

Abstract
The kinetics of inactivation of delayed rectifier K+ current in murine colonic myocytes differed in amphotericin-permeabilized patch and conventional patch clamp. The difference was accounted for by Ca2+ buffering.
Calcium-calmodulin-dependent protein kinase II (CaMKII) inhibitors increased the rate of inactivation and slowed recovery from inactivation of the outward current. This was seen in single steps and in the envelope of the current tails. The effect was largely on the TEA-insensitive component of current.
Dialysis of myocytes with autothiophosphorylated CaMKII slowed inactivation. This effect was reversed by addition of CaMKII inhibitor.
Antibodies revealed CaMKII-like immunoreactivity in murine colonic myocytes and other cells. Immunoblots identified a small protein with CaMKII-like immunoreactivity in homogenates of colonic muscle.
We conclude that CaMKII regulates delayed rectifier K+ currents in murine colonic myocytes. The changes in the delayed rectifier current may participate in the Ca2+-dependent regulation of gastrointestinal motility.
Rhythmic activity consisting of slow waves and spike complexes characterize the electrical activity of colonic smooth muscle in mouse and human (Bywater et al. 1989; Rae et al. 1998; Koh et al. 1999). We have recently shown that a component of delayed rectifier current that inactivates relatively rapidly and is sensitive to 4-aminopyridine (4-AP) plays an important role in regulating rhythmic electrical activity of the murine proximal colon (Koh et al. 1999). Inhibition of this ‘A-like’ current changed the pattern of electrical activity and induced continuous spiking. The molecular identity of this current is unknown but the properties of the current are similar to currents produced by members of the Kv4 family of K+ channels (Koh et al. 1999).
While investigating the regulation of colonic electrical activity we observed that whole-cell currents were sensitive to intracellular Ca2+ buffering. The Ca2+ sensitivity was unlikely to be due to effects on Ca2+-activated K+ channels because charybdotoxin and iberiotoxin were without effect. We hypothesized that Ca2+ may act indirectly on the A-type component of the delayed rectifier current via Ca2+-dependent enzymes. Two candidates that might participate in regulation of the outward current are Ca2+-calmodulin-dependent protein kinase (CaMKII) and Ca2+-dependent protein kinase (PKC).
CaMKII is a multifunctional serine/threonine kinase activated by Ca2+ and calmodulin. This enzyme is widely distributed, but is most highly expressed in brain where it regulates neuronal excitability and synaptic strength (Soderling, 1996). An important mechanism for these actions is the slowing of the rate of N-type inactivation of Kv1.4 channels (Lee et al. 1996; Roeper et al. 1997). CaMKII is expressed by smooth muscle and has been reported to regulate cell migration (Abraham et al. 1997), Ca2+ currents (Edwards et al. 1998), sarcoplasmic reticulum (SR) Ca2+-ATPase (Greenwood et al. 1997), and the Ca2+ sensitivity of smooth muscle myosin light chain kinase (Edwards et al. 1998). Nothing, however, is known about the possible regulation of delayed rectifier channels in smooth muscle by CaMKII. Accordingly, we have investigated the role of CaMKII in the regulation of colonic electrical activity using Western blotting and immunohistochemical techniques to localize CaMKII expression, and voltage-clamp measurements to study the actions of CaMKII and inhibitors of the enzyme on the amplitude and kinetics of the A-like current.
METHODS
Preparation of isolated myocytes
Smooth muscle cells were prepared from colons removed from BALB/c mice. Briefly, mice were anaesthetized with chloroform before cervical dislocation and removal of the colon as approved by the Institutional Animal Care and Use Committee. Colons were cut open along the longitudinal axis, pinned out in a Sylgard-lined dish, and washed with Ca2+-free, phosphate-buffered saline (PBS) containing (mM): 125 NaCl, 5.36 KCl, 15.5 NaOH, 0.336 Na2HPO4, 0.44 KH2PO4, 10 glucose, 2.9 sucrose and 11 Hepes. Mucosa and submucosa were removed using fine-tipped forceps. Pieces of muscle were incubated in a Ca2+-free solution supplemented with 4 mg ml−1 fatty acid-free bovine serum albumin (BSA), 2 mg ml−1 papain, 1 mg ml−1 collagenase and 1 mM dithiothreitol; tissue was incubated at 37°C in this enzyme solution for 8-12 min and then washed with Ca2+-free solution. Tissue pieces were gently agitated to create a cell suspension. Dispersed cells were stored at 4°C in Ca2+-free solution supplemented with minimum essential medium for suspension culture (S-MEM; Sigma) and 0.5 mM CaCl2, 0.5 mM MgCl2, 4.17 mM NaHCO3 and 10 mM Hepes. Experiments were done at room temperature within 6 h of dispersing cells. Cells were allowed to adhere to the bottom of a recording chamber on an inverted microscope for 5 min prior to recording.
The resulting myocytes were from both the longitudinal and the circular smooth muscle layers. Because the circular layer is thicker than the longitudinal layer we expect that most of the cells studied were from the circular layer. As described previously, we found no significant differences in the inactivation kinetics and pharmacology of isolated longitudinal myocytes and circular myocytes (Koh et al. 1999).
Voltage-clamp methods
The whole-cell patch-clamp technique was used to record membrane currents from dissociated murine colonic smooth muscle cells. Currents were amplified with a List EPC-7 (List Electronics) or Axopatch 1A (Axon Instruments). Pipette resistances ranged between 1 and 4 MΩ and uncompensated series resistance averaged 2.9 ± 1 MΩ (n = 12). Thus, voltage errors were typically less than 5 mV. Currents were digitized with a 12-bit A/D converter (Axon Instruments). Data were filtered at 1 kHz and stored on videotape or digitized on-line using pCLAMP software (Axon Instruments).
