

Abstract
The β subunits of voltage-sensitive calcium channels facilitate the incorporation of channels into the plasma membrane and modulate calcium currents. In order to determine whether these two effects of the β subunit are interdependent or independent of each other we studied plasma membrane incorporation of the channel subunits with green fluorescent protein and immunofluorescence labelling, and current modulation with whole-cell and single-channel patch-clamp recordings in transiently transfected human embryonic kidney tsA201 cells.
Coexpression of rabbit cardiac muscle α1C with rabbit skeletal muscle β1a, rabbit heart/brain β2a or rat brain β3 subunits resulted in the colocalization of α1C with β and in a marked translocation of the channel complexes into the plasma membrane. In parallel, the whole-cell current density and single-channel open probability were increased. Furthermore, the β2a isoform specifically altered the voltage dependence of current activation and the inactivation kinetics.
A single amino acid substitution in the β subunit interaction domain of α1C (α1CY467S) disrupted the colocalization and plasma membrane targeting of both subunits without affecting the β subunit-induced modulation of whole-cell currents and single-channel properties.
These results show that the modulation of calcium currents by β subunits can be explained by β subunit-induced changes of single-channel properties, but the formation of stable α1C-β complexes and their increased incorporation into the plasma membrane appear not to be necessary for functional modulation.
Voltage-sensitive calcium channels are multimeric protein complexes formed by the α1 subunit and the auxiliary subunits α2δ, β and γ (Leung et al. 1987; Takahashi et al. 1987; Vaghy et al. 1987). The α1 subunit by itself shows the characteristic properties of a voltage-gated ion channel, i.e. voltage sensing, ion permeation and drug binding. The physiological roles of the auxiliary subunits are currently the subject of intensive investigations. The β subunit modulates calcium currents by increasing the current density and by changing the current kinetics when coexpressed in heterologous expression systems (Lacerda et al. 1991; Varadi et al. 1991; Lory et al. 1992). Furthermore, it has been suggested that the β subunit is involved in the targeting of the α1 subunit to the plasma membrane (Chien et al. 1995; Gregg et al. 1996). Both the α1 and β subunits exist in multiple tissue-specific isoforms, which differ from one another in their primary structure and in their functional properties (Birnbaumer et al. 1994; Isom et al. 1994). The skeletal muscle α1S isoform, for example, shows slow activation and inactivation kinetics compared with the other α1 isoforms. Also, whereas α1C expressed in heterologous expression systems exhibits currents even in the absence of auxiliary subunits (Perez-Garcia et al. 1995), expression of α1S in heterologous systems rarely gives rise to measurable calcium currents (Johnson et al. 1997). The β subunit isoforms differ in their current modulation (Hullin et al. 1992; Sather et al. 1993; Parent et al. 1997) and, when expressed alone, in their subcellular distribution (Chien et al. 1995, 1996; Brice et al. 1997). For example, β2a drastically reduced the speed of inactivation when coexpressed with the neuronal α1E subunit in oocytes (Parent et al. 1997), whereas other β subunit isoforms showed only minor effects on current inactivation. Analogously, β2a differs from most other β isoforms in that it was localized in the plasma membrane when expressed without an α1 subunit in a heterologous expression system (Chien et al. 1995), whereas β1a, β3 and β4 showed a cytoplasmic localization (Brice et al. 1997; Neuhuber et al. 1998b).
A conserved β subunit binding motif has been identified in the cytoplasmic loop between repeats I and II of α1S, α1A, α1B and α1C (Pragnell et al. 1994). Point mutations within this binding motif perturbed α1-β binding and affected calcium current properties when the neuronal α1A isoform was coexpressed with β1b in oocytes. A point mutation (Y366S) within the β subunit binding motif in the I-II linker of the skeletal muscle α1S resulted in the expected loss of α1S-β1a binding, but the probability that tsA201 human embryonic kidney cells cotransfected with α1SY366S and β1a exhibited calcium currents was still increased by the β1a subunit (Neuhuber et al. 1998b). This indicates that stable binding of β1a to the known motif in the I-II linker of α1S is not necessary for the β subunit to increase the frequency of current expression. Thus, association of β with this interaction domain in the cytoplasmic I-II linker plays an important role in β subunit-dependent modulation of calcium currents, but other mechanisms for α1-β interaction may exist. De Waard et al. (1994, 1996) identified a conserved 30 amino acid domain in the β subunit that is complementary to the binding site in the I-II linker on α1A and is involved in the interaction with this subunit. Mutations within this domain of β perturbed binding to α1A and affected modulation of calcium current properties. However, this domain in the β subunit cannot account for all observed modulatory effects of α1A-β interactions, since certain truncated β subunits were only affected in their modulation of inactivation kinetics, not in current stimulation. Therefore, a region of the β subunit other than that interacting with the I-II linker of α1 may be involved in the modulation of α1A (De Waard et al. 1994). Moreover, using chimeras of different β isoforms Olcese et al. (1994) and Qin et al. (1996) have found that regulation of activation and inactivation of α1E channels are two separable functions of the β subunit, suggesting the existence of two separate interaction domains on each of the subunits. Indeed, Tareilus et al. (1997) identified a second β subunit binding domain within the last 277 amino acids of the C-terminus of α1E, and Walker et al. (1998) identified a low affinity binding site in the carboxy-terminal region of α1A that accounts for β4-induced modulation of current inactivation.
Despite this progress in understanding the function of the calcium channel β subunit the mechanism of current modulation by β remains largely unresolved. For example, it is still controversial whether increased insertion of channels into the plasma membrane and modulation of current properties are two independent functions of the β subunit or whether the latter is a direct result of the former. Also, we do not know whether α1 and β form a stable complex in which β serves as a necessary cofactor or mediator of modulatory signals, or whether association and dissociation of the β subunit in itself is the modulatory mechanism. To address these questions we studied the interactions of three different β subunit isoforms with α1C and an α1C mutant with a single amino acid substitution in the β subunit interaction domain of the I-II linker (α1CY467S) using a combination of structural and functional techniques. This approach allowed us to distinguish β isoform-specific effects from common effects of α1C-β interactions and to demonstrate that increased membrane incorporation and modulation of channel properties are two independent effects of β subunits. Single-channel analysis showed that changes in the open probability are sufficiently large to explain the increase in whole-cell current density observed upon coexpression of the β subunit. Further, the comparison of β subunit effects on wild-type and mutant α1C shows that this increase in current density occurs even without the formation of stable α1C-β complexes or their increased incorporation into the plasma membrane.
METHODS
Cell lines
tsA201 cells, a HEK cell subclone stably transfected with the SV40 large T-antigen, were plated and allowed to proliferate in F12 medium (Gibco BRL, Vienna, Austria) containing 10% fetal bovine serum. Cells were grown to 80% confluency before passaging. For structural analysis (green fluorescent protein (GFP) and immunocytochemistry), cells were plated at dilutions of about 1:10 onto poly-L-lysine-coated 13 mm round coverslips and transfected on the following day (see below) when cells reached 30% confluency. For patch-clamp analysis cells were plated on 35 mm culture dishes at a dilution of 1:10. After transfection at about 50% confluency, cells were replated onto poly-L-lysine-coated 25 mm round coverslips at dilutions between 1:5 and 1:10 to get isolated cells for patch-clamp recordings on the following day.
Transfection
Transfections were carried out with a liposomal transfection reagent (DOTAP, Boehringer Mannheim) according to the manufacturer's instructions. The total amount of DNA used per 35 mm culture dish was 10 μg, 5-8 μg of which was specific DNA (expression plasmids encoding α1C constructs, β subunit and GFP), with the rest made up with inert DNA (pUC18; Norrander et al. 1985). In co-transfection experiments two or more expression plasmids were combined at equimolar concentrations. This resulted in coexpression of β with any α1C construct in approximately 70% of β subunit-transfected cells. The liposome-DNA mixture was diluted in 1.5 ml F12 medium and then added to the culture. On the following day the cells were processed for immunocytochemistry or replated for patch-clamp analysis.
Expression plasmids
Details of the expression plasmids are given in Table 1. The coding sequence of rabbit cardiac muscle α1C DNA (Mikami et al. 1989) with an alternative exon in IVS3 (Koch et al. 1990) was inserted into the expression plasmid pcDNA3 (Invitrogen, San Diego, CA, USA) via HindIII and NotIdigestion. The tyrosine to serine substitution in position 467 in α1C (α1CY467S) was introduced by site-directed mutagenesis of α1C-pcDNA3 using the splicing by overlap extension technique. The following mutagenic sense primer was used:
Table 1.
Expression plasmids
| Name | Vector | Insert | Reference |
|---|---|---|---|
| α1C | pcDNA3 | Rabbit cardiac muscle α1 | Mikami et al. 1989; Koch et al. 1990 |
| GFP-α1C | pcDNA3 | GFP-α1C fusion protein | Grabner et al. 1998 |
| α1CY467S | pcDNA3 | Cardiac muscle α1C Y to S substitution | Present study |
| β1a | pcDNA3 | Rabbit skeletal muscle β | Ruth et al. 1989 |
| β1a-GFP | pcDNA3 | β1a-GFP fusion protein | Neuhuber et al. 1998b |
| β2a | pCMV6 | Rabbit heart/brain β | Perez-Reyes et al. 1992 |
| β3 | pCMV6 | Rat brain β | Castellano et al. 1993 |
| GFP(S65T) | pRK5 | Green fluorescent protein | Heim et al. 1995 |
| pUC18 | pUC18 | — | Norrander et al. 1985 |
To facilitate the identification of positive clones, a PvuII site (nucleotide 1376) was eliminated in the mutagenic primers by silent mutation. The mutated α1C fragment was inserted into α1C-pcDNA3 after digestion with BamHI (nucleotide 1263) and EcoRI (nucleotide 2213). The mutation was verified by sequence analysis. In addition, a GFP-α1C fusion protein (Grabner et al. 1998) was used for double fluorescence labelling. The subcellular distribution and the electrophysiological properties of GFP-α1C and α1C were identical.
