

Abstract
G protein-gated K+ channels (KACh channels) in the heart and brain are activated by the βγ subunit of inhibitory G protein. Phosphatidylinositol-4,5-bisphosphate (PIP2) has recently been reported to directly activate KACh channels (GIRK) expressed in oocytes, as well as to support activation by the βγ subunit in the presence of Na+. We examined the effect of Na+, PIP2 and other phospholipids on the KACh channel to understand better their role in KACh channel activation and modulation.
In atrial membrane patches, none of the phospholipids tested including PIP2 caused activation of the KACh channel in either the presence or the absence of 30 mM Na+. PIP2 (3 μM) and other phospholipids (30 μM) blocked acetylcholine-induced activation of the KACh channel.
When KACh channels were first activated with GTPγS, however, all phospholipids (100 μM) tested augmented the KACh channel activity 1·5- to 2-fold. Phosphatidylinositol-4-phosphate (PIP) and PIP2 were an order of magnitude more potent than other phospholipids. The increase in KACh channel activity was the result of a shift in the gating mode of the channel from a short-lived to a longer-lived open state. Such a modulatory effect was qualitatively similar to that produced by intracellular ATP. Trypsin blocked the ATP effect but not the phospholipid effect on the KACh channel kinetics.
The phosphate group linked to the glycerol backbone was important for KACh channel modulation by phospholipids. The higher potency of PIP and PIP2 was due to the presence of inositol phosphates.
Intracellular Na+ (30 mM) increased the frequency of KACh channel opening ≈2-fold if the channels were already active, but did not affect modulation by phospholipids. The effects of Na+ and phospholipids on KACh channel activity were additive.
A low concentration of ATP (20 μM), which had no effect on the KACh channel by itself, potentiated the stimulatory action of phospholipids, indicating that ATP and phospholipids interacted to modulate KACh channel function.
We conclude that exogenously applied PIP2 and other phospholipids block agonist-mediated KACh channel activation. However, if the KACh channel is already activated with GTPγS, phospholipids augment the existing activity by increasing the number of longer-lived channel openings. The evidence for and against the role of PIP and PIP2 in the stimulatory effect of ATP on the KACh channel is presented and discussed.
G protein-gated, inwardly rectifying K+ channels (KACh channels) in the heart and brain are activated by the βγ subunit of Gi/Go family of G proteins (Gilman, 1987; Reuveny et al. 1994; Wickman et al. 1994). Recent studies have demonstrated that the βγ subunit binds directly to the intracellular domains of the KACh channel protein (Huang et al. 1997; Krapivinsky et al. 1998). Binding of the βγ subunit to the channel probably causes a conformational change leading to channel activation. However, the precise molecular mechanism by which the βγ subunit activates the KACh channel is not clearly known.
Recent studies have reported that phospholipids, particularly phosphatidylinositol phosphates, are capable of activating inwardly rectifying K+ channels (Hilgemann & Ball, 1996; Fan & Makielski, 1997; Huang et al. 1998). In the case of G protein-gated K+ channels (GIRK), it was reported that phosphatidylinositol-4,5-bisphosphate (PIP2) applied to the cytoplasmic side of inside-out membrane patches could maximally activate the cardiac G protein-gated K+ channel (GIRK1/4) expressed in Xenopus oocytes (Huang et al. 1998). Furthermore, the βγ subunit failed to activate the KACh channel when the membrane was incubated with phospholipase C in order to deplete PIP2. It was thus concluded that the presence of PIP2 in the membrane was necessary for the βγ subunit to activate the KACh channel. For these effects of PIP2 to be observed, the presence of intracellular Na+ was also important (Huang et al. 1998). These findings suggest that Na+ and PIP2 can activate the KACh channel, and also serve as critical molecules that support KACh channel activation by the βγ subunit. It has been shown earlier that ATP applied to the cytoplasmic side of the membrane can activate the KACh channel in atrial cells in the absence of GTP (Heidbuchel et al. 1990). Therefore, it was proposed that such an effect of ATP on the KACh channel was mediated via PIP2. The observation that PIP2 antibody could reverse the effect of ATP on GIRK provided further support for the role of PIP2 (Huang et al. 1998).
In previous studies, it was observed that intracellular application of ATP activated the KACh channel only minimally under more physiological conditions when intracellular GTP is also present (Kim, 1991; Heidbüchel et al. 1993). This is in keeping with the well-known observation that in the absence of ACh, KACh channels are normally not very active in atrial cells despite the presence of 4-5 mM ATP in the cell. The KACh channels become active when ACh is applied to the cells, due to the markedly increased G protein interaction with the KACh channel protein. Earlier results showed that in inside-out membrane patches, the primary effect of ATP was to augment the KACh channel activity ∼5-fold if the channels were already in the active state (Kim, 1991; Hong et al. 1996), and this was found to be mainly due to the increase in the mean open time. Studies using GIRK1/4 expressed in oocytes also showed that ATP increased the mean open time and the open probability of GIRK1/4 ∼5-fold (Kim et al. 1997), consistent with the changes observed in atrial cells. Therefore, if ATP-induced changes in KACh channel kinetics occur via PIP2 as proposed (Huang et al. 1998), PIP2 should be able to reproduce all the kinetic effects of ATP in inside-out patches. Thus, PIP2 would not be expected to activate the KACh channel but to simply increase the mean lifetime of KACh channel openings that are already active and thereby augment the channel open probability.
To understand the role of PIP2 better, we investigated whether and how PIP2 affects KACh channel function in atrial cells. We then examined the effect of several other phospholipids with different hydrophilic head groups to obtain information on structural requirements for KACh channel modulation and activation. We also studied the interactions among Na+, ATP and PIP2 in KACh channel modulation. The study of the native KACh channel in atrial cells offers an advantage over the cloned K+ channel (GIRK) expressed in a heterologous system such as Xenopus oocytes because the atrial KACh channels have very low basal activity in the absence of agonist. One can then clearly determine whether PIP2 causes true activation of the KACh channel or whether it simply modulates the gating properties.
METHODS
Cell preparation
Hearts from 1-day-old rats (Sprague-Dawley) were dissociated with collagenase and trypsin. Animals were used in accordance with the Guide for the Care and Use of Laboratory Animals (Publication No. (NIH) 85-23). Rats were rapidly decapitated, and right and left atrial tissues from whole hearts were excised and placed in Ca2+-free Hank's medium (Sigma). The tissues were then cut into small pieces (< 1 mm3) with a sharp blade, and placed in Hank's balanced salt medium containing 0.01 % collagenase (Type 2; Worthington) and 0.1 % trypsin (from bovine pancreas; Sigma). Tissues were incubated at 37°C and agitated for 7-8 min. Suspended cells were then removed and added to an equal volume of 50 % fetal calf serum to inhibit the activity of the enzymes. Remaining tissues were incubated in a fresh enzyme solution and allowed to dissociate for another 8 min. This procedure was repeated 5 times. Dissociated cells were collected, centrifuged, and placed in growth medium consisting of culture medium (Dulbecco's modified Eagle's Medium; Sigma), 10 % fetal calf serum and 0.1 % penicillin-streptomycin (Sigma). Cells were plated on glass coverslips and placed in a 37°C incubator gassed with 5 % CO2- 95 % air. Cells were used 1 day after culturing.
Electrophysiology
Gigaseals were formed using Sylgard-coated, thin-walled borosilicate pipettes (Kimax) with ∼4 MΩ resistances. Channel currents were recorded with an Axopatch 200 patch-clamp amplifier (Axon Instruments), digitized with a PCM adapter (VR10, Instrutech, Elmont, NY, USA), and stored on videotape using a videotape recorder. The recorded signal was filtered at 3 kHz using an 8-pole Bessel filter (-3 dB: Frequency Devices, Haverhill, MA) and transferred to a computer (Dell) using the Digidata 1200 interface (Axon Instruments). Continuous single-channel currents were then analysed with the pCLAMP program (version 6.0.3) without further filtering. Data were analysed to obtain duration histograms (only in patches with low channel activity), amplitude histograms, single-channel conductance and channel activity (NPo). N is the number of channels in the patch, and Po is the probability of a channel being open. NPo was determined from 1-2 min of channel recording. Current tracings shown in the figures were filtered at 100 Hz except for expanded traces which were filtered at 1 kHz. Data are expressed as the mean ±s.d. Student's paired t tests were used to test for significance between two values at the level of 0.05.
