

Abstract
In adult rat atrial myocytes, three kinetically distinct Ca2+-independent depolarization-activated outward K+ currents, IK,fast, IK,slow and Iss, have been separated and characterized.
To test directly the hypothesis that different voltage-dependent K+ channel (Kv channel) α subunits underlie rat atrial IK,fast, IK,slow and Iss, the effects of antisense oligodeoxynucleotides (AsODNs) targeted against the translation start sites of the Kv α subunits Kv1.2, Kv1.5, Kv4.2, Kv4.3, Kv2.1 and KvLQT1 were examined.
Control experiments on heterologously expressed Kv α subunits revealed that each AsODN is selective for the subunit against which it was targeted.
Peak outward K+ currents were attenuated significantly in rat atrial myocytes exposed to AsODNs targeted against Kv4.2, Kv1.2 and Kv1.5, whereas AsODNs targeted against Kv2.1, Kv4.3 and KvLQT1 were without effects.
No measurable effects on inwardly rectifying K+ currents (IK1) were observed in atrial cells exposed to any of the Kv α subunit AsODNs.
Kinetic analysis of the currents evoked during long (10 s) depolarizing voltage steps revealed that AsODNs targeted against Kv4.2, Kv1.2 and Kv1.5 selectively attenuate rat atrial IK,fast, IK,slow and Iss, respectively, thus demonstrating that the molecular correlates of rat atrial IK,fast, IK,slow and Iss are distinct.
The lack of effect of the Kv4.3 AsODNs on peak outward K+ currents reveals that Kv4.2 and Kv4.3 do not heteromultimerize in rat atria in vivo. In addition, the finding that Kv1.2 and Kv1.5 contribute to distinct K+ currents in rat atrial myocytes demonstrates that Kv1.2 and Kv1.5 also do not associate in rat atria in vivo.
Depolarization-activated outward potassium (K+) currents play key roles in controlling the amplitudes and durations of cardiac action potentials, and several distinct voltage-gated K+ currents subserving these functions have been identified (Barry & Nerbonne, 1996). This diversity has a functional significance in the heart in that K+ currents with differing time- and voltage-dependent properties, as well as different pharmacological sensitivities, play distinct roles in controlling action potential repolarization (Barry & Nerbonne, 1996). Differences in the types and densities of K+ channels underlie regional variations in action potential waveforms (Barry & Nerbonne, 1996), and these currents are important targets for endogenous neurotransmitters and neurohormones, as well as clinically used antiarrhythmics (Bennett et al. 1993; Barry & Nerbonne, 1996). In addition, considerable evidence has been accumulated demonstrating alterations in the densities and/or properties of voltage-gated K+ channels associated with myocardial damage or disease (Roden & George, 1997; Brown, 1997). As a result, there is considerable interest in identifying the molecular correlates of functional voltage-gated K+ channels, and in delineating the mechanisms involved in the regulation and modulation of these channels.
A number of voltage-gated K+ channel (Kv channel) pore-forming α subunits and accessory β subunits have been cloned from, or shown to be expressed in, mammalian heart (Barry & Nerbonne, 1996; Deal et al. 1996). Heterologous expression of individual Kv α subunits or combination of Kv α and β subunits reveals voltage-gated K+ currents with differing time- and voltage-dependent properties (Barry & Nerbonne, 1996; Deal et al. 1996). Importantly, the molecular cloning has revealed even greater potential for generating functional K+ channel diversity than was expected based on the electrophysiology, and a variety of approaches are being used to explore the relationship(s) between expressed subunits and functional voltage-gated myocardial K+ channels. Recently, for example, HERG (the human ether-á-go-go-related gene) and KvLQT1 have been shown to contribute to two components of delayed rectification in cardiac cells, IKr and IKs (Sanguinetti et al. 1995, 1996; Trudeau et al. 1995; Barhanin et al. 1996). It has also been suggested that the accessory K+ channel subunit minK contributes to both IKr and IKs (Sanguinetti et al. 1996; Barhanin et al. 1996; Splanski et al. 1997), although direct biochemical evidence demonstrating association of minK with either KvLQT1 or HERG in the mammalian heart has not been provided to date. For those channel types that have not been identified by molecular genetics, alternative molecular strategies are being applied. Recently, for example, in experiments using antisense oligodeoxynucleotides, it has been demonstrated that both Kv4.2 and Kv4.3 contribute to the transient outward current (Ito) in rat ventricular myocytes (Fiset et al. 1997; Xu et al. 1997) and that Kv1.5 underlies IKur, the 4-aminopyridine-sensitive, ultrarapid component of delayed rectification in human atrial myocytes (Feng et al. 1997).
Previous electrophysiological studies have identified three K+ currents in adult rat atrial myocytes (Boyle & Nerbonne, 1992), separated and characterized based on differing kinetic properties. Although these currents were originally referred to as IKf, IKs and Iss (Boyle & Nerbonne, 1992), we have modified the terminology slightly here, and refer to IKf and IKs as IK,fast and IK,slow, respectively. This was done to avoid confusion with other distinct types of K+ currents described in other cells (Barry & Nerbonne, 1996). The three components of the rat atrial currents were distinguished based primarily on differences in inactivation kinetics: (1) IK,fast is a rapidly activating and inactivating current that resembles Ito previously described in a variety of other cardiac cells (Barry & Nerbonne, 1996); (2) IK,slow is a novel rapidly activating, slowly inactivating current; and (3) Iss is a rapidly activating, non-inactivating (steady-state) current that resembles IKur in human atrial myocytes (Barry & Nerbonne, 1996). In addition, IK,fast and IK,slow were shown to recover from steady-state inactivation at distinct rates and completely independently, indicating that these must reflect functionally distinct K+ conductance pathways. In pharmacological experiments, IK,fast, IK,slow and Iss were all shown to be 4-aminopyridine sensitive and tetraethylammonium insensitive (Boyle & Nerbonne, 1992). Subsequent work demonstrated that IK,slow is selectively blocked by nanomolar concentrations of α-dendrotoxin and that Iss is selectively suppressed by low concentrations (≤10 μm) of phenylephrine (Van Wagoner et al. 1996). Neither phenylephrine nor the α-dendrotoxin was useful for facilitating further separation or characterization of the atrial K+ currents, however, because both substances provided only partial suppression of the currents, were complicated by time- and voltage-dependent effects, and/or also blocked additional current components at higher concentrations (Van Wagoner et al. 1996). Nevertheless, the results with low concentrations of α-dendrotoxin and phenylephrine are consistent with the kinetic analysis and the computer simulations in suggesting that IK,fast, IK,slow and Iss are functionally distinct K+ currents (Boyle & Nerbonne, 1992). The experiments here were undertaken to test directly the hypothesis that different Kv α channel subunits underlie IK,fast, IK,slow and Iss in rat atrial myocytes. In these experiments, the effects of antisense oligodeoxynucleotides targeted against the translation start sites of several Kv α subunits expressed in rat atria, Kv1.2, Kv1.5, Kv2.1, Kv4.2 and Kv4.3 (Roberds & Tamkun, 1991; Dixon & McKinnon, 1994; Barry et al. 1995; Dixon et al. 1996), were examined. The results presented reveal that distinct Kv channel α subunits, Kv4.2, Kv1.2 and Kv1.5, contribute to IK,fast, IK,slow, and Iss, respectively, in adult rat atrial myocytes.
METHODS
Atrial cell isolation
Atrial cells were isolated from postnatal day 28 (P28) and adult (≥P45) Long-Evans rats using a procedure previously described in detail (Boyle & Nerbonne, 1992). All experiments were conducted according to the guidelines laid down by the Washington University Medical School Animal Use Committee. Briefly, hearts were rapidly excised from anaesthetized (5% halothane-95% O2) animals and attached (by the aorta) to a Langendorff perfusion apparatus. Isolated hearts were retrogradely perfused with 50 ml of a nominally calcium-free Hepes-buffered Earle's balanced salt solution (Gibco BRL), supplemented with 6 mM glucose, amino acids and vitamins (solution A), followed by 50 ml of solution A containing 1-2 mg ml−1 collagenase type II (Worthington Biochemical Corp., Freehold, NJ, USA) and 50 μm calcium (solution B); the temperature of the heart and the perfusate were maintained at 35-37°C. Solution B was filtered (at 5 μm) and recirculated through the heart until the atria were digested (20-35 min), as judged by eye. After perfusion, the atrial appendages were removed, minced and incubated in a fresh solution B for an additional 10 min. The tissue pieces were then transferred to 10 ml of fresh (enzyme-free) solution A supplemented with 1.25 mg ml−1 taurine, 5 mg ml−1 bovine serum albumin (BSA) (Sigma) and 150 μm CaCl2 (solution C) and gently triturated with a fire-polished Pasteur pipette. The resulting suspension was filtered to remove large undissociated tissue fragments. The filtrate was then centrifuged (300-500 r.p.m.; 5-10 min), the supernatant was discarded, and the cells were resuspended in solution C; this step was repeated twice to completely wash out the enzyme and remove unwanted cellular fragments.