GFP and immunofluorescence labelling
Paraformaldehyde-fixed cultures were immuno-stained as previously described (Flucher et al. 1993). For double labelling with GFP, Texas Red-conjugated antibodies (Jackson Immuno Research, West Grove, PA, USA) were used to exclude bleed-through between the red and the green channels. Working dilutions and the sources of primary antibodies are listed in Table 2. Samples were evaluated on a Zeiss Axiovert microscope with epifluorescence and phase-contrast optics and documented on 35 mm high speed black and white film. Controls, such as the omission of primary antibodies and incubation with inappropriate antibodies, were routinely performed. Localization of α1C and GFP-α1C, or β1a and β1a-GFP gave the same results with respect to distribution patterns in all examined conditions.
Table 2.
Antibodies
| Specificity | Code | Type | Dilution | Reference |
|---|---|---|---|---|
| DHP-receptor, α1C | CNC | Affinity purified, rabbit | 1:1500 | Safayhi et al. 1997 |
| DHP-receptor, β | βcom | Affinity purified, rabbit | 1:1200 | Pichler et al. 1997; Neuhuber et al. 1998b. |
Patch-clamp recording
Whole-cell and single-channel recordings were performed as described by Hamill et al. (1981). Cultures grown on 25 mm round coverslips were mounted in a recording chamber and viewed with a × 16 phase-contrast multi-immersion lens on a Zeiss Axiovert microscope. Fluorescent cells (indicating successful transfection with β1a-GFP or GFP) were selected for recording. An Axopatch 200A patch-clamp amplifier controlled by the software pCLAMP 6.0 (Axon Instruments) was used for all recordings.
Whole-cell recordings
The bath solution contained (mM): 40 BaCl2, 100 TEA-Cl and 10 Hepes (adjusted to pH 7.4 using TEA-OH). Patch pipettes, pulled from borosilicate glass and fire polished, were filled with (mM): 130 caesium aspartate, 10 Hepes, 2 Mg-ATP, 2 Cs-EGTA and 0.5 MgCl2 (adjusted to pH 7.4 with CsOH). Resistances of the patch pipettes were between 4 and 7 MΩ. Capacitative currents were compensated using built-in analog circuits (series resistance error was corrected for 80%), and the value for the whole-cell capacity was determined from this adjustment of the amplifier and used to calculate the current density. Leak resistance in the cell-attached mode was normally larger than 8 GΩ. Current data were low-pass Bessel filtered at 2 kHz and sampled at 1 kHz with an IBM compatible PC. In the electrophysiological experiments α1C was used in place of GFP-α1C, which was primarily used in the structural analysis. In control experiments we did not find any difference between calcium currents from cells transfected with GFP-α1C or α1C. I-V curves, obtained by plotting the peak current density, i, against the test potential, V, were fitted by a modified Boltzmann function:
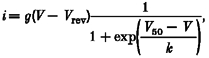 |
where g is the specific conductance (in pA (pF mV)−1), Vrev is the reversial potential (in mV), V50 is the potential of half-maximal activation (in mV), and k is the slope factor (in mV) which determines the steepness of the voltage dependence of activation.
Single-channel recordings
The bath solution contained (mM): 110 potassium aspartate, 20 KCl, 2 MgCl2, 20 Hepes and 2 EGTA (adjusted to pH 7.4 with KOH). Patch pipettes, pulled from borosilicate glass, fire polished and coated with Sigmacote (Sigma), were filled with (mM): 80 BaCl2, 30 TEA-Cl, 15 Hepes and 1 EDTA (adjusted to pH 7.4 with TEA-OH) and had resistances between 5 and 15 MΩ. If not otherwise specified in the text the following procedures for data acquisition and analysis were used. Test pulses of 200 ms duration from -70 to 0 mV were applied at 3 s intervals. Data were low-pass Bessel filtered at 2 kHz, sampled at 10 kHz, and analysed with Fetchan 6.0 (Axon Instruments) after digital Gaussian filtering at 1.5 kHz. The event detection threshold was 0.5 pA. With this event detection threshold events arising from endogenous low voltage-activated small conductance calcium channels, which we sometimes observed in non-transfected tsA201 cells, were excluded from the analysis. This also applies for the low threshold small conductance channels described by Meir & Dolphin (1998) in COS7 cells transfected with α1 subunits. Furthermore, events with a duration of 0.2 ms or shorter were excluded from the analysis. The open probability for a patch, i.e. NPo where N is the number of channels in the patch and Po is the single-channel open probability, was calculated by dividing the sum of the duration of all events, which was obtained from the event list provided by Fetchan 6.0, by the total observation time. This simplified method could be applied since, due to the rare occurrence and short duration of channel openings under our experimental conditions, higher conductance levels were almost never observed. In order to estimate the number of channels in each patch, N, from these NPo values we used two different approaches: (1) a mathematical procedure, which is based on theoretical considerations about the distribution of NPo values for a set of patches with different unknown N values, and (2) an experimental approach, in which the highest conductance level in each patch observed after the addition of the calcium channel agonist (±)-Bay K 8644 to the bath solution was taken as an estimate for the number of channels in the patch (for details, see Results).
RESULTS
Differential localization of individually expressed α1C and β subunits
tsA201 cells were transiently transfected with GFP-α1C, β1a-GFP, β2a or β3 and the subunit localization was determined with either GFP fluorescence or immunofluorescence (Fig. 1). In 79% of 543 examined cells (Table 3) α1C was localized in a dense network of a tubular membrane compartment that extended throughout the entire cytoplasm. The tubular/reticular network was very dense in the perinuclear region but could be resolved in thin regions of the cells (Fig. 1a). Occasionally the nuclear envelope was also labelled. Based on these structural characteristics the α1C-containing compartment was identified as the endoplasmic reticulum. In 15% of the cells α1C was also localized in clusters in the plasma membrane (not shown). There were no differences in the distribution patterns between the wild-type α1C and the mutant α1CY467S. The distribution patterns of the β subunits differed from that of α1C and were different among themselves. When expressed alone, β1a and β3 were both distributed diffusely throughout the cytoplasm of tsA201 cells (Table 4; Fig. 1d and o). In contrast, β2a showed a continuous plasma membrane stain and the cytoplasm was essentially free of immunolabel (Table 4; Fig. 1i).
Figure 1. Changes in subcellular distribution patterns of calcium channel β1a, β2a and β3 subunits coexpressed with α1C or α1CY467S.
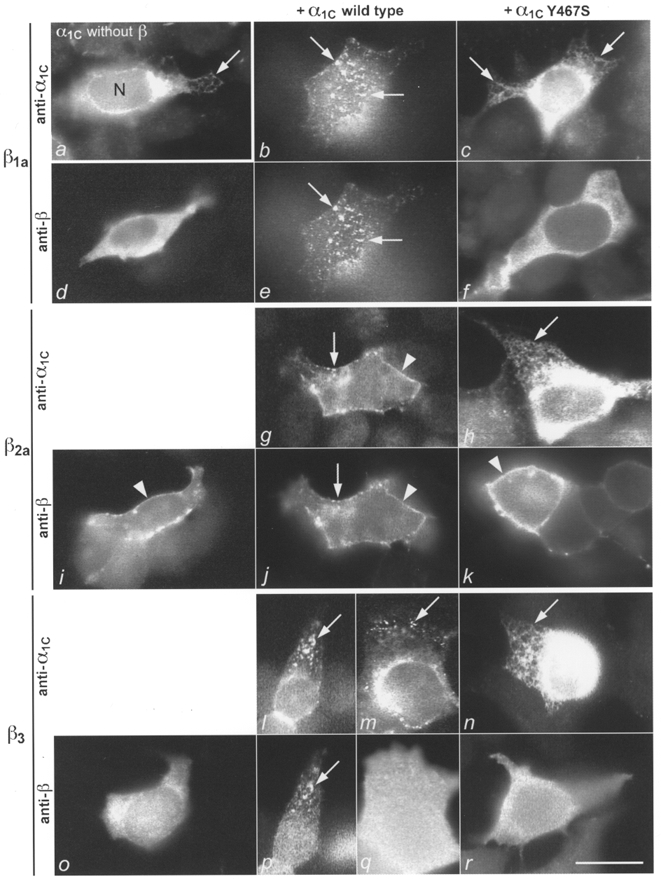
tsA201 cells were transfected with α1C (wild-type or mutant) and β isoforms alone or in different subunit combinations and the subunits were localized with immunofluorescence or GFP labelling. Left column: when expressed alone α1C (a) is localized in a tubular network - presumably the endoplasmic reticulum - that is very dense in the perinuclear region (saturated fluorescence) but can be resolved in the cell periphery (arrows); β1a(d) and β3 (o) are diffusely localized in the cytoplasm; and β2a is localized in the plasma membrane (i; arrowhead). Centre column: when wild-type α1C and β1a are coexpressed, the two colocalize in clusters in the plasma membrane (b and e; examples indicated by arrows); α1C and β2a(g and j) are colocalized throughout the plasma membrane, diffusely (arrowheads) and in clusters (arrows); and α1C and β3 are either colocalized in clusters (l and p; arrows) or β3 remains diffusely distributed in the cytoplasm (m and q). The α1C+β pairs were double labelled by using GFP-α1C (b, g, l and m) and anti-βcom (e, j, p and q). Right column: when coexpressed with the mutant α1CY467S, translocation and colocalization fail and all subunits remain in the same compartments as when expressed individually; α1CY467S in the endoplasmic reticulum (c, h and n; arrows); β1a(f) and β3 (r) in the cytoplasm; and β2a in the plasma membrane (k; arrowhead). N, nucleus; scale bar, 20 μm.
