

Abstract
The outermost charged amino acid of S4 segments in the α subunit of human skeletal muscle sodium channels was mutated to cysteine in domains I (R219C), II (R669C), III (K1126C), and IV (R1448C). Double mutations in DIS4 and DIVS4 (R219C/R1448C), DIIS4 and DIVS4 (R669C/R1448C), and DIIIS4 and DIVS4 (K1126C/R1448C) were introduced in other constructs. Macropatch recordings of mutant and wild-type (hSkM1-wt) skeletal muscle sodium channels expressed in Xenopus oocytes were used to measure deactivation kinetics from open or fast inactivated states.
Conductance (voltage) curves (G (V)) derived from current (voltage) (I (V)) relations indicated a right-shifted G (V) relationship for R669C and for R669C/R1448C, but not for other mutations. The apparent valency was decreased for all mutations. Time-to-peak activation at -20 mV was increased for R1448C and for double mutations.
Deactivation kinetics from the open state were determined from the monoexponential decay of tail currents. Outermost charge-to-cysteine mutations in the S4 segments of domains III and IV slowed deactivation, with the greatest effect produced by R1448C. The deactivation rate constant was slowed to a greater extent for the DIII/DIV double mutation than that calculated from additive effects of single mutations in each of these two domains. Mutation in DIIS4 accelerated deactivation from the open state, whereas mutation in DIS4 had little effect.
Delays in the onset to recovery from fast inactivation were determined to assess deactivation kinetics from the inactivated state. Delay times for R219C and R669C were not significantly different from those for hSkM1-wt. Recovery delay was increased for K1126C, and was accelerated for R1448C.
Homologous charge mutations of S4 segments produced domain-specific effects on deactivation gating from the open and from the fast inactivated state. These results are consistent with the hypothesis that translocations of S4 segments in each domain during deactivation are not identical and independent processes. Non-identical effects of these mutations raise several possibilities regarding deactivation gating; translocation of DIVS4 may constitute the rate-limiting step in deactivation from the open state, DIVS4 may be part of the immobilizable charge, and S4 translocations underlying deactivation in human skeletal muscle sodium channel may exhibit co-operativity.
The flow of sodium ions through voltage-gated sodium channels is responsible for the rising phase of action potentials in excitable cells. Activation of sodium channels is presumed to be mediated in part by movement of charged entities within the channel protein. This model of channel activation was predicted by Hodgkin & Huxley (1952) and experimentally confirmed by Bezanilla & Armstrong (1975). Inherent within the Hodgkin & Huxley (HH) model was the assumption that charged membrane particles were identical and independent. Therefore, the sequence of their movement within the transmembrane electrical field should be random; any one of the particles could move first, second or third.
Functional analysis of channel structure has implicated the S4 membrane-spanning segments as putative voltage sensors. Hydropathy analysis of the deduced sequence of amino acids in the sodium channel α subunit reveals four homologous domains, each of which contains six membrane-spanning segments with a presumed alpha-helical structure (Noda et al. 1984). The fourth segment (S4) in each domain has a relatively high occurrence of positively charged residues at every third position of the helix. The hypothesis that the S4 segments are voltage sensors that translocate in response to alterations in the transmembrane electrical field is supported by the finding that charge neutralizations of S4 segments have marked effects on channel activation (Stuhmer et al. 1989; Papazian et al. 1991; Liman et al. 1992; Logothetis et al. 1993; Fleig et al. 1994; Kontis et al. 1997). In addition, recent experiments have shown that S4 segments in potassium channels (Larsson et al. 1996) as well as in sodium channels (Yang & Horn, 1995; Yang et al. 1997) alter their accessibility to modifying agents in response to changes in the transmembrane electrical field.
In contrast with potassium channels that are comprised of four identical subunits with equivalent charge densities in each S4 segment, the α subunit of sodium channels is comprised of non-identical domains including S4 segments with disparate charge densities. Domain I S4 (DIS4) contains the fewest charged residues, followed in order by DIIS4, DIIIS4, and DIVS4, which contains the greatest number of charged residues (Trimmer et al. 1989). This pattern is consistent for all sodium channel subtypes. In potassium channels, at least some charged residues participate in electrostatic interactions with oppositely charged residues in neighbouring membrane-spanning segments (Papazian et al. 1995). Since similar interactions may exist in sodium channels, it may be simplistic to suppose that total charge content alone dictates particular voltage sensitivities to each of the S4 segments. Nevertheless, it is tempting to speculate that the relative kinetics of S4 translocations for each domain might differ at least partly as a consequence of non-equivalent charge distribution in sodium channels.
Recent experiments have shown that a first charge-to-cysteine (FCC) mutation in DIVS4 (R1441C) of rat skeletal muscle sodium channels (rSkM1) prolongs open-state deactivation (Featherstone et al. 1998). In those experiments, as in others (Ji et al. 1996; Kontis et al. 1997), deactivation was well fitted by a single exponential function suggesting that channel closing can be produced by a single, two-state reaction which may represent the translocation of a single S4. We used single and double FCC mutations in each of the S4 segments of human skeletal muscle sodium channels (hSkM1) to examine the independence of S4 translocations underlying channel deactivation from both open and fast inactivated states. We show that the first charge-to-cysteine mutation in DIVS4 slows deactivation from the open to closed state (O→C transition) to a greater extent than homologous FCC mutations in other domains, but that the delay to recovery from fast inactivation (I→C transition) is accelerated by this mutation. These results raise the possibility that the voltage sensor in DIV may serve as the rate-limiting translocation in open-state deactivation, but may be immobilized and therefore the last to translocate in the I→C transition.
