction pathways mediate any effects observed, and (3) whether adrenergic modulation of IKur,d can alter canine atrial repolarization.
METHODS
Cell isolation
Adult mongrel dogs of either sex weighing 18-25 kg were anaesthetized with pentobarbital (30 mg kg−1i.v.) and their hearts were quickly removed and immersed in room-temperature (20-22°C) Tyrode solution containing 2 mM CaCl2. All solutions used for dissection and perfusion were equilibrated with 100 % O2. The right coronary artery was cannulated and the right atria were dissected free and perfused with Tyrode solution at 37°C until the effluent was clear of blood. Any leaks from arterial branches were stopped by ligation with silk thread to assure adequate perfusion. The tissue was then perfused at 12 ml min−1 with Ca2+-free Tyrode solution for 20 min, followed by perfusion for approximately 40 min with the same solution containing collagenase (100 unit ml−1, CLSII, Worthington Biochemical Corp., Freehold, NJ, USA) and 1 % bovin serum albumin. A small piece of tissue from a well-perfused region was minced and the cells were harvested by trituration. Cells were kept at room temperature in a high-K+ solution before use.
Only quiescent rod-shaped cells showing clear cross-striations were used. A small aliquot of the solution containing the isolated cells was placed in a 1 ml chamber mounted on the stage of an inverted microscope. Five minutes were allowed for cell adhesion to the bottom of the chamber, and then the cells were superfused at 3 ml min−1 with the extracellular solution. Experiments were performed at room temperature.
Solutions
The high-K+ storage solution contained (mM): 20 KCl, 10 KH2PO4, 10 dextrose, 70 glutamic acid, 10 β-hydroxybutyric acid, 10 taurine, 10 EGTA, with 1 % bovine serum albumin, adjusted to pH 7·4 with KOH. The standard Tyrode solution contained (mM): 126 NaCl, 2 CaCl2, 5·4 KCl, 0·8 MgCl2, 0·33 NaH2PO4, 10 dextrose, 10 Hepes, adjusted to pH 7·4 with NaOH, and was used for cell isolation and as the extracellular solution for action potential studies. The extracellular solution for IKur,d recording was similar to Tyrode solution, except for the addition of 200 μM CdCl2 to inhibit ICa.L which would otherwise overlap with and contaminate IKur,d recordings.
The pipette solution for action potential recording contained (mM): 0·1 GTP, 110 potassium aspartate, 20 KCl, 1 MgCl2, 5 Mg2ATP, 10 Hepes, 5 phosphocreatine, 0·05 EGTA, pH adjusted to 7·4 with KOH. The pipette solution for voltage-clamp recording contained (mM): 0·1 GTP, 110 potassium aspartate, 20 KCl, 1 MgCl2, 5 Mg2ATP, 10 Hepes, 5 phosphocreatine, 10 EGTA, pH adjusted to 7·4 with KOH.
Isoproterenol (isoprenaline), 8-bromo-cAMP, and phenylephrine were purchased from Sigma Chemicals and freshly prepared as a 1 or 10 mM stock solution in distilled water. Phenylephrine was always administered along with 1 μM propranolol to prevent any collateral effects mediated by β-adrenergic receptor stimulation. Forskolin (Sigma) and bisindolylmaleimide (Sigma) were dissolved in dimethyl sulphoxide (DMSO) as 5 and 1 mM stock solutions, respectively. The specific protein kinase A (PKA) inhibitor peptide, PKI (Gibco Corp.) and the relatively non-specific protein kinase inhibitor, H7 (Sigma), were prepared as stock solutions in distilled water just before each experiment. Stock solutions (1 mM) of the β- and α-adrenergic receptor antagonists propranolol (Sigma) and prazosin (Sigma) were prepared in distilled water. 4-AP (Sigma) was prepared as a 1 M stock solution, with the pH adjusted to 7·4 with the addition of 1 n hydrochloric acid. TEA (Sigma) was prepared in distilled water as a 2 M stock solution. The protein kinase C (PKC)-stimulating phorbol ester phorbol 12,13-didecanoate (PDD) and its inactive congener 4α-PDD were obtained from Calbiochem-Novobiochem International (La Jolla, CA, USA) and prepared as a 10 mM stock solution in DMSO.
Data acquisition and analysis
The whole-cell patch-clamp technique was used to record ionic currents in the voltage-clamp mode and action potentials were recorded in current-clamp mode. Borosilicate glass electrodes (outer diameter, 1·0 mm) were filled with pipette solution and connected to a patch-clamp amplifier (Axopatch 1-D, Axon Instruments). Electrodes with tip resistances of 1-2 MΩ were used to record whole-cell currents, and 3-5 MΩ electrodes were used to record action potentials. Command pulses were generated by a 12-bit analog-to-digital convertor controlled by pCLAMP software (Axon Instruments). Recordings were low-pass filtered at 10 kHz, and series resistance was compensated. Data were stored on the hard disk of an IBM-compatible personal computer.
Junction potentials (2-8 mV) were zeroed before formation of the membrane-pipette seal in Tyrode solution. Mean seal resistance averaged 9·3 ± 0·8 GΩ. Several minutes after seal formation, the membrane was ruptured by gentle suction to establish the whole-cell configuration for voltage clamping. The series resistance (Rs) was electrically compensated to minimize the duration of the capacitive surge on the current recording. Rs along the clamp circuit was estimated by dividing the time constant obtained by fitting the decay of the capacitive transient by the calculated membrane capacitance (the time integral of the capacitive response to 5 mV hyperpolarizing steps from a holding potential of -60 mV divided by the voltage deflection). Before Rs compensation, the decay of the capacitive surge was expressed by a single exponential having a time constant of 521 ± 45 ms (cell capacitance, 74·6 ± 6·3 pF). Pre-compensation Rs averaged 7·0 ± 0·5 MΩ. After compensation, the time constant was reduced to 149 ± 10 ms (cell capacitance 74·0 ± 3·6 pF) and Rs was reduced to 2·0 ± 0·2 MΩ. Currents recorded during this study rarely exceeded 1·5 nA. The mean maximum voltage drop across series resistance therefore did not exceed 3 mV. Cells with significant leak currents were rejected, and leakage correction algorithms were not applied.
Group data are presented as the means ±s.e.m. unless otherwise stated. Statistical comparisons were made with ANOVA followed by Student's two-tailed t test with Bonferroni correction. P < 0·05 was taken to indicate statistical significance.
RESULTS
Effect of isoproterenol on IKur,d
Figure 1A shows typical IKur,d recordings from one cell. Currents were elicited by 140 ms depolarizations to voltages between -40 and +60 mV, followed by a 60 ms repolarization to -30 mV to record tail currents. A holding potential of -50 mV and an 80 ms prepulse to +30 mV 10 ms before the test pulse were used to suppress transient outward currents (Ito) and elicit selectively IKur,d as previously described (Yue et al. 1996). Typical features of IKur,d were observed, including rapid activation, slow inactivation, distinct tail currents and inward rectification at positive voltages. Isoproterenol (0·1 μM) increased both step and tail currents (Fig. 1B). The increases caused by isoproterenol were reversed by the addition of 1 μM propranolol in the continued presence of isoproterenol (Fig. 1C), returning currents to control levels. Results similar to those shown in Fig. 1A-C were obtained in a total of eight cells. Step currents were quantified as the difference between holding current and the maximum outward current elicited by a depolarizing pulse. The average step current densities at +30 mV under each condition are shown in Fig. 1D. Isoproterenol increased current density from 7·7 ± 0·7 to 10·9 ± 0·8 pA pF−1 (P < 0·001) and propranolol returned the current density to 7·6 ± 0·7 pA pF−1 (n.s. vs. control). Thus, isoproterenol enhances IKur,d via the activation of β-adrenergic receptors.
Figure 1. Effects of isoproterenol on IKur,d.
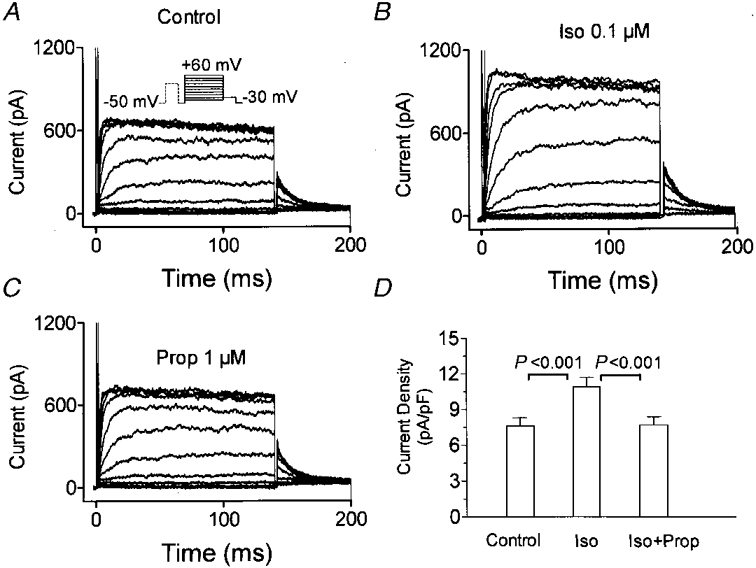
A, currents recorded from a representative canine atrial myocyte with 140 ms depolarizations to various test potentials preceded by an 80 ms prepulse to +30 mV from a holding potential of -50 mV at 0·1 Hz (protocol shown in the inset). B, currents recorded with the same protocol 10 min after the addition of 0·1 μM isoproterenol (Iso). C, currents recorded in the presence of isoproterenol (0·1 μM) and propranolol (1 μM). D, current density (mean ±s.e.m.) at +30 mV in 8 cells.