Table 3.
Subcellular distribution of the calcium channel α1C subunits expressed alone or with β subunit isoforms
| α1C | α1CY467S | |||
|---|---|---|---|---|
| ER | PM | ER | PM | |
| Without β | 79 | 15 | 87 | 7 |
| β1a | 14 | 83 | 87 | 7 |
| β2a | 32 | 64 | 93 | 0 |
| β3 | 49 | 47 | 88 | 6 |
Numbers give percentage of cells in each category (n = 500–800 cells for each condition). Balance to 100% represents cells in which localization could not be unambiguously identified as endoplasmic reticulum (ER) or plasma membrane (PM).
Table 4.
Subcellular distribution of the calcium channel β subunits expressed alone or with α1C
| Without α1C | α1C | α1CY467S | |||||
|---|---|---|---|---|---|---|---|
| Cytoplasm | PM | Cytoplasm | ER* | PM | Cytoplasm | PM | |
| β1a | 98 | 0 | 12 | 19 | 69 | 96 | 0 |
| β2a | 0 | 94 | 0 | 0 | 99 | 0 | 98 |
| β3 | 99 | 0 | 44 | 28 | 28 | 98 | 0 |
Numbers give percentage of cells in each category (n = 400–900 cells for each condition). Balance to 100% represents cells in which localization could not be unambiguously identified as cytoplasm, ER or PM.
ER distribution of β subunits was only observed with α1C and is therefore not included in the other conditions.
Colocalization of coexpressed α1C and β subunits in the plasma membrane
α1C was coexpressed with β1a, β2a or β3 in tsA201 cells and the subcellular distribution of both subunits was determined with GFP fluorescence or immunofluorescence. Upon coexpression with any of the β subunits the localization of α1C changed significantly from that observed when α1C was expressed alone. In all cases the number of cells with an endoplasmic reticulum localization of α1C was strongly reduced, whereas the number of cells with a plasma membrane localization was strongly increased compared with cells in which α1C was expressed alone (Table 3). In parallel, β1a and β3 were no longer found in the cytoplasm but were localized in clusters in the plasma membrane (Fig. 1e and p) and to some extent in the endoplasmic reticulum (Table 4). In both locations α1C and the β subunits were colocalized (Fig. 1b, e, l and p). The degree to which α1C and β were translocated to the plasma membrane was considerably higher for β1a than for β3. Fewer than half of the cells coexpressing α1C and β3 showed a colocalization of the two subunits in plasma membrane clusters, and in the rest of the cells α1C and β3 remained in the endoplasmic reticulum and the cytoplasm, respectively (Fig. 1l, m, p and q; Tables 3 and 4). In contrast to the cytoplasmic β1a and β3 subunits, the plasma membrane-associated β2a subunit showed little change in its distribution pattern when coexpressed with α1C. β2a remained evenly distributed throughout the plasma membrane, where it now was colocalized with α1C (Fig. 1g and j). In addition to the even plasma membrane distribution, the two subunits were frequently colocalized in plasma membrane clusters. The observed changes in the subcellular distribution of the α1C and β subunits upon coexpression indicate that all three examined β subunit isoforms bind to α1C and that the α1C-β complexes become inserted into the plasma membrane.
Translocation and colocalization of α1C and β subunits fails when the β subunit binding domain in the cytoplasmic I-II linker of α1C is mutated
To investigate the mechanism of the direct α1C-β interactions that underlie the observed changes in the subcellular distribution of the channel subunits, we replaced tyrosine in position 467 of the β subunit interaction domain of α1C with serine. The corresponding point mutations in α1A and α1S have been shown to disrupt β1b and β1a binding, respectively (Pragnell et al. 1994; Neuhuber et al. 1998b). Indeed, when α1CY467S was coexpressed with any one of the β subunit isoforms, α1CY467S and all β subunits remained localized in the same subcellular compartment as when expressed alone (Fig. 1). The subunits were not colocalized with one another in the plasma membrane or anywhere else in the cells and no increased expression of α1CY467S clusters occurred. This shows that the translocation of the subunits to the plasma membrane that was observed with GFP fluorescence and immunofluorescence when any one of the β subunits was coexpressed with wild-type α1C depends on an intact β subunit interaction domain in the cytoplasmic I-II linker of α1C.
β subunits modulate current activation in wild-type and mutant α1C channels
To investigate whether the observed structural interactions between α1C and the β subunits are reflected in a modulation of calcium current properties, we performed whole-cell patch-clamp recordings. tsA201 cells were transfected with α1C or α1CY467S with or without one of the β subunit isoforms, plus a plasmid encoding GFP as an expression marker. In the case of β1a, the fusion protein β1a-GFP was used instead of a combination of the two separate plasmids. The fraction of GFP-expressing cells in which high voltage-activated calcium currents could be recorded was 50% for cells transfected with α1C alone and 42% for cells transfected with α1CY467S alone. The percentage of cells exhibiting high voltage-activated calcium currents was about equal or higher when α1C or α1CY467S was coexpressed with a β subunit.
To measure the voltage dependence of activation, 400 ms voltage steps were applied from a holding potential of -80 mV to various test potentials between -40 and +80 mV (Fig. 2). I-V curves were obtained from these measurements by plotting the peak current density against the test potential (Fig. 3A). Coexpression with any one of the β subunits increased the peak current density severalfold. When α1C or α1CY467S was expressed alone the largest peak current was observed at a test potential of +40 mV. Upon coexpression with β2a the test potential at which the largest current was observed shifted to +30 mV. The Y467S point mutation had no effect on the intrinsic current properties of α1C or on the characteristic modulation by the β subunit isoforms. The increase in current density observed with all β subunit isoforms and the shift in the peak of the I-V curve observed with β2a were the same with α1C and α1CY467S. I-V curves were further analysed by fitting them with a modified Boltzmann function (see Methods; Fig. 3B). This analysis revealed that all β subunits increased the specific conductance, g, and that the strongest shift occurred with β1a. The potential of half-maximal activation, V50, and the slope factor, k, were both decreased by coexpression of the β subunits, with β2a showing the strongest effect (with P < 0.001, t test). The value of k was significantly decreased with P < 0.05 by all β subunits. The reversal potential, Vrev, was not significantly altered by coexpression of the β subunits. Interestingly, none of these parameters showed a significant difference between α1C and α1CY467S in any of the examined subunit combinations, suggesting that despite the deficiency in complex formation α1CY467S was still sensitive to modulation by β subunits.
Figure 2. Comparison of the time course of current traces for α1C and α1CY467S expressed without or with β1a, β2a or β3 in tsA201 cells.
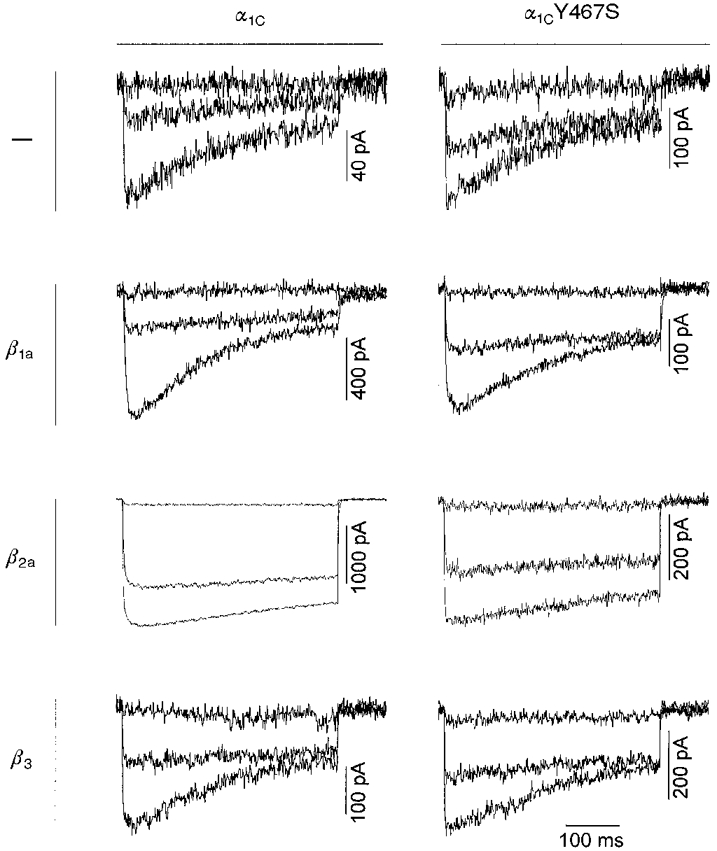
The membrane potential was stepped for 400 ms from a holding potential of -80 mV to 0, +20 or +40 mV (upper, middle and lower traces, respectively). The magnitude of currents varied greatly between individual cells (see also error bars in Fig. 3A and B). Current inactivation was slowed down on coexpression of α1C or α1CY467S with β2a. There was no apparent difference between currents recorded from α1C- or α1CY467S-transfected cells.