METHODS
Site-directed mutagenesis
The hSkM1 mutant R1448C has been described previously (Chahine et al. 1994). Three additional cysteine mutations in hSkM1 corresponding to the outermost positively charged S4 residues in DI (R219C), DII (R669C) and DIII (K1126C) were created using PCR mutagenesis. To construct R219C, a forward primer (5′-GGG ATC TAC ACC TTT GAG TC-3′) spanning nucleotides (nt) 573-592, and a mutagenic reverse primer (5′-GGC CCC CAC GTAT CGT CTT CAG CCC TGG GAT GAC CGT GAT GGT TTT GAG GGC CCG CAG CAC CCG GAA TGT ACA CAG GGC TGA GAT GTT GCC CAA-3′, nt 711-803) were used to create the mutation and incorporate natural restriction sites for Sal I (nt 625) and Pvu I (nt 794). To construct R669C, a mutagenic forward primer (5′-GTA GAG CTA GGC CTG GCC AAC GTA CAG GGA CTG TCT GTG CTC TGC AGC TTC CGT CTG CTG CGG GTC TTC-3′, nt 2040-2108), and a reverse primer (5′-ATG AAC ACG ATG ATA GC-3′, nt 2199-2215) were used to create the mutation and incorporate natural restriction sites for Stu I (nt 2051) and Bst EII (nt 2179). To construct K1126C, a mutagenic forward primer (5′-GGC TAC TCG GAG CTG GGA CCC ATA TGT TCC CTG CGG ACA CTG CGG GCC CTG CGT-3′, nt 3429-3482), and a reverse primer (5′-GTT GAC ACC CAT GAT GC-3′, nt 3601-3618) were used to create the mutation and incorporate natural restriction sites for San DI (nt 3445) and Avr II (nt 3537). Amplifications (20 cycles) were performed using 20 ng of hSkM1 cDNA as template and Taq DNA polymerase. Double mutations were prepared by interchanging the appropriate restriction fragment from single mutants containing the desired mutation for a wild-type fragment in R1448C. For R219C/R1448C and R669C/R1448C, a 2.3 kb Not I-Nsi I fragment was cloned. For K1126C/R1448C and R1448C/QQQ, a 2 kb Bst EII fragment was cloned and mapped for proper orientation. All clones were verified by sequencing. Final products were purified by spin-column chromatography (Qiagen, Santa Clarita, CA, USA), digested with the appropriate restriction endonucleases, and ligated into the corresponding sites in the plasmid pSP64T-hSkM1. The amplified regions were sequenced entirely in the final constructs to verify the mutation and to exclude polymerase errors.
Oocyte preparation
In vitro transcription of α and β1 subunits was performed as described previously (Featherstone et al. 1996; Richmond et al. 1997). RNA for both α and β1 subunits (1:1 volume, α subunit at a concentration of 1 μg μl−1, and β1 subunit at a concentration of 3 μg μl−1) was injected (50.6 nl oocyte−1) into Xenopus oocytes that had been surgically removed from frogs anaesthetized with 0.17 % tricaine (3-aminobenzoic acid ethyl ester; Sigma). Oocytes were separated using 2 mg ml−1 collagenase (Sigma) dissolved in a solution containing (mM): NaCl, 96; KCl, 2; MgCl2, 20; Hepes, 5; pH 7.4. Oocytes were incubated at 18°C for at least 3 days prior to recording in a medium containing (mM): NaCl, 96; KCl, 2; MgCl2, 1; CaCl2, 1.8; Hepes, 5; sodium pyruvate, 2.5; pH 7.4. Before macropatch recording, vitelline membranes were manually removed following brief hyperosmotic treatment in a solution containing (mM): NaCl, 96; KCl, 2; MgCl2, 20; Hepes, 5; mannitol, 400; pH 7.4.
Frogs were allowed to recover following the first five surgeries. After the sixth surgery, frogs were transferred to a freezer while anaesthetized. These procedures were approved by the Institutional Animal Care and Use Committee at Utah State University and are in accordance with NIH regulations.
Electrophysiology
All recordings were from oocytes co-injected with α and β1 sodium channel subunits, using cell-attached macropatch techniques (Featherstone et al. 1998), which allow excellent control of membrane voltage. Pipette solution used was (mM): NaCl, 96; KCl, 4; MgCl2, 1; CaCl2, 1.8; Hepes, 5; pH 7.4. Bath solution used was (mM): NaCl, 9.6; KCl, 88; EGTA, 11; Hepes, 5; pH 7.4. Voltage clamping and data acquisition were performed as described previously (Featherstone et al. 1996; Richmond et al. 1997) using an EPC-9 patch-clamp amplifier (HEKA, Lambrecht, Germany) controlled via Pulse software (HEKA) running on a Power Macintosh 8600/300. Data were acquired at 5 μs per point and low-pass filtered at 5 kHz during acquisition. Experimental bath temperature was maintained at 15 ± 0.2°C for all experiments by using a peltier device controlled by an HCC-100A temperature controller (Dagan, Minneapolis, MN, USA). Holding potential for all experiments was -120 to -150 mV. Leak subtraction was performed automatically by the software using a P/4 protocol. Hardware leak/capacitance subtraction was done upon patch formation and corrected before each voltage clamp experiment. Subsequent analyses and graphing were done using Pulsefit (HEKA) and Igor Pro (Wavemetrics, Lake Oswego, OR, USA), both run on a Power Macintosh G3.
Conductance (voltage) curves were computed using the equation:
where G is conductance, Imax represents the peak test pulse current, Vm the test pulse voltage, and ENa the measured equilibrium potential for sodium. Activation curves were fitted by a Boltzmann distribution, as follows:
where ‘normalized current amplitude’ is measured during a variable-voltage test pulse from a holding potential of -150 mV. Vm is the test pulse/prepulse potential, z is the apparent valency, e0 is the elementary charge, V½ is the midpoint voltage, k is the Boltzmann constant, and T is the absolute temperature.
Descriptions of open-state deactivation rates, given as time constants (τ), were derived from fitting the monoexponential decay of tail currents according to the function:
where I(t) is current amplitude as a function of time, Offset is the plateau amplitude (asymptote), a1 is the amplitude at time 0, and τ is the time constant. The delay to the onset of recovery from fast inactivation was measured to assess deactivation from the inactivated state. A 5 or 21 ms depolarizing voltage step to 0 mV was used to inactivate channels. This was followed by a recovery pulse to voltages that ranged between -190 and -110 mV for durations that ranged from 0.05 to 5 ms in steps of 50 μs. The recovery pulse was followed by a 5 ms test pulse to 0 mV during which the extent of recovery was measured. The recovery in peak amplitude during the test pulse was fitted by a single exponential function as above, over the range of recovery durations from 2 to 5 ms. The exponential function was extrapolated to the time (t) at which current amplitude was 0. This time was taken as the delay to the onset of recovery from fast inactivation. All results are reported as means ±s.e.m. Statistical significance was determined by using Student's t test or, in those cases where there was a statistically significant difference between standard deviations, Welch's alternative t test. Statistical significance of difference was accepted at P values < 0.05.