The concentration dependence of the effects of isoproterenol on IKur,d was evaluated at test potentials from -40 to +60 mV. Figure 2A shows representative recordings at +30 mV (voltage protocol in inset). A clear effect was seen at 1 nM, and the effect increased at 10 and 100 nM. Figure 2B shows average current density-voltage relations for IKur,d in the absence and presence of 1, 10, 50 and 100 nM isoproterenol and after washout in eight cells exposed to all conditions. Statistically significant increases began at an isoproterenol concentration of 1 nM and reached a maximum of 43·8 ± 4·4 % at a concentration of 100 nM. No significant voltage dependence of the action of isoproterenol was detected (ANOVA), as can be appreciated from the plot of percentage change in IKur,d induced by isoproterenol as a function of test potential in Fig. 2C. The concentration-response curve at +30 mV is shown in Fig. 2D. Data were fitted by an equation of the form: B= 100/[1 + (EC50/D)n], where B is the percentage of maximal current increase at drug concentration D, EC50 is the concentration that causes 50 % of maximal increase, and n is the Hill coefficient. The mean value of the isoproterenol EC50 determined in eight experiments was 7·3 ± 0·8 nM, and maximal effects were seen at a concentration of 100 nM.
Figure 2. Concentration-dependent effects of isoproterenol on IKur,d.
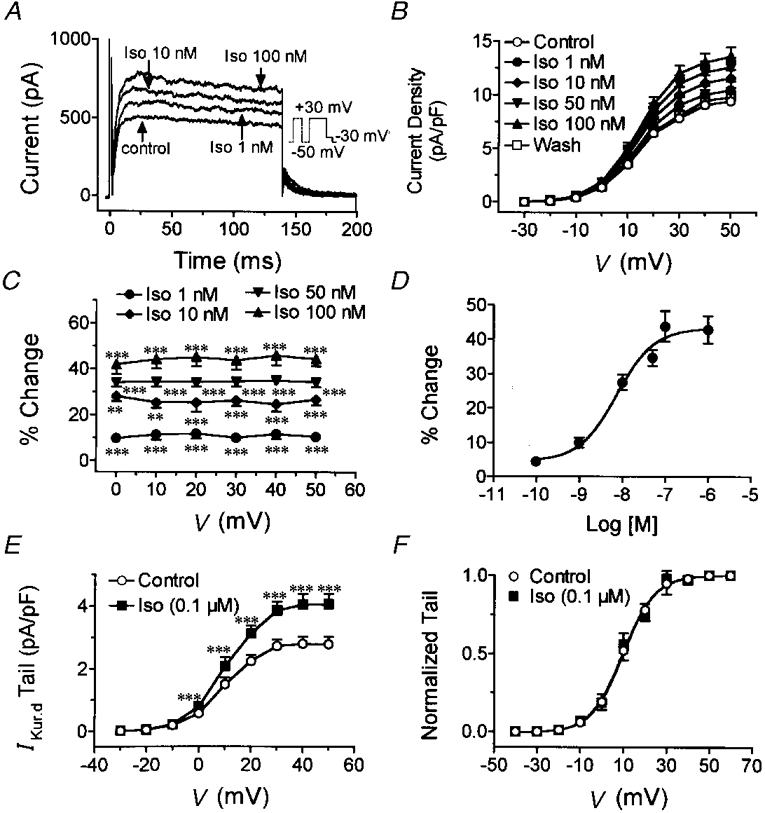
A, currents elicited in a representative cell by depolarizing for 140 ms to +30 mV with the protocol shown in the inset under control conditions, and in the presence of 1, 10 and 100 nM isoproterenol (Iso). B, mean current density-voltage relations from 8 cells under control conditions, in the presence of 1, 10, 50 and 100 nM isoproterenol and after washout. Currents were recorded during 140 ms pulses to the voltages indicated (with a prepulse as shown in A to suppress Ito). C, percentage changes in IKur,d current density (mean ±s.e.m.) induced by 1, 10, 50 and 100 nM isoproterenol at various test potentials. D, concentration-dependent increases in IKur,d induced by isoproterenol (results are mean ±s.e.m. in 8 cells). Curve is best-fit concentration-response equation. E, effects of isoproterenol on IKur,d tail currents recorded at -30 mV following depolarizating step to voltage V with protocol shown in inset of A. F, normalized tail currents and activation curve fitted with Boltzmann distribution. **P < 0·01, ***P < 0·001 vs. control.
To determine whether isoproterenol affects the voltage dependence of IKur,d activation, we analysed tail currents in the absence and presence of 0·1 μM isoproterenol. Figure 2E shows average data for the tail current density-step voltage relation in eight cells. Isoproterenol significantly increased tail currents at all test potentials. Mean tail currents normalized to the value at the most positive potential in each experiment are shown in Fig. 2F. The curves shown are the best-fit Boltzmann distribution equations of the form I/Imax= 1/{1 + exp[(V - V50)/k]}, where I is tail current at an activating voltage V, Imax is tail current at an activating voltage of +60 mV, V50 is the step voltage that produces 50 % of maximal activation and k is a slope constant. When data from each experiment were fitted by this equation, V50 averaged 9·8 ± 0·7 mV under control conditions and 9·5 ± 0·8 mV after the addition of isoproterenol (n.s.). The slope constant averaged 7·3 ± 0·7 mV under control conditions and 7·5 ± 0·7 mV after isoproterenol (n.s.). The data shown in Fig. 2 indicate the concentration dependence and reversibility of the actions of isoproterenol, and show that the drug's effects are not due to a shift in the voltage dependence of the current.
In Figs 1 and 2, we used voltage protocols to separate IKur,d and study the effects of isoproterenol on the current. The possibility remains that some of the effect observed could be due to changes in other currents such as Ito or IK. To evaluate the effects of isoproterenol on IKur,d more specifically, we took advantage of the unusual sensitivity of IKur,d to TEA. TEA inhibits IKur,d in a rapid and reversible fashion with an EC50 in the range of 300 μM and nearly full inhibition at a concentration of 5 mM, at which concentration no other currents in dog atrial myocytes are significantly affected (Yue et al. 1996). IKur,d was first recorded under control conditions, then 5 mM TEA was added to the superfusate and the current was recorded again. TEA was then washed out until the current returned to control values, and isoproterenol (0·1 μM) was added in the absence of TEA. The latter was then added during continued superfusion with isoproterenol. Typical results are shown in Fig. 3. Under control conditions (Fig. 3A), 5 mM TEA virtually eliminated time-dependent step and tail currents. In the presence of isoproterenol, step and tail currents were increased, but the addition of TEA once again eliminated time-dependent currents. TEA-sensitive currents were obtained by digital subtraction of currents in the presence of TEA from those recorded in its absence. As shown in Fig. 3C, isoproterenol strongly increased the TEA-sensitive current. Average current density-voltage relations for TEA-sensitive currents are shown in Fig. 3D. Isoproterenol (100 nM) significantly increased TEA-sensitive currents to an extent similar to that shown in Fig. 2B, indicating that the effects of isoproterenol are specific for IKur,d.
Figure 3. Specificity of isoproterenol effects on IKur,d.