Isolated atrial myocytes were resuspended in solution C, plated on laminin-coated glass coverslips in 35 mm culture dishes and placed in 95% O2-5% CO2 in an incubator at 37°C for 20 min. Ca2+-tolerant rod-shaped atrial myocytes adhered preferentially to the laminin substrate, and damaged cells were removed by replacing solution C, with serum-free Medium-199 (Irvine Scientific, Santa Ana, CA, USA) supplemented with antibiotics (1 unit ml−1 penicillin-streptomycin) 1 h after plating. Electrophysiological recordings were routinely obtained 24-48 h after plating.
HEK-293 and QT-6 cells
HEK-293 cells, obtained from the American Tissue Culture Collection, were maintained in a standard medium containing Opti-MEM (Gibco) supplemented with 10% fetal calf serum (FCS), 1 unit ml−1 penicillin-streptomycin, 36 units ml−1 nystatin; 0.3 mg ml−1 geneticin (G418; Gibco) was added to the medium for the Kv4.2-expressing cell line. QT-6 cells, obtained from the Washington University Tissue Culture Center (TCC), were maintained in a standard QT-6 medium containing Medium 199, 10% TPB (tryptose phosphate buffer; TCC), 5% FCS, 1% DMSO, 1 unit ml−1 penicillin-streptomycin and 36 units ml−1 nystatin. Cells were passaged at confluence (every 3-4 days) by brief trypsinization. The calcium phosphate precipitation method was used to transfect HEK-293 and QT-6 cells.
After passaging, cells were plated on 35 mm dishes coated with cell-Tak (Becton Dickinson, Bedford, MA, USA) at a density of 6-6.5 × 105 cells ml−1 and incubated overnight in normal medium. One hour before transfection, the growth medium was changed to Dulbecco's modified Eagle's medium containing 5% fetal calf serum and 1% DMSO. For transfections, a total 10 μg of DNA (3 μg of the Kv α subunit cDNA, 1 μg Green Lantern (Gibco), which encodes green fluorescent protein (GFP), and 6 μg pSk) was mixed with 100 μl of 2.5 M CaCl2 and 900 μl of 2 × BBS (Bes-buffered saline at pH 6.95), and the mixture was incubated for 20 min at room temperature before addition to the cells. After 15-18 h, the cDNA-containing medium was removed, the cells were rinsed twice with the growth medium and returned to the incubator. Electrophysiological recordings were obtained from GFP-positive cells 18-24 h later. In experiments with antisense oligodeoxynucleotides (AsODNs), cells were exposed to the AsODNs (see below for details) approximately 6 h after removing the cDNA-containing medium, and electrophysiological recordings were obtained 12-18 h later.
Effects of antisense oligodeoxynucleotides
Antisense oligodeoxynucleotides (AsODNs) generated against the translation start sites (nucleotides 4-18) of rat Kv1.2 (5′-TCC GGT AGC CAC TGT-3′), Kv1.5 (5′-CAC CAG GGA GAT CTC-3′), Kv2.1 (5′-CGA GCC ATG CTT CGT-3′), Kv4.2 (5′-TGC AAC ACC GGC TGC 3′), Kv4.3 (5′-TGC AAC TCC TGC CGC-3′), KvLQT1 (5′-TTG GCG CGA TGG GCG-3′), and one directed against nucleotides 24-38 of Kv1.5 (5′-GCA CTG CCA TTC TCC-3′) were obtained from Ransom Hill Bioscience Inc., (Ramona, CA, USA) or Integrated DNA Technologies, Inc. (Coralville, IA, USA). All AsODNs were synthesized with a phosphorothioate backbone and tagged (at the 5′ end) with fluorescein. For experiments, AsODNs (1 μm per dish) were mixed with lipofectamine (Life Technologies Inc., Gaithersburg, MD, USA) (4-8 μg per dish) and incubated at room temperature for ∼30 min prior to addition to the cultures. Approximately 16-20 h later, the oligo-containing medium was removed, and replaced with the normal cell culture medium (see above). The same procedure was used with the Kv α subunit-expressing HEK-293 and QT-6 cells except that the growth medium was changed to Opti-MEM (Gibco) prior to the addition of the AsODN-lipofectamine mixture. Electrophysiological recordings were performed 24-48 h after addition of the AsODNs.
Electrophysiological recordings
The whole-cell variation of the patch-clamp recording technique (Hamill et al. 1981) was used to record Ca2+-independent, voltage-gated outward K+ currents from rat atrial myocytes and from HEK-293 and QT-6 cells using an Axopatch-1D amplifier (Axon Instruments). All recordings were performed at room temperature (22-24°C). The extracellular (bath) solution contained (mM): NaCl, 136; KCl, 4; MgCl2, 2; CaCl2, 1; glucose, 10; Hepes, 10; pH 7.4 (NaOH). For recordings from myocytes, TTX (20 μmol l−1) and CdCl2 (200 μm) were added to suppress voltage-gated Na+ and Ca2+ currents, respectively. Recording pipettes contained (mM): KCl, 135; NaCl, 4; EGTA, 10; Hepes, 10; glucose, 5; Mg-ATP, 3; Na3-GTP, 0.5. pH was adjusted to 7.2 with KOH.
Recording pipettes were fabricated using a horizontal puller (model P-87; Sutter Instruments, Novato, CA, USA), and had resistances (1-3 MΩ) when filled with the standard recording solution. Experiments were controlled by an IBM compatible computer interfaced to the clamp amplifier and using pCLAMP versions 5.5 or 6 (Axon Instruments). Currents were low-pass filtered at 2-5 kHz and digitized at 0.2-10 kHz (test pulses 0.1-10 s). Tip potentials were zeroed before membrane-pipette seals were formed. In each experiment, series resistances (Rs) were electrically compensated. Rs was calculated by dividing the time constant (fit of the decay) of the capacitive transient by the membrane capacitance, Cm (calculated as the time integral of the capacitive response to a 5 mV hyperpolarizing pulse from the holding potential). Voltage errors resulting from uncompensated series resistances were always < 6 mV, and were not corrected. Only data obtained from cells with input resistances ≥1 GΩ were analysed; no linear leakage compensation was performed. Ca2+-independent outward K+ currents were routinely evoked during depolarizing voltage steps to potentials between -50 and +50 mV from a holding potential of -60 mV.
Data analysis
Peak outward currents at each test potential were measured as the maximal amplitude of the current recorded during the first 100 ms of the depolarizing voltage steps. Current densities were obtained by dividing the measured currents by Cm. Exponential fits to decay phases of the currents evoked during long (10 s) depolarizing voltage steps to +30 mV were fitted using the equation y(t) =IK,fastexp(-t/τfast) +IK,slowexp(-t/τslow) +Iss, where t is time and τfast and τslow are the (fast and slow) time constants of inactivation of IK,fast and IK,slow, respectively. All averaged and normalized data are presented as means ±s.e.m. Statistical significance of differences between groups were evaluated using a one-way analysis of variance (ANOVA) and a two-tailed Student's t test was used when comparisons were made with control data; P values are presented in the text.
RESULTS
Outward K+ currents in rat atrial myocytes
In preliminary experiments, isolated adult rat atrial myocytes were exposed to various concentrations of fluorescein-tagged AsODNs in the presence and absence of lipofectamine. In the absence of lipofectamine, no uptake was detected in adult myocytes. When adult myocytes were examined under epifluorescence illumination following incubation in AsODNs at concentrations up to 5 μm in the absence of lipofectamine, no uptake was detected. In the presence of lipofectamine, a few adult atrial cells were labelled, although the intensity of the signals was weak, suggesting that uptake into adult cells is very poor. When similar experiments were completed on atrial myocytes isolated from postnatal day 20-30 animals, it became clear that uptake efficiency, both in terms of the number of cells and fluorescence labelling intensity, varies as a function of age. In myocytes isolated from postnatal day 28 (P28) animals, for example, approximately 50% of the cells are labelled. Importantly, lipofectamine was required for uptake at all ages.