Smooth muscle myocytes were bathed in a Ca2+-free solution containing (mM): 5 KCl, 135 NaCl, 2 MnCl2, 10 glucose, 1.2 MgCl2 and 10 Hepes, adjusted to pH 7.4 with Tris. The pipette solution contained (mM): 130 KCl, 5 MgCl2, 2.7 K2ATP, 0.1 Na2GTP, 2.5 creatine phosphate disodium salt and 5 Hepes, set to pH 7.2 with Tris. In addition, Ca2+ was buffered to varying levels using 0.1 mM EGTA or 10 mM BAPTA. Free Ca2+ levels in these solutions were 81 nM in 0.1 mM EGTA and 13 nM in 10 mM BAPTA, determined by a Ca2+ minielectrode (Baudet et al. 1994). For perforated whole-cell patch-clamp experiments, the composition of the pipette solution was (mM): 140 KCl, 0.5 EGTA and 5 Hepes, adjusted to pH 7.2 with Tris. Amphotericin B (90 mg ml−1, Sigma) was dissolved with DMSO, sonicated, and diluted in the pipette solution to give a final concentration of 270 μg ml−1. External solution for these experiments was the same as for the dialysed whole-cell patch experiments. KN-93, KN-62 and KN-92 (Calbiochem) were dissolved in DMSO and the desired concentrations were obtained by further dilution in extracellular solution. The final concentration of DMSO was less than 0.05 %. In control experiments, 0.05 % DMSO had no effect on delayed rectifier currents of murine colonic myocytes (n = 10). The autothiophosphorylated and boiled CaMKII (a gift from Dr Debra A. Brickey and Dr Tom R. Soderling, Vollum Institute) were added to the pipette solution.
Immunoblotting
Proximal colon and brain were collected from mice, frozen in liquid N2, and stored at -70°C. Crude homogenates were obtained by sonication of the frozen tissue in lysis buffer (25 mM Tris-HCl pH 7.5, 3 mM MgSO4, 2 mM EDTA, 2 mM EGTA, 1 mM dithiothreitol and 1 mM phenylmethylsulfonylfluoride). Protein content was determined by the Bradford assay. Tissue homogenates (50 μg) were separated by SDS-PAGE (12 %) and transferred to nitrocellulose by the method of Towbin et al. (1979). Immunoblotting was performed using a rabbit anti-CaMKII peptide antibody (gift from Dr Harold A. Singer, Geisinger Clinic), followed by horseradish peroxidase-conjugated donkey anti-rabbit IgG (Chemicon), and colorimetric detection using 4-chloro-1-naphthol/H2O2.
Immunohistochemistry
Mouse proximal colon was collected and flushed with PBS (pH 7.4). The tissues were fixed with paraformaldehyde (4 %) in PBS for 20 min. The fixed sections of colon were cryoprotected in increasing gradients of sucrose in PBS (5, 10 and 15 %) for 30 min each and in 20 % sucrose overnight. Tissue was then rapidly frozen by immersion in liquid N2, and embedded in a 1 : 2 solution containing OCT compound (Miles Inc., Elkhart, IN, USA) and 20 % sucrose in PBS. Frozen sections were cut at 8 μm on a cryostat (Leica CM 3050); endogenous peroxide was quenched by incubating in 1 % hydrogen peroxide in PBS for 15 min. The sections were then blocked in 1 % BSA for 1 h at room temperature. Excess blocking serum was removed and sections were incubated with 1 : 100 primary antibody (anti-CaMKII described above) for 24 h at 4°C. Then biotinylated goat anti-rabbit immunoglobulin and horseradish peroxidase-conjugated antibiotin antibodies were applied to the sections for 30 min at room temperature. The sections were washed twice between each step. Peroxidase activity was visualized by applying 3,3′-diaminobenzidine containing 0.05 % hydrogen peroxidase for 4-8 min at room temperature. The sections were rinsed in tap water, dehydrated, cleared and mounted with coverslips. Slides were viewed and photomicrographs were made using a Nikon eclipse E600 microscope incorporating Nomarski optics.
Statistical methods
Data are reported as the mean ± standard error of the mean, and n refers to the number of cells from which recordings were made. Statistical significance was evaluated by Student's t test or ANOVA as appropriate. P values less than 0.05 were considered significant.
RESULTS
Intracellular Ca2+ buffering speeds inactivation of A-like current in murine colonic myocytes
In preliminary experiments we found that delayed rectifier K+ currents of murine colonic myocytes studied with the amphotericin-perforated patch technique inactivated more slowly than currents recorded with conventional patch clamp (Fig. 1). This could be due to the exogenous Ca2+ buffering introduced by conventional patch pipettes. This possibility was supported by experiments comparing the kinetics of inactivation of delayed rectifier currents recorded from cells dialysed with 0.1 mM EGTA or 10 mM BAPTA. Stronger Ca2+ buffering caused the currents to peak earlier and inactivate more rapidly (Fig. 1B). The faster rate of inactivation resulted in a greater loss of current during the 500 ms depolarization. The inactivation of delayed rectifier K+ current in cells dialysed with 10 mM BAPTA or with 0.1 mM EGTA was well described by a single time constant of 49 ± 4 ms (n = 29) in 10 mM BAPTA and 328 ± 24 ms (n = 28) in 0.1 mM EGTA. The time constant of inactivation was 296 ± 89 ms (n = 10) in cells studied with the perforated patch technique. The time constant of inactivation was significantly shorter than this in cells dialysed with 10 mM BAPTA (P < 0.01). Cells dialysed with 0.1 mM EGTA or studied with the perforated patch method displayed time constants that were not significantly different (P > 0.05).