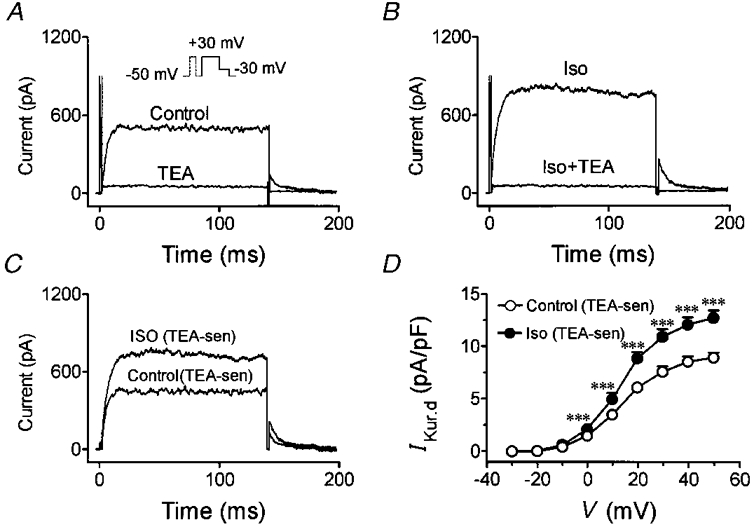
A, representative recordings under control conditions and after addition of 5 mM TEA (voltage protocol in inset). B, currents recorded before and after addition of 5 mM TEA in the presence of 0·1 μM isoproterenol from same cell as shown in panel A. C, TEA-sensitive current obtained by digital subtraction of currents in the presence of TEA from those in its absence under control conditions or in the presence of 0·1 μM isoproterenol (Iso). D, average current density-voltage relations for TEA-sensitive currents for six cells studied under both control conditions and in the presence of 0·1 μM isoproterenol. ***P < 0·001 vs. control at same voltage.
Finally, we performed an additional series of experiments to determine whether β-adrenergic modulation of IKur,d is altered by the absence of strong intracellular Ca2+ buffering. We first studied effects in the presence of 50 μM EGTA in the pipette solution, rather than the 10 mM EGTA we used to prevent Ca2+ overload in most voltage-clamp studies. Under these conditions, IKur,d was increased at +30 mV by 41 ± 2 % (n= 5, P < 0·001) by 1 μM isoproterenol. We also studied the effects of isoproterenol with pipettes containing 1 mM Ca2+ and 10 mM EGTA, which produce cytosolic Ca2+ concentrations comparable to physiological diastolic concentrations. Under these conditions, 1 μM isoproterenol also increased IKur,d by 41 ± 2 % (n= 4, P < 0·01). These results were very similar to results obtained with 10 mM EGTA and no Ca2+ in the pipette solution.
Effects of forskolin and 8-bromo-cAMP on IKur,d
To examine the potential role of cAMP in mediating the effects of isoproterenol on IKur,d, we studied changes produced by forskolin, which activates adenylate cyclase independently of β-adrenergic receptors, and by the cell membrane-permeable form of cAMP, 8-bromo-cAMP (8-Br-cAMP). Figure 4A shows currents recorded from an atrial myocyte under control conditions and then after adding 5 mM TEA. TEA was then washed out until currents returned to control levels. The subsequent addition of 3 μM forskolin substantially increased IKur,d (Fig. 4B) and the addition of 5 mM TEA in the presence of 3 μM forskolin eliminated both step and tail currents. The effect of forskolin on TEA-sensitive currents are shown in Fig. 4C, and indicate a specific stimulatory effect of forskolin on IKur,d. Average current density-voltage relations from six experiments with forskolin are shown in Fig. 4D and indicate that forskolin increased IKur,d over the entire range of voltages positive to 0 mV (n.s. for voltage-dependent action), to an extent similar to that observed with isoproterenol (e.g. at +30 mV forskolin increased IKur,d by 44 ± 3 %). Similar experiments to those shown in Fig. 4 with forskolin were performed with 8-Br-cAMP, with very similar results in six cells exposed to 50 μM 8-Br-cAMP. 8-Br-cAMP increased TEA-sensitive IKur,d in a significant and voltage-independent fashion that was similar to the actions of isoproterenol and forskolin illustrated in Figs 2, 3 and 4. For example, at +30 mV, 8-Br-cAMP increased IKur,d step current density by 40 ± 3 %.
Figure 4. Effect of forskolin on IKur,d.
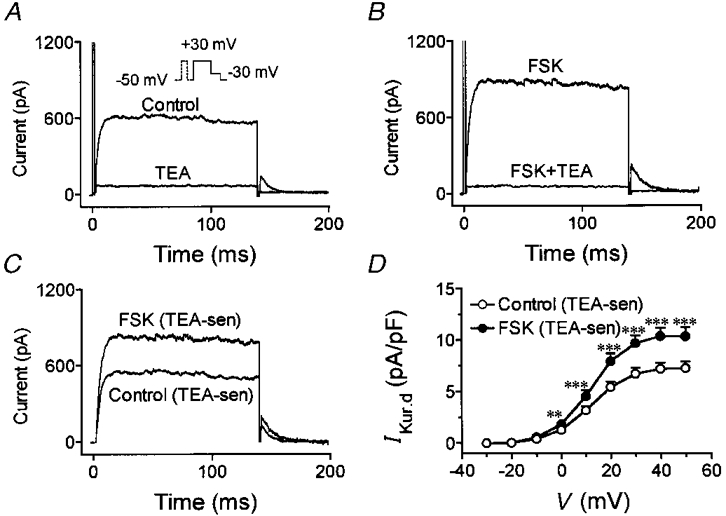
A, currents elicited under control conditions and after addition of 5 mM TEA. TEA was then washed out, and the cell was exposed to 3 μM forskolin (FSK). B, currents recorded from the same cell in presence of 3 μM forskolin before and and after application of 5 mM TEA. C, TEA-sensitive currents. D, average (mean ±s.e.m.) current density-voltage relations of TEA-sensitive currents in six cells studied under both control conditions and in the presence of FSK. **P < 0·01, ***P < 0·001 vs. control at same voltage.
Potential role of protein kinase A in mediating the effects of isoproterenol
The experiments with forskolin and 8-Br-cAMP suggest that the effects of β-adrenergic stimulation on IKur,d are likely to be mediated by increased generation of intracellular cAMP. To determine whether cAMP-dependent protein kinase A (PKA) mediates the actions of isoproterenol on IKur,d, we examined the effects of isoproterenol in the presence of PKA inhibitors. Figure 5A shows representative recordings obtained immediately after membrane rupture with a pipette containing 50 nM PKI, a specific PKA inhibitor. After 20 min of dialysis with PKI-containing pipette solution, IKur,d was unaffected (Fig. 5B). Isoproterenol (1 μM) failed to alter IKur,d in the presence of PKI (Fig. 5C). Mean data from six cells (Fig. 5D) indicate that PKI itself did not alter IKur,d, but that dialysis with PKI fully prevented the effect of isoproterenol.
Figure 5. Effects of the PKA inhibitor peptide PKI in the pipette solution on IKur,d and its response to isoproterenol.
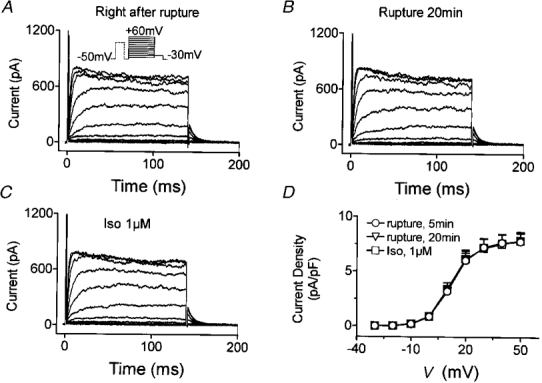
A, currents elicited immediately after membrane rupture with the voltage protocol shown in the inset. B, currents recorded after 20 min of dialysis with PKI-containing solution. PKI (50 nM) did not change IKur,d. C, currents recorded after the addition of 1 μM isoproterenol in the presence of PKI. D, results (means ±s.e.m.) from 6 cells.
Although PKI is an effective and specific inhibitor of PKA (Glass et al. 1989), it has to be included in the pipette solution, making it impossible to compare the effects of isoproterenol before and after the inhibition of PKA in the same cells. We therefore performed additional experiments with the protein kinase inhibitor H7. IKur,d was first recorded under control conditions and then after 10 min of superfusion with 1 μM isoproterenol. Isoproterenol was then washed out and H7 was added to the superfusate at a concentration of 5 μM. Finally, the cell was once more exposed for 10 min to 1 μM isoproterenol. Parallel experiments were performed in the same fashion, but without exposure to H7, to assess the reproducibility of the effects of isoproterenol during consecutive perfusion and washout periods. The results shown in Fig. 6A indicate that repeated exposure to isoproterenol in the absence of H7 produced consistent effects (n= 6). In contrast, a second exposure to isoproterenol in the presence of H7 failed to produce any effect (Fig. 6B).
Figure 6. Effects of 1 μM isoproterenol on IKur,d step current density at +30 mV in the absence and presence of protein kinase inhibitor H7.
