

Abstract
Blockade of uptake carriers of γ-aminobutyric acid (GABA) has been shown to modulate inhibition in cortical slices of experimental animals, although little is known about this mechanism in vivo and, in particular, in humans.
The effects of blockade of GABA uptake were studied using transcranial magnetic stimulation (TMS) in humans. In eight healthy volunteers several measures of cortical excitation and inhibition were obtained before and ≈2 h after ingestion of 5-15 mg of tiagabine (TGB).
After TGB ingestion, the duration of the TMS-induced silent period observable in the electromyogram of the voluntarily contracted target muscle was prolonged. Similarly, paired-pulse inhibition of the motor-evoked potential (MEP), as tested by delivering two magnetic shocks of equal suprathreshold intensities at 160 ms interstimulus interval (ISI), was more pronounced. In apparent contradistinction, paired-pulse inhibition of the MEPs produced by a subthreshold conditioning stimulus delivered 3 ms prior to a suprathreshold stimulus was reduced. Paired-pulse facilitation elicited by the same double-shock protocol at an ISI of 10 ms was increased.
The prolongation of the GABAB receptor-mediated component of the inhibitory postsynaptic potential observed with TGB in in vitro studies probably underlies the increase in cortical silent period duration. The reduction of the paired-pulse inhibition at 3 ms, in turn, probably reflects inhibition of GABAA receptor-mediated inhibition via presynaptic GABAB receptors.
These data provide in vivo evidence of differential modulation of cortical inhibition by blockade of GABA uptake. Presynaptic GABA autoreceptors may be involved in modulating cortical inhibition in the human motor cortex.
Synaptic inhibition in the brain is mainly mediated by γ-aminobutyric acid (GABA) (Biggio, 1992). The effects of GABA are modulated by a powerful uptake system (Dingledine & Korn, 1985) that limits spatial diffusion of GABA and the duration of inhibitory postsynaptic potentials (IPSPs) (Isaacson et al. 1993). If GABA uptake is blocked pharmacologically, profound changes in the shape and duration of stimulus-induced IPSPs can be observed in cortical slices of experimental animals (Dingledine & Korn, 1985; Thompson & Gähwiler, 1992). Tiagabine (TGB), a lipophilic derivative of nipecotic acid, is a novel antiepileptic drug effective in controlling partial seizures (Ben-Menachem, 1995). In in vitro experiments, TGB has been shown to inhibit the uptake of GABA from the synaptic cleft into glial cells and neurons (Suzdak & Jansen, 1995). Because so much is known concerning the mechanism of action of TGB as elucidated in experiments in animals and cortical slice preparations, it may serve as a suitable substance for enabling us to learn more about the organizational principles of cortical inhibition in humans. We used transcranial magnetic stimulation (TMS) to study non-invasively the effects of blocking GABA uptake in the human motor cortex. Although the exact neuronal mechanisms leading to inhibitory or facilitatory phenomena are not known, TMS techniques are now widely used to assess intracortical excitability in certain neurological disorders and under experimental conditions (Rothwell, 1997) including neuropharmacological manipulation (Ziemann et al. 1996b). Our findings, demonstrating distinct and dissociated effects on different measures of intracortical inhibition induced by TGB, also have implications for the interpretation of some of the findings obtained by TMS in the normal and diseased human brain.
METHODS
Subjects
Eight healthy volunteers, including some of the authors, were included in the study (six men and two women, mean age 31.7 years, range 25-41 years). None of the subjects had a history of neurological illness. None of the subjects was on medication at the time of the study. All subjects were non-smokers and refrained from ethanol intake for at least 12 h before the experiments. Approval was obtained from the appropriate ethics committee. All subjects gave their informed, written consent. Studies were performed according to the Declaration of Helsinki.
Substance used
TGB ((R)-N-(4,4-bis (3-methyl-2-thienyl) but-3-en-1-yl) nipecotic acid, Gabatril®, Sanofi-Winthrop, Munich, Germany) is a lipophilic derivative of nipecotic acid. It acts as a selective and potent inhibitor of GABA uptake into neurones and astrocytes (Suzdak & Jansen, 1995). TGB absorption after oral dosing is complete (nearly 100% bioavailability) and rapid (peak concentration at about 90 min) (Mengel, 1994).