Figure 1. The effect of intracellular Ca2+ buffering on inactivation of A-type currents.
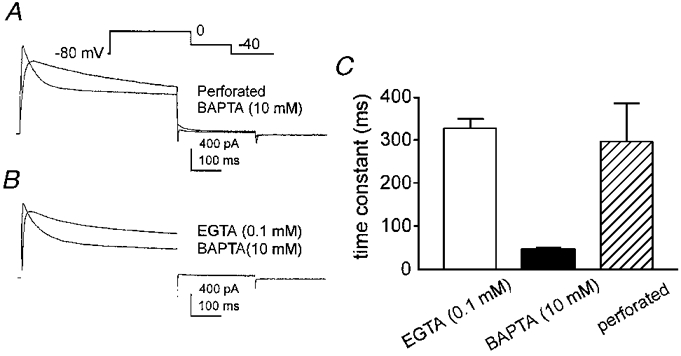
A, membrane currents recorded from two colonic myocytes using either the perforated patch technique or the conventional patch-clamp technique, dialysing the cell with 10 mM BAPTA. Membrane potential was stepped from -80 to 0 mV and then to -40 mV (inset shows voltage-clamp protocol). B, membrane currents recorded from two colonic myocytes dialysed with either 0.1 mM EGTA or 10 mM BAPTA. Voltage protocol is the same as in A. C, summary of time constants obtained by recording delayed rectifier K+ currents in cells dialysed with either 0.1 mM EGTA or 10 mM BAPTA, or studied with the perforated patch technique.
These data demonstrate that the rate of inactivation of the delayed rectifier K+ current in murine colonic myocytes is increased by stronger Ca2+ buffering. Measurements of the free Ca2+ in the pipette solutions suggest that inactivation is regulated by Ca2+ in the range of 10-100 nM. It seemed unlikely this effect could be attributed entirely to direct effects of Ca2+ on delayed rectifier K+ channels, so we investigated the possibility that inactivation might be regulated by Ca2+-dependent enzymes. We first investigated phospholipid and Ca2+-dependent protein kinase (PKC) but found no consistent effect of phorbol esters on the kinetics or amplitude of the delayed rectifier currents (data not shown). This led to an investigation of the involvement of CaMKII.
Inhibition of CaMKII by KN-62 and KN-93 speeds inactivation and slows recovery from inactivation of delayed rectifier K+ current in murine colonic myocytes
Inhibitors of CaMKII (KN-62 and the more potent KN-93) were tested on cells dialysed with 10 mM BAPTA and cells studied with the perforated patch technique. In perforated patch experiments, the time constant of inactivation was significantly reduced by KN-93 in a dose-dependent manner (Fig. 2A and C). The time constant of inactivation in cells dialysed with 10 mM BAPTA was initially shorter than that observed in perforated patch experiments. Exposure to KN-93 reduced the time constant further in a dose-dependent manner (i.e. to 35 ± 5 and 20 ± 1 ms at 1 and 3 μM KN-93, respectively; n = 5; control time constant was 54 ± 8 ms; see Fig. 2B and D). In both protocols and at all concentrations tested, the effects of KN-93 were apparent after a 1 min exposure and responses reached steady state within 5 min. We did not observe a decrease in the effects of KN-93 over the time course of the experiments.
Figure 2. The effect of KN-93 on inactivation time constants of A-type currents.
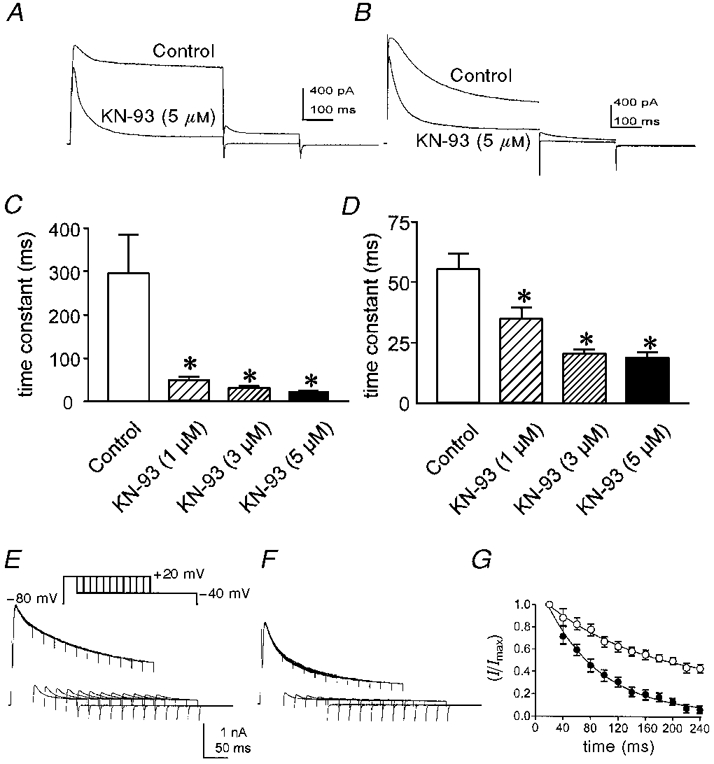
A, membrane currents recorded from a colonic myocyte using the perforated patch technique in control and in the presence of 5 μM KN-93. Voltage protocol is the same as in Fig. 1A. B, membrane currents recorded from a colonic myocyte dialysed with 10 mM BAPTA in control and in the presence of 5 μM KN-93. Voltage protocol is the same as in Fig. 1A. C and D, summary of time constants obtained from cells studied with the perforated patch technique (C) or dialysed with 10 mM BAPTA (D) in control and 1, 3 or 5 μM KN-93. Asterisks denote significant difference from control. E and F, membrane currents recorded from a colonic myocyte dialysed with 10 mM BAPTA in control (E) and in the presence of 5 μM KN-93 (F). Membrane potential was stepped from -80 to +20 mV for increasing durations (20-240 ms in 20 ms increments) before stepping to -40 mV to observe the envelope of delayed rectifier K+ current tails. G, summary of four experiments using the protocol of E and F. Peak tail currents were measured at -40 mV, normalized and plotted as a function of conditioning duration. ○, control; •, in the presence of 5 μM KN-93. The smooth lines are single exponential fits of the data.