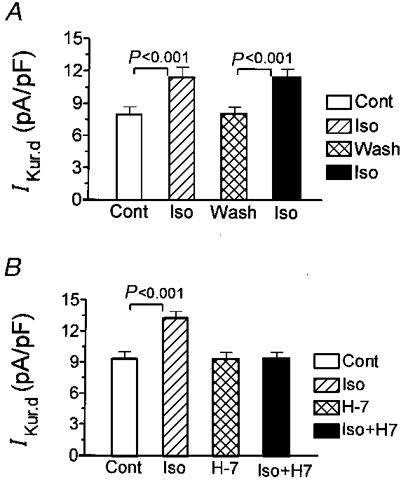
A, IKur,d density (mean ±s.e.m.) indicates that repeated exposure to isoproterenol in the absence of H7 produced consistent effects in 6 cells. B, a second isoproterenol exposure failed to alter IKur,d in the presence of H7 in 6 cells.
The above experiments indicate that isoproterenol increases IKur,d in a reversible fashion via the stimulation of β-adrenergic receptors and cAMP-dependent PKA-mediated mechanisms. We next turned our attention to the potential effects of α-adrenergic receptor stimulation on IKur,d.
Effects of phenylephrine on IKur,d
To evaluate the changes in IKur,d caused by α-adrenergic stimulation, we used phenylephrine as an α-adrenergic agonist, in the presence of 1 μM propranolol to block any potential collateral β-adrenergic effects. Figure 7A shows control IKur,d currents elicited with the protocol shown in the inset, and Fig. 7B shows currents recorded with the same protocol in the same cell after exposure to phenylephrine (PE, 10 μM). The effect of phenylephrine was reversed promptly by the addition of 2 μM prazosin (PZ, Fig. 7C), confirming that the drug's actions on IKur,d were mediated by α1-adrenergic receptors. Average IKur,d step current densities at +30 mV in eight cells are shown in Fig. 7D, and indicate that phenylephrine significantly increased IKur,d density (from 8·1 ± 0·9 to 10·7 ± 0·9 pA pF−1, P < 0·001), an effect reversed by prazosin.
Figure 7. Effects of phenylephrine (PE) on IKur,d.
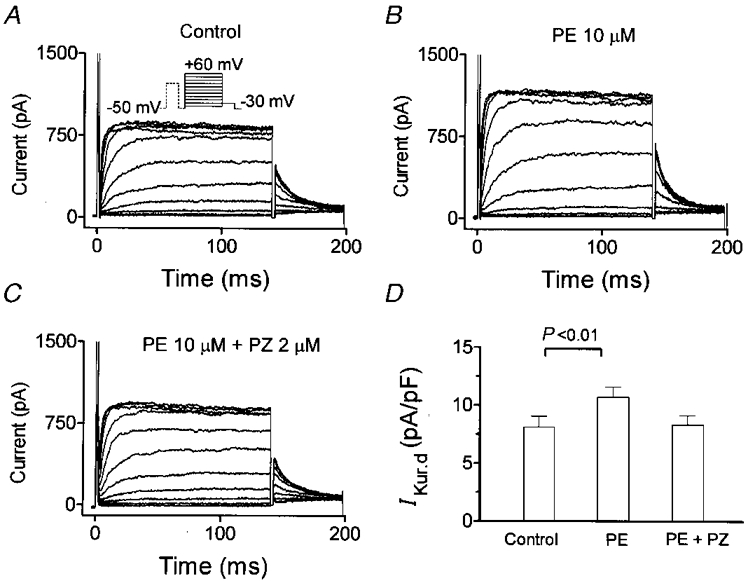
A, recordings under control conditions (voltage protocol in inset). B, currents in the same cell after the addition of 10 μM phenylephrine to the superfusate. C, currents in the same cell after 2 μM prazosin (PZ) was added to the 10 μM phenylephrine-containing superfusate. D, average step current density (mean ±s.e.m.n= 8) at +30 mV.
Figure 8 shows the results of experiments designed to evaluate the concentration and voltage dependence of the actions of phenylephrine. Original IKur,d recordings from one cell shown in Fig. 8A indicate that phenylephrine increased IKur,d step and tail currents in a concentration-dependent manner. Mean current density-voltage relations from eight cells studied under control conditions, in the presence of 0·1, 1 and 10 μM phenylephrine and upon washout are shown in Fig. 8B. Phenylephrine significantly increased IKur,d at a concentration of 0·1 μM and its effects reached a maximum (33·9 ± 3·0 % increase) at a concentration of 100 μM. The effects of phenylephrine did not vary with step potential (Fig. 8C). The concentration- response relation for eight cells at +30 mV is shown in Fig. 8D. The mean value of the phenylephrine EC50 based on non-linear curve fits was 490 ± 56 nM, with near-maximal effects achieved at a concentration of 10 μM.
Figure 8. Concentration- and voltage-dependent effects of phenylephrine on IKur,d.
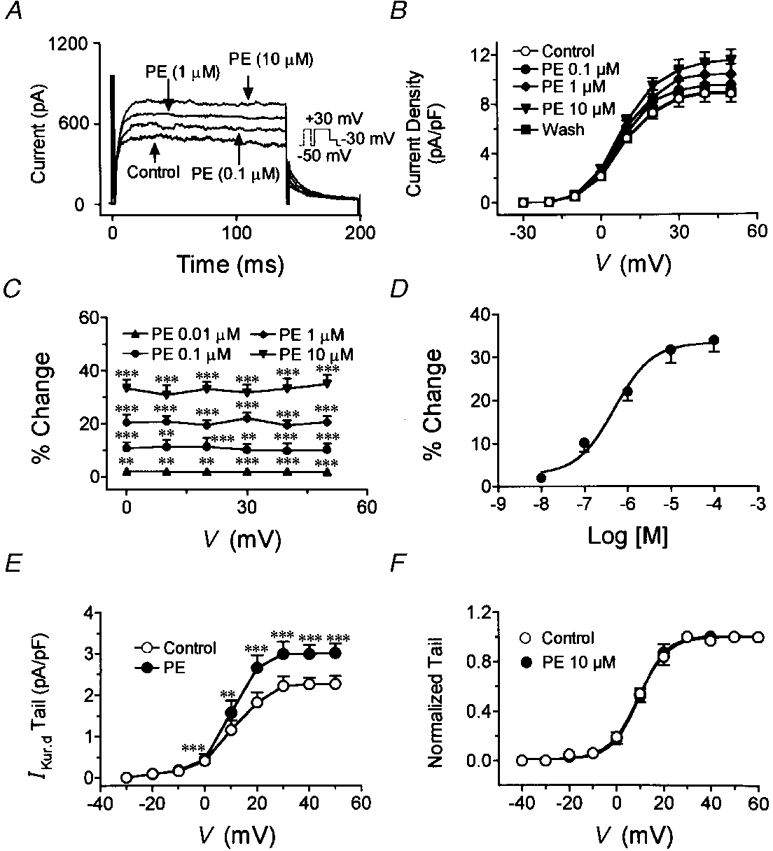
A, representative recordings elicited under control conditions, and in the presence of 0·1, 1 and 10 μM PE in a representative cell. B, mean current density-voltage relations from eight cells under control conditions, in the presence of 0·1, 1 and 10 μM phenylephrine and after washout. C, percentage change (mean ±s.e.m.) in IKur,d current density induced by 0·1, 1 and 10 μM phenylephrine at various test potentials. D, concentration dependence of phenylephrine-induced increases in IKur,d. The curve shown is the best-fit concentration-response curve according to the equation given in text (EC50 from fits in each of eight experiments averaged 0·49 ± 0·06 μM). E, effects of 10 μM phenylephrine on IKur,d tail currents. F, normalized tail currents and activation curve fitted by Boltzmann distribution under control conditions and in the presence of 10 μM PE. **P < 0·01, ***P < 0·001 vs. control.
To assess the effects of phenylephrine on voltage-dependent activation of IKur,d, we analysed the step voltage dependence of tail current in the absence and presence of 10 μM phenylephrine. Figure 8E shows the average tail current density-voltage relation in eight cells. Phenylephrine significantly increased tail currents at all test potentials. Figure 8F shows normalized tail currents (to tail current at maximum step potential), along with the best-fit Boltzmann distribution equations. When data from each experiment were fitted by Boltzmann equations, V50 averaged 9·2 ± 0·5 mV under control conditions and 9·4 ± 0·6 mV after the addition of phenylephrine (n.s.), and the slope constant averaged 6·2 ± 0·4 mV under control conditions and 5·08 ± 0·5 mV after phenylephrine (n.s. vs. control).
To examine more specifically the effects of phenylephrine on IKur,d, TEA-sensitive currents were studied. Figure 9 shows currents from one cell before and after the addition of 5 mM TEA under control conditions (A) and in the presence of 10 μM phenylephrine (B). TEA-sensitive currents are shown in C, and indicate that phenylephrine perceptibly increased IKur,d. Mean TEA-sensitive current density-voltage relations in six cells (Fig. 9D) indicate that phenylephrine increased IKur,d in a significant and voltage-independent fashion. Finally, we evaluated the effects of phenylephrine in the presence of more physiological intracellular Ca2+ concentrations. With 50 μM EGTA in the pipette (in place of the usual EGTA concentration of 10 mM), 10 μM phenylephrine increased IKur,d at +30 mV by 29 ± 6 % (n= 4, P < 0·05). With 1 mM Ca2+ and 10 mM EGTA in the pipette, phenylephrine increased IKur,d by 32 ± 3 % (n= 4, P < 0·01). These results are similar to the effects of phenylephrine obtained with 10 mM EGTA and no Ca2+ in the pipette solution.