Experimental procedures
Surface EMG was recorded from the right first dorsal interosseus muscle. The raw signal was amplified, filtered (time constant of 10 ms, high cut-off filter of 1 kHz), and recorded onto a PC using a 12-bit AD interface (sampling rate of 5 kHz) for off-line analysis. TMS was performed using a figure-of-eight magnetic coil (external loop diameter, 7 cm) connected to two Magstim 200 stimulators via a Bistim module (The Magstim Company, Dyfed, UK). This device allows two magnetic stimulators to discharge through the same coil but attenuates the maximum power output by about 30% (peak magnetic field, 1.5 T). In the text, stimulus intensities are expressed as a percentage of the maximum output when connected via the Bistim unit.
We first determined the optimal coil position over the left motor cortex, with the handle held posteriorly, and laterally at an angle of 45 deg to the sagittal plane, using suprathreshold stimulus intensities. This position was marked directly on the scalp with a soft-tip pen, to ensure accurate repositioning of the coil. At the optimal site, resting motor threshold (RMT) was defined as the stimulator intensity needed to produce a response of more than 50 μV in at least five of ten consecutive trials using an intensity resolution of 1% of the maximal stimulator output.
Several measures of cortical excitability were obtained. Paired-pulse inhibition (PPI3) and facilitation (PPF) were tested at short intervals (see below) and at rest, using the method described by Kujirai and colleagues (1993). In brief, the effect of a conditioning stimulus delivered prior to a second (test) stimulus was investigated. The conditioning stimulus was set at an intensity of 70% of RMT, and, thus, at an intensity known to produce no changes of excitability in the spinal cord (Kujirai et al. 1993; Di Lazzaro et al. 1998). The intensity of the test stimulus was adjusted to evoke unconditioned muscle responses of 1-2 mV peak-to-peak amplitude. The timing of the conditioning shock was varied in relation to the test shock. Single test stimuli and paired stimuli with interstimulus intervals (ISIs) of 3 or 10 ms were delivered 9 s apart in random order generated by the computer. Twenty trials were recorded for each ISI and the control condition (single stimuli). The conditioned response CR3 (or CR10) was defined as the mean amplitude of the conditioned responses at 3 ms (or 10 ms) ISI, expressed as a percentage of the mean amplitude of the unconditioned test responses. The amplitudes of the motor-evoked potentials (MEPs) were measured peak to peak. Resting amplitudes (RAs) were defined as the mean amplitudes of the unconditioned MEPs.
Paired-pulse inhibition was also tested in another protocol using two shocks of equal intensities delivered at an ISI of 160 ms through the same coil (Valls-Soléet al. 1992). The stimulus intensity was 130% of the RMT and twenty trials were sampled at the optimal scalp site while subjects were at rest. CR160 was defined as the mean amplitude of the conditioned (second) responses expressed as a percentage of the mean amplitude of the responses to the first pulse.
Absence of involuntary EMG activity was monitored in all double-shock protocols by audiovisual feedback, and trials with background EMG activity were rejected.
The cortical stimulation-induced silent period (CSSP) was elicited while subjects held a tonic voluntary contraction of approximately 30% of maximum voluntary contraction. Ten trials were conducted at the optimal scalp site using a stimulus intensity of 130% of RMT. The duration of the CSSP was measured in individual trials from stimulus onset to the end of the CSSP, which was defined as the point where the first burst of continuous EMG activity was seen following the period of EMG silence. Measurements of the duration of the CSSP were made, for the most part, in a blinded fashion by a single investigator, and independently confirmed by a second investigator. Active amplitudes (AA) were defined as the mean amplitudes of the MEPs recorded in the same trials and as measured peak to peak.
The silent period induced by peripheral nerve stimulation (PSSP) was studied with the first dorsal interosseus muscle contracted at about 30% of maximal force. Ten electrical stimuli were delivered to the ulnar nerve through a Digitimer D180 electrical stimulator (maximum stimulator output, 750 V, 1 A; Digitimer Ltd, Welwyn Garden City, Hertfordshire, UK) using a stimulus width of 100 μs and a stimulus intensity of 375 V. At this intensity a maximal compound muscle action potential was elicited in all subjects studied. Similar to CSSP, PSSP duration was assessed from the time of stimulation until the first occurrence of continuous EMG activity.