RESULTS
Activation
Activation results from a series of conformational changes involving voltage-sensitive and voltage-insensitive transitions (Patlak, 1991). The voltage-sensitive transitions are thought to arise from translocations of the S4 membrane spanning segments (Yang & Horn, 1995). To determine whether the voltage dependence and kinetics of activation were altered by the FCC mutations, channels were depolarized from a holding potential of -150 mV at 15°C with pulses from -90 to +60 mV. Figure 1 shows the voltage dependence of activation (G/Gmax) for both single and double FCC mutations, compared with hSkM1-wt. Conductance was maximal by 0 mV for all mutations and for wild-type. The two FCC mutations incorporating changes in DIIS4 produced a significant, depolarizing shift in the voltage dependence of activation (R669C, P = 0.0001; R669C/R1448C, P = 0.04). Single and double FCC mutations of S4 in other domains did not alter the voltage dependence of activation. Apparent valency (z in Boltzmann equation: see Methods) was significantly decreased with respect to hSkM1-wt for all FCC mutations (P = 0.03 or less), consistent with the decrease in overall charge expected with such mutations. Time-to-peak activation at -20 mV was increased compared with hSkM1-wt for all FCC mutations involving DIVS4 (R1448C, P = 0.004; R219C/R1448C, P = 0.001; R669C/R1448C, P = 0.001; K1126C/R1448C, P = 0.0001). Single FCC mutations in DIS4, DIIS4 and DIIIS4 did not significantly alter time-to-peak activation. These results are summarized in Table 1.
Figure 1. Normalized conductance curves for hSkM1-wt, single FCC and double mutations.
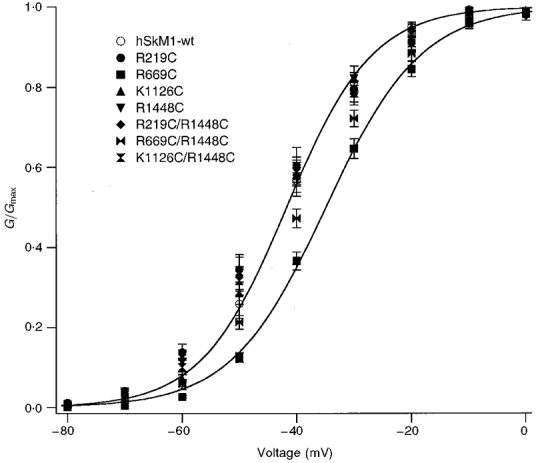
For each, the voltage dependence of activation is shown for the voltage range -80 to 0 mV where maximum conuctance was observed (n = 11-16 experiments for each). Midpoints and apparent valencies were determined from Boltzmann equations describing a two-state reaction; Boltzmann fits for hSkM1-wt (higher) and for R669C (lower) are shown.
Table 1.
Activation properties of hSkM1-wt and FCC mutations
| Mutation | V1/2 (mV) | Apparent valency (z) | Time to peak(μs) | |
|---|---|---|---|---|
| hSkM1-wt | (n = 14) | −42 ± 1 | 3.61 ± 0.06 | 617 ± 41 |
| R219C | (n = 12) | −44 ± 1 | 2.87 ± 0.11* | 685 ± 39 |
| R669C | (n = 16) | −35 ± 1* | 3.13 ± 0.04* | 596 ± 16 |
| K1126C | (n = 11) | −42 ± 1 | 3.35 ± 0.09* | 682 ± 25 |
| R1448C | (n = 13) | −44 ± 2 | 3.26 ± 0.06* | 848 ± 62* |
| R219C/R1448C | (n = 14) | −42 ± 1 | 2.65 ± 0.10* | 1009 ± 61* |
| R669C/R1448C | (n = 12) | −38 ± 1* | 3.01 ± 0.06* | 817 ± 32* |
| K1126C/R1448C | (n = 11) | −41 ± 2 | 2.96 ± 0.10* | 933 ± 55* |
Activation parameters of wild-type, single and double FCC mutations. Midpoint and apparent valency were derived from Boltzmann distributions (see Methods) fitted to the data by a least-squares method. Time-to-peak values were derived by fitting current records with a six-order polynomial waveform and measuring the time between onset of the voltage pulse and the most negative value of the fitted curve. Values are means ± s.e.m.
P < 0.05 compared with hSkM1-wt with Student's t test.
Deactivation from open state
Earlier studies have shown that deactivation is the voltage-dependent transition of channels from the open state to the nearest closed state (Rayner et al. 1993). It is therefore reasonable to speculate that deactivation represents the transition of a single voltage sensor from its depolarization-favoured position into its hyperpolarization-favoured position. Deactivation from the open state is slowed by the analogous DIVS4 FCC mutations R1448C in hSkM1 (Ji et al. 1996) and R1441C in rSkM1 (Featherstone et al. 1998). We sought to determine whether deactivation kinetics were similarly affected by homologous S4 mutations in DI to DIV. Tail currents were elicited by briefly opening channels with 0 mV, 0.25 ms depolarizing pulses from a holding potential of -150 mV, followed by repolarizing pulses ranging from -200 to -50 mV. We restricted our analyses of deactivation time constants (τ) to the voltage range -120 to -70 mV as shown in Figs 2 and 3. Differences in deactivation time constants of wild-type and mutant channels were not evident at voltages more negative than -120 mV. In addition, since fast inactivation is dependent upon the IFM motif (Patton et al. 1992; West et al. 1992), the fraction of current decay due to deactivation versus inactivation at a given command voltage in the tail current protocol was assessed with the IFM1310QQQ mutation (Featherstone et al. 1998). We also used the double mutation R1448C/IFM1310QQQ to test whether the FCC mutation influenced the voltage range over which deactivation versus inactivation prevailed for tail current decay. At 15°C, tail currents completely decayed to baseline at voltages more negative than -60 mV for IFM1310QQQ and for IFM1310QQQ/R1448C.
Figure 2. Open-state deactivation kinetics for single FCC S4 mutations.