In contrast to the increased rate of inactivation, KN-93 (1-3 μM) had no significant effect on the peak outward current. With 5 μM KN-93 there was a modest, but statistically significant reduction in the peak current (16 ± 3 % reduction; P < 0.05). Similar effects on the inactivation time constant were observed with 10 μM KN-62 (data not shown). In contrast, up to 5 μM KN-92, the inactive form of KN-93, had no effect on delayed rectifier current.
The effects of KN-93 were also tested by analysis of tail currents in cells dialysed with 10 mM BAPTA. Depolarizations from -80 to +20 mV for durations of 20-240 ms were followed by repolarization to -40 mV (Fig. 2E and F). The peak amplitudes of the tail currents were plotted as a function of depolarization duration and fitted with single exponentials (Fig. 2G). The decay time constant of the tail current in control conditions was 50 ± 2 ms, identical to the time constant of decay of outward current during a step depolarization (see Fig. 2D). In the presence of KN-93 (3 μM), the time constant of inactivation of the tail current decreased to 27 ± 2 ms (P < 0.05, n = 4), again similar to the effect seen during single depolarizations (see Fig. 2D). Because outward current was reduced by KN-93 at +20 and at -40 mV (potentials positive and negative to the equilibrium potential for Cl−, ECl, in our solutions), these data indicate that the change in membrane current cannot be attributed to a Cl− current.
The effects of KN-93 on the voltage dependence of inactivation of the A-type K+ current were also investigated in cells dialysed with 10 mM BAPTA. Membrane potential was stepped from -80 mV to conditioning potentials between -80 and +20 mV (in 10 mV increments) for 400 ms and then stepped for 100 ms to a test potential of +20 mV before and in the presence of KN-93 (5 μM; Fig. 3A and B). Normalized peak currents elicited by the test potential were plotted as a function of conditioning voltage, and the data were fitted with Boltzmann functions (Fig. 3C). KN-93 had no effect on the half-inactivation voltage (i.e. -46 ± 0.6 mV in control and -45 ± 0.3 mV in the presence of KN-93, n = 5); however, blocking CaMKII resulted in an increase in the amount of inactivation during the 500 ms conditioning potentials. For example, approximately 30 % of the outward current remained at the end of the conditioning potentials positive to -20 mV under control conditions, but after exposure to KN-93 nearly complete inactivation occurred within 500 ms at these potentials. Thus, KN-93 did not affect the voltage dependence of inactivation but increased the degree of inactivation during the conditioning steps.
Figure 3. The effect of KN-93 on the voltage dependence of inactivation of A-type currents.
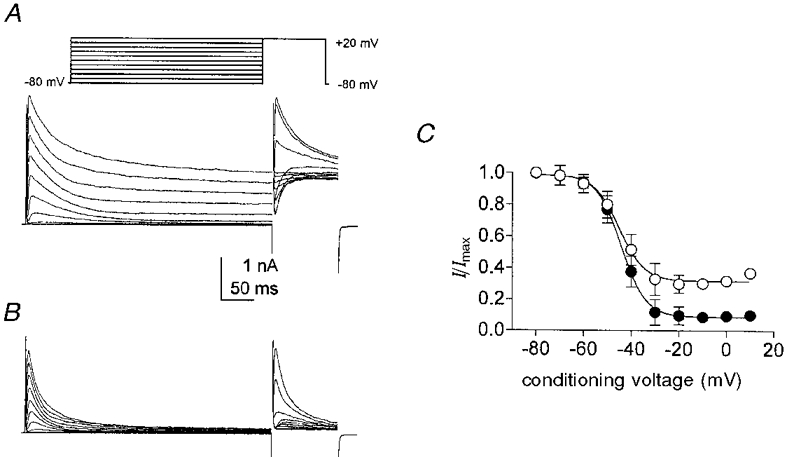
A and B, membrane currents obtained from colonic myocytes dialysed with 10 mM BAPTA. Membrane potential was stepped from -80 mV to conditioning potentials between -80 and +20 mV (10 mV increments) for 400 ms followed by a 100 ms test step to +20 mV (inset). A, control; B, same cell in the presence of 5 μM KN-93. C, summary of inactivation protocol from five cells in control (○) and in the presence of KN-93 (•). Normalized peak test currents are plotted as a function of conditioning voltage. Smooth lines are Boltzmann fits to the data.