In a first series of experiments, the effect of different doses of TGB were investigated in three subjects (all males). Subjects took 5, 10 or 15 mg or an equivalent of 63, 125 or 188 al of 25 μg (kg body weight)−1 of TGB. The order of doses was balanced across subjects. At least 48 h elapsed between any two sessions. Following the baseline (BSL) testing of all variables (RMTBSL, RABSL, AABSL, PSSPBSL, CSSPBSL, CR3BSL, CR10BSL, CR160BSL) subjects were studied again at 90-120 min after oral administration of TGB (RMTTGB, RATGB, AATGB, PSSPTGB, CSSPTGB, CR3TGB, CR10TGB, CR160TGB). The return to baseline levels was ascertained by a separate measurement each time before taking another dose, and on some occasions, on the following day.
In a second series, eight subjects (6 males and 2 females), received an equivalent of approximately 200 μg TGB (kg body weight)−1 (15 mg, 6 males, or 10 mg, 2 females). In this series, RMT, RA, AA, CSSP, CR3 and CR10 were measured. The results of the experiments testing the effect of 15 mg TGB were similar in the three subjects who had already taken 15 mg in the first series. Therefore, in those cases, the mean of the two experiments was used for further analysis.
Analysis
The effect of TGB was analysed for each variable separately using Student's two-tailed paired t test. Differences were considered significant if P < 0.05.
RESULTS
All subjects reported some adverse effects after drug administration such as tiredness, lack of concentration and alertness, dizziness or nausea starting at about 60 min after TGB intake and resolving within 5-7 h. These effects were barely noticeable at 5 mg TGB, and maximal at 15 mg TGB, but did not interfere with the ability of the subjects to complete the study.
Duration of CSSP, PSSP and depth of paired-pulse inhibition at 160 ms ISI
TMS at 1.3 times RMT evoked a CSSP in the voluntarily contracted target muscle of about 150 ms duration. Two hours after intake of TGB, the duration of the CSSP was increased (Fig. 1A). In the three subjects, in whom different doses were tested, the increase of CSSP duration was dose dependent (Fig. 1C) and maximal at 15 mg TGB. At the maximum dosage, duration of the CSSP increased from a baseline of 138 ± 7 ms to 176 ± 35 ms (Fig. 1C). At the same time, CR160 decreased (Fig. 1B) in all three individuals studied, equivalent to an increase of PPI160. The magnitude of the mean decrease of CR160 was similar at all doses of TGB (Fig. 1C). In contrast to CSSP and PPI160, the duration of the PSSP remained unchanged under the influence of TGB at all doses (5 mg: PSSPBSL, 116 ± 5 ms, PSSPTGB, 117 ± 3 ms; 10 mg: PSSPBSL, 120 ± 1 ms, PSSPTGB, 117 ± 1 ms; 15 mg: PSSPBSL, 118 ± 1 ms, PSSPTGB, 119 ± 4 ms; n.s.). In the eight subjects tested at 200 μg TGB (kg body weight)−1, the CSSP durations increased from a baseline of 158 ± 30 ms to 180 ± 29 ms (P < 0.05).
Figure 1.
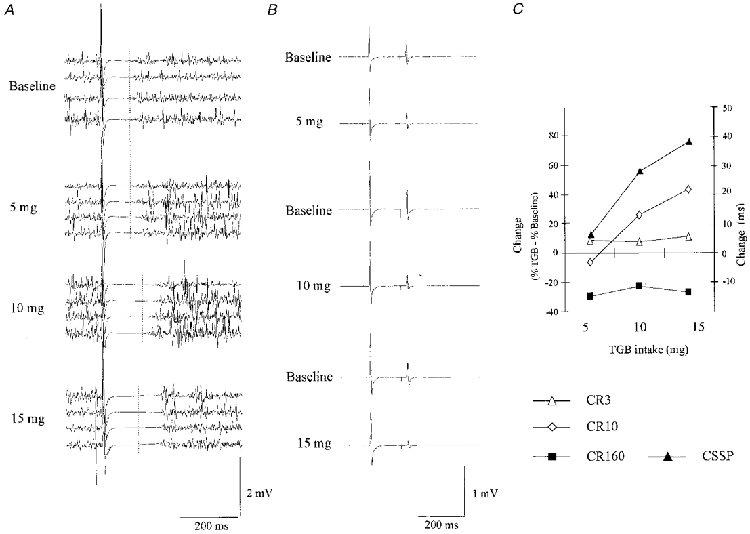
Effects of TGB on CSSP and PPI160 at different dosages
A, example of one subject: four representative trials demonstrating CSSP are shown at baseline (top) and at different doses of TGB, ranging from 5 to 15 mg, as indicated. CSSP duration increased with increasing doses of TGB. Baselines for 10 and 15 mg experiments are indicated as vertical lines. B, averages of 20 trials testing PPI160 are shown. Results are from the same subject. Suppression of the amplitude of the second response was augmented by TGB. C, dose dependency of the effects of TGB on CR160 (▪), CR3 (▵), CR10 (⋄) and CSSP (▴) in three subjects. Effects are expressed as the percentage difference of CR160TGB, CR3TGB or CR10TGB from the baseline condition (no TGB) (left ordinate), or as the duration difference (ms) of CSSPTGB from baseline (right ordinate).