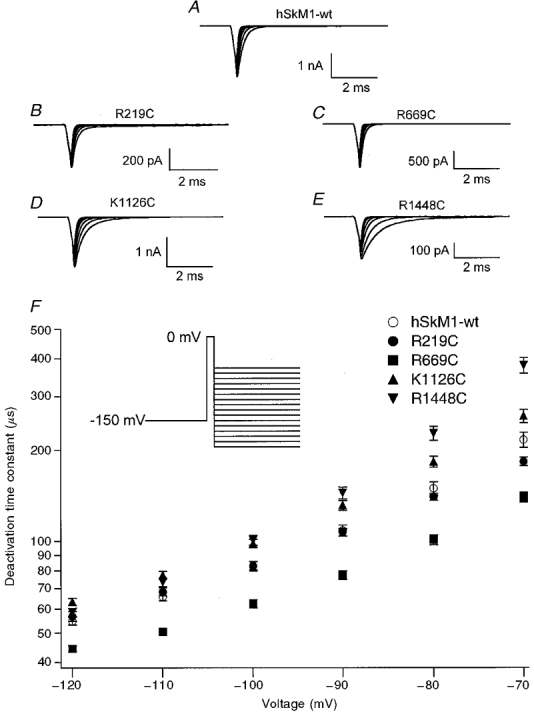
Open-state deactivation kinetics for single FCC S4 mutations as determined by decay of tail currents elicited by a 0 mV, 0.25 ms depolarizing pulse to open channels followed by hyperpolarizing command potentials from -120 to -70 mV. Tail currents are shown for hSkM1-wt (A, n = 31), R219C (B, n = 24), R669C (C, n = 20), K1126C (D, n = 23) and R1448C (E, n = 31); the voltage dependence of the deactivation time constant is shown for each mutation in F. FCC mutations in domains III and IV have slowed deactivation from the open state and FCC mutation in domain II showed a more rapid deactivation from the open state.
Figure 3. Voltage dependence of open-state deactivation kinetics for double FCC S4 mutations.
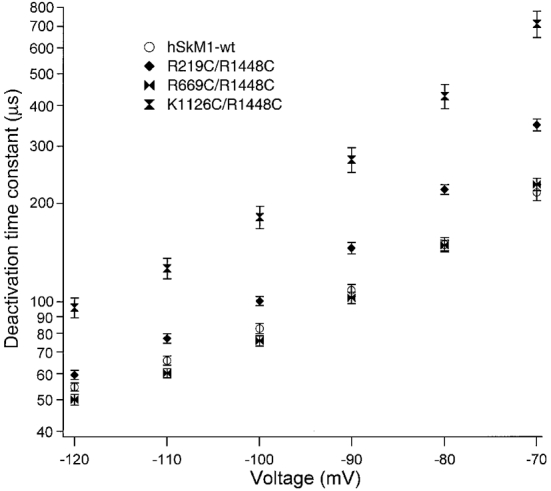
Double FCC S4 mutations in which a single FCC mutation in domain I (R219C/R1448C, n = 7-10), II (R669C/R1448C, n = 8-14) or III (K1126C/R1448C, n = 8-17) is paired with the R1448C mutation for domain IV. Deactivation time constants for double mutations are compared with hSkM1-wt. Deactivation time constants for the double mutation in DIII/DIV were longer than that for either DIII or DIV alone, while time constants for the double mutation in DII/DIV were shorter than that for DIV alone.
Deactivation time constants were determined from the monoexponential decay of tail currents. Figure 2A-E shows tail current traces obtained from macropatches of hSkM1-wt and FCC mutants, and Fig. 2F shows the voltage dependence of deactivation from the open state for these channels. Open-state deactivation time constants for K1126C and R1448C were significantly slower than that for hSkM1-wt over the voltage range from -110 to -70 mV (K1126C versus hSkM1-wt, P = 0.005; R1448C versus hSkM1-wt, P = 0.02). At voltages of -90 and -70 mV, deactivation was most slowed in R1448C. In contrast, R669C significantly accelerated deactivation from the open state over the voltage range shown in Fig. 2 (R669C versus hSkM1-wt, P < 0.0001). R219C did not significantly alter the deactivation time constant.
Double FCC mutations were constructed in which a FCC mutation in DIVS4 was combined with the FCC mutations in DIS4, DIIS4 or DIIIS4. Figure 3 shows the voltage dependence of open-state deactivation kinetics for these mutations. The deactivation time constant was altered by double FCC mutations including R1448C, in the direction predicted by the effects of single FCC mutations. Thus, no significant differences in deactivation time constants were detected between R1448C and R219C/R1448C over the voltage range -120 to -70 mV (P > 0.2). Additionally, the deactivation time constant for R669C/R1448C was less than that observed for R1448C (P = 0.009 or less). Finally, the deactivation time constant for K1126C/R1448C was greater than that observed for R1448C from -120 to -70 mV (P < 0.0001).
If the effects of double mutations on deactivation are independent, then the changes in deactivation rate constants should be additive. Thus, independence between the modified S4s would be revealed by the summed effect of the individual mutations (i.e. the difference between the mutations and hSkM1-wt) being approximately equal to the effect of the double FCC mutation. To determine whether the effects of K1126C/R1448C were additive, we compared the summed differences of the rate constants for the single FCC mutations (relative to hSkM1-wt) with the differences between double FCC mutations and hSkM1-wt. These differences are presented in Table 2, which shows the difference between deactivation rate constants for K1126C and hSkM1-wt (column a), R1448C and hSkM1-wt (column b), and K126C/R1448C and hSkM1-wt (column d). Column c shows the summed differences reported in columns a and b. These are 10- to 15-fold larger than the difference in deactivation rate constants between K1126C/R1448C and hSkM1-wt reported in column d, suggesting co-operativity between the effects of each mutation in the double FCC mutant. Column e reports the ratio of values given in columns c and d, and suggests that there is a slight voltage dependence to the effect of K1126C/R1448C on deactivation.
Table 2.