Figure 9. Specificity of phenylephrine effects on IKur,d.
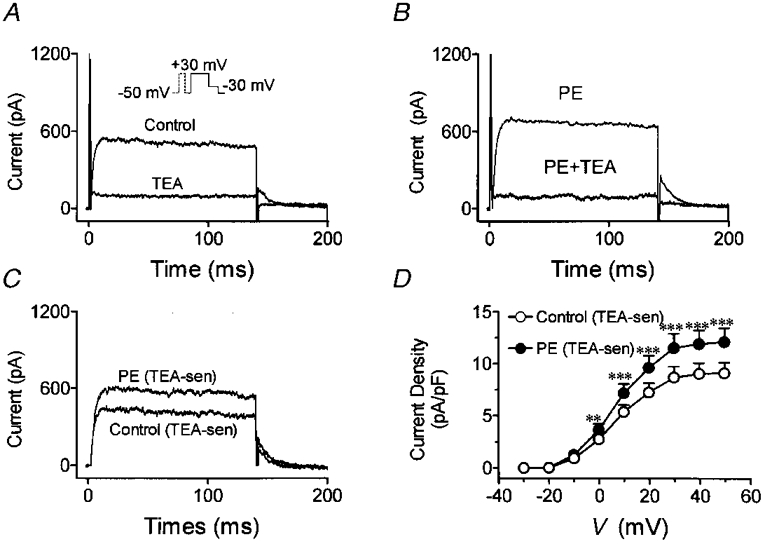
A, representative recordings under control conditions and after the addition of 5 mM TEA (protocol in inset). B, currents recorded before and after 5 mM TEA in the presence of 10 μM PE. C, TEA-sensitive current obtained by digital subtraction of currents in the presence of TEA from those in its absence. D, average TEA-sensitive current density-voltage relations under control conditions and in the presence of phenylephrine. **P < 0·01, ***P < 0·001 vs. control at same voltage.
The potential role of protein kinase C (PKC) in mediating phenylephrine effects was evaluated with the use of the highly selective PKC inhibitor bisindolylmaleimide (Toullec et al. 1991). Cells were first exposed to phenylephrine to quantify the response in the absence of a PKC inhibitor and then re-exposed to phenylephrine in the presence of 50 nM bisindolylmaleimide (Bis). Figure 10A-D shows results from a representative cell under control conditions (A), after the addition of phenylephrine (B), in the presence of bisindolylmaleimide alone (C) and in the presence of bisindolylmaleimide and phenylephrine (D). Parallel experiments were performed in the absence of bisindolylmaleimide to ensure that repeated exposure to phenylephrine produces reproducible effects on IKur,d. Figure 10E shows that the effects on IKur,d of repeated exposure to phenylephrine were reproducible in six cells, whereas the effect of phenylephrine was abolished by co-administration with bisindolylmaleimide (Fig. 10F, n= 6 cells).
Figure 10. Effects of bisindolylmaleimide (Bis), a highly selective PKC inhibitor, on the action of phenylephrine on IKur,d.
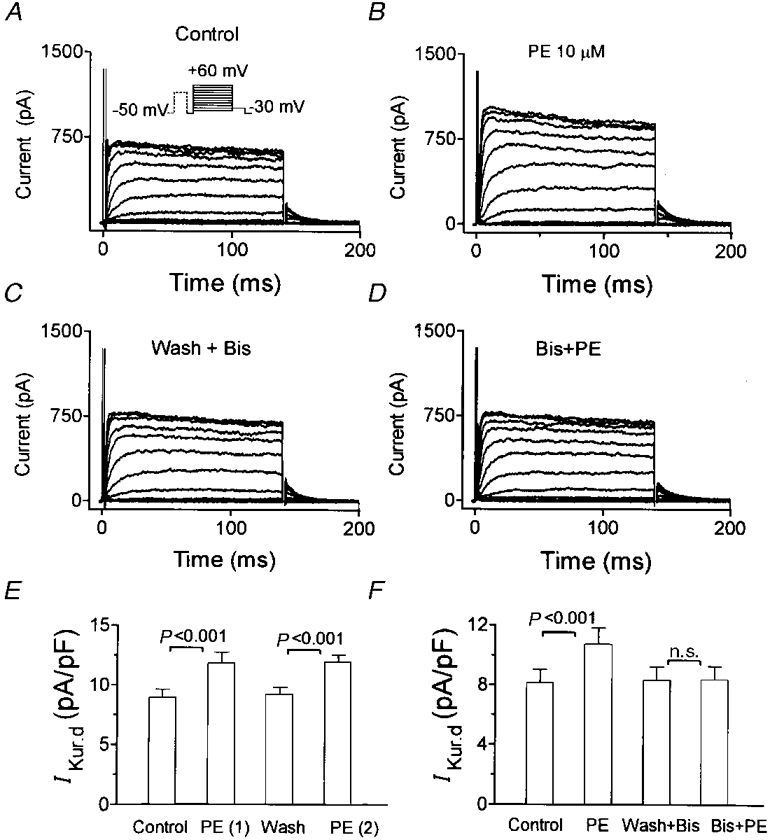
A, control currents. B, the addition of 10 μM phenylephrine increased IKur,d. C, after phenylephrine was washed out, application of 50 nM Bis did not alter IKur,d. D, effects of 10 μM phenylephrine on IKur,d in the presence of Bis. (Results in A-D are in the same cell.) E, average (mean ±s.e.m.) IKur,d step current density at +30 mV shows that repeated exposure to phenylephrine produced consistent effects in the absence of Bis. F, a second exposure to phenylephrine in the presence of Bis failed to increase IKur,d.
If PKC activation mediates the effects of α-adrenergic stimulation on IKur,d, one would expect that direct PKC activation by phorbol esters would also enhance IKur,d. We therefore assessed the effects of the phorbol ester PDD on IKur,d. Figure 11A and B shows currents from a representative cell recorded in the absence and presence of 5 mM TEA in the absence (A) and presence (B) of PDD. TEA-sensitive currents obtained by digital subtraction (Fig. 11C) indicate that PDD produced a substantial increase in IKur,d step and tail currents. Mean TEA-sensitive step current density-voltage relations from six cells (Fig. 11D) demonstrate that PDD significantly increased IKur,d over a broad range of voltages. As shown by the mean percentage changes in IKur,d step current (Fig. 11E), the effect of PDD was voltage independent. The inactive congener 4α-PDD had no effect on IKur,d, as shown by the mean step currents in six cells illustrated in Fig. 11F.
Figure 11. Effects of the PKC activator PDD on IKur,d.
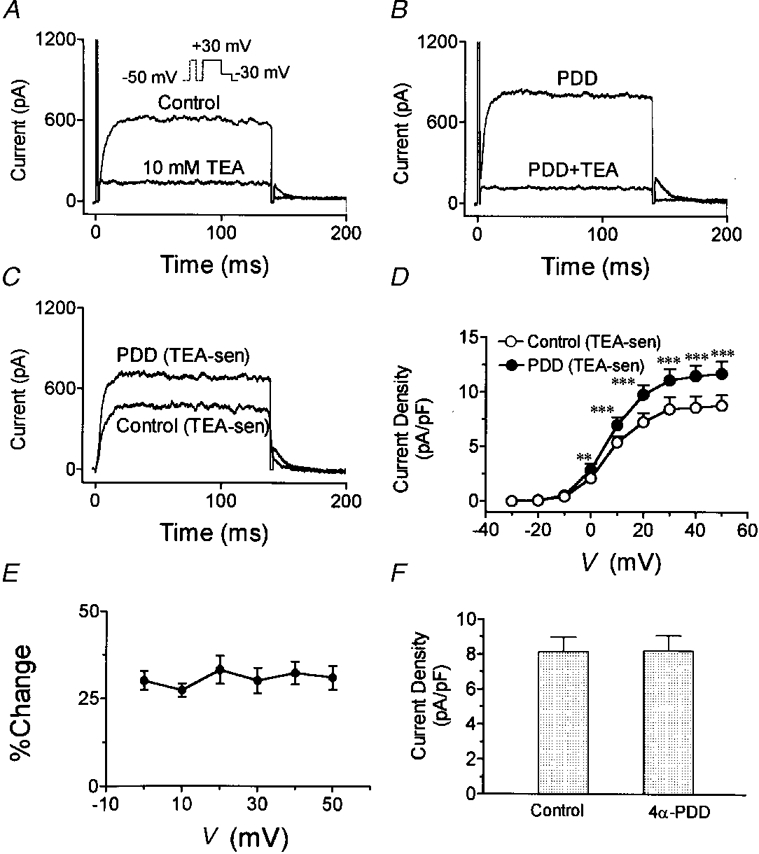
A, control currents at +30 mV before and after addition of TEA. B, currents in presence of 100 nM PDD before and after the addition of TEA (5 mM) to the superfusate in the same cell as in A. C, TEA-sensitive current with and without PDD. D, mean TEA-sensitive current density-voltage relations from 6 cells studied in the presence and absence of PDD. E, PDD-induced percentage change in IKur,d (defined by TEA-sensitive current) as a function of step voltage. F, mean IKur,d density at +30 mV before and after the inactive congener 4α-PDD in 6 cells. **P < 0·01, ***P < 0·001 vs. control.