Paired-pulse inhibition and facilitation
At baseline, the magnitude of a MEP response was reduced if the test shock was preceded by a subthreshold conditioning stimulus at an ISI of 3 ms, and was facilitated at an ISI of 10 ms. After TGB intake, PPI3 was reduced and PPF was increased (Fig. 2A). In the three subjects in whom different doses of TGB were tested, the mean increase of CR3 was independent of the dosage, whereas the CR10 increased with increasing TGB dosage (Fig. 1C). When compared with baseline, CR3 increased in all individuals tested at 200 μg TGB (kg body weight)−1 (CR3BSL: 37.1 ± 21.3%; CR3TGB: 50.4 ± 29.5%; P < 0.01; Fig. 2B). There was a tendency for PPF to increase (CR10BSL: 129.6 ± 41.1%; CR10TGB: 141.8 ± 40.1%), but the increase did not reach significance. CR10 increased in six subjects, and decreased in two subjects (Fig. 2B).
Figure 2.
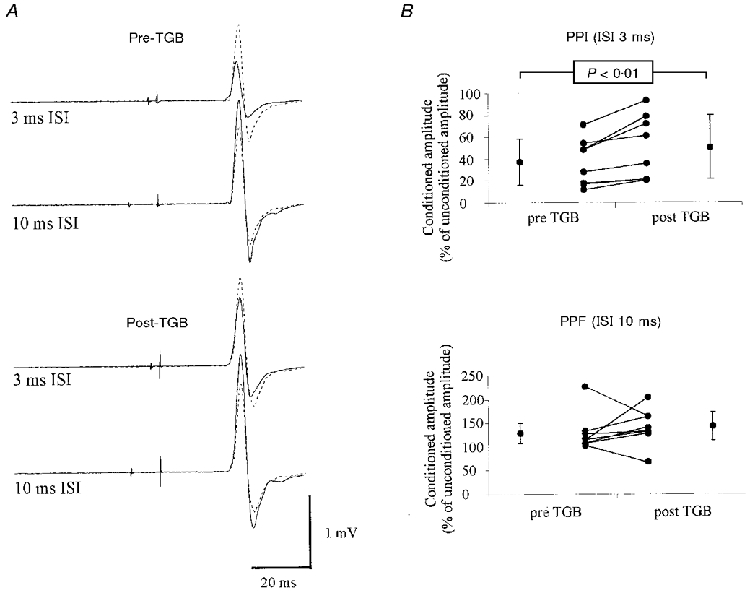
Effects of TGB on PPI3 and PPF
A, results from one subject: each record shows two superimposed traces (average of 10 trials each), the response to the test stimulus given alone (dotted lines), and the response to the test stimulus when given 3 ms (upper traces) or 10 ms (lower traces) after a subthreshold conditioning stimulus (continuous lines). The top two records where obtained prior to, and the lower two records ≈2 h after the administration of 15 mg TGB. B, results from 8 subjects tested at 200 μg kg−1. PPI3 decreased in all subjects and PPF increased in 6 of 8 subjects. Individual results (inner points) as well as group means ±s.d. (outer points) are shown.
Resting motor threshold and size of the MEPs
In the eight subjects studied at 200 μg TGB kg−1, mean motor threshold at rest was 44.9 ± 4.9%. Two hours after administration of TGB, motor thresholds did not differ significantly from baseline (mean 45.8 ± 5.3%, n.s.). There was no significant change in MEP amplitudes under the influence of TGB either at rest (RABSL: 1.4 ± 0.3 mV, RATGB: 1.3 ± 0.5 mV, n.s.) or when the target muscle was active (AABSL: 10.0 ± 5.2 mV, AATGB: 8.4 ± 4.1 mV; n.s.).