Deactivation rate constant (μs−1) differences between FCC mutations and hSkM1-wt
| Voltage (mV) | K1126C - hSkM1-wt (a) | R1448C - hSkM1-wt (b) | Sum of a + b (c) | K1126C/R1448C - hSkM1-wt (d) | Ratio of c/d (e) |
|---|---|---|---|---|---|
| −120 | 0.115 | 0.256 | 0.371 | 0.024 | 15.4 |
| −110 | 0.086 | 0.129 | 0.215 | 0.016 | 13.4 |
| −100 | 0.064 | 0.055 | 0.119 | 0.010 | 11.9 |
| −90 | 0.043 | 0.028 | 0.071 | 0.006 | 11.8 |
| −80 | 0.029 | 0.013 | 0.042 | 0.004 | 10.5 |
Deactivation rate constants were derived from first-order exponential functions (see Methods) fitted to tail currents using a least-squares method. Tail currents were evoked as described in the text and legend to Fig. 2. Rate constants were averaged and FCC averages were subtracted from hSkM1-wt averages to derive values in columns a, b and d.
Delay in onset to recovery from inactivation
Channels recover from fast inactivation almost exclusively by deactivating, rather than passing through the open state (Kuo & Bean, 1994). This finding suggests that the initial delay in the recovery of channels from fast inactivation is due to the time required for inward translocation of the S4 segments in response to hyperpolarization. We used a double pulse protocol to measure deactivation kinetics from the fast inactivated state. The first pulse was used to open and inactivate channels, followed by a hyperpolarizing command interpulse of varying duration and voltage prior to a second depolarizing pulse to test for recovery from inactivation. The delay in recovery was measured by determining the zero current intercept from an exponential function fitted to the recovered peak current amplitudes. Figure 4 shows the results of experiments using a 5 ms depolarizing pulse. Figure 4A-C shows traces for fast inactivation recovery delays with a -130 mV command interpulse for hSkM1-wt, K1126C (delay increased) and R1448C (delay abbreviated). The voltage dependence of delay shown for all single FCC mutations is shown in Fig. 4D. Delay in the onset of recovery was not altered with respect to hSkM1-wt for R219C or for R669C. However, recovery delay was increased for K1126C at -170 mV (P < 0.0001), -150 mV (P < 0.003), -130 mV (P < 0.002) and -110 mV (P < 0.035). In contrast, recovery delay was abbreviated in R1448C at -190 mV (P < 0.002), -170 mV (P < 0.05), -130 mV (P < 0.001) and -110 mV (P < 0.015).
Figure 4. Deactivation kinetics determined by delay in the onset to recovery from fast inactivation.
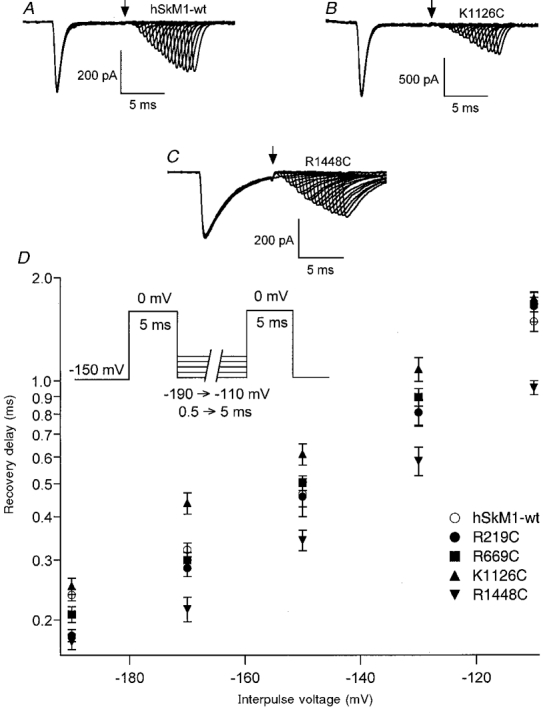
Deactivation kinetics from the inactivated state as determined by the delay in the onset to recovery from fast inactivation. Channels from patches incorporating hSkM1-wt or FCC S4 mutations (n = 7-14 experiments each) were first depolarized to 0 mV for 5 ms, followed by a hyperpolarizing interpulse of varying voltage and duration prior to a second depolarizing pulse to assess channel availability. Traces showing every fifth sweep are shown for hSkM1-wt (A), K1126C (B), and R1448C (C); in each instance the interpulse voltage was -130 mV. Longer delays were observed for K1126C while shorter delays were observed for R1448C. The arrow depicts the time of the second depolarizing pulse for the first sweep. Delays were calculated with a backexponential fit of normalized current amplitudes elicited with the second depolarizing pulse. In D, the voltage dependence of recovery delay on interpulse voltage is shown for hSkM1-wt and for each of the FCC S4 mutations.
We observed that, at 15°C, fast inactivation in R1448C was not always complete with a 5 ms initial depolarizing pulse. As a result of this we also used a 21 ms initial depolarizing pulse. Figure 5 compares the voltage dependence of recovery delay for hSkM1-wt with that for R1448C with inactivation complete, and shows a similar result of abbreviated delay in the DIVS4 FCC mutant. Other FCC mutations were also tested with the longer first pulse protocol, and delays for these mutants are also depicted. Recovery delay was not affected in R219C or in R669C except at -190 mV (R219C, P < 0.0015, n = 8-10; R669C, P < 0.045, n = 14-16). By contrast, delay was longer for K1126C over the voltage range from -190 to -130 mV (P = 0.02, n = 9-15) but was abbreviated for R1448C at all voltages tested (P = 0.02, n = 11-14). Thus, the effect of a single FCC mutation in DIII prolonged both deactivation from the fast inactivated state and deactivation from the open state, whereas the effect on recovery delay of a single FCC mutation in DIV was in the opposite direction to its effect on deactivation from the open state.
Figure 5. Voltage dependence of delay in onset to recovery from fast inactivation.