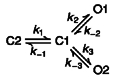
As every patch contained multiple openings, it was not possible to determine accurate measurements of mean lifetimes of open states using the pCLAMP programs. Therefore, a maximum likelihood algorithm was used to determine single-channel kinetic parameters from idealized patch clamp data (Qin et al. 1996). This method (QuB single-channel analysis) can be used on data containing multiple channel openings, and estimates all transition rates between states. For KACh channels activated with GTPγS alone, the open time duration histogram can be fitted well to a single exponential function. Therefore, two linear gating schemes (C2-C1-O1 and C2-O1-C1) were tested. The two schemes were equally good and produced similar transition rates, and therefore the first scheme (Scheme 1) was used to analyse all channels activated with GTPγS alone. For channels activated in the presence of ATP or phospholipids, we tested several different gating schemes that included two or three closed and open states. The highest log likelihood was obtained with the gating scheme (Scheme 2) shown above. Therefore, all channel data were modelled according to the two gating schemes shown above and transition rates obtained using the QuB(MIL) program (Qin et al. 1996). The mean lifetime for each open state was calculated as the value equal to 1 divided by the transition rate leading away from the state. Using the determined transition rates, we also obtained simulated single-channel data using the SIMU program (Qin et al. 1996) in order to compare with the actual current recording.
Scheme 1.

Scheme 2.
Solutions
The pipette and bath solutions contained 140 mM KCl, 0.5 mM MgCl2, 10 mM Hepes and 5 mM EGTA (pH 7.2). To change solutions perfusing the cytosolic surface of the inside-out patches, the pipette with the attached membrane was brought to the mouth of the polypropylene tubing through which flowed the desired solution at a rate of ∼1 ml min−1. For studies using ATP, amounts of MgCl2 and ATP were determined to produce desired concentrations of free Mg2+ and MgATP using EQCAL software (Biosoft, Milltown, NJ, USA). Free Mg2+ concentration in solutions was always kept constant at 0.5 mM. For solutions containing Ca2+, no EGTA was used. All experiments were performed at 24-26°C.
Materials
Acetylcholine (ACh), GTP, GTPγS, ATPγS, ATP and AMP-PNP (adenylyl-imidophosphate) were purchased from Boehringer Mannheim Chemicals. Trypsin (porcine pancreas, Type 2) was purchased from Sigma. Purified bovine βγ subunit was purchased from Calbiochem. All phospholipids were purchased from Sigma. PIP2 was also purchased from Calbiochem. Diacylglycerols (1-stearoyl-2-arachidonoyl-sn-glycerol and 1-stearoyl-2-linoleoyl-sn-glycerol) were purchased from Biomol (Plymouth Meeting, PA, USA), and dissolved in DMSO just before use. All phospholipids (phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidic acid (PA), phosphatidylinositol (PI), phosphatidylinositol-4-phosphate (PIP) and phosphatidylinositol-1,4-bisphosphate (PIP2)) were first dissolved in chloroform and kept at -70°C. The chloroform was then evaporated off and bath solution added to give the desired concentration of the phospholipid. The mixture was then sonicated (Heat Systems-Ultrasonics, Inc., W-380, Farmingdale, NY, USA) in pulses in ice bath until the solution became clear (∼10 min). The phospholipid solution was used immediately after sonication.
RESULTS
Inhibition of ACh-induced activation of the KACh channel by phospholipids
We first wished to confirm the recently reported finding that PIP2 activated G protein-gated K+ channels (GIRK1/4) expressed in oocytes. We used rat atrial cells in which the KACh channels have been well characterized and shown to have well-defined kinetics (Kim, 1991; Kurachi, 1995). As shown in Fig. 1, KACh channels were activated when cell-attached atrial patches were formed with 10 μM ACh in the pipette. The membrane potential was held at -60 mV to record inward currents. Upon formation of inside-out patches, the KACh channel activity decreased to basal levels (NPo < 0.01) due to washout of cellular GTP. Application of PIP2 (50 μM) to the cytosolic side of the membrane failed to activate KACh channels for up to 10 min of exposure (n = 6; Fig. 1A). Application of GTPγS, which normally results in activation of the KACh channels, failed to do so after PIP2 treatment. However, purified bovine βγ subunit (∼50 nM) was able to activate the KACh channels, indicating that the channels were still functional and responsive to the G protein subunit.
Figure 1. Lack of activation of KACh channels by inositol-containing phospholipids and PE in inside-out patches.
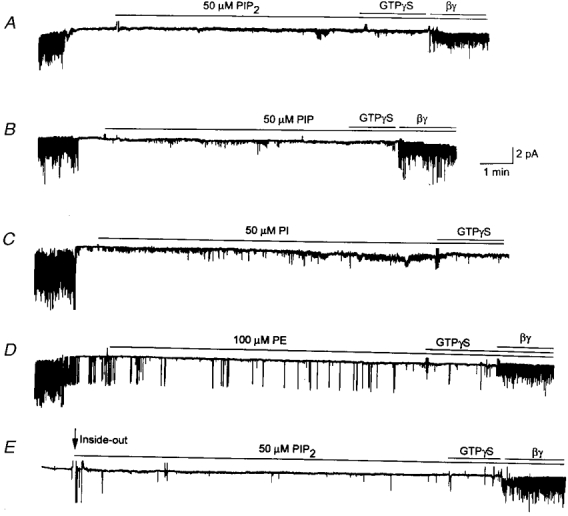
A-D, cell-attached patches were formed with 10 μM ACh in the pipette. Channels were activated when ACh was present in the pipette. Upon formation of inside-out patches, channels closed immediately. PIP2 (50 μM), PIP (50 μM), PI (50 μM) or PE (100 μM) was then applied to the cytoplasmic side of the membrane for ≈10 min. GTPγS (10 μM) was then applied for ≈1 min, and then purified bovine βγ subunit (50 nM) applied subsequently. Although not shown in C, the βγ subunit activated the channels when applied ≈5 min after the GTPγS application. In D, occasional openings of one KATP channel can be seen. In E, no ACh was added to the pipette solution. Membrane potential was held at -60 mV. All tracings were filtered at 100 Hz.
When PI and PIP (50 μM) were tested similarly, both failed to activate KACh channels. The βγ subunit, but not GTPγS, could activate KACh channels when applied subsequently to the same membrane patches (Fig. 1B and C). Another phospholipid, PE (100 μM), also failed to activate KACh channels (Fig. 1D). Similar to that observed with PIP2, only the βγ subunit could activate the KACh channels after PE treatment. Although not shown, two other phospholipids, PS and PC (100 μM), also failed to activate KACh channels (n = 3 each). These results showed that phospholipids in general did not activate KACh channels. Rather, they inhibited agonist (ACh)-induced activation of the KACh channel, suggesting that either the receptor has been uncoupled from the G protein or that the G protein subunits somehow failed to dissociate to release free βγ subunits. In patches in which ACh was not added to the pipette, channel activity was near zero in the cell-attached state (Fig. 1E). After formation of inside-out patches, application of 50 μM PIP2 also failed to activate the KACh channels, although the βγ subunit could open the channels when applied at the end of the experiment (n = 5). Similarly, PIP (50 μM) also failed to activate the KACh channels when ACh was not present in the pipette (not shown).
The results that phospholipids blocked ACh-induced activation of the KACh channel led us to examine in more detail their inhibitory effect on the KACh channel. Using inside-out patches in which the KACh channels were first activated with 10 μM ACh in the pipette and 100 μM GTP in the bath solution, we studied the concentration-dependent effect of several phospholipids on KACh current. PE produced an inhibition of KACh channel activity starting at ∼10 μM and complete inhibition was observed at 100 μM (Fig. 2A). Again, the βγ subunit, but not GTPγS, was able to activate the channel when applied at the end of the experiment. On average, phospholipids such as PE, PS and PC produced a half-maximal inhibition of channel activity at ∼30 μM. PI, PIP and PIP2 showed an inhibitory potency an order of magnitude greater than that of PE (Fig. 2B-D). Thus, the channel activity decreased markedly even at 3 μM. The data summarized in Fig. 2F show that the concentration range at which inositol-containing phospholipids inhibit the KACh channels is extremely narrow (1-3 μM) compared with those produced by PE (3-100 μM). We have not tested the possibility that the inhibitory effect becomes greater after a longer exposure (> 3 min) to a concentration of a phospholipid. Nevertheless, these results clearly showed that, at low micromolar concentrations, PIP2 blocked activation of KACh channels by ACh when applied directly to membrane patches.
Figure 2. Inhibitory effect of phospholipids on the KACh channel activated by GTP.

After formation of inside-out patches with ACh in the pipette, GTP (100 μM) was applied to activate KACh channels. A-E, in the presence of GTP, different phospholipids were applied starting at 1 or 3 μM. GTPγS or the βγ subunit was applied at the end of the experiment in some patches. The phospholipids tested were PE, PI, PIP and PIP2. F, the KACh channel activity in the presence of a phospholipid was calculated as a fraction of the KACh channel activity determined in the absence of a phospholipid (control, first bar). Each bar represents the mean ±s.d. of 5 determinations. Asterisk above a bar indicates a significant difference from the control value (P < 0.05).