Figure 3. Voltage dependence of current activation for wild-type and mutant α1C expressed with and without β subunits.
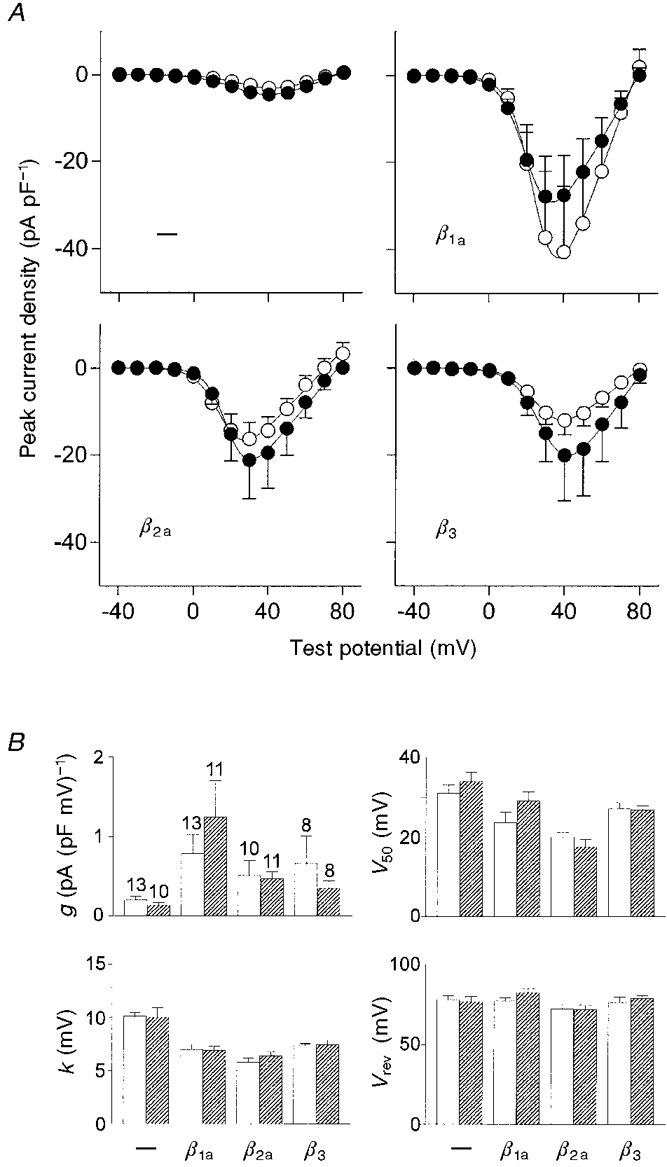
A, I-V curves obtained by plotting the peak current density from voltage step measurements as shown in Fig. 2 against the test potential. Coexpression of α1C (•) and α1CY467S (○) with any one of the β subunits increased the current amplitude severalfold (means and s.e.m.). B, I-V curves for each individual recording were fitted by a modified Boltzmann function (see Methods) and the obtained parameters were subsequently averaged. □, α1C;  , α1CY467S; error bars are s.e.m., and the number of cells recorded is shown. The specific conductance, g, was increased on β subunit coexpression whereas V50 and k were decreased by β subunit coexpression. The decrease in k on coexpression with each of the β subunits compared with α1C or α1CY467S expression alone was significant with P < 0.05. The reversal potential, Vrev, was not significantly altered by the β subunits.
, α1CY467S; error bars are s.e.m., and the number of cells recorded is shown. The specific conductance, g, was increased on β subunit coexpression whereas V50 and k were decreased by β subunit coexpression. The decrease in k on coexpression with each of the β subunits compared with α1C or α1CY467S expression alone was significant with P < 0.05. The reversal potential, Vrev, was not significantly altered by the β subunits.
Current kinetics are modulated by the β subunits
As a measure of the activation kinetics the time from the onset of the voltage step to 70% of the total rise in current amplitude, τ70%, was determined (Fig. 4A). Whereas β3 had no effect, β1a decreased the speed of activation and β2a slightly accelerated activation. The activation kinetics of α1CY467S were always somewhat faster than those of wild-type α1C, regardless of whether they were expressed alone or together with a β subunit. This suggests that tyrosine in position 467 of α1C is involved in determining the channel activation kinetics. The acceleration of activation kinetics by the mutation was statistically significant (P < 0.05) for coexpression with β1a and β2a, but not for β3 and expression of α1C alone.
Figure 4. Calcium current kinetics in tsA201 cells transfected with α1C or α1CY467S with and without β subunits.
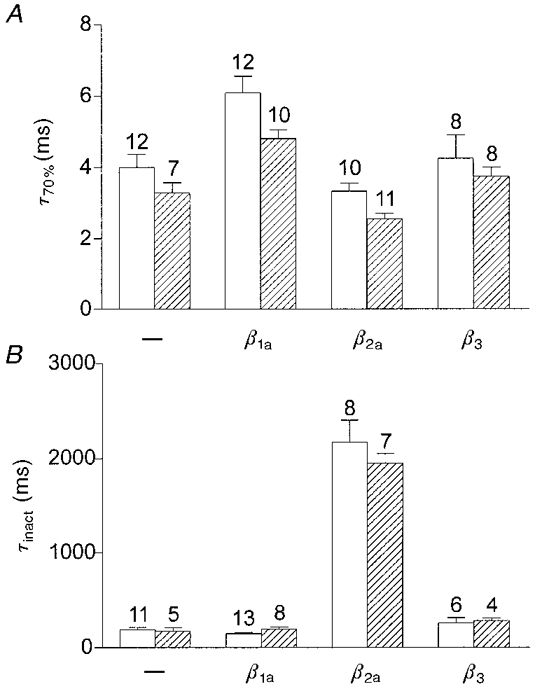
□, α1C; , α1CY467S. A, activation kinetics of calcium currents are expressed as the time from the onset of a voltage step to +40 mV (holding potential, -80 mV) to 70% of the total rise in current amplitude, τ70%. β1a slowed down current activation, β2a accelerated it and β3 did not affect activation kinetics. In all cases α1CY467S was slightly faster than the wild-type α1C. B, inactivation kinetics were determined by fitting a single exponential function with a time constant τinact to the decay phase of the current during a voltage step to +40 mV from a holding potential of -80 mV. For α1C and α1CY467S coexpressed with β2a, test pulse duration was 4 s; for all other conditions test pulse duration was 400 ms. Only β2a severely slowed down current inactivation, by a factor of ≈10.
The most dramatic modulatory effect of a β subunit appeared in the inactivation kinetics (Fig. 4B). Current inactivation was determined by fitting a single exponential function to the decaying phase of the current (for cells cotransfected with β2a, 4 s-long test pulses were used instead of 400 ms pulses). Figure 4B shows the inactivation time constants for test pulses to +40 mV. Whereas coexpression with β1a or β3 had only small effects (P > 0.04), β2a slowed current inactivation by a factor of approximately 10 (P < 0.0001). The inactivation kinetics of the mutant α1CY467S were indistinguishable from those of wild-type α1C.
Steady-state inactivation is not significantly modulated by the β subunits
Steady-state inactivation was determined by measuring the current amplitude at a test potential of +40 mV (holding potential, -100 mV) after 10 s prepulses of different potentials (2 ms interpulse interval). Figure 5A shows the normalized current amplitudes as a function of the prepulse potential. The data were fitted by a Boltzmann function, the parameters of which are shown in Fig. 5B. The analysis did not indicate any modulatory effects of the β subunits on steady-state inactivation, or any significant effects of the point mutation in α1C. Only the potential of half-maximal inactivation, V50, was different, being about 7 mV higher for α1CY467S than for α1C, when the α subunits were not coexpressed with a β subunit, but this effect was not significant (P > 0.05). Although coexpression with the β subunits resulted in small changes of V50, statistically there was no significant difference from the V50 values for α1C and α1CY467S expressed alone. The slope factor, k, was the same for all subunit combinations tested. Thus, steady-state inactivation of α1C was not subject to modulation by any one of the examined β subunit isoforms.
Figure 5. Steady-state inactivation in tsA201 cells transfected with α1C or α1CY467S with and without β subunits.