Preliminary experiments also suggested that steady-state uptake of AsODNs was achieved in ∼16 h. As reported previously by others (Boyle & Nerbonne, 1992; Feng et al. 1996, 1997), the morphology of atrial cells maintained in vitro change over time. Specifically, striations become less clearly evident and some of the cells adopt a spherical (from rod-shaped) morphology. To assess the electrophysiological properties of these spherical cells, recordings were obtained from P28 rod-shaped (P28R) and spherical (P28S) cells. Comparisons of the current records obtained revealed that the waveforms of the currents in P28S (Fig. 1C) and P28R (Fig. 1B) are indistinguishable. The mean ±s.e.m. peak outward current density-voltage relations in P28R and P28S cells and adult cells are also indistinguishable (Fig. 1D). Taken together, these results suggest that in spite of changes in cell shape, the properties of the K+ currents in P28 rat atrial cells are unaffected over at least the first 48 h in vitro.
Figure 1. Current-voltage relations of peak outward K+ currents in isolated adult and postnatal day 28 (P28) rat atrial myocytes are indistinguishable.
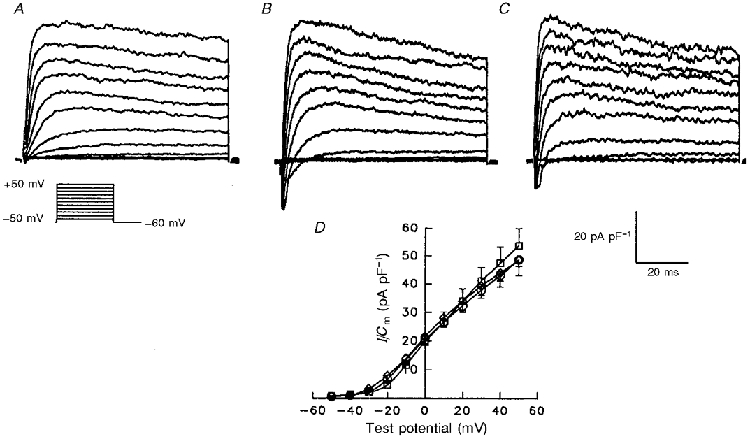
Outward currents, evoked during 100 ms depolarizing voltage steps to potentials from -50 to +50 mV from a holding potential of -60 mV, were recorded in individual cells and normalized to the whole-cell membrane capacitance, Cm (determined in the same cell). Representative current waveforms, normalized for difference in cell sizes, recorded in adult (A) and in postnatal day 28 rod-shaped (P28R) (B) and spherical (P28S) (C) rat atrial myocytes are displayed. D, mean ±s.e.m. peak outward current density for the currents recorded in P28S (n = 25), P28R (n = 9) and adult (n = 11) cells are plotted as a function of test potential. One-way analysis of variance (ANOVA) revealed no significant differences in peak current-voltage relations in adult (○), P28S (⋄) and P28R (□) atrial myocytes.
As noted in the introduction, three components of the total depolarization-activated outward K+ currents have been distinguished in adult rat atrial myocytes based on differences in rates of inactivation and recovery from inactivation (Boyle & Nerbonne, 1992). These are: (1) a rapidly activating and inactivating current, IK,fast; (2) a rapidly activating, slowly inactivating current, IK,slow; and (3) a rapidly activating, non-inactivating (steady-state) current, Iss. Analyses of the currents evoked during long (10 s) depolarizing voltage steps in P28 rat atrial myocytes (Fig. 2) also revealed that the decay phases of the currents were well-described by the sum of two exponential components and that there is a component of the current that is non-inactivating (Iss) (Fig. 2B). The inactivation time constants derived from these fits differ by approximately an order of magnitude, and neither time constant displays any appreciable voltage dependence (Fig. 2C); the mean ±s.e.m. (n = 22) inactivation time constants for IK,fast and IK,slow in P28 cells were 188 ± 17 ms and 2111 ± 109 ms (Table 3), respectively. These values are not significantly different from the mean ±s.d. decay time constants of 181 ± 124 ms and 3006 ± 1016 ms reported previously for IK,fast and IK,slow in adult rat atrial myocytes (Boyle & Nerbonne, 1992). In P28 rat atrial myocytes, IK,fast, IK,slow and Iss contribute 51 ± 4, 35 ± 3 and 16 ± 2%, respectively, to the peak outward K+ currents (Fig. 2D). These values are also similar to the mean distributions reported for IK,fast, IK,slow, and Iss, respectively in adult rat atrial myocytes (Boyle & Nerbonne, 1992; Van Wagoner et al. 1996). The waveforms, densities and properties of the outward K+ currents in P28 atrial myocytes, therefore, are indistinguishable from those in adult cells (Barry & Nerbonne, 1992; Van Wagoner et al. 1996).
Figure 2. Three components of the peak outward currents in P28 rat atrial myocytes.
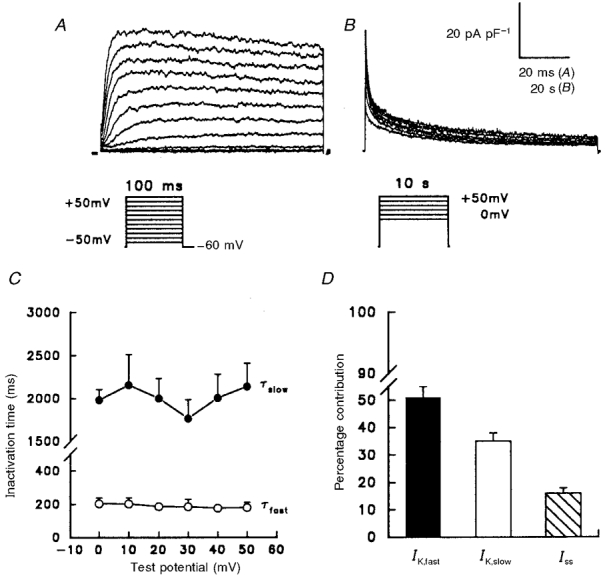
Outward currents were evoked as described in the legend of Fig. 1 during 100 ms (A) and 10 s (B) depolarizing voltage steps; for the records presented in B, the interpulse interval was 60 s. The records in A and B were obtained from the same cell; note also that, in B, the data are plotted as points and the continuous lines reflect double exponential fits to the decay phases of the currents (see text). C, mean ±s.e.m. (n = 6) time constants for the fast (○) and slow (•) components of peak outward decay, determined from double exponential fits (as in B) to the decay phases of the outward currents evoked during 10 s depolarizations to potentials between 0 and +50 mV. D, mean ±s.e.m. (n = 22) percentage contribution of IK,fast, IK,slow and Iss to the peak outward K+ currents in P28 rat atrial myocytes at +30 mV (see text).
Table 3.
Selective effects of antisense oligodeoxynucleotides on atrial IK,fast, IK,slow and Iss
| Control | Kv4.2 AsODN | Kv1.2 AsOD | Kv1. 5 AsODN | Kv2.1 AsODN | |
|---|---|---|---|---|---|
| Time constants | |||||
| τfast (ms) | 188 ± 17 | 149 ± 25 | 191 ± 49 | 159 ± 14 | 216 ± 45 |
| τslow (ms) | 2111 ± 109 | 2222 ± 217 | 2008 ± 191 | 2114 ± 356 | 2488 ± 266 |
| Current density* | |||||
| IK,fast/Cm | 15.7 ± 2.1 | 5.0 ± 0.8** | 9.4 ± 1.7 | 11.6 ± 1.7 | 11.5 ± 3.4 |
| IK,slow/Cm | 11.2 ± 1.5 | 8.6 ± 2.0 | 2.7 ± 0.4** | 8.7 ± 2.0 | 10.2 ± 3.0 |
| Iss/Cm | 5.0 ± 0.7 | 6.2 ± 1.9 | 6.4 ± 0.8 | 1.6 ± 0.3*** | 5.0 ± 1.7 |
| Peak IoutCm | 31.9 ± 3.4 | 19.8 ± 1.5 | 18.5 ± 2.0 | 21.9 ± 3.4 | 26.7 ± 3.7 |
| n | 22 | 8 | 6 | 11 | 7 |
Determined at +30 mV.
Values significantly different at the P < 0.01 level
values significantly different at the P < 0.001 level (Student's two-tailed t test vs. control); n number of cells.
K+ currents are not affected by lipofectamine
As noted above, lipofectamine was required for detectable uptake of the AsODNs into P28 rat atrial myocytes. Control experiments were completed, therefore, to assess directly the effects of lipofectamine on the membrane properties of these cells. As illustrated in Fig. 3, the waveforms of (the outward and inward) K+ currents recorded under control conditions (Fig. 3A) and following 24 h incubation in the presence of (2-8 μg ml−1) lipofectamine (Fig. 3B) were indistinguishable. Analysis of results obtained in many experiments revealed no significant effect of lipofectamine on the normalized current density-voltage relations for peak outward K+ currents (Fig. 3C). The mean ±s.e.m. peak outward K+ current density at +30 mV, for example, was 39.2 ± 4.3 pA pF−1 (n = 23) in the absence and 37.8 ± 1.8 pA pF−1 (n = 25) in the presence of lipofectamine. Similarly, IK1 was unaffected by lipofectamine; the mean ±s.e.m.IK1 densities (at -120 mV) were 7.1 ± 0.7 pA pF−1 (n = 18) and 7.0 ± 0.6 pA pF−1 (n = 23) in the absence and the presence of lipofectamine, respectively.