Phospholipids increase KACh channel activity if already activated with GTPγS
The observation that the purified βγ subunit activated KACh channels in the presence of a phospholipid indicated that the channel itself did not lose the ability to interact with the βγ subunit. To study the direct effect of phospholipids on the KACh channel function, we activated the channel irreversibly with GTPγS before applying a phospholipid. We first chose PE and tested its intracellular effect on the KACh channel. Inside-out patches were formed and 10 μM GTPγS applied to activate KACh channels maximally (Fig. 3A). Concentrations of GTPγS up to 500 μM did not cause any further increase in channel activity in these atrial membrane patches. As soon as PE (100 μM) was applied, the KACh channel activity increased to a new steady-state level within a few seconds. This can be seen from the recording itself as well as from the plot where NPo was averaged every second (Fig. 3B). The average increase in KACh channel activity produced by PE was 62 % above control (n = 5). In our earlier studies, intracellular ATP was found to augment the KACh channel activity ∼5-fold above that produced by GTP or GTPγS (Kim, 1991; Hong et al. 1996). Therefore, we tested whether ATP could still produce such an increase in channel activity in the presence of PE. Addition of 1 mM ATP produced a typical ∼5-fold increase in KACh channel activity, indicating that PE did not affect the action of 1 mM ATP on the KACh channel (Fig. 3C). Figure 3D shows that the stimulatory effect of PE begins to take effect at ∼10 μM and increases up to 100 μM. Above 100 μM, PE produced an inhibitory effect but the true effect could not be determined due to incomplete solubility.
Figure 3. Stimulatory effect of PE and ATP on the KACh channel activity.
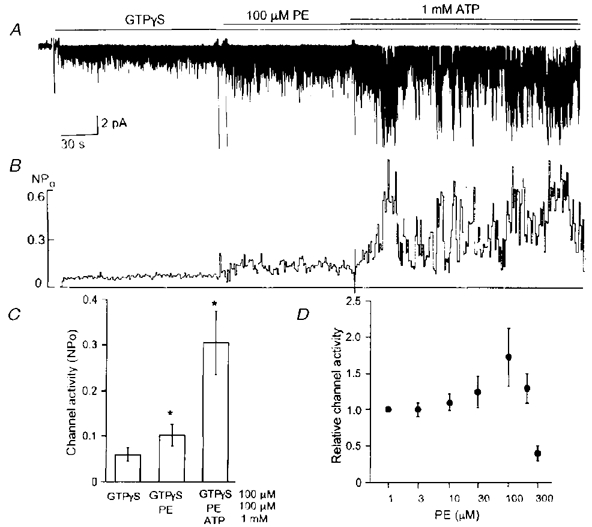
A, after applying GTPγS (10 μM) to activate the KACh channels in an inside-out patch, 100 μM PE was further applied. After ≈2 min, 1 mM ATP was added to the solution containing GTPγS and PE. B, channel activity (NPo) was averaged every second and plotted as a function of time. C, channel activities averaged over 40 s periods in the presence of GTPγS, GTPγS + PE and GTPγS + PE + ATP were determined. Each bar represents the mean ±s.d. of 5 determinations. Asterisk above a bar indicates a significant difference from the control, GTPγS value (P < 0.05). D, a concentration-effect relationship for PE is shown. Each point is the mean ±s.d. of 5 values.
The observation that PE could increase the KACh channel activity led us to test the effect of other phospholipids with various hydrophilic head groups. Again, KACh channels in inside-out patches were first activated with GTPγS, and a desired concentration of phospholipid was applied subsequently. We used 100 μM PC, PS and PA, and 50 μM PI, PIP and PIP2, the highest concentrations achievable for each phospholipid without solubility problems. Typical current tracings before and after application of phospholipids are shown in Fig. 4A. All phospholipids tested (PC, PS, PA, PI, PIP and PIP2), regardless of the type of hydrophilic head group, produced significant increases in KACh channel activity (Fig. 4B). We then examined the concentration- effect relationships for PC, PE, PIP and PIP2, by testing each concentration on separate patches. Channel activity (NPo) was plotted as a function of phospholipid concentration (Fig. 4C). Only the rising phases of the stimulatory effects of phospholipids could be obtained, as they became insoluble at higher concentrations. Concentration-effect relationships showed that PIP and PIP2 were most potent, with their stimulatory effect starting at ∼3 μM. PE and PC required 10-fold higher amounts to start augmenting the KACh channel activity. We found no significant difference between PIP and PIP2 in their effects on the KACh channel activity. These results showed that phospholipids, particularly PIP and PIP2, could augment the KACh channel activity, if the channels were already activated with GTPγS.
Figure 4. Stimulatory effect of phospholipids on the KACh channel activity.
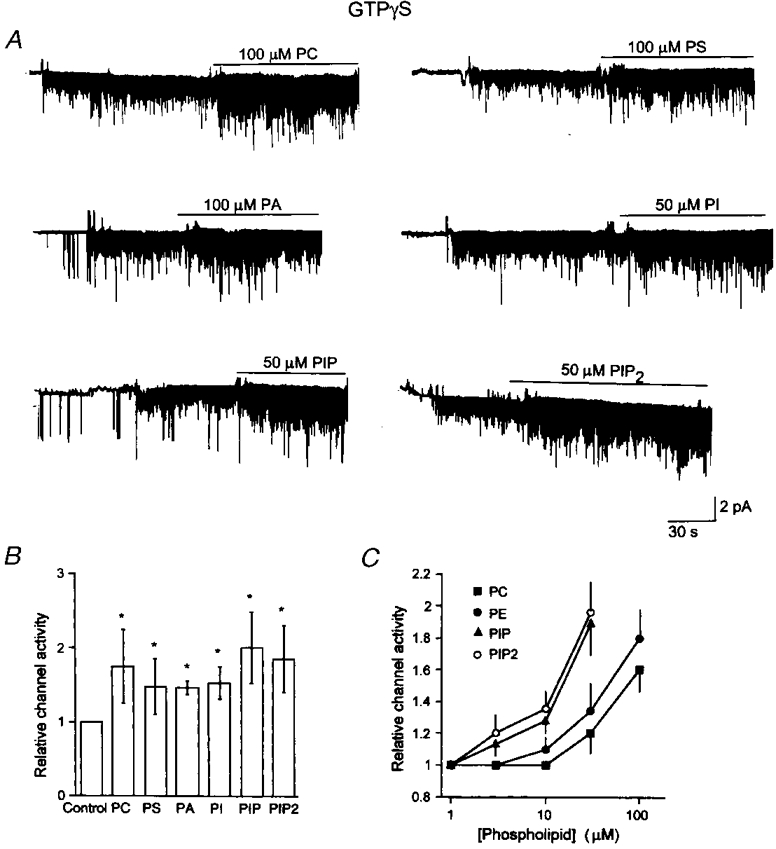
A, inside-out patches were formed and GTPγS (10 μM) applied to activate KACh channels. A phospholipid was then applied to the patch together with GTPγS. The concentrations tested were 100 μM for PC, PS and PA, and 50 μM for PI, PIP and PIP2. B, channel activities were determined for each phospholipid, and represented as a relative value of that determined in the presence of GTPγS alone (control). Each bar represents the mean ±s.d. of 5 determinations. Asterisk indicates a significant difference from the control value (P < 0.05). C, effects of a range of concentrations of PC, PE, PIP and PIP2 on KACh channel activity were determined using inside-out patches. Each concentration was tested on separate patches. Each point represents the mean ±s.d. of 3 determinations from patches containing comparable channel activity. Channel activity (NPo) was determined from ≈1 min of recording.
Phospholipids modify the gating mode of the KACh channel
During the course of our experiments, we observed that phospholipids tended to prolong the open time duration of the KACh channel. Figure 5A shows time-expanded current recordings in the presence of GTPγS alone (left panel) and after application of a phospholipid (right panel) in the same patch. To determine the effect of phospholipids on the mean lifetimes of open states, channel currents were idealized and modelled using two kinetic schemes as described above. For GTPγS-activated (control) channels, a simple linear scheme consisting of two closed and one open state was used. For phospholipid-treated channels, a scheme consisting of two closed and two open states was used, as shown in Fig. 5B. Transition rates between states were derived from the maximum likelihood estimation using the QuB analysis programs (Qin et al. 1997). The rates and the calculated mean lifetimes for each open state are summarized in Table 1. It is clear from these results that phospholipids have shifted the mode of channel gating such that an additional open state with longer-lived openings is now present. Figure 5B shows examples of simulated single-channel data generated using the transition rates obtained for KACh channels activated with GTPγS alone or with GTPγS and PE or PIP.
Figure 5. Effect of phospholipids on the KACh channel openings in inside-out patches.