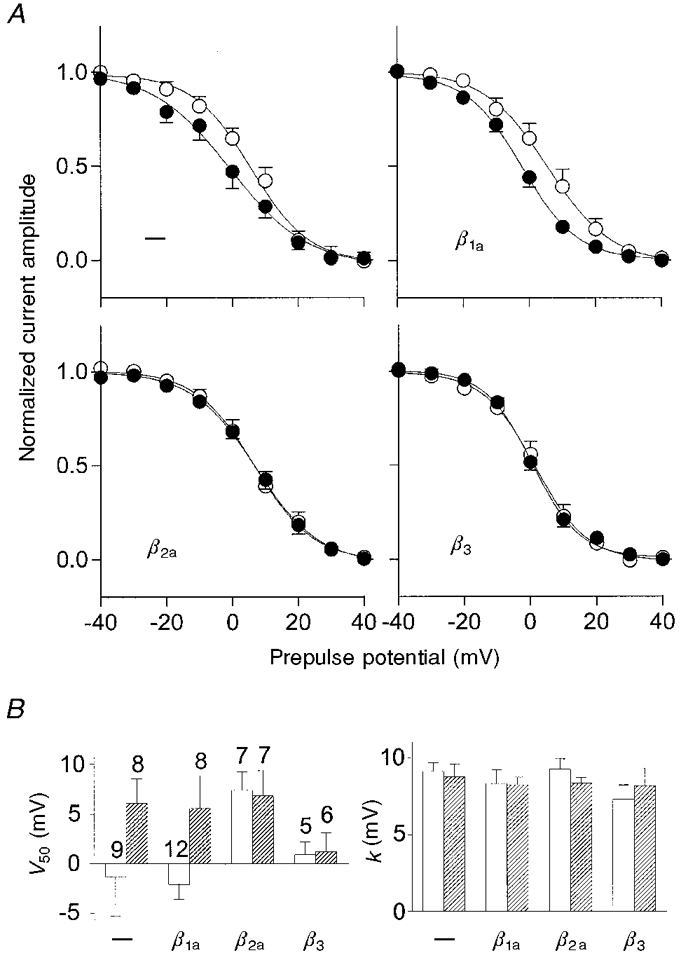
A, normalized current amplitude during a voltage step from -100 to +40 mV after 10 s prepulses to various potentials with an interpulse interval of 2 ms (•, α1C; ○, α1CY467S; means and s.e.m. of individual recordings are shown). There were only minor differences in steady-state inactivation between the different subunit combinations. B, steady-state inactivation curves for each individual recording were fitted by a Boltzmann function: i = 1/(1 + exp((V - V50)/k)), where i is the test pulse current amplitude, V is the prepulse potential, V50 is the prepulse potential of half-maximal inactivation and k is the slope factor, and the obtained parameters were subsequently averaged (□, α1C; , α1CY467S; error bars are s.e.m.). The apparent difference in the slopes of the averaged curves shown in the upper left figure in A is not reflected in the slope factor, k, shown in the right panel of B, due to the large variability of V50 for α1C expression (see error bar in left panel of B). This analysis does not indicate statistically significant differences in steady-state inactivation parameters between cells transfected with different subunit combinations.
Single-channel open probability of α1C is increased by β1a
The data presented above suggest that the increase in whole-cell current density observed when α1C or α1CY467S was coexpressed with a β subunit was not necessarily linked to an increase in plasma membrane localization of the α subunit, because the latter was not observed with α1CY467S. Thus, the possibility that the β subunit-induced increase in current density is due to changes of single-channel properties, such as a higher open probability or single-channel conductance, needed to be addressed directly. Therefore we performed single-channel recordings of α1C or α1CY467S alone, and α1C or α1CY467S coexpressed with β1a (Figs 6 and 7), the β isoform that induced the largest current increase in whole-cell recordings. The amplitude of single-channel opening events, i, was about the same in all examined subunit combinations, showing that the single-channel conductance was not modulated by β1a (i = 0.90± 0.05 pA for α1C, 1.00 ± 0.04 pA for α1C+β1a, 1.00 ± 0.16 pA for α1CY467S, and 0.80 ± 0.03 pA for α1CY467S +β1a (means ±s.e.m.)). In contrast, the frequency and the duration of channel openings increased significantly when α1C or α1CY467S was coexpressed with β1a (Fig. 6). The open-state dwell time histograms (Fig. 6) show that coexpression of β1a increased the mean open time 2-fold. The observation that the mutation itself did not change single-channel properties, together with the results from the whole-cell experiments, shows that the Y467S substitution does not mimic the effects of β subunit coexpression. The fraction of null sweeps was highly variable from cell to cell but on average no significant differences between α1C+β1a (51%) and α1CY467S +β1a (55%) were observed.
Figure 6. Single-channel recordings from tsA201 cells transfected with α1C (A), α1C+β1a (B), α1CY467S (C) and α1CY467S +β1a (D).
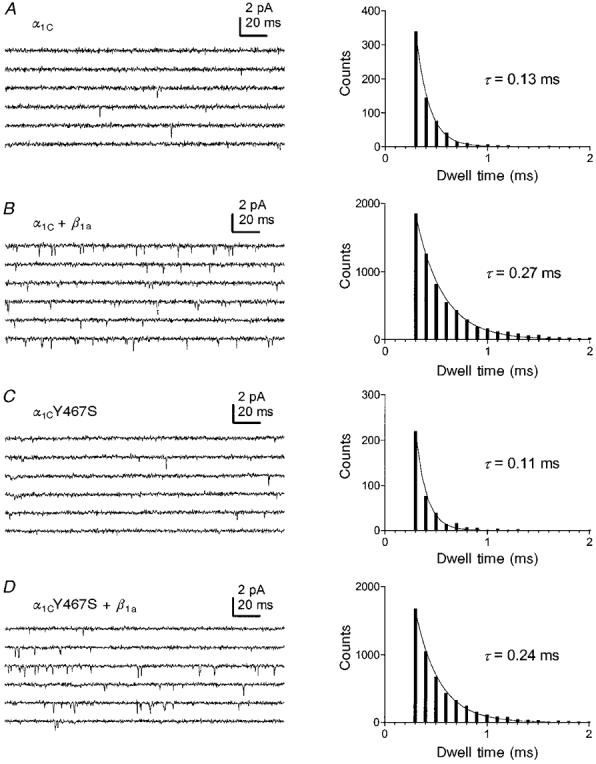
Voltage pulses of 200 ms duration from a holding potential of -70 mV to a test potential of 0 mV were applied every 3 s. Coexpression of α1C or α1CY467S with the β1a subunit resulted in an increased frequency of channel openings. The right panel shows open-state dwell time histograms. The mean open times, τ, obtained by fitting single exponential functions to the histograms, were increased by a factor of about 2 by β1a coexpression.
Figure 7. Determination of the number of channels in a patch, N, and the single-channel open probability, Po.
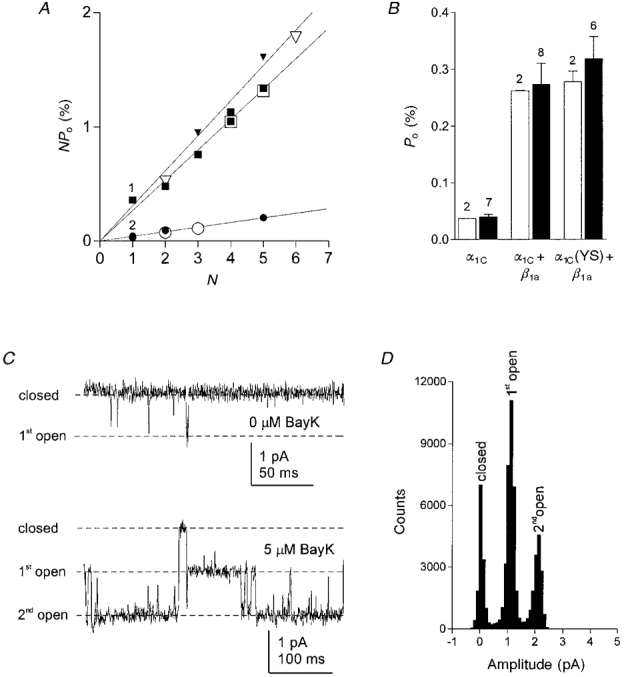
Open probabilities for individual patches, NPo, were calculated from recordings from tsA201 cells transfected with α1C (○), α1C+β1a (□), or α1CY467S +β1a (▿) and plotted against the number of channels in each patch, N. N values were determined by two different approaches. (i) Open symbols in A represent recordings in which at the end of an experiment 5 μm (±)-Bay K 8644 was added to the bath solution to visualize simultaneous openings of multiple channels in the patch. C shows an example of the short and long openings before and after addition of (±)-Bay K 8644, respectively, in a cell transfected with α1CY467S and β1a (upper trace, test pulse duration 200 ms at 0 mV; lower trace, test pulse duration 400 ms at +10 mV). The all-points amplitude histogram in D corresponding to the lower trace in C shows the number of conductance levels. (ii) Filled symbols in A represent recordings for which N values were estimated by finding the best fit of all NPo values in one experimental group to a linear regression crossing the abscissa at zero. Multiple data points with the same or similar NPo marked with 1 (▪, ▾, ▾) and 2 (•, •). The slope corresponds to the mean single-channel open probability. B, the Po values determined experimentally with (±)-Bay K 8644 (□) closely match the values from the fitting procedure (▪; error bars give standard deviation). Coexpression of β1a resulted in a 7- to 8-fold increase of Po.
In order to determine the single-channel open probability, Po, it was necessary to determine the number of channels, N, in each patch. The usual approach is to estimate N from the maximum number of conductance levels observed. This is reasonable only when Po is sufficiently high and the mean open time is sufficiently long to observe simultaneous openings of multiple channels. Due to the rare occurrence and short mean open time of events under the conditions used here, simultaneous openings of multiple channels are extremely unlikely and were almost never observed. Therefore we applied two different approaches to estimate N for each patch. In the first approach, we added the calcium channel agonist (±)-Bay K 8644 to the bath solution to a final concentration of 5 μm after we had recorded several hundred sweeps without (±)-Bay K 8644 for the analysis of NPo. With (±)-Bay K 8644 in the bath solution and depolarizations to +10 mV, channel openings lasting up to several hundred milliseconds occurred, allowing the observation of simultaneous openings of multiple channels in the patch (Fig. 7C). An all-points amplitude histogram of these recordings gives the number of conductance levels and thus the number of channels in the patch (Fig. 7D). With the value determined for N it was then possible to calculate Po (open symbols in Fig. 7A and open bars in Fig. 7B). The second approach to estimate the unknown N for each NPo value was a procedure based on theoretical considerations. This method does not require the application of the calcium channel agonist, allowing us to include recordings in the analysis that had not been tested for the number of channels with (±)-Bay K 8644, e.g. when the patch was not stable for long enough. The fitting procedure is based on the supposition that under the same experimental conditions the differences between the NPo values from different patches can only be due to different numbers of channels in the patches. Thus, the NPo values should always be an integer multiple (N) of the smallest possible value Po. In other words, when plotting NPo against N all data points should lie on a straight line crossing the abscissa at zero. According to this supposition, we estimated the unknown value N by assigning those values of N to each patch that resulted in the best linear regression through all data points and zero (Fig. 7A). The values for the single-channel open probability estimated with this method gave the same results as those obtained by the experimental approach (cf. open and filled bars in Fig. 7B). This indicates that the fitting method is a useful approach to estimate the number of channels in a patch, and thus to determine the single-channel open probability, when simultaneous openings of events are too rare to be detected or when the mean open time cannot be increased sufficiently by application of a channel agonist.
