

Abstract
Sigma receptors bind a diverse group of chemically unrelated ligands, including pentazocine, apomorphine (a dopamine receptor agonist) and haloperidol (a dopamine receptor antagonist). Although sigma binding sites are widely distributed, their physiological roles are poorly understood. Here, the whole-terminal patch-clamp technique was used to demonstrate that sigma receptors modulate K+ channels in rodent neurohypophysis.
Previous work suggested that dopamine type 4 (D4) receptors modulate neurohypophysial K+ current, so this study initially tested the role of dopamine receptors. Experiments using transgenic mice lacking D2, D3 or D4 receptors indicated that the reduction of K+ current by PPHT and U101958 (ligands thought to be selective for dopamine receptors) is not mediated by dopamine receptors. The sensitivity of the response to U101958 (a drug that binds to D4 receptors) was the same in both wild-type and D4 receptor-deficient mice.
Experiments with other ligands revealed a pharmacological signature inconsistent with any known dopamine receptor. Furthermore, dopamine itself (at 100 μm) had no effect. Thus, despite the activity of a number of putative dopamine receptor ligands, dopamine receptors play no role in the modulation of neurohypophysial K+ channels.
Because of the negative results regarding dopamine receptors, and because some of the dopamine receptors ligands used here are known to bind also to sigma receptors, experiments were conducted to test for the involvement of sigma receptors. In rat neurohypophysis the sigma receptor ligands SKF10047, pentazocine, and ditolylguanidine all reversibly inhibited K+ current in a concentration-dependent fashion, as did haloperidol and apomorphine (ligands that bind to both dopamine and sigma receptors). The activity of these and other ligands tested here matches the reported binding specificity for sigma receptors.
Fifteen candidate endogenous sigma receptor ligands, including biogenic amines (e.g dopamine and serotonin), steroids (e.g. progesterone), and peptides (e.g. neuropeptide Y), were screened for activity at the sigma receptor. All were without effect.
Haloperidol reduced K+ current proportionally at all voltages without shifting the voltage dependence of activation and inactivation. Sigma receptor ligands inhibited current through two distinct K+ channels, the A-channel and the Ca2+-dependent K+ channel. In rat, all drugs reduced current through both channels proportionally, suggesting that both channels are modulated by a single population of sigma receptors. In contrast, mouse peptidergic nerve terminals either have two receptors which are sensitive to these drugs, or a single receptor that is differentially coupled to ion channel function.
The inhibition of voltage-activated K+ current by sigma receptors would be expected to enhance the secretion of oxytocin and vasopressin from the neurohypophysis.
Nerve terminals are an important site of action for neurotransmitters. Presynaptic receptors can exert either positive or negative control over neurosecretion, by mechanisms that involve either the modulation of ion channels in the nerve terminal plasma membrane or the modification of proteins mediating exocytosis (Thompson et al. 1993; Jackson, 1995). The neurohypophysis contains peptidergic nerve terminals amenable to intracellular recording techniques (Lemos & Nowycky, 1989; Bourque, 1990; Jackson et al. 1991) and, as such, provides a powerful system for investigating the chemical regulation of neurosecretion. The nerve terminals of the neurohypophysis release oxytocin and vasopressin, and these two neuropeptides define two populations of nerve terminals present in roughly equal numbers. A number of substances have been shown to modulate the release of neurohypophysial peptides (Bondy et al. 1989; Crowley & Armstrong, 1992), and the ion channels of the neurohypophysis present a clear target for the regulation of secretion. K+ channel inactivation has been implicated in the broadening of action potentials and the frequency-dependent facilitation of release (Jackson et al. 1991). Ca2+ entry through Ca2+ channels and subsequent activation of Ca2+-activated K+ channels raises the threshold for action potential generation and can contribute to the use-dependent depression of release (Bielefeldt & Jackson, 1993).