Experiments to characterize the effects of KN-93 on the rate of recovery from inactivation were also performed. Cells dialysed with 10 mM BAPTA were held at -80 mV and then stepped to a conditioning potential of 0 mV for 500 ms. Then the cells were returned to the holding potential for variable recovery periods before stepping to a test potential of 0 mV. This protocol was repeated before and in the presence of KN-93 (5 μM; Fig. 4A and B). The amplitudes of the currents elicited by the test potentials were plotted as a function of recovery time (Fig. 4C). The normalized control peak current recovered with a half-time of 35 ± 4 ms (n = 4). Treatment with KN-93 resulted in faster inactivation during the conditioning step and slower recovery from inactivation (i.e. half-time of recovery was 104 ± 5 ms; n = 4).
Figure 4. The effect of KN-93 on recovery from inactivation of A-type currents.
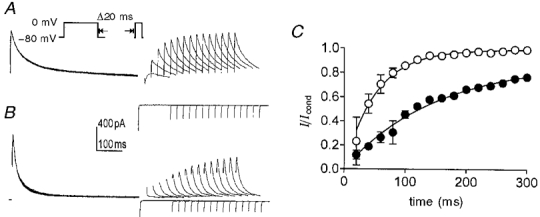
A and B, typical membrane currents obtained from colonic myocytes dialysed with 10 mM BAPTA. Membrane potential was stepped from -80 mV to a conditioning potential of 0 mV for 500 ms followed by a variable recovery period at -80 mV and a test step to 0 mV. Recovery intervals ranged from 20 to 300 ms in increments of 20 ms (inset). A, control; B, same cell in the presence of 5 μM KN-93. C, summary of recovery of delayed rectifier K+ currents in four cells. Peak test currents were normalized to the peak conditioning currents and plotted as a function of recovery interval in control (○) and in the presence of KN-93 (•). Smooth lines are single exponential fits to the data.
Effects of dialysis with autothiophosphorylated CaMKII on A-type K+ current
We further investigated the effect of CaMKII on the A-type K+ current in colonic myocytes by introducing Ca2+-independent, autothiophosphorylated CaMKII into cells via the patch pipettes. Cells were held at -80 mV and repetitively stepped to 0 mV for a period of 200 s after whole-cell conditions were established (i.e. during the period of cell dialysis). Inclusion of active CaMKII resulted in a time-dependent increase in the inactivation time constant (Fig. 5A and B). The time constant of inactivation increased from 100 ms immediately after the start of recording to 400 ms after 200 s of dialysis (Fig. 5B). Addition of KN-93 after dialysis with CaMKII reversed the slowing of inactivation caused by exogenous enzyme (Fig. 5C and D). As a control, four cells were dialysed with boiled autothiophosphorylated CaMKII, and this resulted in a slight, time-dependent reduction in the time constant of inactivation of the A-type current during depolarizations to 0 mV (Fig. 5A). Similar effects were observed when the enzyme was omitted (data not shown).
Figure 5. The effect of dialysis with autothiophosphorylated CaMKII on A-type currents.
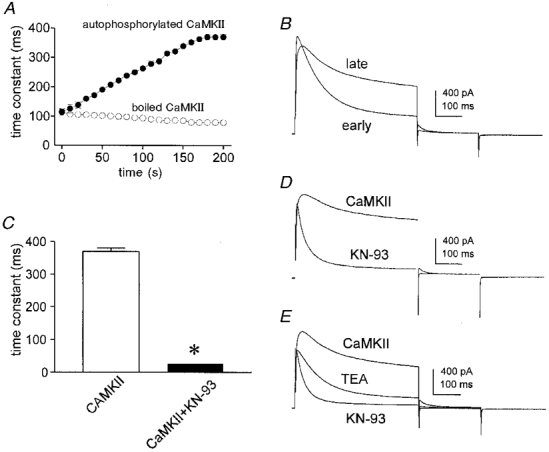
A, time course of change in inactivation time constant following the start of whole-cell recording (n = 3). Membrane potential was stepped from -80 to 0 mV for 500 ms every 10 s for determination of the inactivation time constant. •, cells dialysed with 10 mM BAPTA plus 10 nM autothiophosphorylated CaMKII; ○, cells dialysed with 10 mM BAPTA plus boiled enzyme. B, membrane currents obtained from a cell dialysed with 10 mM BAPTA plus 10 nM autothiophosphorylated CaMKII immediately upon establishing whole-cell recording (early) and after 200 s of dialysis (late). C, summary of steady-state effects of KN-93 on the time constant of inactivation of delayed rectifier current in cells dialysed with 10 nM autothiophosphorylated CaMKII. The time constants were 370 ± 10 ms before and 26 ± 1 ms (n = 3) during exposure to KN-93. Asterisk denotes significant difference from control. D, membrane currents obtained from a cell dialysed with 10 mM BAPTA plus 10 nM autothiophosphorylated CaMKII before and during exposure to KN-93. Voltage protocol as in Fig. 1A. E, membrane currents recorded from a cell dialysed with 10 mM BAPTA plus 10 nM autothiophosphorylated CaMKII alone, during exposure to 10 mM TEA, and during exposure to 10 mM TEA plus 5 μM KN-93.