Open probabilities determined for each condition according to these procedures were approximately 7- to 8-fold higher in cells coexpressing α1C or α1CY467S with β1a than for cells expressing the α subunit alone. These single-channel data show that the β1a-induced 4- to 5-fold increase in the specific conductance, g, seen in whole-cell recordings (Fig. 3B) can be fully explained by the increase in single-channel open probability without the need for an increased number of functional channels in the plasma membrane.
DISCUSSION
In the present study we used heterologous expression of α1C with three different β subunit isoforms in tsA201 cells to describe isoform-specific effects of the β subunit on the subcellular distribution of the calcium channel subunits and on the modulation of current properties. The role of the β subunit interaction domain in the cytoplasmic I-II linker of α1C was analysed by comparing the properties of wild-type α1C with those of an α1C construct in which tyrosine in position 467 was replaced with serine. Immunofluorescence and GFP fluorescence were used to determine the subcellular distribution patterns of the α1C and β subunits, and current properties were studied with whole-cell and single-channel patch-clamp recordings. The combination of structural and functional analyses revealed β isoform-specific effects on the targeting of both channel subunits and the modulation of calcium currents, but also showed that the two effects of the β subunit are not causally linked.
Isoform-specific effects of β1a, β2a and β3 on targeting of α1C and on current properties
Coexpression of α1C with one of the β subunit isoforms resulted in changes of the subcellular distribution of both subunits and in changes in the current properties. Both structural and functional effects of the β subunit on α1C showed differences depending on the β isoform. When expressed alone, only β2a was localized in the plasma membrane, whereas β1a and β3 were diffusely distributed in the cytoplasm. This is consistent with previous studies (Chien et al. 1995; Brice et al. 1997; Neuhuber et al. 1998b) and can be explained by the intrinsic ability of β2a to associate with the plasma membrane via a palmitoylation site in its N-terminus. Without a β subunit, α1C accumulated in the endoplasmic reticulum of tsA201 cells. In only a small fraction of transfected cells could clusters of α1C be visualized in the plasma membrane, even though calcium currents revealed the presence of functional channels in the majority of transfected cells. Apparently, without coexpression of the β subunit the density of functionally expressed channels in the plasma membrane is too low to be detected with immunocytochemistry or GFP labelling. A rough estimate of the density of functional α1C subunits in the plasma membrane based on the single-channel properties and the whole-cell data yielded a channel density of 21 μm−2 (channel density =IpeakC/iPo= (0.72 × 0.01)/(0.9 × 0.00039) where Ipeak (at 0 mV) is in pA pF−1, i is in pA, and the specific membrane capacity (C) is in pF μm−2, assuming a value of C of 1 μF cm−2= 0.01 pF μm−2). This value is far below the density of approximately 2200 channels μm−2 found in skeletal muscle triads where calcium channels can be reliably detected with immunocytochemistry.
When α1C was coexpressed with any one of the tested β isoforms, α1C became detectable with immunocytochemistry and GFP labelling in the plasma membrane, where it was now colocalized with the β subunit. This was accompanied by an increase in current density for all subunit combinations, indicating that α1C and β formed complexes that facilitated the incorporation of channels into the plasma membrane. However, the results of the single-channel recordings show that the current increase observed with the β subunits can be explained by changes of the single-channel properties, i.e. an increase in open channel probability. Thus the number of functional channels in the plasma membrane did not change during coexpression of α1C with β. Neely et al. (1993) reached the same conclusion for experiments in which β subunit coexpression led to an increase in whole-cell currents without a parallel increase in gating charges. However, other studies suggested that the increase in whole-cell currents was entirely or in part due to an increase in the number of channels in the plasma membrane (Wakamori et al. 1993; Kamp et al. 1996; Josephson & Varadi, 1996). Our observation that the massive incorporation of α1C-β complexes into the plasma membrane was not accompanied by an increased number of functional channels suggests that some additional factor is limiting the expression of functional channels. If such a factor is saturated to different degrees in different experimental systems, it could explain why some investigators observed a β subunit-induced increase in channel density while others did not.
The ability of the different β isoforms to facilitate incorporation of α1C-β complexes into the plasma membrane differed qualitatively and quantitatively. Coexpression of the plasma membrane-associated β2a isoform resulted in an even distribution of α1C throughout the plasma membrane, suggesting that the intrinsic distribution of β2a also determined the distribution of α1C. In addition, clusters of both subunits in the plasma membrane could be observed. The cytoplasmic β1a and β3 subunits were not homogeneously expressed throughout the plasma membrane when coexpressed with the α1 subunit, but instead were only found colocalized with α1C in distinct clusters. This indicates that their own translocation from the cytoplasm to the plasma membrane is dependent on association with α1C and that an intrinsic predisposition of α1C to aggregate in clusters may determine the distribution of the α1C-β complexes in the plasma membrane. Our observation that only about half of the cells coexpressing β3 and α1C showed α1C-β complexes in the plasma membrane, and in the other cells α1C and β3 remained individually localized in the endoplasmic reticulum and the cytoplasm, respectively, suggests that β3 binds to α1C with a somewhat lower affinity than β1a and β2a. This is consistent with data from De Waard et al. (1995) who showed that β3 bound to α1A with lower affinity than β1b, β2a or β4.
The isoform-specific modes of membrane targeting of the calcium channel α1C and β subunits were paralleled by the isoform-specific modulation of whole-cell calcium currents. Whereas all three β isoforms caused an increase of current density due to an increased specific conductance, a shift in the voltage dependence of activation was most pronounced with β2a, and only β2a caused a dramatic decrease in the rate of inactivation. A specific effect of β2a on current inactivation has previously been shown for α1A and α1E when expressed in oocytes (Sather et al. 1993; Qin et al. 1996; Parent et al. 1997), indicating that the distinct effects of β2a shown in the present study are not the result of combining the two native subunit partners of cardiac muscles; instead this property must reside on β2a itself. The observation that β2a differs from β1a and β3 in its targeting properties as well as in its modulatory ability suggests that these properties may be related. This interpretation is supported by a recent finding by Qin et al. (1998) showing that removal of the palmitoylation site in β2a reversed several β2a-specific modulatory effects on α1C and α1E. Thus, the subcellular organization of isolated calcium channel complexes has a profound influence on the functional characteristics of the channel.
Dissociation of effects of the β subunit on targeting of α1C and on calcium currents
The substitution of a single residue (Y467S) in the β subunit interaction domain of α1C disrupted the formation and incorporation of α1C-β complexes into the plasma membrane but not the modulatory effects of β on calcium currents. Pragnell et al. (1994) identified a motif of nine conserved amino acids that is critical for binding the β subunit, in the cytoplasmic loop between repeats I and II of the α1 subunit. Mutations within this motif of α1A perturbed β subunit binding (Pragnell et al. 1994). Here we have shown that α1CY467S and a coexpressed β subunit did not colocalize in the plasma membrane but remained localized in distinct subcellular compartments. α1CY467S was concentrated in the endoplasmic reticulum, β1a and β3 were diffusely distributed in the cytoplasm, and β2a was evenly distributed in the plasma membrane - all as when wild-type α1C and the β subunits were expressed individually. Therefore we conclude that the stable association of α1CY467S and β failed and that this had a dramatic effect on the incorporation of all subunits except β2a into the plasma membrane. Previously we have shown that the stable association of the skeletal α1S isoform and β1a can occur in the endoplasmic reticulum upon coexpression, as seen by translocation of β1a from the cytoplasm to the endoplasmic reticulum, and that this translocation fails when the β subunit interaction domain in α1S is mutated at the corresponding tyrosine residue (Y366S) (Neuhuber et al. 1998b). Here we observed in some cells the colocalization of the cytoplasmic β subunits with α1C (but not with α1CY467S) in the endoplasmic reticulum, indicating that in these cells the two subunits formed a complex but were not efficiently transported to the plasma membrane. Therefore, if the mutation in the cardiac α1CY467S subunit had not perturbed binding, we would expect to see the colocalization of α1CY467S and β in the endoplasmic reticulum, even if the export of α1CY467S from the endoplasmic reticulum had failed for a reason other than the lack of α1C-β complex formation; but instead α1CY467S and the β subunits remained in distinct compartments. Moreover, α1CY467S expressed alone generated calcium currents of the same magnitude as wild-type α1C, indicating that α1CY467S is a fully functional channel. Thus, the lack of colocalization of α1CY467S and the β subunits indicates that none of the three examined β isoforms form stable complexes with the mutated α1CY467S subunit.