The catecholamine dopamine is known to modulate neurotransmitter release in the brain (Grace & Bunney, 1985). The effects of dopamine on release from the neurohypophysis have been studied, but the results are conflicting (Crowley & Armstrong, 1992). Dopamine has been reported both to enhance (Bridges et al. 1976) and inhibit (Barnes & Dyball, 1982) oxytocin release from isolated neurointermediate lobe; enhancement of oxytocin release appeared to be under the control of a dopamine type 1 (D1) receptor (Crowley et al. 1991). Dopamine-containing fibres from the arcuate and periventricular nuclei descend along the neural stalk and terminate in the posterior and intermediate lobes of the pituitary, providing an anatomical basis for dopamine action at the nerve terminals of the neurohypophysis (Baumgarten et al. 1972; Holzbauer & Racke, 1985). Drugs with activity at dopamine receptors influence circulating levels of neurohypophysial hormones in animals (Melis et al. 1990; Crowley et al. 1991; Crowley & Armstrong, 1992). However, the interpretations of these results are complicated by the different potencies displayed by these drugs on different dopamine receptor subtypes, and by the difficulty of distinguishing direct actions on peptidergic nerve terminals from actions within the integrative circuitry of the hypothalamus.
Recent work from this laboratory has shown that certain dopamine receptor ligands inhibit voltage-activated K+ current (IK) within the nerve terminals of the rat neurohypophysis (Wilke et al. 1998). In an attempt to identify the receptor, a number of drugs were tested. The broad-spectrum D2/D3/D4 receptor agonist 2-(N-phenylethyl-N-propyl) amino-5-hydroxytetralin (PPHT) was effective, but the D1/D5 receptor agonist SKF38393 was not. The response to PPHT was blocked by the broad-spectrum dopamine receptor antagonist eticlopride. Moreover, the finding that RBI257, a recently developed D4 receptor-specific antagonist, blocked responses to PPHT appeared to support the hypothesis that inhibition of IK resulted from the activation of a D4 receptor (Wilke et al. 1998). However, another compound U101958, also developed as a D4 receptor antagonist (TenBrink et al. 1996), was found to show agonist activity. Furthermore, the dopamine receptor agonist quinpirole (at 100 μm) had no effect (Wilke et al. 1998).
The pharmacological profile of the receptor mediating these responses is not consistent with results from cell lines transfected with the human D4 receptor cDNA (Chabert et al. 1994; Liu et al. 1996; Watt & Neve, 1998). These observations raised questions about the identity of the receptor, and led to the consideration of factors that could complicate the interpretation of experiments with dopamine receptor ligands. One important problem is that many of the pharmacological probes used to investigate dopamine receptor function also bind to sigma receptors. Although the sigma receptor was initially classified as a novel opioid receptor (Martin et al. 1976), subsequent studies demonstrated that the sigma binding site represents a distinct pharmacological entity (Su, 1982). Sigma receptor ligands comprise a diverse group of chemically unrelated compounds (Su, 1993), including ditolylguanidine (DTG), pentazocine, and N-allylnormetazocine (SKF10047; the drug for which this receptor was initially named). One of the most widely used sigma receptor ligands, haloperidol, is also well known as a dopamine receptor antagonist. Other drugs known to bind to both sigma and dopamine receptors include apomorphine, chlorpromazine, and PPHT. The rat pituitary expresses sigma receptor binding sites (Wolfe et al. 1989), thus making plausible the hypothesis that the inhibition of neurohypophysial K+ current is mediated by sigma receptors.
We pursued these issues first by conducting experiments to rule out the involvement of dopamine receptors. Drug actions were tested in genetically altered mice which lack D2, D3, or D4 receptors. The results of these experiments are presented here, and show that wild-type mice and mice lacking dopamine receptors responded similarly to putative dopamine receptor agonists. With dopamine receptors thus ruled out, a series of experiments were conducted to test the role of sigma receptors. The actions of a large number of ligands were tested and found to inhibit neurohypophysial IK. The pharmacological signature of this response was consistent with known sigma receptor selectivity. These experiments support the conclusion that rat and mouse neurohypophyses contain sigma receptors coupled to K+ channels, implying that sigma receptor activation can modulate the release of oxytocin and vasopressin.
METHODS
Animals
Experiments were carried out in accordance with the National Institutes of Health guide for the care and use of laboratory animals. Animals were housed under a 12 h light-dark cycle with free access to water and food. Experiments were performed on Sprague-Dawley rats (males weighing ∼250 g) and wild-type (C57BL/6J) and transgenic mice. Transgenic mice were prepared by homologous recombination. Mice lacking the D4 receptor have recently been described by Rubinstein et al. (1997). D2 receptor-deficient mice have been described by Kelly et al. (1997). D3 receptor-deficient mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Appropriate wild-type strains were examined as controls for each of these mutants.