Figure 3. Lipofectamine alone has no effects on outward or inward K+ currents in rat atrial myocytes.
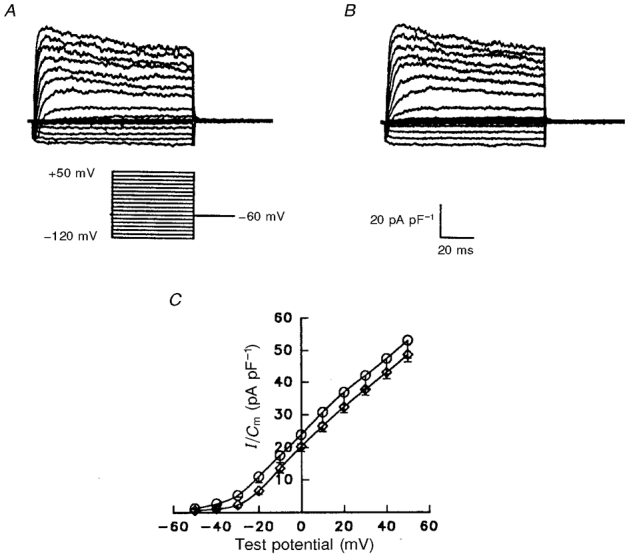
Representative normalized K+ currents recorded from P28 rat atrial cells in the absence (A) and presence (B) of lipofectamine are displayed. Currents were recorded as described in the legend to Fig. 1, except that the test potential range was -120 to +50 mV. C, mean ±s.e.m. peak current density-voltage relations in the absence (○, n = 23) and presence (⋄, n = 25) of lipofectamine are not significantly different (Student's t test).
Antisense oligodeoxynucleotides are Kv α subunit specific
Oligodeoxynucleotides with the phosphorothioate backbone are more resistant to degradation by nucleases than their O-linked counterparts, bind RNA efficiently and target this double-stranded RNA for degradation by RNase H (Davis, 1994). Nevertheless non-specific effects could result from the propensity of phosphorothioate AsODNs to bind to intracellular proteins and to components of the extracellular matrix (Chrisey et al. 1995; Krieg & Stein, 1995). To examine the specificity and the potential for cross-reactivity, the effects of AsODNs on heterologously expressed Kv α subunits were examined. Representative current recordings from HEK-293 cells expressing the various Kv α subunits in the presence and absence of AsODNs are presented in Figs 3 and 4, and the results of experiments completed on many cells are tabulated in Table 2.
Figure 4. Effects of AsODNs targeted against Kv4.3 and Kv4.2 are specific.
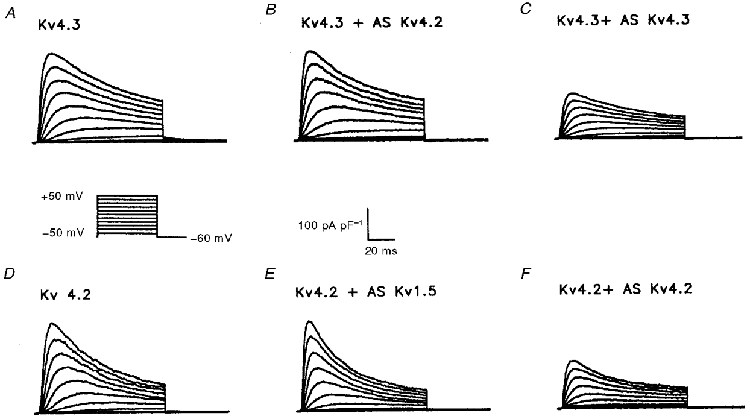
Representative normalized K+ currents recorded from HEK-293 cells expressing Kv4.3 (A) or Kv4.2 (D) are displayed; currents were recorded as described in the legend to Fig. 1. The Kv4.3-induced K+ currents (A) were decreased following incubation with the Kv4.3 AsODN (C), but not after exposure to the Kv4.2 AsODN (B). Similarly, the currents produced on expression of Kv4.2 (D) were significantly attenuated by the Kv4.2 AsODN (F), whereas the Kv1.5 AsODN did not have any measurable effects (E).
Table 2.
Effects of antisense oligodeoxynucleotides on heterologously expressed Kv α subunits
| Peak current density at +30 mV (pA pF−1) | ||||||
|---|---|---|---|---|---|---|
| Kv α subunit | No AsODN | Kv1.2 AsODN | Kv1. 5 AsODN | Kv2.1 AsODN | Kv4.2 AsODN | Kv4.3 AsOD |
| 1.2 | 28 ± 5 (14) | 7 ± 1 (8)* | 26 ± 6 (9) | n.d. | n.d. | n.d. |
| 1. 5 | 36 ± 10 (12) | 23 ± 8 (7) | 9 ± 1 (11)* | n.d. | n.d. | n.d. |
| 2.1 | 133 ± 35 (11) | n.d. | 128 ± 38 (9) | 19 ± 4 (10)* | n.d. | n.d. |
| 4.2 | 188 ± 33 (26) | n.d. | 180 ± 42 (9) | n.d. | 90 ± 15 (21)* | n.d. |
| 4.3 | 433 ± 101 (11) | n.d. | n.d. | n.d. | 374 ± 144 (7) | 84 ± 37 (9)* |
n. d., not determined.
Value significantly different from control at the P < 0.01 level (Student's two-tailed t test). The number of cells is given in parentheses.
As illustrated in Fig. 4A, rapidly activating and inactivating currents are evident in HEK-293 cells expressing Kv4.3. The amplitudes of the currents are markedly reduced following exposure to the Kv4.3 AsODN (Fig. 4C), whereas no significant differences between the Kv4.3-induced currents recorded under control conditions (Fig. 4A) and in the presence of the Kv4.2 AsODN (Fig. 4B) were observed (Table 2). Similarly, outward K+ current amplitudes in a stable Kv4.2-expressing HEK-293 cell line (Fig. 4D) were markedly reduced in the presence of the Kv4.2 AsODN (Fig. 4F), but not in the presence of the Kv1.5 AsODN (Fig. 4E). The mean ±s.e.m. peak outward current density (at +30 mV) in Kv4.2-expressing HEK-293 cells, for example, was reduced significantly (P < 0.01) from 188 ± 33 pA pF−1 (n = 26) to 90 ± 15 pA pF−1 (n = 21) in cells exposed to Kv4.2 AsODN (Table 2). Similar results were obtained for transient expression of Kv4.2 in QT-6 cells (a quail fibroblast cell line). The mean ±s.e.m. peak outward current density (at +30 mV) was reduced from 231 ± 31 pA pF−1 (n = 9) in control cells to 113 ± 26 pA pF−1 (n = 10) in cells exposed to the Kv4.2 AsODN and no effects on the Kv4.2-induced K+ currents were evident in cells exposed to the Kv4.3 AsODN (not shown). Similar specificity was seen for the AsODNs targeted against the translation start sites of Kv2.1, Kv1.2 and Kv1.5 (Fig. 5, Table 2).
Figure 5. The effects of AsODNs targeted against Kv2.1 and Kv1.2 are also subunit specific.
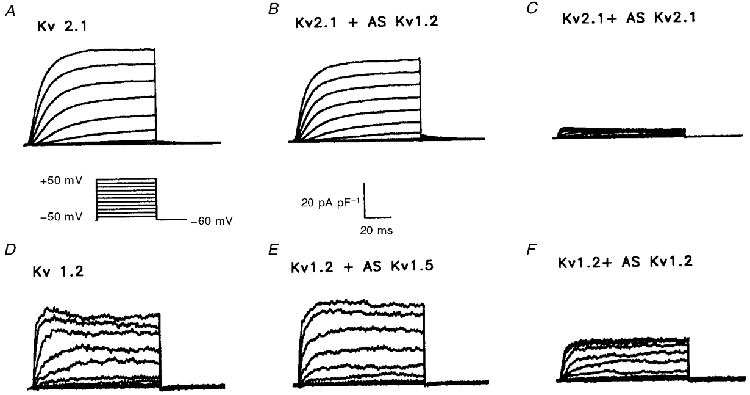
Representative normalized currents recorded from HEK-293 cells expressing Kv2.1 (A) and Kv1.2 (D) are shown. Currents were recorded as described in the legend to Fig. 1. The Kv2.1-induced K+ currents (A) were decreased following incubation with the Kv2.1 AsODN (C), but not after exposure to the Kv1.2 AsODN (B). The Kv1.2-induced currents (D) were significantly attenuated by the Kv1.2 AsODN (F), whereas the Kv1.5 AsODN did not have any measurable effect (E).