Nevertheless, the increase in current density, the modulation of current kinetics, and the increase in single-channel open probability upon coexpression of β were equally observed with wild-type α1C and the mutant α1CY467S. This shows first that the increased plasma membrane localization of α1C, observed when α1C was coexpressed with β, is not a prerequisite for the increased current density, and second that the stable association of α1 with a β subunit is not required for their functional interaction. Apparently, the majority of visible channels in the plasma membrane are not functional and unfortunately it cannot be unambiguously verified that the population of functional channels does not form complexes with the β subunit as well. However, if the large visible fraction of α1CY467S channels fails to associate with β subunits one can expect that the functional fraction will equally fail to do so and, consequently, their functional modulation by β subunits most probably does not depend on the stable association of α1CY467S with a β subunit. Our finding that upregulation of current density by β is due to a mechanism other than the increased channel density, such as an increased channel open probability, is in agreement with several previous studies in which β subunit-induced effects on single-channel properties have been reported (Neely et al. 1993; Wakamori et al. 1993; Shistik et al. 1995). In the present study we have shown an increase in dwell time and open probability of α1C and α1CY467S upon coexpression with β1a. Whereas the analysis of the number of functional channels per patch gave no indication of a strong increase on coexpression with β, single-channel open probability increased about 7- to 8-fold - more than enough to explain the increase in whole-cell current density. Consequently, the simultaneously occurring increase of detectable channels in the plasma membrane must be a parallel but not causally linked effect of the β subunit; only in this way can one fail without affecting the other. In a recent study in Xenopus oocytes, Yamaguchi et al. (1998) showed similar separate effects of β3 on the functional modulation of α1C and on increased membrane incorporation. They achieved a temporal dissociation of the two effects by injecting a β3 fusion protein into α1C-expressing oocytes and were able to block membrane incorporation without affecting β subunit-induced modulation of current kinetics and voltage dependence. Together, the study by Yamaguchi et al. (1998) and the present study provide strong evidence for dual and independent roles of the accessory β subunit in targeting and modulation of the α1 subunit.
To what extent these functions contribute to the normal incorporation and function of the channels in the excitation-contraction coupling apparatus in muscle is not clear. Expression of the skeletal α1S isoform with the analogous mutation in the β subunit interaction domain in skeletal myotubes of the dysgenic mouse also resulted in the disruption of α1S-β1a complexes, although without perturbing the specific targeting of α1S into the triads and the reconstitution of normal function (Neuhuber et al. 1998a). This is consistent with the interpretation of the results presented here that β subunits can exert their functions without forming stable α1-β complexes. However, because in the muscle expression system calcium currents and excitation-contraction coupling could not be analysed in the absence of the endogenous β subunit, functional modulation of calcium currents by β could not be directly tested with α1S. Our present result that specific modulatory effects of three different β subunits can all be observed with a mutated α1 subunit (α1CY467S), which fails to colocalize with the β isoforms, provides strong evidence that the formation of α1C-β complexes is not required for functional interactions between the two calcium channel subunits.
Dual mode of α1C-β interactions
We observed two effects of the β subunit on α1C: an increased incorporation of α1C into the plasma membrane which is dependent on the formation of stable α1C-β complexes, and the modulation of current properties which is independent of the formation of stable α1C-β complexes. The β subunit interaction domain in the cytoplasmic loop between repeats I and II of α1C is clearly responsible for the stable association of α1C and β, and for the increased incorporation of the complex into the plasma membrane. In contrast, it may or may not be the site of functional α1C-β interactions. Either the point mutation lowered the affinity of this β subunit interaction domain on α1C or a second, low affinity interaction domain exists apart from that in the I-II linker. In either case high concentrations of free β subunit in the cytoplasm or in the plasma membrane could interact with such a low affinity binding site and thus cause functional modulation without forming a stable complex. Distinct domains for the functional modulation of α1 have been described on the β subunit (Olcese et al. 1994; Cens et al. 1998). Furthermore, a second low affinity binding domain in α1E that mediates modulation of inactivation kinetics has been described (Walker et al. 1998). However, these results differ from the present data in that other functional effects of the β subunit on α1E, like the increase in current amplitude, were mediated by the I-II linker, whereas in our study the loss of binding to the I-II linker of α1C did not perturb any modulatory effects of β on current properties. Finally, it has been suggested that more than one β subunit can interact simultaneously with different binding sites on the α1 subunit. Our results are consistent with such a model in that structural and functional effects of β subunit coexpression appeared to be independent. However, one can also envisage a mechanism for α1C-β interactions by which binding of a β subunit to the β subunit interaction domain in the I-II linker brings β into a position to interact with the functionally important residues on α1C, but that this binding is not necessarily required.
In all, the results of this study show that the β subunits of calcium channels serve a dual function, firstly in the formation of a complex with α1C and its incorporation into the plasma membrane and secondly in the modulation of single-channel calcium current properties, but it also shows that these two functions of the β subunit are independent of each other.
Acknowledgments
We thank Drs M. Grabner and E. Perez-Reyes for their generous gifts of expression plasmids. We thank Dr J. Hoflacher for excellent help with experiments, Dr P. Dietl for valuable discussion of the manuscript, and Dr H. Glossmann for generously providing laboratory space and support for the pursuit of this project. This work was supported in part by the Fonds zur Förderung der wissenschaftlichen Forschung, Austria, grants P12653-MED (to B. E. F.), P12667 (to K. G.) and P12641-MED (to J. S.), and the European Commission's Training and Mobility of Researchers Network Grant ERBFMRXCT960032 (to B. E. F.).
References
- Birnbaumer L, Campbell KP, Catterall WA, Harpold MM, Hofmann F, Horne WA, Mori Y, Schwartz A, Snutch TP, Tanabe T. The naming of voltage-gated calcium channels. Neuron. 1994;13:505–506. doi: 10.1016/0896-6273(94)90021-3. [DOI] [PubMed] [Google Scholar]
- Brice NL, Berrow NS, Campbell V, Page KM, Brickley K, Tedder I, Dolphin AC. Importance of the different β subunits in the membrane expression of the α1A and α2 calcium channel subunits: studies using a depolarization-sensitive α1A antibody. European Journal of Neuroscience. 1997;9:749–759. doi: 10.1111/j.1460-9568.1997.tb01423.x. [DOI] [PubMed] [Google Scholar]
- Castellano A, Wei X, Birnbaumer L, Perez-Reyes E. Cloning and expression of a neuronal calcium channel β subunit. Journal of Biological Chemistry. 1993;268:12359–12366. [PubMed] [Google Scholar]
- Cens T, Restituito S, Vallentin A, Charnet P. Promotion and inhibition of L-type Ca2+ channel facilitation by distinct domains of the β subunit. Journal of Biological Chemistry. 1998;273:18308–18315. doi: 10.1074/jbc.273.29.18308. [DOI] [PubMed] [Google Scholar]
- Chien AJ, Carr KM, Shirokov RE, Rios E, Hosey MM. Identification of palmitoylation sites within the L-type calcium channel β2a subunit and effects on channel function. Journal of Biological Chemistry. 1996;271:26465–26468. doi: 10.1074/jbc.271.43.26465. [DOI] [PubMed] [Google Scholar]
- Chien AJ, Zhao X, Shirokov RE, Puri TS, Chang CF, Sun D, Rios E, Hosey MM. Roles of membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. Journal of Biological Chemistry. 1995;270:30036–30044. doi: 10.1074/jbc.270.50.30036. 10.1074/jbc.270.50.30036. [DOI] [PubMed] [Google Scholar]
- De Waard M, Pragnell M, Campbell KP. Ca2+-channel regulation by a conserved β subunit domain. Neuron. 1994;13:495–503. doi: 10.1016/0896-6273(94)90363-8. 10.1016/0896-6273(94)90363-8. [DOI] [PubMed] [Google Scholar]
- De Waard M, Scott VE, Pragnell M, Campbell KP. Identification of critical amino acids involved in α1-β interaction in voltage-dependent Ca2+ channels. FEBS Letters. 1996;380:272–276. doi: 10.1016/0014-5793(96)00007-5. 10.1016/0014-5793(96)00007-5. [DOI] [PubMed] [Google Scholar]
- De Waard M, Witcher DR, Pragnell M, Liu H, Campbell KP. Properties of the α1-β anchoring site in voltage-dependent Ca2+ channels. Journal of Biological Chemistry. 1995;270:12056–12064. doi: 10.1074/jbc.270.20.12056. 10.1074/jbc.270.20.12056. [DOI] [PubMed] [Google Scholar]