DISCUSSION
In healthy subjects, the ingestion of a single dose of TGB prolonged the duration of the CSSP evoked by TMS in the isometrically contracted target muscle. Similarly, inhibition, as tested in the resting target muscle using a paired-pulse protocol with a long interstimulus interval, was increased. In apparent contradistinction, cortical excitability was found to be increased when measured using a paired-pulse technique with short interstimulus intervals. On the basis of what is known about the mechanism of action of TGB, both findings suggest that GABA uptake carriers play an important role in modulating intracortical inhibition in the human motor cortex. As outlined below, our data indicate that the organizational principles of GABAergic intracortical inhibition as observed in vivo in humans, are in remarkable agreement with those elaborated in in vitro studies in cortical slices from experimental animals.
Effects observed at long latencies (160-200 ms)
In vitro studies in brain slices of experimental animals have shown that TGB inhibits GABA uptake from the synaptic cleft (Suzdak & Jansen, 1995). The effect of GABA released by the presynaptic axon terminals of inhibitory interneurons depends on the type of receptor on the postsynaptic cell membrane that it interacts with: the GABAA receptor mediates a short-lasting Cl−-dependent component of stimulation-induced inhibitory postsynaptic potentials (IPSPs), whereas a long-lasting K+-dependent component results from activation of postsynaptic GABAB receptors (McCormick, 1992). The latter component can only be detected when inhibitory interneurons are strongly activated, suggesting that activation of GABAB receptors requires a higher GABA concentration or a longer exposure to GABA than that necessary for the activation of GABAA receptors (Otis & Mody, 1992). It has been proposed that with heavy release of GABA from inhibitory interneurons, the capacity of the GABA uptake enzymes may be insufficient to remove GABA quickly from the synaptic cleft, thereby favouring postsynaptic GABAB receptor activation (Thompson & Gähwiler, 1992). Through a similar mechanism, TGB may prolong the stimulation-induced IPSPs produced in organotypic cultures of hippocampal neurones (Thompson & Gähwiler, 1992). The TMS-induced silent period detectable in surface EMG recordings of voluntarily contracted muscles is generated cortically, at least in its later part (Hallett, 1995). Because of the similar time course of the GABAB receptor-induced IPSPs and the CSSP, it has been proposed that the CSSP reflects activation of postsynaptic GABAB receptors (Roick et al. 1993). This view is strongly supported by the observation that the prototypic GABAB agonist, baclofen, when given intrathecally, markedly prolongs the CSSP in a dose-dependent fashion (Siebner et al. 1998). The present in vivo results indicate that GABAB-induced IPSPs are strongly controlled by active GABA uptake systems in the human motor cortex. Additionally, they substantiate the view that the CSSP is closely related to GABAB receptor-mediated IPSPs in the human cortex. In support of this view, TGB did not alter the silent period induced by peripheral nerve stimulation, which is known to depend on spinal mechanisms (Shahani & Young, 1973).
Evidence suggests that similar to CSSP, the double-shock protocol, using two magnetic stimuli of equal size at long (i.e. > 100 ms) interstimulus intervals, tests intracortical inhibition (Inghilleri et al. 1993). Because TGB increased the magnitude of the inhibition of the test response amplitude in the relaxed target muscle at an ISI of 160 ms, we consider it likely that this inhibition is related to GABAB receptor-dependent IPSPs. Since the GABAB receptor-dependent IPSP peaks at 150-200 ms (Mott & Lewis, 1994), our findings suggest that, in addition to prolonging the duration of the GABAB receptor-mediated IPSPs, TGB also increases their maximum amplitude. TGB is known to have no effect on the maximum amplitude of the GABAA receptor-dependent IPSP component (Thompson & Gähwiler, 1992). In our sample of three subjects, an increase in paired-pulse inhibition at ISIs of 160 ms was already present at 5 mg TGB, and no further increase was noted with increasing doses of TGB. In contrast, the duration of the CSSP was dose dependent. This may indicate that the maximum amplitude of the GABAB receptor-mediated IPSP is saturated at a relatively minor increase in GABA concentration.