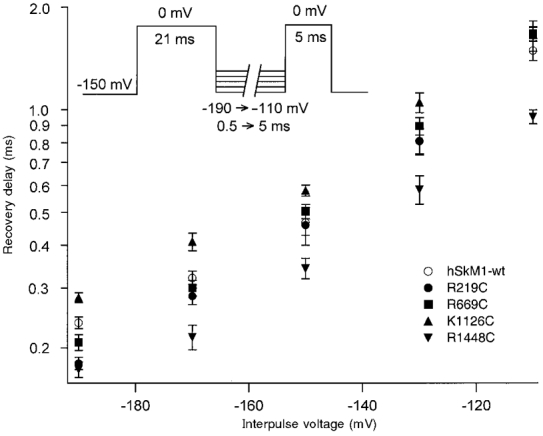
Voltage dependence of the delay in onset to recovery from fast inactivation assessed using a 21 ms first depolarizing pulse to fully inactivate channels for hSkM1-wt (n = 15-17) and R1448C (n = 11-14). The FCC mutation in DIVS4 resulted in an abbreviated recovery delay, as also observed in Fig. 4 in which fast inactivation was not complete. Recovery delays in R219C (n = 8-10) and R669C (n = 14-16) were not significantly affected except at -190 mV. By contrast, delay was longer for K1126C over the voltage range from -190 to -130 mV (n = 9-15).
Figure 6 shows the voltage dependence of delay in the onset of recovery from fast inactivation for double mutations incorporating the FCC mutation of DIVS4 with FCC mutations in DIS4 (R219C/R1448C), in DIIS4 (R669C/R1448C) and in DIIIS4 (K1126C/R1448C). Double mutations produced effects on recovery delay consistent with the effects of single FCC mutations. For example, R219C/R1448C recovery delay was not significantly different from that for R1448C at any voltage, but delays were significantly less than those observed with hSkM1-wt over the entire voltage range tested (P = 0.03). In contrast, recovery delays for K1126C/R1448C were significantly longer than those observed for R1448C at -190 to -110 mV (P = 0.02) but were not significantly different from recovery delays observed for hSkM1-wt over the entire voltage range tested. Finally, R669C/R1448C recovery delays were longer than those observed for R1448C at -170 mV (P < 0.02) and at -130 mV (P < 0.002; data not shown), consistent with the effect of R669C to slightly increase recovery delay with respect to hSkM1-wt at these voltages (see Fig. 4C and D).
Figure 6. Delay in recovery for double FCC S4 mutations.
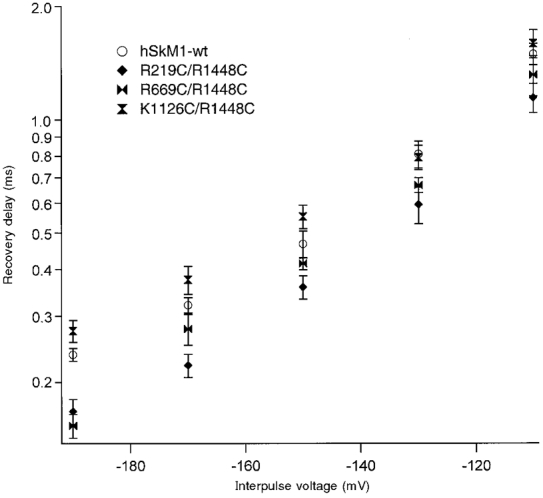
The voltage dependence of delay is shown for hSkM1-wt (n = 15-17) and for the double FCC S4 mutations in DI/DIV (R219C/R1448C, n = 8-11), DII/DIV (R669C/R1448C, n = 7-14) and DIII/DIV (K1126C/R1448C, n = 9-10). Delay times for these mutations are consistent with predicted additive effects based on deactivation kinetics relative to hSkM1-wt of single FCC mutations shown in Figs 4 and 5 (inactivated-state deactivation) and on deactivation kinetics depicted in Fig. 2 (open-state deactivation).
DISCUSSION
This paper explores the effects of homologous mutations in each domain of the human skeletal muscle sodium channel on the kinetics and voltage dependencies of activation, deactivation, and the delay to the onset of recovery from open-state fast inactivation. In contrast to the multiexponential waveform of sodium channel activation, deactivation from the open state is readily fitted by a single exponential function. The simplest assumption based on these observations is that channel closing is accomplished by a single conformational change, whereas activation requires a series of conformational changes in protein structure. Since the rate of deactivation is steeply voltage dependent and follows a time course fitted by a single exponential function (Bezanilla & Armstrong, 1977; Ji et al. 1996; Kontis et al. 1997; Featherstone et al. 1998), it is reasonable to presume that the conformational change leading to channel closing is likely to be the movement of a single voltage sensor. The rate of this reaction can be approximated from the exponential decay of current following a depolarizing pulse sufficiently brief so as to avoid significant open-state fast inactivation. Closed-state fast inactivation, as produced by depolarizing prepulses, should not appreciably affect tail current kinetics since channels inactivated from the closed state are unlikely to recover and thus contribute to deactivation.
Longer depolarizations, by contrast, will permit channels to fast inactivate. Since tail currents cannot be readily recorded from the inactivated state, the I→C transition may best be measured from the delay to the onset of recovery from fast inactivation (Kuo & Bean, 1994). This delay should be limited by immobilization of the gating particles (Armstrong & Bezanilla, 1977), i.e. one or more of S4 segments, the putative voltage sensors. Although the mechanism of charge immobilization has not been elucidated, a reasonable hypothesis is that immobilization is achieved via interactions between the DIII-DIV linker (which has been demonstrated to comprise an important component of the fast inactivation mechanism; see Patton et al. 1992; West et al. 1992) and S4-S5 linkers in DIII (Smith & Goldin, 1997) and in DIV (Lerche et al. 1997). These interactions might impose constraints on the I→C transition that could differentiate its reaction rate from that of the O→C transition.
We investigated the role of each of the four S4s in the O→C and I→C transitions with homologous FCC mutations. These mutations produced non-identical effects on the voltage sensitivity and kinetics of activation, and on kinetics of the O→C and I→C transitions. For example, the FCC mutation in DIS4 (R219C) had no significant effect on any of these processes with the exception of changing the apparent valency of activation, as did all single and double FCC mutations. In contrast, the FCC mutation in DIIS4 (R669C) shifted the G (V) midpoint and accelerated the O→C transition without affecting the I→C transition. Activation kinetics were unaffected by the FCC mutation in DIIIS4 (K1126C). In contrast to the effect of R669C, K1126C slowed the O→C transition as well as the I→C transition. An increase in time-to-peak activation was observed in DIVS4 (R1448C), but not in any other FCC mutations. R1448C slowed the rate of the O→C transition as did K1126C, but accelerated the I→C transition, unlike K1126C. Our observations, therefore, are in agreement with previous studies that have shown non-identical behaviour of the putative voltage sensors in sodium channels (Chahine et al. 1994; Chen et al. 1996; Kontis & Goldin, 1997; Kontis et al. 1997), and that have demonstrated dissimilar effects of R1448C on O→C and I→C transitions (Ji et al. 1996). Results of the present and earlier investigations (Stuhmer et al. 1989; Kontis et al. 1997) suggest that, in contrast to the predictions of the Hodgkin & Huxley (HH) formulation, the S4 voltage sensors respond neither identically nor independently to changes in the electrical field. Thus, each S4 may play a different role in activation and in the link between activation and fast inactivation.