- Flucher BE, Andrews SB, Fleischer S, Marks AR, Caswell AH, Powell JA. Triad formation: organization and function of the sarcoplasmic reticulum calcium release channel and triadin in normal and dysgenic muscle in vitro. Journal of Cell Biology. 1993;123:1161–1174. doi: 10.1083/jcb.123.5.1161. 10.1083/jcb.123.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner M, Dirksen RT, Beam KG. Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-L-type Ca2+ channels expressed in dysgenic myotubes. Proceedings of the National Academy of Sciences of the USA. 1998;95:1903–1908. doi: 10.1073/pnas.95.4.1903. 10.1073/pnas.95.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg RG, Messing A, Strube C, Beurg M, Moss R, Behan M, Sukhareva M, Haynes S, Powell JA, Coronado R, Powers PA. Absence of the β subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the α1 subunit and eliminates excitation-contraction coupling. Proceedings of the National Academy of Sciences of the USA. 1996;93:13961–13966. doi: 10.1073/pnas.93.24.13961. 10.1073/pnas.93.24.13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Hullin R, Singer-Lahat D, Freichel M, Biel M, Dascal N, Hofmann F, Flockerzi V. Calcium channel β subunit heterogeneity: functional expression of cloned cDNA from heart, aorta and brain. EMBO Journal. 1992;11:885–890. doi: 10.1002/j.1460-2075.1992.tb05126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL, DeJongh KS, Catterall WA. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Brousal JP, Peterson BZ, Gallombardo PA, Hockerman GH, Lai Y, Scheuer T, Catterall WA. Modulation of the cloned skeletal muscle L-type Ca2+ channel by anchored cAMP-dependent protein kinase. Journal of Neuroscience. 1997;17:1243–1255. doi: 10.1523/JNEUROSCI.17-04-01243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson IR, Varadi G. The β subunit increases Ca2+ currents and gating charge movements of human cardiac L-type Ca2+ channels. Biophysical Journal. 1996;70:1285–1293. doi: 10.1016/S0006-3495(96)79685-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp TJ, Pérez-García MT, Marban E. Enhancement of ionic current and charge movement by coexpression of calcium channel β1A subunit with α1C subunit in a human embryonic kidney cell line. The Journal of Physiology. 1996;492:89–96. doi: 10.1113/jphysiol.1996.sp021291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WJ, Ellinor PT, Schwartz A. cDNA cloning of a dihydropyridine-sensitive calcium channel from rat aorta. Evidence for the existence of alternatively spliced isoforms. Journal of Biological Chemistry. 1990;265:17786–17791. [PubMed] [Google Scholar]
- Lacerda AE, Kim HS, Ruth P, Perez-Reyes E, Flockerzi V, Hofmann F, Birnbaumer L, Brown AM. Normalization of current kinetics by interaction between the α1 and β subunits of the skeletal muscle dihydropyridine-sensitive Ca2+ channel. Nature. 1991;352:527–530. doi: 10.1038/352527a0. [DOI] [PubMed] [Google Scholar]
- Leung AT, Imagawa T, Campbell KP. Structural characterization of the 1,4-dihydropyridine receptor of the voltage-gated calcium channel from rabbit skeletal muscle. Evidence for two distinct high molecular weight subunits. Journal of Biological Chemistry. 1987;262:7943–7946. [PubMed] [Google Scholar]
- Lory P, Varadi G, Schwartz A. The β subunit controls the gating and dihydropyridine sensitivity of the skeletal muscle Ca2+ channel. Biophysical Journal. 1992;63:1421–1424. doi: 10.1016/S0006-3495(92)81705-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir A, Dolphin AC. Known calcium channel α1 subunits can form low threshold small conductance channels with similarities to native T-type channels. Neuron. 1998;20:341–351. doi: 10.1016/s0896-6273(00)80461-4. [DOI] [PubMed] [Google Scholar]
- Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, Numa S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature. 1989;340:230–233. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E. Potentiation by the β subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 1993;262:575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- Neuhuber B, Gerster U, Döring F, Glossmann H, Tanabe T, Flucher BE. Association of calcium channel α1S and β1a subunits is required for the targeting of β1a but not of α1S into skeletal muscle triads. Proceedings of the National Academy of Sciences of the USA. 1998a;95:5015–5020. doi: 10.1073/pnas.95.9.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhuber B, Gerster U, Mitterdorfer J, Glossmann H, Flucher BE. Differential effects of Ca2+ channel β1a and β2a subunits on complex formation with α1S and on current expression in tsA201 cells. Journal of Biological Chemistry. 1998b;273:9110–9118. doi: 10.1074/jbc.273.15.9110. [DOI] [PubMed] [Google Scholar]
- Norrander J, Kempe T, Messing J. Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene. 1985;26:101–106. doi: 10.1016/0378-1119(83)90040-9. [DOI] [PubMed] [Google Scholar]
- Olcese R, Qin N, Schneider T, Neely A, Wei X, Stefani E, Birnbaumer L. The amino terminus of a calcium channel β subunit sets rates of channel inactivation independently of the subunit's effect on activation. Neuron. 1994;13:1433–1438. doi: 10.1016/0896-6273(94)90428-6. [DOI] [PubMed] [Google Scholar]
- Parent L, Schneider T, Moore CP, Talwar D. Subunit regulation of the human brain α1E calcium channel. Journal of Membrane Biology. 1997;160:127–140. doi: 10.1007/s002329900302. [DOI] [PubMed] [Google Scholar]
- Perez-Garcia MT, Kamp TJ, Marban E. Functional properties of cardiac L-type calcium channels transiently expressed in HEK293 cells. Roles of α1 and β subunits. Journal of General Physiology. 1995;105:289–306. doi: 10.1085/jgp.105.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, Wei X, Birnbaumer L. Cloning and expression of a cardiac/brain β subunit of the L-type calcium channel. Journal of Biological Chemistry. 1992;267:1792–1797. [PubMed] [Google Scholar]
- Pichler M, Cassidy TN, Reimer D, Haase H, Kraus R, Ostler D, Striessnig J. β subunit heterogeneity in neuronal L-type Ca2+ channels. Journal of Biological Chemistry. 1997;272:13877–13882. doi: 10.1074/jbc.272.21.13877. [DOI] [PubMed] [Google Scholar]
- Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- Qin N, Olcese R, Zhou J, Cabello OA, Birnbaumer L, Stefani E. Identification of a second region of the β-subunit involved in regulation of calcium channel inactivation. American Journal of Physiology. 1996;271:C1539–1545. doi: 10.1152/ajpcell.1996.271.5.C1539. [DOI] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Costantin JL, Stefani E, Birnbaumer L. Unique regulatory properties of the type 2a Ca2+ channel β subunit caused by palmitoylation. Proceedings of the National Academy of Sciences of the USA. 1998;95:4690–4695. doi: 10.1073/pnas.95.8.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruth P, Röhrkasten A, Biel M, Bosse E, Regulla S, Meyer HE, Flockerzi V, Hofmann F. Primary structure of the β subunit of the DHP-sensitive calcium channel from skeletal muscle. Science. 1989;245:1115–1118. doi: 10.1126/science.2549640. [DOI] [PubMed] [Google Scholar]
- Safayhi H, Haase H, Kramer U, Bihlmayer A, Roenfeldt M, Ammon HP, Froschmayr M, Cassidy TN, Morano I, Ahlijanian MK, Striessnig J. L-type calcium channels in insulin-secreting cells: biochemical characterization and phosphorylation in RINm5F cells. Molecular Endocrinology. 1997;11:619–629. doi: 10.1210/mend.11.5.9922. [DOI] [PubMed] [Google Scholar]
- Sather WA, Tanabe T, Zhang J-F, Mori Y, Adams ME, Tsien RW. Distinctive biophysical and pharmacological properties of class A (BI) calcium channel subunits. Neuron. 1993;11:291–303. doi: 10.1016/0896-6273(93)90185-t. [DOI] [PubMed] [Google Scholar]
- Shistik E, Ivanina T, Puri T, Hosey M, Dascal N. Ca2+ current enhancement by α2/δ and β subunits in Xenopus oocytes: contribution of changes in channel gating and α1 protein level. The Journal of Physiology. 1995;489:55–62. doi: 10.1113/jphysiol.1995.sp021029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Seagar MJ, Jones JF, Reber BF, Catterall WA. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proceedings of the National Academy of Sciences of the USA. 1987;84:5478–5482. doi: 10.1073/pnas.84.15.5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareilus E, Roux M, Qin N, Olcese R, Zhou J, Stefani E, Birnbaumer L. A Xenopus oocyte β subunit: Evidence for a role in the assembly/expression of voltage-gated calcium channels that is separate from its role as a regulatory subunit. Proceedings of the National Academy of Sciences of the USA. 1997;94:1703–1708. doi: 10.1073/pnas.94.5.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaghy PL, Striessnig J, Miwa K, Knaus H-G, Itagaki K, McKenna E, Glossmann H, Schwartz A. Identification of a novel 1,4-dihydropyridine- and phenylalkylamine-binding polypeptide in calcium channel preparations. Journal of Biological Chemistry. 1987;262:14337–14342. [PubMed] [Google Scholar]
- Varadi G, Lory P, Schultz D, Varadi M, Schwartz A. Acceleration of activation and inactiviaion by the β subunit of the skeletal muscle calcium channel. Nature. 1991;352:159–162. doi: 10.1038/352159a0. [DOI] [PubMed] [Google Scholar]
- Wakamori M, Mikala G, Schwartz A, Yatani A. Single-channel analysis of a cloned human heart L-type Ca2+ channel α1 subunit and the effects of a cardiac β subunit. Biochemical and Biophysical Research Communications. 1993;196:1170–1176. doi: 10.1006/bbrc.1993.2374. [DOI] [PubMed] [Google Scholar]
- Walker D, Bichet D, Campbell KP, De Waard M. A β4 isoform specific interaction site in the carboxyl-terminal region of the voltage-dependent Ca2+ channel α1A subunit. Journal of Biological Chemistry. 1998;273:2361–2367. doi: 10.1074/jbc.273.4.2361. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Hara M, Strobeck M, Fukasawa K, Schwartz A, Varadi G. Multiple modulation pathways of calcium channel activity by a β subunit. Direct evidence of β subunit participation in membrane trafficking of the α1C subunit. Journal of Biological Chemistry. 1998;273:19348–19356. doi: 10.1074/jbc.273.30.19348. [DOI] [PubMed] [Google Scholar]