Physiological relevance of the adrenergic modulation of IKur,d
To determine whether the adrenergic modulation of IKur,d can mediate effects on the action potential, we evaluated the effects of phenylephrine on action potential duration (APD) in the presence and absence of IKur,d inhibition with 1 mM TEA. Figure 12A shows that in the absence of TEA, phenylephrine (10 μM in the presence of 1 μM propranolol) substantially shortened APD. On the other hand, in the presence of TEA (Fig. 12B), no clear effect of phenylephrine was observed. Mean data for the effects of phenylephrine on APD for six cells studied in the absence of TEA and six in the presence of TEA are shown in Table 1. Phenylephrine significantly shortened all phases of the action potential in the absence of TEA, but had no effect in the presence of TEA concentrations that selectively inhibit IKur,d.
Figure 12. Potential role of adrenergic modulation of IKur,d in action potential regulation.
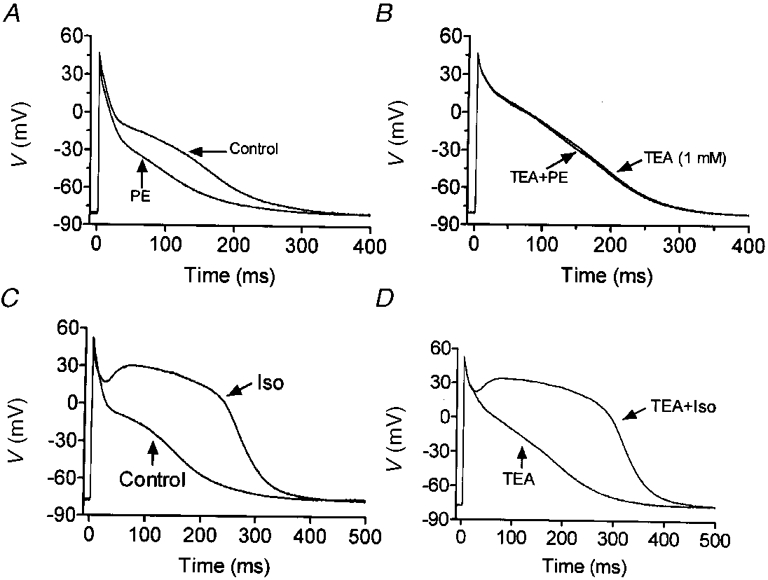
A, action potentials of a representative cell at 0·1 Hz before and after exposure to 10 μM phenylephrine (in the presence of 1 μM propranolol). B, action potentials of a different cell in the presence of 1 mM TEA, before and after exposure to 10 μM phenylephrine (in the presence of 1 μM propranolol). C, action potentials obtained from one cell before and after 1 μM isoproterenol (Iso). D, action potentials obtained from a cell exposed first to 1 mM TEA and then 1 mM TEA plus 1 μM isoproterenol.
Table 1.
Effects of phenylephrine (10 μm) and isoproterenol (1 μm) on action potential duration (ms) at 0.1 Hz in the absence and presence of 1mm TEA
| Effects of phenylephrine | Effects of isoproterenol | |||||||
|---|---|---|---|---|---|---|---|---|
| Control | Phenylephrine | TEA | TEA + phenylephrine | Control | Isoproterenol | TEA | TEA + isoproterenol | |
| APD20 | 9 ± 1 | 5 ± 1*** | 12 ± 1 | 14 ± 1 | 9 ± 1 | 170 ± 12*** | 11 ± 1 | 240 ± 18*** |
| APD50 | 69 ± 5 | 37 ± 3** | 114 ± 11 | 113 ± 11 | 65 ± 5 | 260 ± 22*** | 108 ± 9 | 321 ± 28*** |
| APD90 | 207 ± 18 | 143 ± 12*** | 263 ± 24 | 261 ± 22 | 186 ± 21 | 342 ± 32*** | 249 ± 21 | 404 ± 34*** |
APD20, APD50, APD90, action potential duration (ms) to 20, 50, and 90 % respectively. Results shown are for 6 cells each exposed to control and then phenylephrine or isoproterenol, or to TEA and then TEA plus isoproterenol or TEA plus phenylephrine.
P < 0.01
P < 0.001, vs. appropriate control group (‘control’ for cells studied in absence of TEA; ‘TEA’ for cells studied in presence of TEA before and after drug administration).
The effects of isoproterenol in the absence and presence of TEA are illustrated in Fig. 12C and D, respectively. Isoproterenol prolonged APD under both conditions, but the prolongation appeared to be greater in the presence of TEA. The mean data shown in Table 1 support this observation, which is most striking for the earlier phases of the action potential. For example, in the absence of TEA, isoproterenol increased APD20 from 9 ± 1 to 170 ± 12 ms, whereas in the presence of TEA isoproterenol increased APD20 from 11 ± 1 to 240 ± 18 ms.
DISCUSSION
We have demonstrated that β- and α1-adrenergic receptor agonists increase IKur,d reversibly and with high potency in dog atrial myocytes. The effects of β-adrenergic stimulation appeared to be mediated by cyclic AMP-induced activation of PKA, whereas the effects of α-adrenergic stimulation were mediated by the activation of PKC. Action potential recordings suggested the physiological relevance of the adrenergic modulation of IKur,d.
Comparison with previous studies of adrenergic actions on other K+ channels
Adrenergic activation is well known to modulate the properties of a variety of K+ channels. Since the classic study of Tsien et al. (1972), it has been well recognized that β-adrenergic stimulation enhances IK by a cyclic AMP-dependent mechanism. Studies of α-adrenergic effects on IK have provided variable results, with an increase having been noted in guinea-pig ventricular myocytes (Tohse et al. 1992) and a decrease in rabbit sino-atrial node cells (Satoh & Hashimoto, 1988). Several other K+ currents including Ito (Apkon & Nerbonne, 1988; Fedida et al. 1990), IK1 (Fedida et al. 1991) and IKACh (Braun et al. 1992) are also inhibited by α-adrenergic stimulation. On the other hand, some workers have noted activation of IKACh by α-adrenergic stimulation in the presence of atropine (Kurachi et al. 1989).
The human ultrarapid delayed rectifier IKur is increased by β-adrenergic stimulation but decreased by α-adrenergic stimulation (Li et al. 1996). Phenylephrine also inhibits ultrarapid delayed rectifier-type currents in rat atrium (Van Wagoner et al. 1996). In the present study, isoproterenol and phenylephrine enhanced IKur,d through α1- and β-adrenergic receptor activation respectively. The increase induced by 100 nM isoproterenol, the concentration that achieved maximum effects, averaged 44 ± 4 %, of the same order as the effect of isoproterenol on human atrial IKur (Li et al. 1996). Unlike human atrium, dog atrial IKur was enhanced by α-adrenergic stimulation.
Adrenergic enhancement of IK has been reported to be voltage dependent (Tsien et al. 1972), with β-adrenergic stimulation shifting the activation curve to more negative potentials. PKA, a β-adrenergic mediator, also causes a negative shift in the activation voltage dependence of IK, whereas PKC primarily changes the slope of the activation curve without substantially altering the half-activation voltage. In the present study, we found that neither α- nor β-adrenergic stimulation affect the voltage-dependent activation of IKur,d. This finding is similar to the lack of effect of adrenergic stimulation on the voltage dependence of human IKur noted in previous work (Li et al. 1996).
Mechanisms of adrenergic modulation of IKur,d
The activation of PKA and PKC are important potential signal transduction pathways through which adrenoceptors can modulate ion channel function (Walsh & Kass, 1988; Fedida et al. 1993). Both β- and α-adrenergic stimulation can also affect ion channels by mechanisms independent of PKA and PKC (Brown & Birnbaumer, 1988; Freeman et al. 1992). In the present study, we obtained evidence for a central role of cAMP-dependent PKA activation and PKC activation in mediating β-adrenergic and α-adrenergic effects on IKur,d, respectively.