Tissue preparation
Pituitary slice preparation followed previously published methods (Jackson et al. 1991; Jackson, 1993) for both rat and mouse. Animals were rendered unconscious by a rising concentration of CO2 and then decapitated. The brain was removed and discarded. The cranium was immediately filled with ice-cold artificial cerebrospinal fluid (ACSF), consisting of (mM): 115 NaCl, 4.0 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 1 MgCl2, and 10 glucose, saturated with 95% O2-5% CO2. With the aid of a binocular dissecting microscope, the entire pituitary was removed from the calvarium and glued to a plastic block. The posterior portion of the gland was sliced at a thickness of 75 μm with a Lancer Vibratome, and then stored at 21-24°C in 95% O2-5% CO2-saturated ACSF. All tissue was used within 3 h of harvest. Because of the smaller size of the mouse pituitary compared with the rat, only two or three slices were obtained per animal.
Electrophysiology
Recordings were made from tissue slices superfused with 95% O2-5% CO2-bubbled ACSF. Individual nerve terminals at the slice surface were located with an upright differential interference contrast microscope (Diastar, Reichert Jung) equipped with a Zeiss × 40 water immersion, long working distance objective lens. The structures from which recordings were made are roughly spherical and range in diameter from 5 to 15 μm. Neurobiotin labelling in this preparation has shown that these structures are large axonal swellings (Jackson, 1993), and capacitance measurements revealed Ca2+-triggered exocytosis (Hsu & Jackson, 1996) to demonstrate a defining functional property of nerve terminals.
Slices prepared from mice had large nerve terminals, as in rat, but these were seen less frequently, thus making patch recording somewhat more difficult. Patch pipettes were fabricated from thin-walled aluminosilicate glass, and the pipette shanks were coated with Sylgard to reduce electrode capacitance (Hamill et al. 1981). Recordings were made with an EPC-9 patch-clamp amplifier interfaced to a Macintosh computer. Data acquisition, voltage control, and analysis were carried out with the computer program Pulse + Pulse Fit (Instrutech, Inc., Great Neck, NY, USA).
Whole-terminal IK was recorded under voltage-clamp mode, using patch pipettes filled with 130 mM KCl, 10 mM EGTA, 2 mM MgCl2, 4 mM MgATP, 300 μm NaGTP, and 10 mM Hepes, at pH 7.3. Prior to contact with the cell membrane, pipette resistances ranged from 3 to 6 MΩ. Immediately after breaking in, cell capacitance and series resistance were determined with the transient cancellation feature of the EPC-9. In cases where the initial series resistance exceeded 15 MΩ, this value was partially compensated electronically.
Drug application
All drugs used in these experiments were obtained from either Sigma or Research Biochemicals International. Compounds were dissolved in ACSF at the desired concentrations and either superfused onto the preparation at a rate of 2-4 ml min−1 with a simple gravity-feed system, or added directly to the bathing solution by pipette injection (see figure legends). Prior to adding drugs, IK was recorded at 15 s intervals for 1-3 min to verify the stability of the baseline. Current was also recorded after drug removal to demonstrate reversibility. In experiments with highly lipophilic drugs like PPHT, the agent was first dissolved in DMSO, and then diluted into ACSF to obtain the desired drug concentration. The final concentration of DMSO never exceeded 0.1%. Vehicle (0.1% DMSO in ACSF) has been tested and found to have no effect on whole-terminal IK.
Data analysis
Current recordings were analysed on a Macintosh computer with Pulse + PulseFit. This program was used to fit current decays to a double exponential function. Concentration-response and current- voltage plots were fitted using Origin software (Micro-Cal, Northampton, MA, USA) on a personal computer. Simple statistical analyses were performed on data exported from Pulse, using Microsoft Excel software. When arithmetic means were computed they were presented with the standard error of the mean. Probabilities for statistical significance were based on Student's t test, with P < 0.05 taken as statistically significant.
RESULTS
Modulation of K+ channels in mouse neurohypophysis
All nerve terminals tested in neurohypophysial slices prepared from C57BL/6J wild-type or mutant mice displayed a prominent IK in response to depolarizing pulses under voltage clamp (Fig. 1). A voltage step from -80 mV to 10 mV rapidly activated this current. IK then declined with a double exponential decay while the test potential was held constant at 10 mV for 500 ms. The temporal characteristics of mouse IK resembled those described previously in rat neurohypophysial nerve terminals (Bielefeldt et al. 1992). Pharmacological and biophysical studies in the rat established that this current is a flux of K+ through three distinct types of K+ selective channels. Current through two of these channels is readily distinguished on the basis of the rate of inactivation at positive potentials. The rapid component of decay reflects inactivation of an A current (IA), and the slow component of decay reflects inactivation of a Ca2+-activated K+ current (IBK) (Bielefeldt et al. 1992). (Current through the neurohypophysial Ca2+-activated K+ channel can be seen even with 10 mM EGTA in the pipette filling solution because this channel has an unusually high sensitivity to Ca2+ (Bielefeldt & Jackson, 1993).)