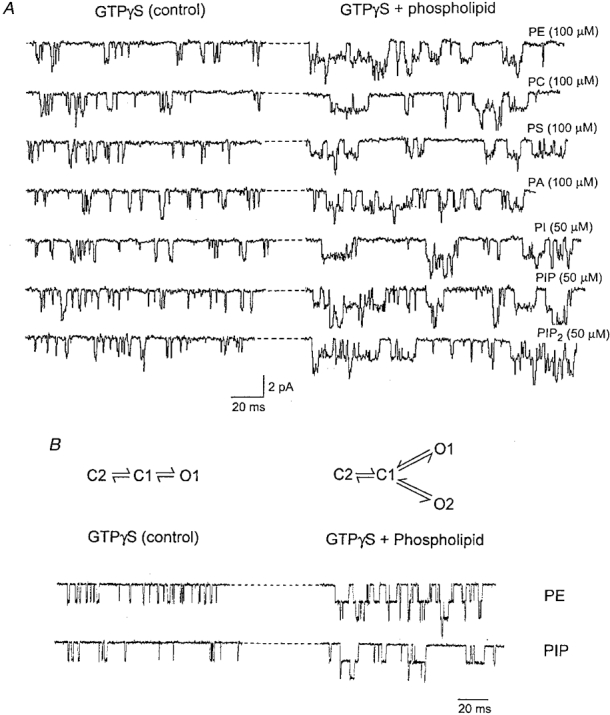
A, the current tracings were taken from experiments such as those shown in Fig. 4A and shown on an expanded time scale. The left portion shows channel openings in the presence of GTPγS alone, and the right portion shows the effect of adding a phospholipid to the same inside-out patch. The increase in the open time duration can be discerned easily for all phospholipids. B, a gating scheme for channels activated by GTPγS alone and a scheme for channels activated by GTPγS and a phospholipid were used to obtain transition rates between states after idealization of real data. Channel currents were simulated using these rates. A simulated current with GTPγS, and a current with GTPγS and 100 μM PE or 50 μM PIP are shown for comparison with actual data in A.
Table 1.
Effect of phospholipids and ATP on the single KAch channel parameters
| Substance | k−2 (s−1) | k−3 (s−1) | T1 (ms) | T2 (ms) |
|---|---|---|---|---|
| GTPγS alone | 884 ± 86 | — | 1.1 ± 0.2 | — |
| PC (100 μm) | 839 ± 60 | 354 ± 40 | 1.2 ± 0.1 | 2.9 ± 0.3 |
| PE (100 μm) | 851 ± 75 | 327 ± 23 | 1.2 ± 0.1 | 3.1 ± 0.3 |
| PS (100 μm) | 828 ± 67 | 345 ± 49 | 1.2 ± 0.1 | 3.0 ± 0.4 |
| PA (100 μm) | 779 ± 27 | 373 ± 31 | 1.3 ± 0.1 | 2.7 ± 0.2 |
| PI (50 μm) | 826 ± 59 | 336 ± 38 | 1.2 ± 0.1 | 3.0 ± 0.4 |
| PIP (50 μm) | 778 ± 94 | 299 ± 19 | 1.3 ± 0.2 | 3.4 ± 0.2 |
| PIP2 (50 μm) | 768 ± 87 | 318 ± 38 | 1.3 ± 0.1 | 3.2 ± 0.4 |
| ATP (1 mm) | 786 ± 44 | 151 ± 33 | 1.3 ± 0.1 | 6.9 ± 1.3 |
The gating scheme used to obtain the transition rates between states was C2-C1-O1 for GTPγS alone, and C2-C1-O1(C1-O2) for all phospholipids and ATP in the presence of GTPγS (see Methods). Only transition rates leading away from the open states are shown (mean ± s.d.; n = 4 – 5).
The modulation of the KACh channel gating produced by phospholipids can be compared with that produced by ATP which also prolonged the open time duration of the KACh channel (Kim, 1991; Hong et al. 1996). We show in Fig. 6 the effect of 1 mM ATP on the GTPγS-activated KACh channel in a patch with low channel activity and showing mainly one level of opening. ATP had no effect on the single-channel conductance (34 ± 1 vs. 35 ± 2 pS; n = 3). When durations of openings only to the first level were initially used to obtain the histogram using the pCLAMP analysis program, the channel openings in the presence of ATP could be well fitted with two exponential functions with time constants of 1.0 ms (34 % of total area under the curve) and 8.6 ms (66 %). Since all patches contained multiple channel openings, we repeated the analysis using the QuB program. When KACh channel openings in four patches were analysed and the mean lifetimes of each open state calculated from the transitions rates, the open time constants were 1.3 ± 0.1 and 6.9 ± 1.3 ms (see Table 1). Figure 6D shows simulated single-channel currents obtained using the transition rates determined from fitting the current data shown in Fig. 6A to the gating schemes described in Fig. 5B. These results, together with those shown in Fig. 5, showed that both phospholipids and ATP modulated the gating of the KACh channel such that a second open state with a longer-lived open duration was present. However, the effect of ATP on the mean lifetime of the new open state was much greater than those produced by all of the phospholipids we have tested, suggesting that the underlying mechanisms of action may be different. In patches in which the KACh channels were activated by GTPγS, application of ATPγS or AMP-PNP (1 mM) again had no significant effect on the KACh channel activity and did not produce a second open state (data not shown), similar to results reported earlier (Kim, 1991).
Figure 6. Modification of the KACh channel gating by ATP.
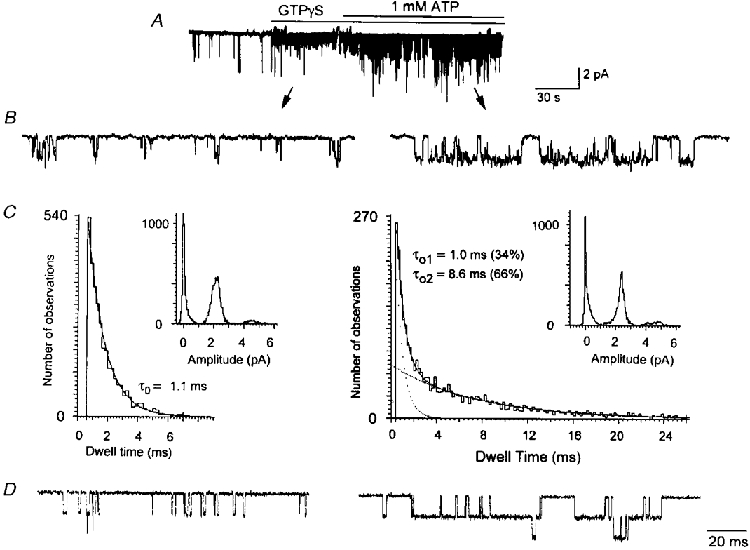
A, GTPγS was applied to an inside-out patch to activate KACh channels and then 1 mM ATP was applied ≈1 min later. B, expanded current tracings show the effect of ATP on the open time duration. C, amplitude and duration histograms obtained from only the channel openings to the first level are shown. ATP did not affect the amplitude but caused a marked prolongation of open time duration in a fraction of the population. D, two gating schemes were then used to model the data, and transition rates between states determined as described in the Methods. Simulated single-channel openings were obtained using these rates. The rates used to obtain the simulated currents are (s−1): for GTPγS (C2-C1-O1); 19 (k1), 184 (k-1), 186 (k2) and 860 (k-2): for GTPγS + ATP; 12.2 (k1), 309 (k-1), 255 (k2), 1043 (k-2), 344 (k3) and 111 (k-3). See Methods for the gating schemes.
Trypsin inhibits ATP- but not phospholipid-induced effect on the KACh channel
In our recent study, we found that intracellular application of trypsin prevented the increase in channel activity and open time duration normally produced by ATP (Pleumsamran et al. 1998). Although we do not know at what step(s) in the pathway trypsin blocked the ATP effect, it was of interest to examine whether the similar modulatory effects of phospholipids on the KACh channel activity and open time duration could also be blocked by trypsin. Inside-out patches were formed and a solution containing GTPγS and trypsin was applied to the bath solution. Addition of 1 mM ATP ∼3 min later failed to produce the typical increase in channel activity observed in the absence of trypsin. In the continuous presence of ATP, application of 100 μM PE, 40 μM PI, or 40 μM PIP resulted in significant increases in channel activity, similar to those observed without trypsin treatment (Fig. 7). In Fig. 7C, trypsin was applied alone without GTPγS, as the enzyme can slowly and spontaneously activate the KACh channel by an unknown mechanism (Kirsch & Brown, 1989). In such patches, ATP failed to augment channel activity. Thus, trypsin blocked the KACh channel modulation by ATP but not that by PE, PI or PIP. We also examined the effect of PC and found that trypsin could not block the stimulatory effect of PC (Fig. 7D). Therefore, these results raise the possibility that phospholipids and ATP may be modulating the KACh channel gating via distinct signalling pathways.
Figure 7. Trypsin inhibits ATP- but not phospholipid-mediated increase in channel activity.