The effect on the O→C transition induced by R1448C was significantly greater than that observed for any other single FCC mutation. This result leads us to suggest that DIVS4 may be the rate-limiting step in the O→C transition. That is, the S4 transmembrane segment in DIV may be the first to translocate from its depolarized-favoured position to its hyperpolarized-favoured position when channels close directly from the open state. Thus, in deactivation, DIVS4 could have the highest probability of moving first, as opposed to the random sequence predicted by the HH model. An extension to this line of thinking suggests that DIIIS4 might either translocate next, or have the second highest probability of moving first, since its effect to increase the deactivation time constant was second greatest. Although it may be coincidental, it is at least noteworthy that S4 charge inequalities for hSkM1 are consistent with these putative differences in reactivities to changes in the transmembrane electrical field; in hSkM1, DIVS4 has eight positively charged residues, DIIIS4 has six, DIIS4 has five, and DIS4 has four (George et al. 1992).
It has been elegantly demonstrated that not all charges respond similarly to changes in the electrical field (Papazian et al. 1995). Results from those and other studies (Auld et al. 1990; Kontis et al. 1997) have suggested that, in addition to charge content of an individual residue, amino acid structure contributes to overall S4 movement. Therefore, allosteric effects of channel mutations cannot be discounted as contributors to the results of the present and other studies using site-directed mutagenesis to investigate structure- function relationships. Consistent with this idea, we found that one of the FCC mutations, R669C, accelerated rather than slowed O→C deactivation. Nevertheless, our results are consistent with the hypothesis that the putative voltage sensor with the greatest number of charges (DIVS4) might be the most reactive, accelerate most quickly and, therefore, be the rate-limiting step in deactivation (O→C) gating. Experiments in progress are investigating the effects of charge neutralization of each of the basic amino acid residues in DIVS4 in rSkM1 on deactivation gating, since FCC mutations of this domain produced the most dramatic effects.
In contrast to its slowing of O→C deactivation, R1448C significantly accelerated the delay to onset of recovery from fast inactivation. We measured the delay by fitting an exponential curve to the recovery from fast inactivation and extrapolating to determine when recovery first begins, thereby avoiding the measurement of ionic currents that could be obscured by electrical noise. Using a similar double pulse protocol, Kuo & Bean (1994) demonstrated that sodium channels must deactivate before they can recover from fast inactivation. The delay in the onset of recovery from fast inactivation, therefore, in part represents deactivation. In fact, we observed a similar effect on deactivation from the open state and from the fast inactivated state for FCC mutations in DIS4, in DIIS4 and in DIIIS4. By contrast, the mutation in DIVS4 resulted in a decreased rate of deactivation from the open state, but an abbreviated onset to the delay of recovery from fast inactivation. Thus, the effects of this mutation were both different from those observed for FCC mutations in other domains, and differential with respect to the state from which deactivation occurred.
Several possibilities might explain these findings with the FCC mutation in DIVS4. First, deactivation from the open state and from the fast inactivated state may not be closely related, and mutations of charge may not be expected to affect open-state deactivation in a manner similar to the effect on recovery delay. However, if deactivation from the open state and delay in recovery from fast inactivation do in fact represent similar S4 translocations as suggested by Kuo & Bean (1994), our results suggest an alternative hypothesis. Charge immobilization, first measured by Armstrong & Bezanilla (1977), accompanies fast inactivation. Although, as noted above, the exact mechanism of charge immobilization has yet to be elucidated, it seems reasonable to postulate that the translocation of one or more S4s must be restricted, and charge immobilization must, therefore, be overcome before deactivation can occur and channels can recover from fast inactivation. The I→C transition might thus involve a sequence of recovery from charge immobilization followed by S4 translocation. Our results showing that FCC in DIVS4 accelerates I→C suggests the possibility that this transmembrane segment is influenced by charge immobilization, and that FCC in DIVS4 reduces the extent of immobilization. Our findings, taken in the light of these previous studies, are consistent with the hypothesis that DIVS4 could comprise the immobilizable component of gating charge movement (Armstrong & Bezanilla, 1977; Chen et al. 1996). Gating current and fluorescence measurements of single S4 translocations support this hypothesis (Cha et al. 1999). In addition, experiments are in progress to examine more closely the relationship between deactivation from the open state and delay in the onset to the recovery from fast inactivation in DIV.
The results of tail current measurements using double FCC mutations suggest a co-operative interaction between S4 segments in different domains. If S4 translocations in DIIIS4 and DIVS4 were independent, then the summed difference of deactivation time constants for K1126C from hSkM1-wt and for R1448C from hSkM1-wt should be the same as that for the double FCC mutation from hSkM1-wt. However, open-state deactivation in the DIIIS4/DIVS4 double mutant was affected to a significantly greater extent than by these summed effects of K1126C and of R1448C on the deactivation time constant at all voltages tested from -120 to -70 mV (see Fig. 2E and Table 2). Previous studies have suggested subunit co-operativity in potassium channels (Tytgat & Hess, 1992), and our results extend the idea of synergism to the inter-domain level.
Our results with double FCC mutations are consistent with the suggestion that tail currents and delay to the onset of recovery from fast inactivation may be closely related (Kuo & Bean, 1994). Thus, the O→C transition was slowed by both K1126C and R1448C, and the K1126C/R1448C double mutant slowed O→C deactivation to a greater extent, as noted above. The I→C transition was accelerated by R1448C and delayed by K1126C, and the double mutant was not statistically different from hSkM1-wt. In other words, the effects on I→C of these two FCC mutations appeared to effectively cancel one another's individual effects. The synergistic effects of DIIIS4/DIVS4 double mutants on both O→C and I→C suggest that these transitions may be related, having in common the translocation of voltage sensors into their hyperpolarization-favoured positions.