In myocytes dialysed with 1 mM EGTA, TEA (10 mM) reduced the delayed rectifier current by an amount equal to the current flowing at the end of a 500 ms depolarization, i.e. the outward current inactivated to zero (Koh et al. 1999). This observation led us to identify two components of the delayed rectifier in these cells. One component inactivated and was apparently insensitive to TEA, whereas the other component showed little inactivation and was inhibited by TEA. It was of interest to re-examine this issue in view of our new observations that the kinetics and amount of inactivation of the outward current were sensitive to CaMKII. Figure 5E shows records from a myocyte dialysed with 10 mM BAPTA plus autothiophosphorylated CaMKII. Inactivation in this condition was characteristically slow and incomplete as just described. TEA (10 mM) reduced the outward current in a manner similar to that seen in cells dialysed with 1 mM EGTA: the kinetics of inactivation were relatively unchanged while the current was reduced. However, the outward current in the presence of TEA did not fully inactivate in cells dialysed with 10 mM BAPTA plus autothiophosphorylated CaMKII. We hypothesized that this might be due to incomplete inactivation of the TEA-insensitive channels in the presence of CaMKII. This idea is supported by the observation that inhibition of CaMKII by KN-93 results in currents that inactivate completely (Fig. 5E). In addition, the action of KN-93 in the presence of TEA implies that CaMKII modulates the TEA-insensitive component of delayed rectifier current.
Colonic smooth muscle myocytes contain CaMKII
CaMKII is present in several smooth muscle types, as demonstrated by immunochemical studies and CaMKII activity assays of tissue lysates (Schworer et al. 1993). Figure 6 shows an immunoblot of proteins isolated from murine brain and proximal colon probed with an antibody raised against a peptide sequence common to all CaMKII isoforms (Schworer et al. 1993). Immunoreactive bands of 50 and 60 kDa (corresponding to the α and β subunits) were obtained from murine brain homogenates (Soderling, 1996). The antibody detected a band of CaMKII-like immunoreactivity of approximately 45 kDa in the homogenates from the proximal colon.
Figure 6. Immunodetection of CaMKII.

Western blot of murine brain and proximal colon tissue lysates. Immunoblotting was carried out as described in Methods. The protein standards used were phosphorylase, 94 kDa; bovine serum albumin, 67 kDa; ovalbumin, 43 kDa; carbonic anhydrase, 30 kDa; soybean trypsin inhibitor, 20 kDa; and lysozyme, 14 kDa. The immunoblot detected a 43 kDa band in colonic lysate.
The antibodies used for immunoblotting were also used to localize CaMKII-like immunoreactivity in cryostat cross-sections of proximal colon. CaMKII-like immunoreactivity was observed throughout the tunica muscularis of the proximal colon (Fig. 7). Smooth muscle cells in the circular and longitudinal muscle layers and within the muscularis mucosae contained CaMKII-like immunoreactivity. Enteric ganglia were also strongly immunopositive.
Figure 7. CaMKII-like immunoreactivity in mouse proximal colon.
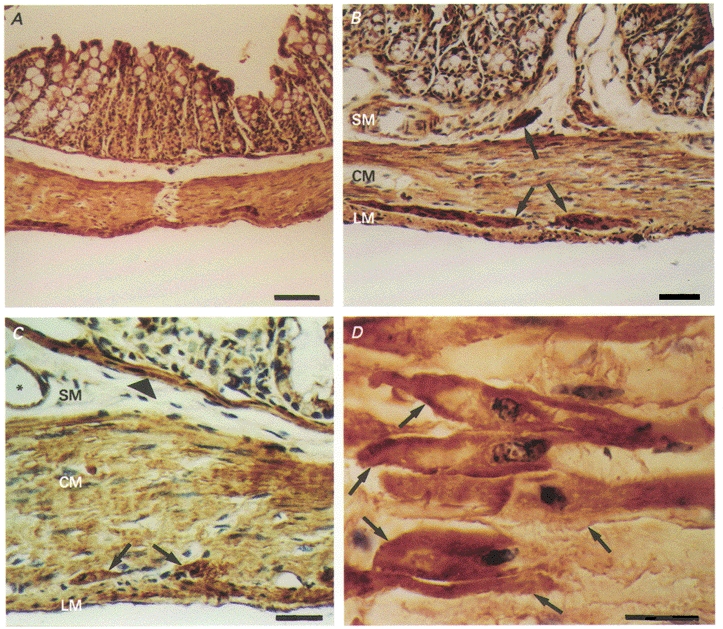
Haematoxylin counterstain. A, CaMKII-like immunoreactivity (in brown) exhibited throughout the entire external muscularis (scale bar = 100 μm). B, CaMKII-like immunoreactivity (in brown) within both longitudinal and circular muscle layers. Also, more intense staining of enteric ganglia within the deep muscular plexus of the submucosa and in the myenteric plexus between the longitudinal and circular muscle layers (arrows) (scale bar = 50 μm). C, CaMKII-like immunoreactivity (in brown) again showing positive staining of the longitudinal and circular muscle layer and enteric ganglia. Also, positive reactivity with the muscularis mucosa (arrowhead) and blood vessels (*) (scale bar = 20 μm). D, CaMKII-like immunoreactivity (in brown) of individual smooth muscle cells of the circular muscularis (arrows). Staining appears more intense at the level of the cell membrane (scale bar = 20 μm). SM, submucosa; CM, circular muscularis; LM, longitudinal muscularis.