Effects observed at short latencies (3 and 10 ms)
A subthreshold conditioning cortical magnetic stimulus inhibits or facilitates the amplitude of the MEP following a test stimulus, depending on the interstimulus interval (Kujirai et al. 1993). This modulation has been shown to reflect intracortical interneuronal phenomena (Kujirai et al. 1993). PPI produced with this protocol at 3 ms ISI is mainly the result of the suppression of late descending volleys (‘I-waves’) that are generated by trans-synaptic activation of pyramidal tract neurons (Nakamura et al. 1997). Recently, elegant evidence has been presented, showing that the depression of these late I-waves lasts longer than 20 ms (Hanajima et al. 1998), linking it directly to the component of the IPSP that is produced by activation of GABAA receptors and that is known to last for tens of milliseconds (McCormick, 1992). In accordance with these studies, pharmacological stimulation of GABAA receptors, e.g. by the benzodiazepine lorazepam (Ziemann et al. 1996a), attenuates cortical excitability as assessed by PPI3 or PPF. A decrease of PPI3, as present in all subjects exposed to TGB, has not been shown to be associated with any anti-epileptic drug, and, in particular, not with those modulating inhibitory neurotransmission (note, however, the decrease of PPI3 by 13% of the control amplitude induced by Vigabatrin, an inhibitor of GABA transaminase, as described by Ziemann et al. 1996b). To explain this finding it is important to note that GABAB receptors are also present presynaptically on axon terminals of inhibitory interneurons (Bowery, 1980). In slices of rat dentate gyrus, activation of these presynaptically located GABAB receptors by a prior conditioning stimulus, or by chemically induced inhibitory interneuronal barrage, led to suppression of stimulation-induced IPSPs in the postsynaptic neuron (Otis & Mody, 1992). We consider it likely that the inhibition of the GABAA receptor-mediated component of the IPSP by presynaptic stimulation of GABAB receptors also underlies the reduction of PPI3 found in the present study.
PPF, another measure of excitability dependent on intracortical mechanisms, was increased by TGB in six of eight subjects, although the increase did not quite reach significance when considering the entire group of subjects. The fact that it was dose dependent in our sample of three subjects, however, provides additional support for the contention that TGB also exerted an effect on the facilitation at 10 ms ISI. The increase in PPF may result from an increase of the excitatory drive onto the pyramidal output cell, or from a decrease of an ongoing profound inhibitory activity impinging on the same cell by the same mechanism proposed for the reduction of PPI3. The latter hypothesis, favoured by us, would be consistent with the fact that the GABAA receptor-dependent component of the IPSP peaks at 10-20 ms (McCormick, 1992).
The effects of TGB on cortical excitability as tested by PPI3 and PPF were greatest at the maximum dose, but were also noticeable at the lowest dose (Fig. 2C). This finding suggests that even small changes in GABA uptake may profoundly influence the balance of excitation and inhibition. Thus GABA uptake is a powerful mechanism, probably of fundamental importance also under physiological conditions.
The RMT has been shown to reflect changes in membrane excitability rather than neurotransmission at synapses (Ziemann et al. 1996b). In line with this concept, the RMT was unchanged by TGB in our study. Resting amplitudes, as well as active amplitudes did not show a significant decrease under the influence of TGB. This finding indicates that the recruitment pattern of the neuronal elements activated by TMS remains essentially unaltered both in the relaxed, as well as in the active state. In view of the differential changes demonstrated in parameters reflecting intracortical inhibition, this conclusion suggests that the recruitment pattern is equally modulated by both types of inhibition.
Significance for paired-pulse TMS studies and anti-epileptic properties of tiagabine
The present results may influence the interpretation of numerous studies investigating intracortical excitability in various neurological disorders. Insufficient suppression of the test amplitude by a prior conditioning shock at short intervals has usually been taken as evidence for a plainly deficient intracortical inhibition. Our findings, demonstrating that intracortical excitability as tested by PPI3 and PPF increases under the influence of prolonged exposure to, or increased concentrations of GABA, raise the additional possibility that an increased presynaptic inhibition underlies the decrease of PPI3 or increase of PPF in some pathophysiological conditions.