In contrast to potassium channels, which are made up of four identical subunits, sodium and calcium channels provide a unique opportunity to study differences in S4 contributions to gating mechanisms. Our observations of human skeletal muscle sodium channels suggest that the putative voltage sensors do not respond identically to changes in the electrical field, and both confirm and extend previous reports of a unique role for DIVS4 in the interaction between activation and fast inactivation.
Acknowledgments
The authors thank J. Olsen for performing some of the experiments in this study and J. Abbruzzese and L. Walter for help with data analysis and with the figures. This work was supported by a Muscular Dystrophy Association grant and PHS grant R01-NS29204 to P.C.R., a Harvey Mudd College Faculty Research Grant to J.R.G, and PHS NS32387 to A.L.G.
References
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. Journal of General Physiology. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld VJ, Goldin AL, Krafte DS, Catterall WA, Lester HA, Davidson N. A neutral amino acid change in segment IIS4 dramatically alters the gating properties of the voltage-dependent sodium channel. Proceedings of the National Academy of Sciences of the USA. 1990;87:323–327. doi: 10.1073/pnas.87.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Properties of the sodium channel gating current. Philosophical Transactions of the Royal Society. 1975;B270:449–458. doi: 10.1098/rstb.1975.0022. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. Journal of General Physiology. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha A, Ruben PC, George AL, Fujimoto E, Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are coupled to Na+ channel fast inactivation. Neuron. 1999;22:73–87. doi: 10.1016/s0896-6273(00)80680-7. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Zhou A, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Chen LQ, Santarelli V, Horn R, Kallen RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. Journal of General Physiology. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Fujimoto E, Ruben PC. A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. The Journal of Physiology. 1998;506:627–638. doi: 10.1111/j.1469-7793.1998.627bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Richmond JE, Ruben PC. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophysical Journal. 1996;71:3098–3109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleig A, Fitch JM, Goldin AL, Rayner MD, Starkus JG, Ruben PC. Point mutations in IIS4 alter activation and inactivation of rat brain IIA Na+ channels in Xenopus oocyte macropatches. Pflügers Archiv. 1994;427:406–413. doi: 10.1007/BF00374254. [DOI] [PubMed] [Google Scholar]
- George AL, Jr, Komisarof J, Kallen RG, Barchi RL. Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Annals of Neurology. 1992;31:313–337. doi: 10.1002/ana.410310203. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. The Journal of Physiology. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji S, George AL, Jr, Horn R, Barchi RL. Paramyotonia congenita mutations reveal different roles for segments S3 and S4 of domain D4 in hSkM1 sodium channel gating. Journal of General Physiology. 1996;107:183–194. doi: 10.1085/jgp.107.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis KJ, Goldin AL. Sodium channel inactivation is altered by substitution of voltage sensor positive charges. Journal of General Physiology. 1997;110:403–413. doi: 10.1085/jgp.110.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis KJ, Rounaghi A, Goldin AL. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. Journal of General Physiology. 1997;110:391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Bean BP. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12:819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Larsson HP, Dhillon DS, Isacoff EY. Transmembrane movement of the Shaker K+ channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- Lerche H, Peter W, Fleischhauer R, Pika-Hartlaub U, Malina T, Mitrovic N, Lehmann-Horn F. Role in fast inactivation of the IV/S4-S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. The Journal of Physiology. 1997;505:345–352. doi: 10.1111/j.1469-7793.1997.345bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman E, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Kammen BF, Lindpaintner K, Bisbas D, Nadal-Ginard B. Gating charge differences between two voltage-gated K+ channels are due to the specific charge content of their respective S4 regions. Neuron. 1993;10:1121–1129. doi: 10.1016/0896-6273(93)90060-5. [DOI] [PubMed] [Google Scholar]
- Noda M, Shizimu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, Kangawa K, Matsuo H, Raftery MA, Hirose T, Inayama S, Hayashida H, Miyata T, Numa S. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- Papazian DM, Shao XM, Seoh S, Mock AF, Huang Y, Wainstock DH. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 1995;14:1–20. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- Papazian DM, Timpe LC, Jan YN, Jan LY. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature. 1991;349:305–310. doi: 10.1038/349305a0. [DOI] [PubMed] [Google Scholar]
- Patlak J. Molecular kinetics of voltage-dependent Na+ channels. Physiological Reviews. 1991;71:1047–1080. doi: 10.1152/physrev.1991.71.4.1047. [DOI] [PubMed] [Google Scholar]
- Patton DE, West JW, Catterall WA, Goldin AL. Amino acid residues required for fast Na+ channel inactivation: charge neutralizations and deletions in the III-IV linker. Proceedings of the National Academy of Sciences of the USA. 1992;89:10905–10909. doi: 10.1073/pnas.89.22.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner MD, Starkus JG, Ruben PC. Hydration forces in ion channel gating. Comments in Molecular and Cellular Biophysics. 1993;8:155–187. [Google Scholar]
- Richmond JE, Van De Carr D, Featherstone DE, George AL, Jr, Ruben PC. Defective fast inactivation recovery and deactivation account for sodium channel myotonia in the I1160V mutant. Biophysical Journal. 1997;73:1896–1903. doi: 10.1016/S0006-3495(97)78220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophysical Journal. 1997;73:1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–604. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Trimmer JS, Cooperman SS, Tomiko SA, Zhou J, Crean SM, Boyle MB, Kallen RG, Sheng Z, Barchi RL, Sigworth FJ, Goodman RH, Agnew WS, Mandel G. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 1989;3:33–49. doi: 10.1016/0896-6273(89)90113-x. [DOI] [PubMed] [Google Scholar]
- Tytgat J, Hess P. Evidence for cooperative interactions in potassium channel gating. Nature. 1992;359:420–421. doi: 10.1038/359420a0. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proceedings of the National Academy of Sciences of the USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Horn R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Yang N, George AL, Jr, Horn R. Probing the outer vestibule of a sodium channel voltage sensor. Biophysical Journal. 1997;73:2260–2268. doi: 10.1016/S0006-3495(97)78258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]