DISCUSSION
We have previously characterized the components of voltage-dependent outward current expressed by myocytes of the murine proximal colon (Koh et al. 1999) and identified two components of delayed rectifier current. One is an A-type current that inactivates relatively rapidly (over hundreds of milliseconds); this component is sensitive to 4-AP and insensitive to TEA. The other component is a more typical smooth muscle delayed rectifier K+ current that inactivates slowly with sustained depolarization (over seconds); this component is insensitive to 4-AP and inhibited by TEA. The molecular nature of these currents is unclear. However, qualitative PCR amplification of mRNA isolated from murine colonic myocytes identified transcripts encoding delayed rectifier K+ channel subunits Kv1.6, Kv4.1, Kv4.2, Kv4.3 and Kvβ1.1, while mRNA encoding Kv1.4 was not detected (Koh et al. 1999). Accordingly, we proposed that one or more members of the Kv4 family may underlie the A-type current in these cells.
In the present study, we characterized the regulation of delayed rectifier currents by CaMKII. Regulation by CaMKII was observed in the presence of 10 mM TEA (which blocks the sustained component of outward current in murine colonic myocytes; see Koh et al. 1999). Thus, CaMKII modulates the 4-AP-sensitive, A-type current that remains in the presence of TEA. From the present experiments it is difficult to determine whether CaMKII also affected the TEA-sensitive component of the delayed rectifier. However, when the A-type current was pre-inactivated by holding cells at -40 mV, we could not detect a significant effect of CaMKII (data not shown).
We hypothesized that a Ca2+-dependent enzyme might regulate the inactivation process because intracellular Ca2+ buffering affected the rate of inactivation of the A-type current. Activators of PKC had no effect on inactivation, but several drugs and conditions that affect the activity of CaMKII dramatically changed the inactivation of the delayed rectifier current. In particular, specific inhibitors of CaMKII (KN-93 and KN-62) caused a dose-dependent increase in the rate of inactivation with little or no effect on the amplitude of the peak current or the voltage dependence of inactivation. KN-92, an inactive analogue that does not inhibit CaMKII (Peretz et al. 1998), was without effect. These observations imply activity of endogenous CaMKII in our isolated myocytes. Interestingly, we detected this activity in cells studied with amphotericin-perforated patches and in cells dialysed with 10 mM BAPTA, a high concentration of a fast Ca2+ buffer that set free Ca2+ in the pipette solution to 13 nM. Observation of inhibition of CaMKII in the dialysed cells implies that CaMKII did not diffuse out of the cells during recording and that the CaMKII activity did not require Ca2+ and calmodulin (Soderling, 1996). The Ca2+ independence of autophosphorylated CaMKII is well established and the presence of this protein could account for the CaMKII activity observed in our dialysed myocytes.
Although previous immunochemical and molecular studies have localized CaMKII isoforms in vascular smooth muscle (Schworer et al. 1993) this enzyme had not been identified in gastrointestinal smooth muscle. We addressed this issue using a rabbit anti-CaMKII peptide antibody that recognizes all known isoforms of the protein. We identified CaMKII-like immunoreactivity in immunoblots of proteins from murine colon lysates and localized CaMKII-like immunoreactivity in longitudinal and circular colonic myocytes and in several other cell types within the tunica muscularis and muscularis mucosae. Interestingly, the molecular weight of the protein from colon identified in our immunoblots was lower than the molecular weight of brain CaMKII. This might represent a novel CaMKII isoform or a proteolytic fragment, and we are currently investigating this issue.
In addition to experiments examining the action of endogenous CaMKII, we dialysed cells with constitutively active autothiophosphorylated CaMKII, and obtained similar results. Our observations using CaMKII inhibitors as well as exogenous enzyme provide strong evidence that CaMKII activity is a potent regulator of the inactivation of delayed rectifier current in murine colonic myocytes.
A-type K+ currents are expressed in neurons and are regulated by neurotransmitters and second messengers (Kaczmarek & Strumwasser, 1984). Some investigators have suggested that modulation of A-type currents may contribute to the changes in neuronal activity involved in learning and memory (Nelson et al. 1990). Currents with similar characteristics have been observed in the gastrointestinal tract; however, the physiological significance of A-type currents in gastrointestinal smooth muscles is not completely understood. The murine colon typically fires bursts of several action potentials that take off from potentials near -50 mV, and this activity is followed by periods of electrical quiescence with resting potentials near -60 mV (Lomax et al. 1996). We reported previously that 4-AP reduces the TEA-insensitive current in isolated murine colonic myocytes in the voltage range near the threshold for spiking, and triggers continuous spiking in intact tissues (Koh et al. 1999). The present observations that CaMKII is a potent regulator of the TEA-insensitive current component suggest a mechanism by which intracellular Ca2+ can regulate the pattern of electrical activity and thus the pattern of phasic contractions. Specifically, a rise in cytosolic Ca2+ during bursts of action potentials could activate Ca2+- and Ca2+-CaM-dependent signalling pathways causing a slowing of inactivation of the A-type current and reduced smooth muscle excitability. As intracellular Ca2+ is restored between bursts of action potentials, inactivation of A-type currents would be reduced and excitability would tend to increase. This proposal is consistent with earlier observations using 4-AP (Koh et al. 1999).