Effects of antisense oligodeoxynucleotides on rat atrial K+ currents
Having documented specificity, subsequent experiments were focused on examining the effects of the AsODNs on the depolarization-activated K+ currents in P28 rat atrial myocytes. Representative current waveforms evoked in response to 100 ms voltage steps to test potentials between -120 and +50 mV in control cells and in cells exposed to the various AsODNs are presented in Fig. 6. As is evident, the outward K+ currents are markedly reduced in cells exposed to the AsODNs targeted against Kv4.2, Kv1.2 and Kv1.5 (Fig. 6B-D). In contrast, outward K+ currents recorded in atrial myocytes treated with the Kv2.1, Kv4.3 and KvLQT1 AsODNs (Fig. 6E-G) appear indistinguishable from controls (Fig. 6A). To quantify the effects of the AsODNs, peak outward K+ currents recorded at each test potential were measured in individual cells and normalized to the whole-cell membrane capacitance (in the same cell); mean ±s.e.m. peak outward K+ current densities are plotted as a function of test potential in Fig. 7. These analyses revealed that in cells exposed to AsODNs targeted against Kv4.2, Kv1.2 and Kv1.5, mean ±s.e.m. peak outward K+ current densities are reduced significantly (P < 0.01; Student's t test) at all test voltages positive to -10 mV. In contrast, the mean ±s.e.m. peak outward K+ current densities in cells treated with the Kv2.1, Kv4.3 and KvLQT1 AsODNs are not significantly different from control K+ current densities (Fig. 7).
Figure 6. Peak outward K+ currents in atrial myocytes are reduced following exposure to Kv1.2, Kv4.2 and Kv1.5 AsODNs.
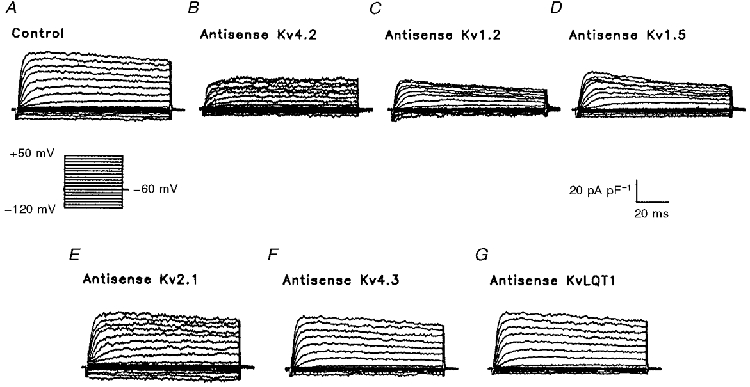
Representative normalized K+ currents, recorded from atrial myocytes following exposure to lipofectamine alone (A) or lipofectamine and one of the Kv α subunit AsODNs (B-G), are displayed. Currents were recorded as described in the legend to Fig. 2; the voltage clamp protocol is illustrated under record A. When compared with control records (A), peak outward K+ currents were decreased significantly in the presence of AsODNs targeted against Kv4.2 (B), Kv1.2 (C) and Kv1.5 (D), whereas the AsODNs targeted against Kv2.1 (E), Kv4.3 (F) and KvLQT1 (G) were without effect. No measurable effects of any of the AsODNs on IK1 were evident (see also Table 1).
Figure 7. Effect of AsODNs on the peak atrial outward K+ current density-voltage relations.
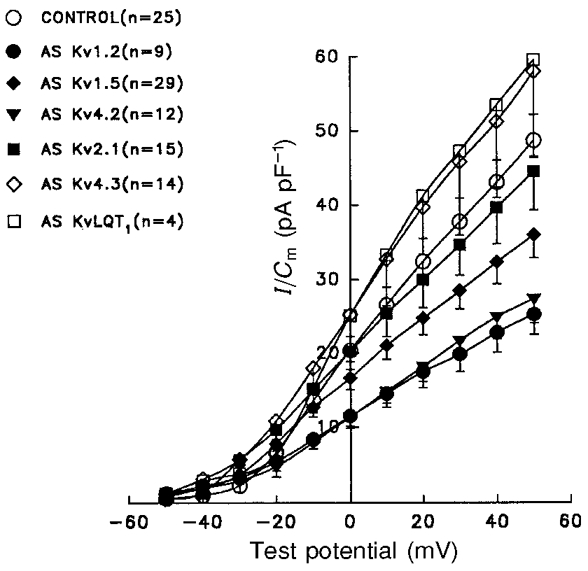
Outward currents, evoked during 100 ms depolarizing voltage steps to potentials from -50 to +50 mV from a holding potential of -60 mV, were recorded in P28 rat atrial myocytes under control conditions or following exposure to Kv α subunit AsODNs (see Fig. 5). Peak outward currents were measured in individual cells and normalized to the whole-cell membrane capacitance, Cm (determined in the same cell). Mean ±s.e.m. peak outward K+ currents densities are plotted here as a function of test potential. Peak outward K+ current densities were reduced significantly (P < 0.01) in cells exposed to the Kv1.2, Kv1.5 and Kv4.2 AsODNS, whereas no significant effects on the currents were evident in cells treated with Kv2.1, Kv4.3 or KvLQT1 AsODNs.
Table 1.
Lack of effects of antisense oligodeoxynucleotides on IK1
| Experiment | IK1 (pA pF−1)* | n |
|---|---|---|
| Control | 7.1 ± 0.7 | 18 |
| Lipofectamine | 7.0 ± 0.6 | 23 |
| Kv1.2 AsODN | 7.6 ± 0.9 | 9 |
| Kv1. 5 AsODN† | 7.4 ± 1.3 | 11 |
| Kv1. 5 AsODN‡ | 7.6 ± 1.0 | 6 |
| Kv2.1 AsODN | 8.3 ± 1.2 | 9 |
| Kv4.2 AsODN | 6.9 ± 1.0 | 10 |
| Kv4.3 AsODN | 8.7 ± 1.2 | 14 |
| KvLQT1 AsODN | 7.5 ± 1.8 | 5 |
Mean ± s.e.m. peak current density determined at −120 mV; n, number of cells.
AsODN against nucleotides 4–18 of rat Kv1. 5
AsODN against nucleotides 24–38 of rat Kv1. 5.
Selective effects of the Kv1.2, Kv4.2 and Kv1.5 AsODNs
The experiments described above revealed significant decreases in peak outward K+ current densities in P28 rat atrial myocytes exposed to AsODNs targeted against Kv1.2, Kv 4.2 and Kv1.5. Because three components of the total depolarization-activated K+ currents in rat atrial myocytes, IK,fast, IK,slow and Iss, have been distinguished (Boyle & Nerbonne, 1992), subsequent experiments were focused on determining if the effects of the Kv1.2, Kv1.5 and/or Kv4.2 AsODNs might be selective for IK,fast, IK,slow and/or Iss. For this purpose, outward K+ currents, evoked during long (10 s) depolarizing voltage steps, were recorded from control P28 rat atrial myocytes and from cells exposed to the various AsODNs. As illustrated in Fig. 2C, the time constants of inactivation (τdecay) of IK,fast and IK,slow do not vary measurably with voltage (Boyle & Nerbonne, 1992). Detailed analyses of the decay phases of the currents evoked during 10 s depolarizations, therefore, were completed for currents recorded at one test potential, +30 mV. Importantly, preliminary experiments revealed that exposure to AsODNs has no detectable effects on the kinetic properties of the K+ currents in rat atrial cells (see also below); only the amplitudes of the currents are reduced (Fig. 7).
Figure 8 illustrates representative outward K+ current waveforms evoked during 10 s depolarizations to +30 mV from a holding potential of -60 mV from a P28 control rat atrial myocyte (A) and from cells exposed to the Kv1.5 (A, *), Kv4.2 (B) or the Kv1.2 (C) AsODN; note that the currents were scaled to the same peak current amplitude to facilitate comparisons of the waveforms by eye. In Fig. 8D, the records in panels A, B and C are superimposed for comparison purposes. When this is done, the marked attenuation of the fast component of current decay, IK,fast, is clearly evident in the cell exposed to the Kv4.2 AsODN, whereas the relative amplitude of the slow component of current decay, IK,slow, is reduced in the cell exposed to the Kv1.2 AsODN (Fig. 8D) and the relative amplitude of the steady-state current, Iss, is selectively attenuated in the cells exposed to the Kv1.5 AsODN (Fig. 8A and D). Analysis of the current records in panel B revealed that neither the τdecay for IK,fast nor the τdecay for IK,slow is affected by the Kv4.2AsODN (Table 3). The amplitude (density) of IK,fast, however, is reduced significantly following exposure to the Kv4.2 AsODN; similar results were obtained in many experiments, and mean ±s.e.m.IK,fast density is significantly (P < 0.01) lower in cells exposed to the Kv4.2 AsODN compared with control cells (Table 3). In contrast, IK,slow and Iss densities are unaffected by the Kv4.2 AsODN (Table 3).