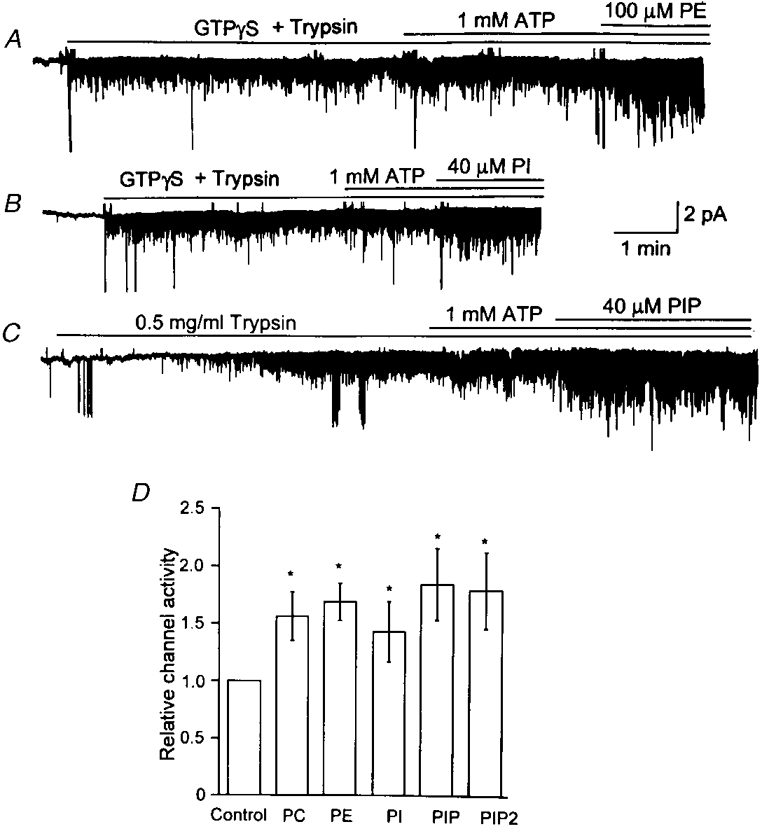
A, GTPγS (10 μM) and trypsin (0.5 mg ml−1) were applied together to an inside-out patch, resulting in channel activation. Application of 1 mM ATP failed to increase channel activity whereas PE (100 μM) produced the typical increase. B, similarly, the stimulatory effect of PI (40 μM) was not blocked by trypsin. C, in some patches, KACh channels were allowed to spontaneously activate in the presence of trypsin alone. Under this condition, ATP still failed to affect KACh channels, but PIP (40 μM) was able to elicit a 1.8-fold increase in channel activity. D, summary of results where each bar represents the mean ±s.d. of 4 determinations. The effects of PC (100 μM) and PIP2 (40 μM) were also studied and the results added to the bar graph (n = 3 each). Asterisk indicates a significant difference from the control value (P < 0.05).
Role of Na+ in KACh channel modulation by phospholipids
Recent studies have reported that the presence of intracellular Na+ may be necessary to observe activation of GIRKs expressed in oocytes by ATP or PIP2 (Huang et al. 1998; Sui et al. 1998). Therefore, we examined the effect of Na+ on the ATP- and phospholipid-induced effects on the KACh channel in atrial cells to determine whether Na+ is indeed necessary for the normal function of KACh channels, and whether Na+ affects the responsiveness of the channel to activators and modulators. These studies were first performed in the absence of ACh in the pipette. When a cell-attached patch was formed, only a basal level of activity (one opening every few seconds) was present. After formation of an inside-out patch, the bath solution was changed to that containing 110 mM K+ and 60 mM sucrose. Application of ATP (1 mM) to the bath solution did not cause activation of the KACh channel and the basal level was maintained (Fig. 8A). Addition of 30 mM Na+ (and no sucrose) in the presence of ATP resulted in a small sustained increase in channel activity (NPo, 0.03 ± 0.01; n = 5). When GTPγS was applied after ∼2 min to the same solution, a marked increase in channel activity was observed (NPo, 2.02 ± 0.94; n = 5). Thus, although ATP in the presence of Na+ could activate the KACh channel, this was minimal and only ∼2 % of the activation by GTPγS under identical conditions (Fig. 8A). When 30 mM Na+ was applied first and then ATP (1 mM) added subsequently to inside-out patches, the channel activity after addition of ATP was 0.04 ± 0.02 (n = 4; data not shown). Upon further application of GTPγS, the channel activity increased quickly to 2.41 ± 0.87 (n = 4).
Figure 8. Effect of Na+ on the KACh channel in the presence of ATP or PIP2.
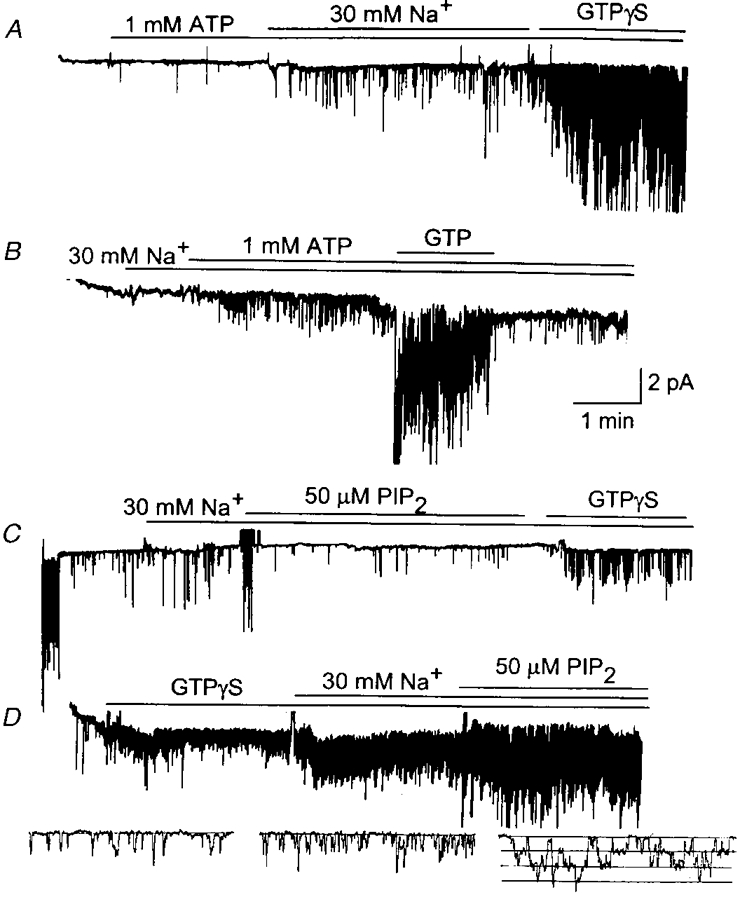
A, an inside-out patch was formed without ACh in the pipette in solution containing 110 mM KCl. After a few minutes, the bath solution was changed to that containing 110 mM KCl and 1 mM ATP. In the presence of ATP, 30 mM Na+ was applied and then GTPγS (100 μM) added. B, ACh (10 μM) was present in the pipette. Same experiment as in A except that the order of application of Na+ and ATP has been switched. C, current recording from a cell-attached patch with ACh in the pipette is shown initially. After forming an inside-out patch, and in the presence of 30 mM Na+, PIP2 (50 μM) was applied first and then GTPγS applied after ≈5 min. Activation by GTPγS was minimal compared with the activation observed in the cell-attached patch shown at the beginning of the recording. D, KACh channels were first activated with GTPγS in Na+-free solution. Addition of 30 mM Na+ resulted in an ≈2-fold increase in channel activity. Additional application of PIP2 (50 μM) resulted in a significant increase (1.9 (± 0.4)-fold, n = 3) in channel activity above that obtained in the presence of 30 mM Na+. Expanded current tracings (shown below) also show the clear stimulatory effects of Na+ and PIP2.
Similar experiments were also performed with 10 μM ACh in the pipette. After formation of an inside-out patch, bath solution containing 140 mM K+ was switched to a solution containing 110 mM K+ and 30 mM Na+. This substitution alone did not cause activation of KACh channels in any of the patches tested. Application of 1 mM ATP in the presence of 30 mM Na+ resulted in a small activation (NPo, 0.03 ± 0.01; n = 5). Upon application of GTP in the same solution, the channel activity increased markedly to 1.35 ± 0.44 (n = 5), resulting in a 45-fold increase in channel activity (Fig. 8B). We also tested the effect of applying ATP first and then adding Na+ to membrane patches in the presence of ACh in the pipette. ATP (1 mM) itself was not effective in activating the KACh channel, but caused a small activation when 30 mM Na+ was applied together with ATP (NPo, 0.02 ± 0.01; n = 3). Further addition of GTP again resulted in a large increase in channel activity (NPo, 1.38 ± 0.62; n = 3), similar to the results observed without ACh in the pipette. Thus, regardless of the order of application of ATP and Na+, the magnitude of KACh channel activation by ATP/Na+ was very small compared with that produced by GTP or GTPγS in either the presence or the absence of ACh.