As noted earlier, members of the Kv4 family are candidates for mediating the A-type current in murine colonic myocytes. Kv4.1 and Kv4.3 have consensus sequences for CaMKII phosphorylation of Thr53 near the N-terminus of the channel protein. Phosphorylation of this site could mediate direct regulation of the Kv4 channels by CaMKII as has been suggested by Roeper et al. (1997) for Kv1.4 channels. These channels undergo N-type inactivation similar to that shown by Kv4 channels and Roeper et al. (1997) found that phosphorylation of Ser123 of Kv1.4 slowed inactivation and accelerated recovery from inactivation in agreement with our results. Thus, the mechanisms that underlie the regulation of N-type inactivation of cloned Kv channels and native smooth muscle delayed rectifier K+ currents may be similar. This issue remains to be resolved in experiments identifying the molecular basis of smooth muscle delayed rectifier K+ currents and their regulation in heterologous expression systems. We also point out that the possibility remains that CaMKII regulates murine colonic K+ currents indirectly.
In summary, we found that endogenous CaMKII slows the rate of inactivation of the delayed rectifier K+ current of murine colonic myocytes, and that this effect can be observed in cells dialysed with Ca2+ buffers. We have further localized CaMKII-like immunoreactivity to colonic myocytes (and other cell types in the murine colon). These findings strongly support the conclusion that CaMKII is an important regulator of colonic motility, linking intracellular Ca2+ to the activation of contraction.
Acknowledgments
This study was supported by a Program Project Grant from the National Institutes of Health, DK41315, and by NS36318 from NINDS. We are grateful to Dr Debra A. Brickey and Dr Tom R. Soderling (Vollum Institute) for the gift of truncated, autothiophosphorylated CaMKII, and Dr Harold A. Singer (Geisinger Clinic) for the gift of the anti-CaMKII antibody. We thank Ms Rebecca Walker for the Ca2+ electrode measurements.
References
- Abraham ST, Benscoter HA, Schworer CM, Singer HA. A role for Ca2+/calmodulin-dependent protein kinase II in the mitogen-activated protein kinase signaling cascade of cultured rat aortic vascular smooth muscle cells. Circulation Research. 1997;81:575–584. doi: 10.1161/01.res.81.4.575. [DOI] [PubMed] [Google Scholar]
- Baudet S, Hove-Madsen L, Bers DM. How to make and use calcium-specific mini- and microelectrode. Methods in Cell Biology. 1994;40:93–113. [PubMed] [Google Scholar]
- Bywater RA, Small RC, Taylor GS. Neurogenic slow depolarizations and rapid oscillations in the membrane potential of circular muscle of mouse colon. The Journal of Physiology. 1989;413:505–519. doi: 10.1113/jphysiol.1989.sp017666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RA, Walsh MP, Sutherland C, Vogel HJ. Activation of calcineurin and smooth muscle myosin light chain kinase by Met-to-Leu mutants of calmodulin. Biochemical Journal. 1998;331:149–152. doi: 10.1042/bj3310149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Helliwell RM, Large WA. Modulation of Ca2+-activated Cl− currents in rabbit portal vein smooth muscle by an inhibitor of mitochondrial Ca2+ uptake. The Journal of Physiology. 1997;505:53–64. doi: 10.1111/j.1469-7793.1997.053bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek LK, Strumwasser F. A voltage-clamp analysis of currents underlying cyclic AMP-induced membrane modulation in isolated peptidergic neurons of Aplysia. Journal of Neurophysiology. 1984;52:340–349. doi: 10.1152/jn.1984.52.2.340. [DOI] [PubMed] [Google Scholar]
- Koh SD, Ward SM, Dick GM, Epperson A, Bonner HA, Sanders KM, Horowitz B, Kenyon JL. Contribution of delayed rectifier potassium currents to the electrical activity of murine colonic smooth muscle. The Journal of Physiology. 1999;515:475–487. doi: 10.1111/j.1469-7793.1999.475ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TE, Philipson LH, Nelson DJ. N-type inactivation in the mammalian Shaker K+ channel Kv1.4. Journal of Membrane Biology. 1996;151:225–235. doi: 10.1007/s002329900073. 10.1007/s002329900073. [DOI] [PubMed] [Google Scholar]
- Lomax A, Harney SC, Jackson VM, Ward SM. Interaction of inhibitory transmitters regulate electrical and mechanical activity in the murine colon. Gastroenterology. 1996;110:A709. [Google Scholar]
- Nelson TJ, Collin C, Alkon DL. Isolation of a G protein that is modified by learning and reduces potassium currents in Hermissenda. Science. 1990;247:1479–1483. doi: 10.1126/science.247.4949.1479. [DOI] [PubMed] [Google Scholar]
- Peretz A, Abitbol I, Sobko A, Wu CF, Attali B. A Ca2+/calmodulin-dependent protein kinase modulates drosophila photoreceptor K+ currents: A role in shaping the photoreceptor potential. Journal of Neuroscience. 1998;18:9153–9162. doi: 10.1523/JNEUROSCI.18-22-09153.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae MG, Fleming N, McGregor DB, Sanders KM, Keef KD. Control of motility patterns in the human colonic circular muscle layer by pacemaker activity. The Journal of Physiology. 1998;510:309–320. doi: 10.1111/j.1469-7793.1998.309bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeper J, Lorra C, Pongs O. Frequency-dependent inactivation of mammalian A-type K+ channel KV1.4 regulated by Ca2+/calmodulin-dependent protein kinase. Journal of Neuroscience. 1997;17:3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schworer CM, Rothblum LI, Thekkumkara TJ, Singer HA. Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. Journal of Biological Chemistry. 1993;268:14443–14449. [PubMed] [Google Scholar]
- Soderling TR. Structure and regulation of calcium/calmodulin-dependent protein kinases II and IV. Biochimica et Biophysica Acta. 1996;1297:131–138. doi: 10.1016/s0167-4838(96)00105-7. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]