Figure 8. AsODNs targeted against Kv4.2, Kv1.2 and Kv1.5 attenuate different components of the outward K+ currents in atrial cells.
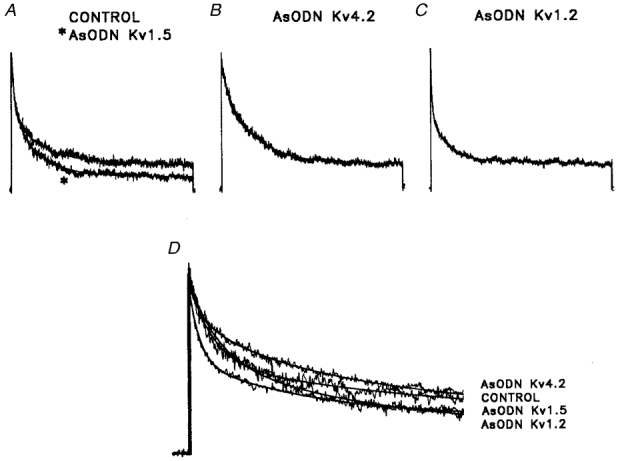
A-C, outward K+ currents, evoked during 10 s depolarizing voltage steps to +30 mV from a holding potential of -60 mV, were recorded from control atrial myocytes (A) and from cells exposed to the Kv1.5 (A, asterisk), Kv4.2 (B) and Kv1.2 (C) AsODNs. The records obtained from cells exposed to AsODNs (in A-C) were scaled to the peak amplitude of the current in the control cell (A) to facilitate comparison of the current waveforms. As is evident, the waveforms of the outward currents evoked during 10 s voltage steps in the presence of the Kv1.5 (A), Kv4.2 (B) and Kv1.2 (C) AsODNs are distinct from the control (A). To determine the amplitudes of the fast (IK,fast), the slow (IK,slow) and the steady-state (Iss) current components, the decay phases of the currents were fitted to the sum of two exponentials (see Methods). Representative fits (plotted as lines) to the decay phases of the currents in a control P28 atrial myocyte and in P28 atrial cells exposed to the Kv1.5, the Kv4.2 or the Kv1.2 AsODN are illustrated in D. Note that only the first 2 s of the (10 s) depolarizing voltage steps are illustrated for clarity, and (as in A-C) that the peak outward currents are scaled (to the control cell). The Kv1.5, Kv4.2 and Kv1.2 AsODNs did not affect the time constants of IK,fast or IK,slow decay; only the amplitudes of the currents were reduced. Similar experiments were completed on many cells, and mean ±s.e.m. normalized data are presented in Table 3.
Similar experiments were completed on cells exposed to the AsODNs targeted against Kv1.2 and Kv1.5, which also resulted in attenuation of the peak outward K+ currents in P28 rat atrial myocytes (Figs 6 and 7). Analysis of the waveforms of the depolarization-activated outward currents recorded from cells following exposure to the Kv1.2 and Kv1.5 AsODNs revealed effects quite distinct from those observed in cells exposed to the Kv4.2 AsODN (Fig. 8A and D, Table 3). Specifically, exposure to the Kv1.2 AsODN, selectively reduces IK,slow (Fig. 8C and D) without affecting IK,fast or Iss (Table 3), and exposure to the Kv1.5 AsODN affects only Iss (Fig. 8A and D, Table 3). Similar to the results with the Kv4.2 AsODN, there were no detectable effects of the Kv1.2 or Kv1.5 AsODNs on the kinetic properties of the currents, only IK,slow and Iss amplitudes (densities) were reduced following AsODN treatment (Table 3).
DISCUSSION
Effects of AsODNs on atrial K+ currents
The results of the experiments presented here demonstrate that AsODNs targeted against three of the Kv α subunits expressed in rat heart Kv1.2, Kv1.5 and Kv4.2 (Roberds & Tamkun, 1991; Dixon & McKinnon, 1994; Barry et al. 1995; Xu et al. 1996) attenuate peak outward K+ current amplitudes recorded in isolated P28 rat atrial myocytes. Although current amplitudes (densities) in control and AsODN-treated P28 rat atrial myocytes vary considerably among cells, on average, peak outward current densities were reduced approximately 30% in cells exposed to the AsODNs (Table 3). The magnitude of the Kv1.2, Kv1.5 and Kv4.2 AsODN effects observed here are qualitatively similar to those reported previously in studies documenting the effects of AsODNs targeted against Kv1.5 and Kir2.1 on potassium currents in human atrial (Feng et al. 1997) and rat ventricular (Nakamura et al. 1998) myocytes.
In contrast to the effects of the Kv1.2, Kv1.5 and Kv4.2 AsODNs, no significant effects on outward K+ currents were observed in atrial cells exposed to AsODNs targeted against three other endogenous Kv α subunits, Kv2.1, Kv4.3 or KvLQT1 (Roberds & Tamkun, 1991; Dixon & McKinnon, 1994; Barry et al. 1995; Dixon et al. 1996; Xu et al. 1996; Takimoto et al. 1997). Importantly, control experiments, completed on heterologously expressed Kv α subunits, revealed that each AsODN was selective for the Kv α subunits against which it was targeted; no cross-reactivity was detected. In addition, the inwardly rectifying K+ current in rat atrial myocytes, IK1, was not affected by any of the Kv α subunit AsODNs. Taken together, these results suggest that Kv1.2, Kv1.5 and Kv4.2 contribute to the formation of functional depolarization-activated K+ channels in adult rat atrial myocytes, whereas Kv2.1, Kv4.3 and KvLQT1 do not. It is certainly possible, however, that other, yet to be identified, pore-forming Kv α subunits also contribute to the formation of functional voltage-gated K+ channels in rat atrial cells. Accessory β subunits may also play a role; further experiments will be necessary to test this hypothesis directly.
Separation of the three components of the total depolarization-activated outward K+ currents in adult rat atrial myocytes, IK,fast, IK,slow and Iss (Boyle & Nerbonne, 1992), further revealed that the effects of the Kv1.2, Kv1.5 and Kv4.2 AsODNs are distinct. Specifically, the AsODN targeted against Kv4.2 significantly (P < 0.01) attenuated IK,fast without affecting either IK,slow or Iss, whereas exposure to the Kv1.2 and Kv1.5 AsODNs significantly (P < 0.01, P < 0.001 respectively) reduced IK,slow and Iss, respectively, and had no detectable effects on IK,fast (Table 2). These results demonstrate that the three K+ conductance pathways in rat atrial myocytes, IK,fast, IK,slow and Iss, separated previously based on differences in inactivation and recovery kinetics (Boyle & Nerbonne, 1992), are distinct molecular entities. Interestingly, the finding that the Kv1.2 AsODN attenuates IK,slow selectively, whereas the Kv1.5 AsODNs is specific for Iss, suggests that Kv1.2 and Kv1.5 do not coassemble to form heteromultimeric K+ channels in rat atria in vivo. Similarly, the fact that the Kv4.2 AsODN attenuates IK,fast, whereas the Kv4.3 AsODN does not, suggests that these two subunits are also not associated in rat atria in vivo.
Relationship to previous studies on the molecular correlates of Ito
Previous studies, focused on examining Kv α subunit mRNA and protein expression levels led to suggestions that Kv α subunits of the Kv4 subfamily, Kv4.2 and/or Kv4.3, underlie Ito in ventricular cells (Dixon & McKinnon, 1994; Barry et al. 1995; Dixon et al. 1996), and considerable evidence has accumulated recently in support of this hypothesis. Using an adenoviral construct encoding a truncated Kv4.2 subunit (Kv4.2ST) that functions as a dominant negative, for example, Johns and coworkers (1997) reported that Ito in rat ventricular myocytes is selectively attenuated. A pore mutant of Kv4.2 (Kv4.2W362F), which also functions as a dominant negative, has been expressed in transgenic mice and shown to result in the functional knockout of Ito (now referred to as Ito, fast or Ito,f; Xu et al. 1999) in ventricular myocytes, increased action potential durations, and prolongation of the QT interval (Barry et al. 1998). In addition, reductions in Ito density are observed in rat ventricular myocytes exposed to AsODNs targeted against either Kv4.2 or Kv4.3 (Fiset et al. 1997). These, and similar results reported by Xu et al. (1997), suggest that both Kv4.2 and Kv4.3 contribute to rat ventricular Ito. The findings presented here, however, demonstrate that Kv4.2 contributes to IK,fast in rat atrial myocytes, whereas Kv4.3 does not. These differences in subunit composition probably underlie the distinct kinetic properties of rat atrial IK,fast and rat ventricular Ito (Apkon & Nerbonne, 1991; Boyle & Nerbonne, 1992). Nevertheless, the similarities in the properties and now in the molecular correlates of the transient outward K+ currents in rat atrial and ventricular myocytes suggest that IK,fast (Boyle & Nerbonne, 1992) would more appropriately be called (rat atrial) Ito (Nerbonne, 1998).