According to these results, PIP2 would be expected to produce a small activation in the presence of Na+ if PIP2 were to mediate the effect of ATP. When 50 μM PIP2 was applied to an inside-out patch in solution containing 30 mM Na+, no activation was observed (Fig. 8C; n = 5). Washout of PIP2 and addition of GTPγS resulted in minimal or no activation. Therefore, even in the presence of 30 mM Na+, PIP2 caused block of channel activation by GTPγS, in keeping with the results shown in Fig. 2, but in contradiction to the results that Na+ and PIP2 can activate the KACh channels (Huang et al. 1998; Sui et al. 1998). Since PIP2 augmented the KACh channel activity when the channel was first activated with GTPγS, we tested whether such an effect of PIP2 could still occur in the presence of 30 mM Na+. An inside-out patch was formed and GTPγS applied to active the KACh channels. Under this condition, application of 30 mM Na+ to the cytosolic side of the membrane caused a 1.8 (± 0.2)-fold increase in channel activity (n = 3). This was due to an increase in the frequency of opening without any significant changes in the open time duration (0.9 ± 0.1 vs. 0.8 ± 0.1 ms; n = 3; Fig. 8D). When PIP2 was applied to the bath solution in the presence of 30 mM Na+, it produced a 1.9 (± 0.4)-fold (n = 3) increase in KACh channel activity above that observed in the presence of 30 mM Na+. We also tested the effects of PE (100 μM), and PIP (40 μM) in the presence of 30 mM Na+, and found significant stimulatory effects essentially similar to those obtained in the absence of Na+ (1.6 (± 0.2)- and 1.7 (± 0.3)-fold increase, respectively; n = 3 each). Thus, the stimulatory effects of Na+ and phospholipids on the KACh channel activity were additive if they were applied to GTPγS-activated channels.
Potentiation of the phospholipid effect by a low concentration of MgATP
As phospholipids produced an effect on the KACh channel qualitatively similar to that of ATP, i.e. an increase in channel activity due to an increase in mean open time, we investigated whether their effects were additive. When KACh channels in inside-out patches were fully stimulated with GTPγS and 1 mM ATP, addition of PE (100 μM), PIP (40 μM) or PIP2 (40 μM) resulted in 12 ± 10, 6 ± 12 and 4 ± 6 % changes, respectively, in channel activity (n = 3 each). The lack of effect of phospholipids could be because ATP had already produced the maximal possible increase in channel activity. We therefore used a threshold concentration of ATP. When patches were exposed to GTPγS and 20 μM ATP, a concentration of the nucleotide that had a minimal effect on the KACh channel activity, application of 100 μM PC or 50 μM PIP now produced an ∼4-fold increase in channel activity (Fig. 9A and B). These results have been summarized in Fig. 9C and show that an ineffective concentration of ATP can potentiate the action of phospholipids on the KACh channel activity. Thus, although 20 μM ATP had no discernible effect on channel kinetics, the nucleotide must have caused some other effects that allowed phospholipids to act on the KACh channels with a greater efficacy. These results further indicated that there was a functional interaction between ATP and phospholipids in the modulation of KACh channel gating.
Figure 9. Potentiation of the stimulatory effect of phospholipids by ATP.
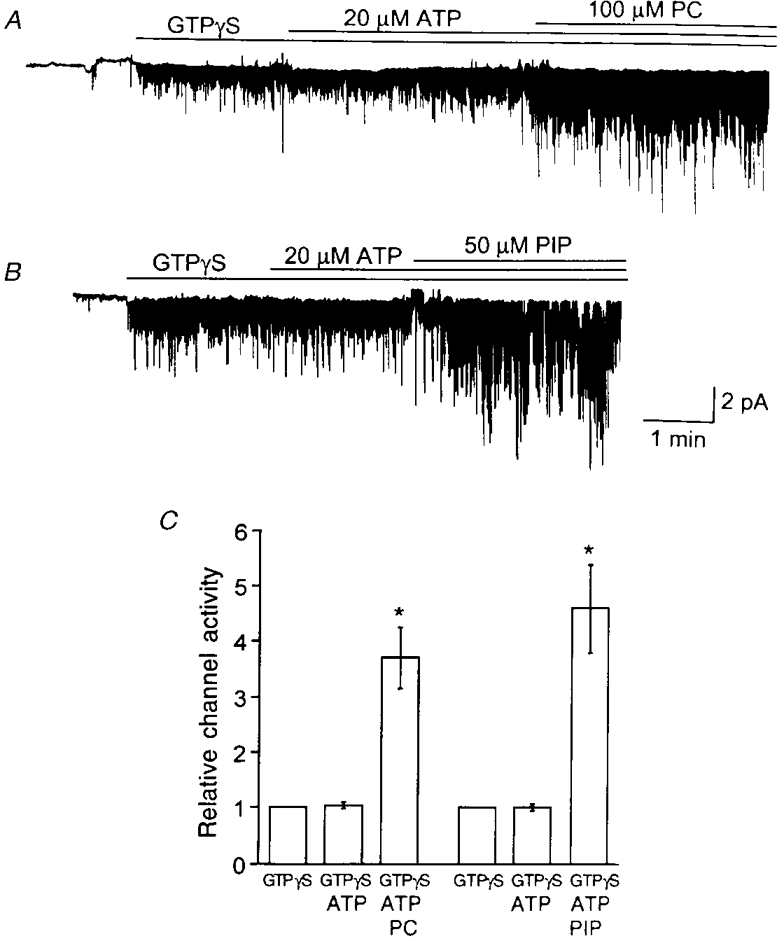
GTPγS was applied to an inside-out patch and 20 μM ATP added subsequently. This concentration of ATP had a minimal effect on KACh channel activity. When PC (A; 100 μM) or PIP (B; 50 μM) was also applied to the patches, the increase in KACh channel activity was ≈4-fold. C, summary of results shows the marked effect of PC and PIP on NPo in the presence of 20 μM ATP (n = 3). Asterisk indicates a significant difference from the corresponding control value (P < 0.05).
Effect of PIP2 on the ATP-sensitive K+ channel (KATP channel)
Due to the amphipathic nature of phospholipids and their high lipid solubility, it was important to be sure that the phospholipids used in this study were of good quality. In several earlier studies, PIP2 has been shown to activate KATP channels that have run down (Hilgemann & Ball, 1996; Fan & Makielski, 1997). Therefore, we tested whether a similar effect occurred under our experimental conditions. A cell-attached patch was formed on an adult rat ventricular cell and an inside-out patch was subsequently formed, which resulted in the opening of ∼10 KATP channels. Quick rundown was produced by applying a solution containing 100 μM Ca2+ to the perfusion bath for ∼20 s. After washing off Ca2+, 30 μM PIP2 was applied to the cytoplasmic side of the membrane. As shown in Fig. 10, PIP2 started to reactivate KATP channels and the activation was maintained as long as PIP2 was present. Repeat of Ca2+ and PIP2 application resulted in a similar rundown and reactivation of the KATP channels. In one patch in which the channel was allowed to run down spontaneously without using Ca2+, the subsequent action of PIP2 was faster and a greater percentage (∼80 %) of channels was reactivated. PIP2 solutions left at room temperature for more than an hour were generally ineffective in reactivating the KATP channels. Thus, these effects of PIP2 on KATP channels were similar to those of earlier studies (Hilgemann & Ball, 1996; Fan & Makielski, 1997).
Figure 10. Reactivation of KATP channels by PIP2.
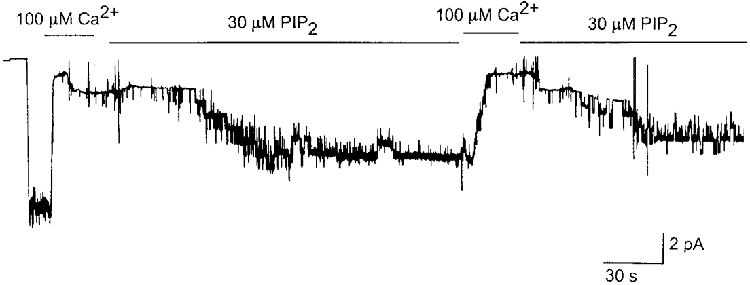
A cell-attached patch was formed first on a rat ventricular cell. Upon formation of an inside-out patch, many KATP channels were activated. Bath solution was changed to that containing 100 μM Ca2+ to cause fast rundown of the channels. After washing off Ca2+, PIP2 (30 μM) was applied to the patch, which resulted in reactivation of the channels. Applications of Ca2+ and PIP2 were repeated in the same patch after PIP2 was found to activate the KATP channels. Similar results were obtained in two other patches.