Failure of GABA-dependent inhibition is a well-established mechanism in many models of epilepsy (Fisher, 1989). In general, reduction of GABAA receptor-mediated inhibition has been found to be the main pathophysiological factor. However, loss of the GABAB receptor-dependent component of the IPSP may also play a role in the generation of seizures, since, in the presence of experimentally impaired GABAA receptor-mediated inhibition, additional pharmacological blockade of GABAB receptors leads to an exacerbation of the seizure activity (Sutor & Luhmann, 1998). Therefore, enhancing the GABAB receptor-dependent component of the IPSP by TGB may well explain its anti-epileptic properties, which have been demonstrated in several clinical studies (Ben-Menachem, 1995). Reduction of PPI3, if related to presynaptic inhibition of GABAA receptor-mediated IPSP, would, however, predict that TGB may also have a pro-epileptic effect, in addition to its anti-epileptic properties. Interestingly, TGB has been observed to induce absence status epilepticus or generalized myoclonic seizures with generalized EEG seizure patterns in some patients with focal epilepsy (Eckardt & Steinhoff, 1998; J. Classen and K. J. Werhahn, unpublished observations). Furthermore, TGB provoked repetitive hypersynchronous EEG discharges in rats, suggesting increased cortical excitability (Lancel et al. 1998).
In summary, our findings indicate that inhibition in the human motor cortex is distinctly influenced by GABA uptake carriers. Additionally, we have provided in vivo evidence to suggest that blockade of GABA uptake: (a) increases the duration and magnitude of the GABAB receptor-mediated IPSP component, and (b) decreases the GABAA receptor-mediated inhibition, probably through activation of presynaptic GABAB receptors.
Acknowledgments
We wish to thank J. Ditterich for constructing part of the equipment, and M. Johnson for editorial comments. This work was supported by Deutsche Forschungsgemeinschaft grant Cl 95/3-1 to J. C.
References
- Ben-Menachem E. International experience with tiagabine add-on therapy. Epilepsia. 1995;36(suppl. 6):S14–21. doi: 10.1111/j.1528-1157.1995.tb06010.x. [DOI] [PubMed] [Google Scholar]
- Biggio G. Gabaergic Synaptic Transmission: Molecular, Pharmacological, and Clinical Aspects. New York: Raven Press; 1992. [Google Scholar]
- Bowery NG, Hill DR, Hudson AL, Doble A, Middlemiss DN, Shaw J, Turnbull M. (-)Baclofen decreases neurotransmitter release in the mammalian CNS by an action at a novel GABA receptor. Nature. 1980;283:92–94. doi: 10.1038/283092a0. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Restuccia D, Oliviero A, Profice P, Ferrara L, Insola A, Mazzone P, Tonali P, Rothwell JC. Magnetic transcranial stimulation at intensities below active motor threshold activates intracortical inhibitory circuits. Experimental Brain Research. 1998;119:265–268. doi: 10.1007/s002210050341. 10.1007/s002210050341. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Korn SJ. γ-Aminobutyric acid uptake and the termination of inhibitory synaptic potentials in the rat hippocampal slice. The Journal of Physiology. 1985;366:387–409. doi: 10.1113/jphysiol.1985.sp015804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckardt KM, Steinhoff BJ. Non-convulsive status epilepticus in two patients receiving tiagabine treatment. Epilepsia. 1998;39:671–674. doi: 10.1111/j.1528-1157.1998.tb01438.x. [DOI] [PubMed] [Google Scholar]
- Fisher RS. Animal models of the epilepsies. Brain Research Reviews. 1989;14:245–278. doi: 10.1016/0165-0173(89)90003-9. 10.1016/0165-0173(89)90003-9. [DOI] [PubMed] [Google Scholar]
- Hallett M. Transcranial magnetic stimulation: negative effects. In: Fahn S, Hallett M, Lüders HO, Marsden CD, editors. Negative Motor Phenomena. Philadelphia, USA: Lippincott-Raven; 1995. pp. 107–114. [Google Scholar]
- Hanajima R, Ugawa Y, Terao Y, Sakai K, Furubayashi T, Machii K, Kanazawa I. Paired-pulse magnetic stimulation of the human motor cortex: differences among I waves. The Journal of Physiology. 1998;509:607–618. doi: 10.1111/j.1469-7793.1998.607bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inghilleri M, Berardelli A, Cruccu G, Manfredi M. Silent period evoked by transcranial stimulation of the human cortex and cervicomedullary junction. The Journal of Physiology. 1993;466:521–534. [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. 10.1016/0896-6273(93)90308-E. [DOI] [PubMed] [Google Scholar]