The studies presented here and others noted above clearly demonstrate that members of the Kv4 subfamily underlie Ito (Ito,f) in mouse and rat heart. Nevertheless, it certainly seems possible that other Kv α subunits contribute to the transient outward currents in other cell types and/or species. The properties of Ito in rabbit heart, for example, are distinct from those of Ito in mouse and rat heart (as well as Ito in other species) in that inactivation is slow and biexponential, and recovery (from steady-state inactivation) is very slow, proceeding with time constants in the range of 5-8 s (Clark et al. 1988; Giles & Imaizumi, 1988; Fermini et al. 1992). Recently, the presence of a slowly inactivating and slowly recovering (from steady-state inactivation) transient outward current was identified in cells isolated from the mouse left ventricular septum (Xu et al. 1999). This current was referred to as Ito,slow (or Ito,s) to distinguish it from the rapidly inactivating and inactivating transient outward current (Ito,fast or Ito,f), that is prominent in cells isolated from the apex (Xu et al. 1999) and is eliminated in all ventricular myocytes isolated from Kv4.2W362F-expressing transgenic mice (see above and Barry et al. 1998). Interestingly, the properties of Ito in rabbit myocytes and of Ito,s in mouse septal cells are similar to those of heterologously expressed Kv1.4 (Tseng-Crank et al. 1990; Petersen & Nerbonne, 1999), suggesting the intriguing possibility that Kv1.4 does play a role in the generation of Ito in rabbit and of Ito,s in mouse. Recently, it was also reported that the time- and voltage-dependent properties of Ito in ferret left ventricular epicardial and endocardial myocytes are distinct (Brahmajothi et al. 1998). In addition, in situ hybridization and immunohistochemical data reveal regional differences in the expression of Kv1.4 and Kv4.2/Kv4.3 in ferret heart, suggesting that Kv1.4 and Kv4.2/Kv4.3 underlie Ito in ferret left ventricular endocardial and epicardial myocytes, respectively (Brahmajothi et al. 1998). It has also been suggested that Kv1.4 underlies Ito in early postnatal (day 1-3) rat ventricular myocytes (Wickenden et al. 1997). Experiments aimed at determining directly if Kv1.4 contributes to the formation of ‘slow’Ito channels will clearly be of interest.
Relationship to previous studies on the molecular correlates of IK
Heterologous expression of Kv1.5 yields rapidly activating, non-inactivating K+ currents that are similar to rat atrial Iss (Boyle & Nerbonne, 1992; Van Wagoner et al. 1996). A similar current, IKur (for ultrarapid) has been described in human and canine atrial myocytes (Wang et al. 1993; Yue et al. 1996). In rat and human heart, Kv1.5 is expressed at the message (Tamkun et al. 1991; Roberds & Tankun, 1991; Dixon & McKinnon 1994) and the protein (Barry et al. 1995; Mays et al. 1995) levels, leading to the suggestion that Kv1.5 underlies Iss and IKur (Fedida et al. 1993; Wang et al. 1993; Barry et al. 1995; Mays et al. 1995; Van Wagoner et al. 1996; Yue et al. 1996). Direct support for a role for Kv1.5 was provided with the demonstration that IKur is reduced in human atrial cells exposed to Kv1.5 AsODNs (Feng et al. 1997). The results presented here reveal that rat atrial Iss is selectively attenuated by Kv1.5 AsODNs, consistent with the hypothesis that rat atrial Iss and human/canine atrial IKur reflect the same conductance pathway (Barry & Nerbonne, 1996). We suggest, therefore, that for simplicity, rat atrial Iss also be referred to as IKur.
Several recent studies have also provided important insights into the molecular identities of several other components of delayed rectification in the mammalian heart, including IKr and IKs. Heterologous expression of HERG, which has been identified as the locus of one form of long QT syndrome, LQT2 (Curran et al. 1995), for example, reveals K+-selective channels that are similar to the rapid component of cardiac delayed rectification, IKr (Sanguinetti et al. 1995; Trudeau et al. 1995). Another K+ channel α subunit, KvLQT1, has been identified as the locus of mutations leading to LQT1 (Wang et al. 1996). Coexpression of KvLQT1 with IminK produces slowly activating K+ currents that are similar to the slow component of delayed rectification in the heart, IKs (Barhanin et al. 1996; Sanguinetti et al. 1996), suggesting that functional IKs channels are heteromeric, comprising the protein products of KvLQT1 and IminK. The experiments completed here, however, reveal that KvLQT1 does not contribute to rat atrial IK,slow channels. Rather, the results demonstrate that Kv1.2 underlies rat atrial IK,slow. There is no relationship, therefore, between rat atrial IK,slow and IKs in other cells (Barry & Nerbonne, 1996). It will be of interest to determine if currents similar to rat atrial IK,slow are expressed in atrial (or other) cells in other species and if Kv1.2 also plays a role in the generation of these currents.
Acknowledgments
The authors thank Sacha Malin for kindly providing the Kv4.2-expressing HEK-293 cell line, and Dr Haodong Xu for many helpful discussions and suggestions throughout the course of this work. In addition, the financial support provided by the National Institutes of Health (R01 HL 34161 to J. M. N.) and the American Heart Association (National Affiliate Grant in Aid to J. M. N. and Missouri Affiliate Postdoctoral Fellowship to E. B.) is gratefully acknowledged.