DISCUSSION
Cloning of G protein-gated K+ channel subunits has helped to determine the regions of the K+ channel that interact with G proteins (Dascal et al. 1993; Kubo et al. 1993), and to identify the βγ subunit of the G protein as the activator of the G protein-gated K+ channels in the heart and brain (Reuveny et al. 1994; Wickman et al. 1994; Huang et al. 1995; Kofuji et al. 1995). The results of a recent study showing that PIP2 can activate the KACh channels (GIRK) expressed in oocytes even in the absence of the βγ subunit, and that PIP2 is needed for fast activation by the βγ subunit were quite unexpected and suggested that inositol-containing phospholipids may serve as important second messengers in this signalling pathway (Janmey, 1994; Hilgemann, 1997). The study further indicated that PIP2 was the final mediator of ATP, which also activated GIRK1/4 expressed in oocytes. In earlier studies using atrial cells, however, it was shown clearly that ATP was not necessary for KACh channel activation by G proteins (Heidbüchel et al. 1993; Kurachi, 1995). In isolated membrane patches containing active KACh channels (i.e. in the presence of GTP and an agonist), ATP was found to simply augment the existing KACh channel activity primarily by prolonging the open time duration (Kim, 1991). Therefore, we investigated the effects of PIP2 and other phospholipids in KACh channel activation and modulation using native KACh channels in atrial cells.
What is the real effect of PIP2 on the KACh channel current?
Our results show that PIP2 does not activate the KACh channel in either the presence or the absence of ACh as the agonist, or of 30 mM Na+ on the cytosolic side. These findings in atrial cells therefore do not support the earlier results that PIP2 and Na+ activate G protein-gated K+ channels (Huang et al. 1998; Sui et al. 1998). How can PIP2 inhibit KACh channel activation by G protein in atrial cells but appear to activate GIRK1/4 or GIRK2 expressed in oocytes? The most likely explanation is that in the oocyte system, the basal GIRK1/4 activity is already very high (0.5-2 μA) due to activation by endogenous βγ subunit even in the absence of a receptor agonist, as shown by many earlier studies (Lesage et al. 1994; Hedin et al. 1996; Kofuji et al. 1996; Tucker et al. 1996). In on-cell patches used by Huang et al. (1998), the basal GIRK current observed in the absence of receptor stimulation or exogenous βγ subunit was ∼10 nA. This situation is similar to an atrial membrane patch in which the βγ subunit (or GTPγS) was applied first to activate the KACh channel. Under this condition, PIP2 probably augmented GIRK1/4 activity by increasing the duration of the open state, as our results showed that exogenously applied PIP2 prolonged the open time duration of KACh channels, provided that the KACh channels were already in the activated state. The presence of Na+ very likely helped to record a larger basal GIRK current in oocytes as Na+ increased the opening frequency of already activated GIRK channels, as also observed previously (Lesage et al. 1995).
It is well known that different types of phospholipids form integral components of membrane lipids and are asymmetrically distributed across the thickness of the bilayer (Cullis & Hope, 1991). Do any of these phospholipids modulate KACh channels in atrial cells under normal physiological conditions? This seems unlikely for the following reason. Exogenously applied phospholipids induced a fraction of channel openings to shift to longer-lived openings, resulting in two open kinetic states. However, in cell-attached patches or inside-out patches, regardless of whether the KACh channel was activated by GTP (with ACh), GTPγS or the βγ subunit, only a single open time distribution is present at steady state (Kim, 1991; Kurachi, 1995). Therefore, we are inclined to believe that phospholipids in the membrane do not participate in KACh channel modulation under normal steady-state conditions in atrial cells. If they did, we would expect the channel kinetics to be described by two open states, with one state having longer-lived channel openings, especially in the cell-attached state where [ATP] in the cell is high. Thus, PIP2 or any other phospholipids that exist in the membrane are unlikely to serve as second messengers of G protein signal transduction involving the KACh channel. However, if certain phospholipids were endogenously generated near the channel in response to a stimulus, they could potentially modulate the KACh channel function.
Evidence for and against the role of PIP2 in KACh channel modulation by ATP
Our single-channel measurements tend to support the proposal that PIP2 mediates the ATP effect, since PIP2-induced changes in KACh channel kinetics are qualitatively similar to those produced by ATP, although the extent to which PIP2 increased the mean open time and channel activity was less than that produced by ATP. If PIP2 were indeed the mediator of ATP on the KACh channel, then why is the effect of PIP2 much less than that of ATP? One possible explanation is that endogenously generated PIP2 is much more effective than exogenously applied compounds. Endogenously generated PIP2 may be in the right compartment in a correct structural configuration and have full access to the interaction site on the KACh channel molecule at the right concentration, events that may not occur with exogenous application of lipophilic substances.
In a recent study, when cardiac membrane vesicles were incubated with ATP, synthesis of PIP was present, with little production of PIP2 (Berberian et al. 1998). It was speculated that the synthesis of PIP and PIP2 from PI was mediated via lipid kinases that use ATP as well as ATPγS as the phosphate donor. The presence of 1 μM Ca2+ was found to be essential for PIP2 production from PIP. In our study, ATP was able to exert its effect fully in solution containing a very low Ca2+ concentration (< 20 nM). Therefore, PIP could be a more important modulator of the KACh channel under low Ca2+ conditions. Our results show that PIP and PIP2 are equipotent in augmenting KACh channel activity, and thus suggest that both may be important in KACh channel modulation by ATP.
Despite these considerations that support the view that PIP/PIP2 may transduce the signal produced by ATP, several pieces of evidence argue against it. (1) PIP/PIP2 inhibited ACh-induced activation of the KACh channel (with GTP) whereas ATP augmented the KACh channel activity. (2) Trypsin did not block the PIP/PIP2 effect whereas it blocked the ATP effect. (3) The effect of PIP/PIP2 was not specific, since all phospholipids tended to produce similar effects on the KACh channel kinetics. (4) The ability of a low concentration of ATP (20 μM) to enhance the effect of PC and PIP (and presumably other phospholipids) is inconsistent with the view that PIP2 mediates the ATP effect. Our results suggest that ATP may have a dual effect on the KACh channel. At low concentrations, ATP may be modifying the channel (for example via phosphorylation) such that it becomes more sensitive to phospholipids. At high concentrations, ATP increases Po maximally, in addition to sensitizing the channel to phospholipids. (5) Another strong piece of evidence against the role of PIP2 in the ATP effect is that ATPγS does not mimic the effect of ATP in atrial cells (Kim, 1991), although ATPγS has been shown to be as effective as ATP in generating PIP and PIP2 in cardiac cell membranes (Hilgemann, 1997; Berberian et al. 1998). (6) After complete rundown of ATP-sensitive K+ channels, a process that has been attributed to depletion of PIP/PIP2 in the membrane (Hilgemann & Ball, 1996; Fan & Makielski, 1997), we could easily activate KACh channels with GTP (with ACh in the pipette) or GTPγS. In support of this, it was reported recently that KACh channels could be activated via G protein even in the complete absence of ATP and Na+ (Otero et al. 1998). Together, these observations suggest strongly that ATP and PIP/PIP2 use separate signalling pathways to modulate the G protein-gated K+ channel. Therefore, we should be cautious in accepting the earlier proposal that PIP/PIP2 is the membrane messenger for the ATP-induced increase in KACh channel activity, until a specific inhibitor of a membrane lipid kinase is found to block the effect of ATP.
Structural requirements for KACh channel modulation by phospholipids
Since all phospholipids tested were capable of producing similar effects on the KACh channel, there seems to be no critical structural requirement for particular hydrophilic head groups. The effect was similar despite differences in the net charge of the molecule. For example, PC and PE have no net charge whereas PS, PA and PI are anionic. Therefore, the effects of phospholipids on the KACh channel seem to be clearly different from their effects on the KATP channel. PI (50 μM), PS, PC and PE (100 μM) were all ineffective whereas PIP and PIP2 (5-25 μM) were effective in reactivating the rundown KATP channels in ventricular cells and HIT-T15 β-cells (Fan & Makielski, 1997; Shyng & Nichols, 1998) and in reducing the blocking effect of ATP (Shyng & Nichols, 1998; Baukrowitz et al. 1998). The stimulatory effect of PA on the KACh channel, but lack of effect of two diacylglycerols (1-stearoyl-2-arachidonoyl-glycerol (SAG) and 1-stearoyl-2-linoleoyl-glycerol (SLG)), indicate that the phosphate group on the third position of the glycerol backbone, not the long-chain fatty acids, is important for KACh channel modulation. The hydrophilic head groups such as choline, ethanolamine and serine certainly do not seem to be important. However, inositol-containing phospholipids were nearly an order of magnitude more potent than other phospholipids in modulating the KACh channel. The structural requirements for a potent modulatory effect on the KACh channel thus include the inositol phosphates linked to the phosphate group on the third carbon of the glycerol backbone. Esters of long chain fatty acids such as those present in PIP and PIP2 may also be required for membrane insertion and stability, although not directly involved in channel modulation.