- Kujirai T, Caramia MD, Rothwell JC, Day BL, Thompson PD, Ferbert A, Wroe S, Asselman P, Marsden CD. Corticocortical inhibition in human motor cortex. The Journal of Physiology. 1993;471:501–519. doi: 10.1113/jphysiol.1993.sp019912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancel M, Faulhaber J, Deisz RA. Effect of the GABA uptake inhibitor tiagabine on sleep and EEG power spectra in the rat. British Journal of Pharmacology. 1998;123:1471–1477. doi: 10.1038/sj.bjp.0701769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex. Journal of Clinical Neurophysiology. 1992;9:212–223. doi: 10.1097/00004691-199204010-00004. [DOI] [PubMed] [Google Scholar]
- Mengel H. Tiagabine. Epilepsia. 1994;35(suppl. 5):S81–84. doi: 10.1111/j.1528-1157.1994.tb05976.x. [DOI] [PubMed] [Google Scholar]
- Mott DD, Lewis DV. The pharmacology and function of central GABAB receptors. International Review of Neurobiology. 1994;36:97–223. doi: 10.1016/s0074-7742(08)60304-9. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Kitagawa H, Kawaguchi Y, Tsuji H. Intracortical facilitation and inhibition after transcranial magnetic stimulation in conscious humans. The Journal of Physiology. 1997;498:817–823. doi: 10.1113/jphysiol.1997.sp021905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Mody I. Differential activation of GABAA and GABAB receptors by spontaneously released transmitter. Journal of Neurophysiology. 1992;67:227–235. doi: 10.1152/jn.1992.67.1.227. [DOI] [PubMed] [Google Scholar]
- Roick H, von Giesen HJ, Benecke R. On the origin of the postexcitatory inhibition seen after transcranial magnetic brain stimulation in awake human subjects. Experimental Brain Research. 1993;94:489–498. doi: 10.1007/BF00230207. [DOI] [PubMed] [Google Scholar]
- Rothwell JC. Techniques and mechanisms of action of transcranial stimulation of the human motor cortex. Journal of Neuroscience Methods. 1997;74:113–122. doi: 10.1016/s0165-0270(97)02242-5. 10.1016/S0165-0270(97)02242-5. [DOI] [PubMed] [Google Scholar]
- Shahani BT, Young RR. Studies of the normal human silent period. In: Desmedt JE, editor. New Developments in Electromyography and Clinical Neurophysiology. Basel: Karger; 1973. pp. 589–602. [Google Scholar]
- Siebner HR, Dressnandt J, Auer C, Conrad B. Continuous intrathecal baclofen infusions induced a marked increase of the transcranially evoked silent period in a patient with generalized dystonia. Muscle and Nerve. 1998;21:1209–1215. doi: 10.1002/(sici)1097-4598(199809)21:9<1209::aid-mus15>3.0.co;2-m. 10.1002/(SICI)1097-4598(199809)21:9<1209::AID-MUS15>3.3.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Sutor B, Luhmann HJ. Involvement of GABAB receptors in convulsant-induced epileptiform activity in rat neocortex in vitro. European Journal of Neuroscience. 1998;10:3417–3427. doi: 10.1046/j.1460-9568.1998.00351.x. 10.1046/j.1460-9568.1998.00351.x. [DOI] [PubMed] [Google Scholar]
- Suzdak PD, Jansen JA. A review of the preclinical pharmacology of tiagabine: a potent and selective anticonvulsant GABA uptake inhibitor. Epilepsia. 1995;36:612–626. doi: 10.1111/j.1528-1157.1995.tb02576.x. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Effects of the GABA uptake inhibitor tiagabine on inhibitory synaptic potentials in rat hippocampal slice cultures. Journal of Neurophysiology. 1992;67:1698–1701. doi: 10.1152/jn.1992.67.6.1698. [DOI] [PubMed] [Google Scholar]
- Valls-Solé J, Pascual-Leone A, Wassermann E, Hallett M. Human motor evoked responses to paired transcranial magnetic stimuli. Electroencephalography and Clinical Neurophysiology. 1992;81:355–364. doi: 10.1016/0168-5597(92)90048-g. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Lönecker S, Steinhoff BJ, Paulus W. The effect of lorazepam on the motor cortical excitability in man. Experimental Brain Research. 1996a;109:127–135. doi: 10.1007/BF00228633. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Lönnecker S, Steinhoff BJ, Paulus W. Effects of antiepileptic drugs on motor cortex excitability in humans: A transcranial magnetic stimulation study. Annals of Neurology. 1996b;40:367–378. doi: 10.1002/ana.410400306. [DOI] [PubMed] [Google Scholar]