Phosphorylation by PKA and PKC can alter various properties of K+ channels, including current amplitude, voltage-dependent activation and inactivation, and kinetics. Drain et al. (1994) demonstrated that the application of phosphatases to the cytoplasmic side of Shaker channels in excised inside-out patches slows N-type inactivation, implying a role for phosphorylation in regulating N-type inactivation. The current density of both Ito and Kv4 channels is decreased by PKC (Nakamura et al. 1997). PKA increases Kv1.2 channel activity by increasing its open probability, an effect requiring the intactness of a single PKA consensus site near the amino terminus (Huang et al. 1994). PKC activation has biphasic effects on transient outward currents carried by Kv1.4, causing an initial small augmentation followed by a significant decrease (Murray et al. 1994).
We have found that many properties of IKur,d are similar to those reported for Kv3.1 cloned channels (Yue et al. 1996). Neuronal Kv3.1 currents are reduced by PKC activating phorbol esters and not affected by PKA (Kanemasa et al. 1995; Moreno et al. 1995). Variations in the amino acid sequence encoding otherwise similar channels in different tissues or species are known to result in discrepancies in modulation by adrenergic stimulation or protein kinases (Honore et al. 1991; Zhang et al. 1994). The molecular basis for IKur,d remains unclear for the moment, although we have preliminary unpublished data consistent with the presence of Kv3.1 in dog atrium. Cloning of the channel α-subunit and identification of potential associated subunits would be helpful in order to establish potential PKA and PKC phosphorylation sites on the pore-forming α-subunit, and to determine whether adrenergic enhancement is due to α-subunit phosphorylation or to interactions with accessory or regulatory proteins. If the role of Kv3.1 in carrying IKur,d is confirmed, this might provide a molecular basis for the divergent response to α-adrenergic stimulation of the canine current compared with human IKur, which is carried by Kv1.5 channel subunits.
Physiological relevance
In different tissues and species, varying effects of adrenergic stimulation on APD have been reported. Both α-adrenergic stimulation and PKC activation appear to decrease APD in guinea-pig papillary muscles (Dirksen & Sheu, 1990), although no change in APD has also been noted (Siems & Brasch, 1995). In rabbit and rat hearts, phenylephrine prolongs APD, apparently by inhibiting Ito (Apkon & Nerbonne, 1988; Hescheler et al. 1988; Braun et al. 1992). In the present study, we found that α-adrenergic stimulation reduces canine atrial APD.
Adrenergic stimulation may play an important role in controlling canine atrial refractoriness, with β-adrenergic stimulation abbreviating the refractory period (Liu & Nattel, 1997). In action potential studies, we found that phenylephrine abbreviates canine atrial APD, and that this action is abolished by relatively small concentrations of TEA (1 mM) that are highly selective for IKur,d. Thus, the degree of IKur,d enhancement that is caused by α-adrenergic stimulation is sufficient to exert significant effects on canine atrial repolarization. The effects of isoproterenol on the action potential were more complex, and probably involved multiple currents. IKur,d increases caused by isoproterenol were quantitatively greater than the maximal effect we noted for α-adrenergic stimulation, which was sufficient to significantly affect repolarization. It is therefore likely that the IKur,d enhancing effect of isoproterenol counterbalances APD-prolonging actions of β-adrenergic stimulation (such as increased ICa), resulting in less APD prolongation than would be caused in the absence of IKur,d. This concept is supported by the greater APD prolongation by isoproterenol in the presence of TEA than in its absence (Fig. 12).
Potential limitations
Since IKur,d is a rapidly activating current, it is easier to resolve and study at room temperature (Yue et al. 1996); we therefore performed all the whole-cell voltage-clamp experiments in the present study at room temperature. Adrenergic changes in some currents, such as classical IK, are temperature dependent (Walsh et al. 1989). Larger effects might have been observed had we performed the studies at body temperatures.
One of the difficulties in studying protein kinase mediation of drug action is the limited availability of highly specific inhibitors for use as probes. Bisindolylmaleimide is a highly selective inhibitor of PKC (Toullec et al. 1991). Although PKI is a specific inhibitor of PKA, the fact that it has to be applied by intracellular dialysis makes it impossible to evaluate the response to isoproterenol before and after PKA inhibition in the same cell. Therefore, we did complementary experiments in which the response to isoproterenol was assessed before and after exposure to H7. H7 inhibits both PKA and PKC, with an IC50 for PKA (3 μM) that is about half the IC50 for PKC (6 μM). At a concentration that was almost double the IC50 for PKA and below the IC50 for PKC, 5 μM, H7 completely prevented the response to isoproterenol. The results with H7, along with those obtained with the peptide PKA inhibitor, point to a central role for PKA as a mediator of β-adrenergic actions on IKur,d.
Conclusions
Both β- and α-adrenergic stimulation enhance IKur,d, acting via cAMP-mediated stimulation of PKA and by activation of PKC, respectively. These actions are likely to contribute to the adrenergic control of canine atrial repolarization, and point to the importance of ultrarapid delayed rectifier currents in the regulation of cardiac repolarization.
Acknowledgments
This work was supported by the Medical Research Council of Canada, the Quebec Heart Foundation and the Fonds de Recherche de l'Institut de Cardiologie de Montréal. L. Y. is supported by a Canadian Heart Foundation Studentship and Z. W. by a Research Scholarship Award of the Fonds de Recherche en Santé de Québec.
References
- Apkon M, Nerbonne JM. α1-Adrenergic agonists selectively suppress voltage-dependent K+ currents in rat ventricular myocytes. Proceedings of the National Academy of Sciences of the USA. 1988;85:8756–8760. doi: 10.1073/pnas.85.22.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkon M, Nerbonne JM. Characterization of two distinct depolarization-activated K+ currents in isolated adult rat ventricular myocytes. Journal of General Physiology. 1991;97:973–1011. doi: 10.1085/jgp.97.5.973. 10.1085/jgp.97.5.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backx PH, Marban E. Background potassium current active during the plateau of the action potential in guinea pig ventricular myocytes. Circulation Research. 1993;72:890–900. doi: 10.1161/01.res.72.4.890. [DOI] [PubMed] [Google Scholar]
- Boyle WA, Nerbonne JM. A novel type of depolarization-activated K+ current in isolated adult rat atrial myocytes. American Journal of Physiology. 1991;260:H1236–1247. doi: 10.1152/ajpheart.1991.260.4.H1236. [DOI] [PubMed] [Google Scholar]
- Braun AP, Fedida D, Giles WR. Activation of alpha 1-adrenoceptors modulates the inwardly rectifying potassium currents of mammalian atrial myocytes. Pflügers Archiv. 1992;421:431–439. doi: 10.1007/BF00370253. [DOI] [PubMed] [Google Scholar]
- Brown AM, Birnbaumer L. Direct G protein gating of ion channels. American Journal of Physiology. 1988;254:H401–410. doi: 10.1152/ajpheart.1988.254.3.H401. [DOI] [PubMed] [Google Scholar]
- Bruckner R, Mugge A, Scholz H. Existence and functional role of alpha 1-adrenoceptors in the mammalian heart. Journal of Molecular and Cellular Cardiology. 1985;17:639–645. doi: 10.1016/s0022-2828(85)80063-8. [DOI] [PubMed] [Google Scholar]
- Dirksen RT, Sheu SS. Modulation of ventricular action potential by α1-adrenoceptors and protein kinase C. American Journal of Physiology. 1990;258:H907–911. doi: 10.1152/ajpheart.1990.258.3.H907. [DOI] [PubMed] [Google Scholar]
- Drain P, Dubin AE, Aldrich RW. Regulation of Shaker K+ channel inactivation gating by the cAMP-dependent protein kinase. Neuron. 1994;12:1097–1109. doi: 10.1016/0896-6273(94)90317-4. [DOI] [PubMed] [Google Scholar]
- Duan D, Fermini B, Nattel S. Alpha-adrenergic control of volume-regulated Cl− currents in rabbit atrial myocytes. Characterization of a novel ionic regulatory mechanism. Circulation Research. 1995;77:379–393. doi: 10.1161/01.res.77.2.379. [DOI] [PubMed] [Google Scholar]
- Fedida D, Braun AP, Giles WR. Alpha 1-adrenoceptors reduce background K+ current in rabbit ventricular myocytes. The Journal of Physiology. 1991;441:673–684. doi: 10.1113/jphysiol.1991.sp018772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedida D, Braun AP, Giles WR. Alpha 1-adrenoceptors in myocardium: functional aspects and transmembrane signaling mechanisms. Physiological Reviews. 1993;73:469–487. doi: 10.1152/physrev.1993.73.2.469. [DOI] [PubMed] [Google Scholar]