Summary
We have studied the effects of PIP2, and other phospholipids on the KACh channel activation. Our results showed that none of the phospholipids were capable of activating the KACh channel in the presence or absence of Na+. Rather, they were found to block agonist-mediated activation of the KACh channel. Inositol-containing phospholipids were particularly potent in G protein inhibition. However, if the KACh channels were already activated with GTPγS, phospholipids increased channel open probability mainly by increasing the number of long-lived openings. Inositol-containing phospholipids were most potent in augmenting the KACh channel activity under such conditions. The phosphate group and its linkage to the inositol phosphates together appear to be the critical components in modulating the KACh channel gating by PIP and PIP2. Despite the qualitatively similar effects of ATP and PIP/PIP2 on the KACh channel kinetics, our results do not support the proposal that PIP2 is the mediator of ATP. Our results support the view that PIP and PIP2 do not play a significant role in the activation of the KACh channel by G proteins under steady-state physiological conditions.
Acknowledgments
This work was supported by a grant from the National Institutes of Health.
References
- Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP, Fakler B. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- Berberian G, Hidalgo C, DiPolo R, Beauge L. ATP stimulation of Na+/Ca2+ exchange in cardiac sarcolemmal vesicles. American Journal of Physiology. 1998;274:C724–733. doi: 10.1152/ajpcell.1998.274.3.C724. [DOI] [PubMed] [Google Scholar]
- Cullis PR, Hope MJ. Physical properties and functional roles of lipids in membranes. In: Vance DE, Vance J, editors. New Comprehensive Biochemistry; Biochemstry of Lipids, Lipoproteins and Membranes. Vol. 20. London: Elsevier; 1991. pp. 1–40. [Google Scholar]
- Dascal N, Schreibmayer W, Lim NF, Wang L, Chavkin C, Dimagno L, Labarca C, Kieffer BL, Gaveriaux-Ruff C, Trollinger D, Lester HA, Davidson N. Atrial G protein-activated K+ channel: Expression cloning and molecular properties. Proceedings of the National Academy of Sciences of the USA. 1993;90:10235–10239. doi: 10.1073/pnas.90.21.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Makielski JC. Anionic phospholipids activate ATP-sensitive potassium channels. Journal of Biological Chemistry. 1997;272:5388–5395. doi: 10.1074/jbc.272.9.5388. 10.1074/jbc.272.9.5388. [DOI] [PubMed] [Google Scholar]
- Gilman AF. G proteins: transducers of receptor-generated signals. Annual Review of Biochemistry. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Hedin K, Lim N, Clapham D. Cloning of a Xenopus laevis inwardly rectifying K+ channel subunit that permits GIRK1 expression of IKACh currents in oocytes. Neuron. 1996;16:423–429. doi: 10.1016/s0896-6273(00)80060-4. 10.1016/S0896-6273(00)80060-4. [DOI] [PubMed] [Google Scholar]
- Heidbüchel H, Callewaert G, Vereecke J, Carmeliet E. ATP-dependent activation of atrial muscarinic K+ channels in the absence of agonist and G-nucleotides. Pflügers Archiv. 1990;416:213–215. doi: 10.1007/BF00370246. [DOI] [PubMed] [Google Scholar]
- Heidbüchel H, Callewaert G, Vereecke J, Carmeliet E. Acetylcholine-mediated K+ channel activity in guinea-pig atrial cells is supported by nucleoside diphosphate kinase. Pflügers Archiv. 1993;422:316–324. doi: 10.1007/BF00374286. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW. Cytoplasmic ATP-dependent regulation of ion transporters and channels: Mechanisms and messengers. Annual Review of Physiology. 1997;59:193–220. doi: 10.1146/annurev.physiol.59.1.193. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW, Ball R. Regulation of cardiac Na+, Ca2+ exchange and KATP potassium channels by PIP2. Science. 1996;273:956–959. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- Hong SG, Pleumsamran A, Kim D. Regulation of atrial muscarinic K+ channel activity by a cytosolic protein via G protein-independent pathway. American Journal of Physiology. 1996;270:H526–537. doi: 10.1152/ajpheart.1996.270.2.H526. [DOI] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Huang CL, Jan YN, Jan LY. Binding of the G protein βγ subunit to multiple regions of G protein-gated inward-rectifying K+ channels. FEBS Letters. 1997;405:291–298. doi: 10.1016/s0014-5793(97)00197-x. [DOI] [PubMed] [Google Scholar]
- Huang C-L, Slesinger PA, Casey PJ, Jan YN, Jan LY. Evidence that direct binding of Gβγ to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- Janmey PA. Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annual Review of Physiology. 1994;56:169–191. doi: 10.1146/annurev.ph.56.030194.001125. [DOI] [PubMed] [Google Scholar]
- Kim D. Modulation of acetylcholine-activated K+ channel function in rat atrial cells by phosphorylation. The Journal of Physiology. 1991;437:133–155. doi: 10.1113/jphysiol.1991.sp018588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Watson M, Indyk V. ATP-dependent regulation of a G protein-coupled K+ channel (GIRK1/GIRK4) expressed in oocytes. American Journal of Physiology. 1997;272:H195–206. doi: 10.1152/ajpheart.1997.272.1.H195. [DOI] [PubMed] [Google Scholar]
- Kirsch GE, Brown AM. Trypsin activation of atrial muscarinic K channels. American Journal of Physiology. 1989;257:H334–338. doi: 10.1152/ajpheart.1989.257.1.H334. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Davidson N, Lester HA. Evidence that neuronal G-protein-gated inwardly rectifying K+ channels are activated by Gβγ subunits and function as heteromultimers. Proceedings of the National Academy of Sciences of the USA. 1995;92:6542–6546. doi: 10.1073/pnas.92.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Doupnik CA, Davidson N, Lester HA. A unique P-region residue is required for slow voltage-dependent gating of a G protein-activated inward rectifier K+ channel expressed in Xenopus oocytes. The Journal of Physiology. 1996;490:633–645. doi: 10.1113/jphysiol.1996.sp021173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Kennedy ME, Medina NJ, Krapivinsky L, Clapham DE. Gβγ binding to GIRK4 subunit is critical for G protein-gated channel activation. Journal of Biological Chemistry. 1998;273:16946–16952. doi: 10.1074/jbc.273.27.16946. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Reuveny E, Slesinger PA, Jan YN, Jan LY. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature. 1993;364:802–806. doi: 10.1038/364802a0. [DOI] [PubMed] [Google Scholar]
- Kurachi Y. G protein regulation of cardiac muscarinic potassium channel. American Journal of Physiology. 1995;269:C821–830. doi: 10.1152/ajpcell.1995.269.4.C821. [DOI] [PubMed] [Google Scholar]
- Lesage F, Duprat F, Fink M, Guillemare E, Coppola T, Lazdunski M, Hugnot J-P. Cloning provides evidence for a family of inward rectifier and G-protein coupled K+ channels in the brain. FEBS Letters. 1994;353:37–42. doi: 10.1016/0014-5793(94)01007-2. [DOI] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Find M, Duprat F, Heurteaux C, Fosset M, Romey G, Barhanin J, Lazdunski M. Molecular properties of neuronal G-protein-activated inwardly rectifying K+ channels. Journal of Biological Chemistry. 1995;270:28660–28667. doi: 10.1074/jbc.270.48.28660. [DOI] [PubMed] [Google Scholar]
- Otero AS, Xu L, Ni Y, Szabo G. Receptor-independent activation of atrial muscarinic potassium channels in the absence of nucleotides. Journal of Biological Chemistry. 1998;273:28868–28872. doi: 10.1074/jbc.273.44.28868. [DOI] [PubMed] [Google Scholar]
- Pleumsamran A, Wolak M, Kim D. Inhibition of ATP-induced increase in muscarinic K+ current by trypsin, alkaline pH and anions. American Journal of Physiology. 1998;275:H751–759. doi: 10.1152/ajpheart.1998.275.3.H751. [DOI] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. Estimating single channel kinetic parameters from idealized patch-clamp data containing missed events. Biophysical Journal. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iñiguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY. Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature. 1994;370:143–146. doi: 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- Shyng S-L, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Sui JL, Petit-Jacques J, Logothetis DE. Activation of the atrial KACh channel by the βγ subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proceedings of the National Academy of Sciences of the USA. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Pessia M, Adelman JP. Muscarinic-gated K+ channel: subunit stoichiometry and structural domains essential for G protein stimulation. American Journal of Physiology. 1996;271:H349–385. doi: 10.1152/ajpheart.1996.271.1.H379. [DOI] [PubMed] [Google Scholar]
- Wickman KD, Iñiguez-Lluhi JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, Gilman AG, Clapham DE. Recombinant G-protein βγ-subunits activate the muscarinic-gated atrial potassium channel. Nature. 1994;368:255–257. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]