References
- Apkon M, Nerbonne JM. Characterization of two distinct depolarization-activated K+currents in isolated adult rat ventricular myocytes. Journal of General Physiology. 1991;97:973–1011. doi: 10.1085/jgp.97.5.973. 10.1085/jgp.97.5.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(v)LQT1 and IsK (minK) proteins associate to form the I-Ks cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Barry DM, Nerbonne JM. Myocardial potassium channels: Electrophysiological and molecular diversity. Annual Review of Physiology. 1996;58:363–394. doi: 10.1146/annurev.ph.58.030196.002051. [DOI] [PubMed] [Google Scholar]
- Barry DM, Trimmer JS, Merlie JP, Nerbonne JM. Differential expression of voltage-gated potassium channel subunits in adult rat heart: relation to functional potassium channels? Circulation Research. 1995;77:361–369. doi: 10.1161/01.res.77.2.361. [DOI] [PubMed] [Google Scholar]
- Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, Long QT syndrome and cardiac remodelling in mice expressing a dominant negative Kv4 α subunit. Circulation Research. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- Bennett PB, Snyders DJ, Tamkun MM. Molecular and functional diversity of cloned potassium channels. Cardiovascular Drugs and Therapy. 1993;7:585–592. doi: 10.1007/BF00877624. [DOI] [PubMed] [Google Scholar]
- Boyle WA, Nerbonne JM. Two functionally distinct 4-aminopyridine sensitive outward potassium currents in rat atrial myocytes. Journal of General Physiology. 1992;100:1041–1067. doi: 10.1085/jgp.100.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmajothi MV, Campbell DL, Rasmusson RL, Morales MJ, Nerbonne JM, Trimmer JS, Strauss HC. Biophysical and immunolocalization analysis of two distinct Ito phenotypes in ferret left ventricular epicardial and endocardial myocytes. Biophysical Journal. 1998;74:A209. abstract 10.1016/S0301-4622(98)00180-X. [Google Scholar]
- Brown AM. Cardiac potassium channels in health and disease. Trends in Cardiovascular Medicine. 1997;7:118–124. doi: 10.1016/S1050-1738(97)00002-9. [DOI] [PubMed] [Google Scholar]
- Chrisey LA, Parizendeh M, Liss HS. Nonsequence-specific inhibition of bacterial luminescence by phosphorothioate oligodeoxyribonucleotides. Antisense Research and Development. 1995;5:261–269. doi: 10.1089/ard.1995.5.261. [DOI] [PubMed] [Google Scholar]
- Clark RB, Giles WR, Imaizumi Y. Properties of the transient outward current in rabbit atrial cells. The Journal of Physiology. 1988;405:147–168. doi: 10.1113/jphysiol.1988.sp017326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: herg mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- Davis AR. Current potential of antisense oligodeoxynucleotide as therapeutic drugs. Trends in Cardiovascular Medicine. 1994;4:51–55. doi: 10.1016/1050-1738(94)90009-4. 10.1016/1050-1738(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Deal KK, England SK, Tamkun MM. Molecular physiology of cardiac potassium channels. Physiological Reviews. 1996;76:49–67. doi: 10.1152/physrev.1996.76.1.49. [DOI] [PubMed] [Google Scholar]
- Dixon JE, McKinnon D. Quantitative analysis of mRNA expression in atrial and ventricular muscles of rats. Circulation Research. 1994;75:252–260. doi: 10.1161/01.res.75.2.252. [DOI] [PubMed] [Google Scholar]
- Dixon JE, Shi WS, Wang HS, McDonald C, Yu H, Wymore RS, Cohen IS, McKinnon D. Role of the Kv4.3 potassium channel in ventricular muscle - a molecular correlate for the transient outward current. Circulation Research. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- Fedida D, Wible B, Wang Z, Fernini B, Faust F, Nattel S, Brown AM. Identity of a novel delayed rectifier current from human heart with a cloned potassium channel current. Circulation Research. 1993;73:210–216. doi: 10.1161/01.res.73.1.210. [DOI] [PubMed] [Google Scholar]
- Feng JL, Fermini B, Nattel S. Properties of sodium and potassium currents of cultured adult human atrial myocytes. American Journal of Physiology. 1996;270:H1676–1686. doi: 10.1152/ajpheart.1996.270.5.H1676. [DOI] [PubMed] [Google Scholar]
- Feng JL, Wible B, Li GR, Wang ZG, Nattel S. Antisense oligodeoxynucleotides directed against Kv 1.5 mRNA specifically inhibit ultrarapid delayed rectifier potassium current in cultured adult human atrial myocytes. Circulation Research. 1997;80:572–579. doi: 10.1161/01.res.80.4.572. [DOI] [PubMed] [Google Scholar]
- Fermini B, Wang Z, Duan D, Nattel S. Differences in rate dependence of the transient outward current in rabbit and human atrium. American Journal of Physiology. 1992;263:H1747–1754. doi: 10.1152/ajpheart.1992.263.6.H1747. [DOI] [PubMed] [Google Scholar]
- Fiset C, Clark RB, Shimoni Y, Giles WR. Shal-type channels contribute to the Ca2+-independent transient outward potassium current in rat ventricle. The Journal of Physiology. 1997;500:51–64. doi: 10.1113/jphysiol.1997.sp021998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles WR, Imaizumi Y. Comparison of potassium currents in rabbit atrial and ventricular cells. The Journal of Physiology. 1988;405:123–145. doi: 10.1113/jphysiol.1988.sp017325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Johns DC, Nuss HB, Marban E. Suppression of neuronal and cardiac transient outward currents by viral gene transfer of dominant negative Kv4.2 constructs. Journal of Biological Chemistry. 1997;272:31598–31603. doi: 10.1074/jbc.272.50.31598. [DOI] [PubMed] [Google Scholar]
- Krieg AM, Stein CA. Phosphorothioate oligodeoxynucleotides: antisense or antiprotein? Antisense Research and Development. 1995;5:241. doi: 10.1089/ard.1995.5.241. editorial. [DOI] [PubMed] [Google Scholar]
- Mays DJ, Foose JM, Philipson LH, Tamkun MM. Localization of the Kv1.5 potassium channel protein in explanted cardiac tissue. Journal of Clinical Investigations. 1995;96:282–292. doi: 10.1172/JCI118032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TY, Artman M, Rudy B, Coetzee WA. Inhibition of rat ventricular IK1 with antisense oligonucleotides targeted to Kir2.1 mRNA. American Journal of Physiology. 1998;274:H892–900. doi: 10.1152/ajpheart.1998.274.3.H892. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM. Regulation of voltage-gated K+ channel expression in the developing mammalian myocardium. Journal of Neurobiology. 1998;37:37–59. doi: 10.1002/(sici)1097-4695(199810)37:1<37::aid-neu4>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Petersen KR, Nerbonne JM. Expression environment determines K+ currents properties: Kv 1 and Kv 4 α subunit-induced K+ currents in mammalian cell lines and cardiac myocytes. Pflügers Archiv. 1999;437:381–392. doi: 10.1007/s004240050792. [DOI] [PubMed] [Google Scholar]
- Qin D, Zhang Z-H, Caref EB, Boutjdir M, Jain P, El-Sherif N. Cellular and ionic basis of arrhythmias in postinfarction remodeled ventricular myocardium. Circulation Research. 1996;79:461–473. doi: 10.1161/01.res.79.3.461. [DOI] [PubMed] [Google Scholar]
- Roberds SL, Tamkun MM. Cloning and tissue-specific expression of five voltage-gated potassium channel cDNAs expressed in rat heart. Proceedings of the National Academy of Sciences of the USA. 1991;88:1798–1802. doi: 10.1073/pnas.88.5.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, George AL. Structure and function of cardiac sodium and potassium channels. American Journal of Physiology. 1997;42:H511–525. doi: 10.1152/ajpheart.1997.273.2.H511. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(v)LQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Snyder DJ, Tamkun MM, Bennett PB. A rapidly activating and slowly inactivating potassium channel cloned from human heart. Journal of General Physiology. 1993;101:513–543. doi: 10.1085/jgp.101.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splanski I, Tristani-Firouzi M, Lehman MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nature Genetics. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- Takimoto K, Li D, Hershman KM, Li P, Jackson EK, Levitan ES. Decreased expression of Kv4.2 and novel Kv4.3 potassium channel subunit mRNAs in ventricles of Renovascular hypertensive rats. Circulation Research. 1997;81:533–539. doi: 10.1161/01.res.81.4.533. [DOI] [PubMed] [Google Scholar]
- Tamkun MM, Knoth KM, Walbridge JA, Kroemer H, Roden DM, Glover DM. Molecular cloning and characterization of two voltage-gated K+ channel cDNAs from human ventricle. FASEB Journal. 1991;5:331–337. doi: 10.1096/fasebj.5.3.2001794. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetsky B, Robertson GA. H-erg, a human inward rectifier with structural and functional homology to voltage-gated potassium channels. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Tseng-Crank JCL, Tseng GN, Schwartz A, Tanouye MA. Molecular cloning and functional expression of a potassium channel cDNA isolated from a rat cardiac library. FEBS Letters. 1990;268:63–68. doi: 10.1016/0014-5793(90)80973-m. [DOI] [PubMed] [Google Scholar]
- Van Wagoner DR, Kirian M, Lamorgese M. Phenylephrine suppresses outward potassium currents in rat atrial myocytes. American Journal of Physiology. 1996;271:H937–946. doi: 10.1152/ajpheart.1996.271.3.H937. [DOI] [PubMed] [Google Scholar]
- Wang K, Curran ME, Splawski I, Burn TC, Millholland JM, Vanray TJ, Shen J, Timothy KW, Vincent GM, Dejager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KvLQT1 mutations cause cardiac arrhythmias. Nature Genetics. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes: evidence for a novel delayed rectifier potassium current similar to Kv1.5 cloned channel currents. Circulation Research. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Kaprielan R, Parker TG, Jones OT, Backx PH. Effects of development and thyroid hormone on K+ currents and K+ channel gene expression in rat ventricle. The Journal of Physiology. 1997;504:271–286. doi: 10.1111/j.1469-7793.1997.271be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HD, Barry DM, Nerbonne JM. Molecular correlates of functional Ito and IK channels in rat ventricular myocytes probed with potassium channel toxins and antisense oligodeoxynucleotides. Circulation. 1997;96:I-421. suppl. [Google Scholar]
- Xu H, Dixon JE, Barry DM, Trimmer JS, Merlie JP, McKinnon D, Nerbonne JM. Developmental analysis reveals mismatches in the expression of K+channel α subunits and voltage-gated K+ channel currents in rat ventricular myocytes. Journal of General Physiology. 1996;108:405–419. doi: 10.1085/jgp.108.5.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Guo W, Nerbonne JM. Four kinetically-distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. Journal of General Physiology. 1999;113 doi: 10.1085/jgp.113.5.661. in the Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue LX, Feng JL, Li GR, Nattel S. Characterization of an ultrarapid delayed rectifier potassium channel involved in canine atrial repolarization. The Journal of Physiology. 1996;496:647–662. doi: 10.1113/jphysiol.1996.sp021716. [DOI] [PMC free article] [PubMed] [Google Scholar]