- Fedida D, Shimoni Y, Giles WR. Alpha-adrenergic modulation of the transient outward current in rabbit atrial myocytes. The Journal of Physiology. 1990;423:257–277. doi: 10.1113/jphysiol.1990.sp018021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Wible B, Li GR, Wang Z, Nattel S. Antisense oligonucleotides directed against Kv1.5 mRNA specifically inhibit ultrarapid delayed rectifier potassium current in cultured adult human atrial myocytes. Circulation Research. 1997;80:572–579. doi: 10.1161/01.res.80.4.572. [DOI] [PubMed] [Google Scholar]
- Fiset C, Clark RB, Larsen TS, Giles WR. A rapidly activating sustained K+ current modulates repolarization and excitation-contraction coupling in adult mouse ventricle. The Journal of Physiology. 1997;504:557–563. doi: 10.1111/j.1469-7793.1997.557bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman LC, Kwok WM, Kass RS. Phosphorylation-independent regulation of cardiac IK by guanine nucleotides and isoproterenol. American Journal of Physiology. 1992;262:H1298–1302. doi: 10.1152/ajpheart.1992.262.4.H1298. [DOI] [PubMed] [Google Scholar]
- Gintant GA, Liu DW. Beta-adrenergic modulation of fast inward sodium current in canine myocardium. Syncytial preparations versus isolated myocytes. Circulation Research. 1992;70:844–850. doi: 10.1161/01.res.70.4.844. [DOI] [PubMed] [Google Scholar]
- Glass DB, Cheng HC, Mende-Mueller L, Reed J, Walsh DA. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. Journal of Biological Chemistry. 1989;264:8802–8810. [PubMed] [Google Scholar]
- Harvey RD, Hume JR. Autonomic regulation of a chloride current in heart. Science. 1989;244:983–985. doi: 10.1126/science.2543073. [DOI] [PubMed] [Google Scholar]
- Hescheler J, Nawrath H, Tang M, Trautwein W. Adrenoceptor-mediated changes of excitation and contraction in ventricular heart muscle from guinea-pigs and rabbits. The Journal of Physiology. 1988;397:657–670. doi: 10.1113/jphysiol.1988.sp017024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore E, Attali B, Romey G, Heurteaux C, Ricard P, Lesage F, Lazdunski M, Barhanin J. Cloning, expression, pharmacology and regulation of a delayed rectifier K+ channel in mouse heart. EMBO Journal. 1991;10:2805–2811. doi: 10.1002/j.1460-2075.1991.tb07829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XY, Morielli AD, Peralta EG. Molecular basis of cardiac potassium channel stimulation by protein kinase A. Proceedings of the National Academy of Sciences of the USA. 1994;12:624–628. doi: 10.1073/pnas.91.2.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaki K, Sukhatme VP, Shubeita HE, Chien KR. Alpha- and beta-adrenergic stimulation induces distinct patterns of immediate early gene expression in neonatal rat myocardial cells. fos/jun expression is associated with sarcomere assembly; Egr-1 induction is primarily an alpha 1-mediated response. Journal of Biological Chemistry. 1990;265:13809–13817. [PubMed] [Google Scholar]
- Jeck CD, Boyden PA. Age-related appearance of outward currents may contribute to developmental differences in ventricular repolarization. Circulation Research. 1992;71:1390–1403. doi: 10.1161/01.res.71.6.1390. [DOI] [PubMed] [Google Scholar]
- Kanemasa T, Gan L, Perney TM, Wang LY, Kaczmarek LK. Electrophysiological and pharmacological characterization of a mammalian Shaw channel expressed in NIH 3T3 fibroblasts. Journal of Neurophysiology. 1995;74:207–217. doi: 10.1152/jn.1995.74.1.207. [DOI] [PubMed] [Google Scholar]
- Koumi S, Backer CL, Arentzen CE, Sato R. Beta-adrenergic modulation of the inwardly rectifying potassium channel in isolated human ventricular myocytes. Alteration in channel response to beta-adrenergic stimulation in failing human hearts. Journal of Clinical Investigation. 1995;96:2870–2881. doi: 10.1172/JCI118358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi Y, Ito H, Sugimoto T, Shimizu T, Miki I, Ui M. Alpha-adrenergic activation of the muscarinic K+ channel is mediated by arachidonic acid metabolites. Pflügers Archiv. 1989;414:102–104. doi: 10.1007/BF00585635. [DOI] [PubMed] [Google Scholar]
- Li GR, Feng J, Wang Z, Fermini B, Nattel S. Adrenergic modulation of ultrarapid delayed rectifier K+ current in human atrial myocytes. Circulation Research. 1996;78:903–915. doi: 10.1161/01.res.78.5.903. [DOI] [PubMed] [Google Scholar]
- Liu L, Nattel S. Differing sympathetic and vagal effects on atrial fibrillation in dogs: role of refractoriness heterogeneity. American Journal of Physiology. 1997;273:H805–816. doi: 10.1152/ajpheart.1997.273.2.H805. [DOI] [PubMed] [Google Scholar]
- Moreno H, Kentros C, Bueno E, Weiser M, Hernandez A, Vega-Saenz de Miera E, Ponce A, Thornhill W, Rudy B. Thalamocortical projections have a K+ channel that is phosphorylated and modulated by cAMP-dependent protein kinase. Jounal of Neuroscience. 1995;15:5486–5501. doi: 10.1523/JNEUROSCI.15-08-05486.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KT, Fahrig SA, Deal KK, Po SS, Hu NN, Snyders DJ, Tamkun MM, Bennett PB. Modulation of an inactivating human cardiac K+ channel by protein kinase C. Circulation Research. 1994;75:999–1005. doi: 10.1161/01.res.75.6.999. [DOI] [PubMed] [Google Scholar]
- Nakamura TY, Coetzee WA, Vega-Saenz de Miera E, Artman M, Rudy B. Modulation of Kv4 channels, key components of rat ventricular transient outward K+ current, by PKC. American Journal of Physiology. 1997;273:H1775–1786. doi: 10.1152/ajpheart.1997.273.4.H1775. [DOI] [PubMed] [Google Scholar]
- Noble D, Tsien RW. Outward membrane currents activated in the plateau range of potentials in cardiac Purkinje fibres. The Journal of Physiology. 1969;200:205–231. doi: 10.1113/jphysiol.1969.sp008689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. Journal of General Physiology. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Hashimoto K. Effect of alpha 1-adrenoceptor stimulation with methoxamine and phenylephrine on spontaneously beating rabbit sino-atrial node cells. Naunyn-Schmiedeberg's Archives of Pharmacology. 1988;337:415–422. doi: 10.1007/BF00169533. [DOI] [PubMed] [Google Scholar]
- Siems T, Brasch H. Influence of phorbol esters on contractile force, action potential and calcium current of isolated guinea-pig heart tissues. Basic Research in Cardiology. 1995;90:459–466. doi: 10.1007/BF00788538. [DOI] [PubMed] [Google Scholar]
- Tohse N, Nakaya H, Kanno M. Alpha 1-adrenoceptor stimulation enhances the delayed rectifier K+ current of guinea pig ventricular cells through the activation of protein kinase C. Circulation Research. 1992;71:1441–1446. doi: 10.1161/01.res.71.6.1441. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhammel L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. Journal of Biological Chemistry. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Tsien RW, Giles W, Greengard P. Cyclic AMP mediates the effect of adrenaline on cardiac Purkinje fibers. Nature New Biology. 1972;240:181–183. doi: 10.1038/newbio240181a0. [DOI] [PubMed] [Google Scholar]
- Van Wagoner DR, Kirian M, Lamorgese M. Phenylephrine suppresses outward K+ currents in rat atrial myocytes. American Journal of Physiology. 1996;271:H937–946. doi: 10.1152/ajpheart.1996.271.3.H937. [DOI] [PubMed] [Google Scholar]
- Walsh KB, Begenisich TB, Kass RS. β-Adrenergic modulation of cardiac ion channels. Differential temperature sensitivity of potassium and calcium currents. Journal of General Physiology. 1989;93:841–854. doi: 10.1085/jgp.93.5.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh KB, Kass RS. Regulation of a heart potassium channel by protein kinase A and C. Science. 1988;242:67–69. doi: 10.1126/science.2845575. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circulation Research. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- Yue L, Feng J, Li GR, Nattel S. Characterization of an ultrarapid delayed rectifier potassium channel involved in canine atrial repolarization. The Journal of Physiology. 1996;496:647–662. doi: 10.1113/jphysiol.1996.sp021716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, Jurkiewicz NK, Folander K, Lazarides E, Salata JJ, Swanson R. K+ currents expressed from the guinea-pig cardiac IsK protein are enhanced by activators of protein kinase C. Proceedings of the National Academy of Sciences of the USA. 1994;91:1766–1770. doi: 10.1073/pnas.91.5.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipes DP. Sympathetic stimulation and arrhythmias. New England Journal of Medicine. 1991;325:656–657. doi: 10.1056/NEJM199108293250911. [DOI] [PubMed] [Google Scholar]