Figure 1. Outward IK in voltage-clamped mouse neurohypophysial nerve terminals of wild-type C57BL/6J mice.
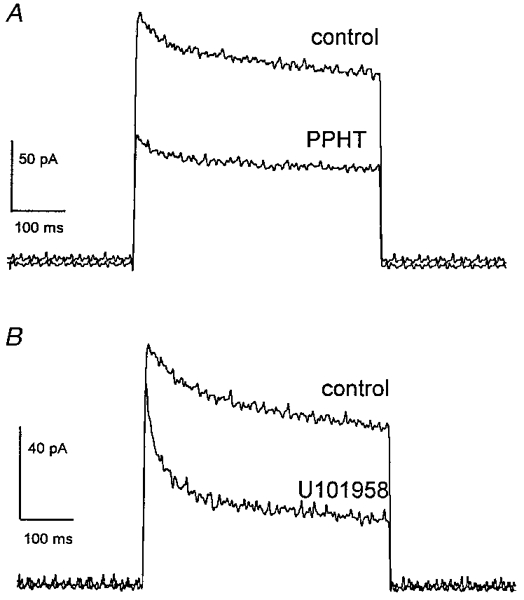
A holding potential of -80 mV was maintained before and after voltage steps of 500 ms to 10 mV. In each set of tracings, a prominent outward current was rapidly activated, and then decayed over time. A, current recorded before (control) and after a 3 min exposure to PPHT (30 μm). B, current recorded before and after a 3 min exposure to U101958 (100 μm). PPHT reduced both peak and sustained IK. U101958 reduced sustained IK, but had little effect on peak IK.
Both PPHT (an agonist at the closely related D2/D3/D4 receptors; Kebabian et al. 1997) and U101958 (a D4 receptor-specific ligand; TenBrink et al. 1996) inhibited neurohypophysial IK (Fig. 1). PPHT inhibited both peak and sustained (final) components of IK, while U101958 mainly reduced the sustained component. Since the action of these two drugs appeared different, the inhibition of the two principal types of K+ channels underlying IK was quantified by fitting current decays to a double exponential function:
| (1) |
where I denotes current, I0 denotes sustained current at the endpoint of inactivation, I1 and τ1 denote the amplitude and time constant of the rapidly decaying current, respectively, and I2 and τ2 denote the amplitude and time constant of the slowly decaying current, respectively. The time constants of decay remained essentially unchanged in 30 μm PPHT, but the amplitudes were reduced by similar percentages. The fast component, I1, was reduced to 50 ± 8% of control and the slow component, I2, was reduced to 56 ± 8% of control (n = 4). In contrast, 100 μm U101958 inhibited the slow component to 26 ± 2% of control, but left the fast component essentially unchanged at 119 ± 14% of control (n = 5).
The different effects of PPHT and U101958 on peak and sustained IK (Fig. 1) are consistent with the different effects on the rapidly and slowly decaying exponential components. The percentage reductions in peak and sustained current induced by these drugs are shown in Fig. 2. PPHT reduced both peak and sustained IK, whereas U101958 reduced only sustained IK. Both peak IK and I1 primarily reflect the rapidly inactivating IA. The failure of U101958 to reduce peak IK or I1 reflects the fact that this drug does not modulate IA. In contrast, sustained IK and I2 reflect current flowing primarily through the Ca2+-activated K+ channel, and the reduction of sustained IK and I2 by both drugs is consistent with an effect of both drugs on IBK. The different actions of these two drugs suggest that there are two receptors, distinguished by their ligand specificity and their coupling to different K+ channels. A single receptor mediating responses to both drugs may also be possible, but this would require variable coupling to two different transduction pathways, depending on the choice of agonist. The differential effect of these drugs stands in contrast with results in rat, where several drugs including PPHT and U101958 reduced both components of IK in parallel (see results below and Wilke et al. 1998).
Figure 2. Dopamine agonist selectivity profile in wild-type mice.
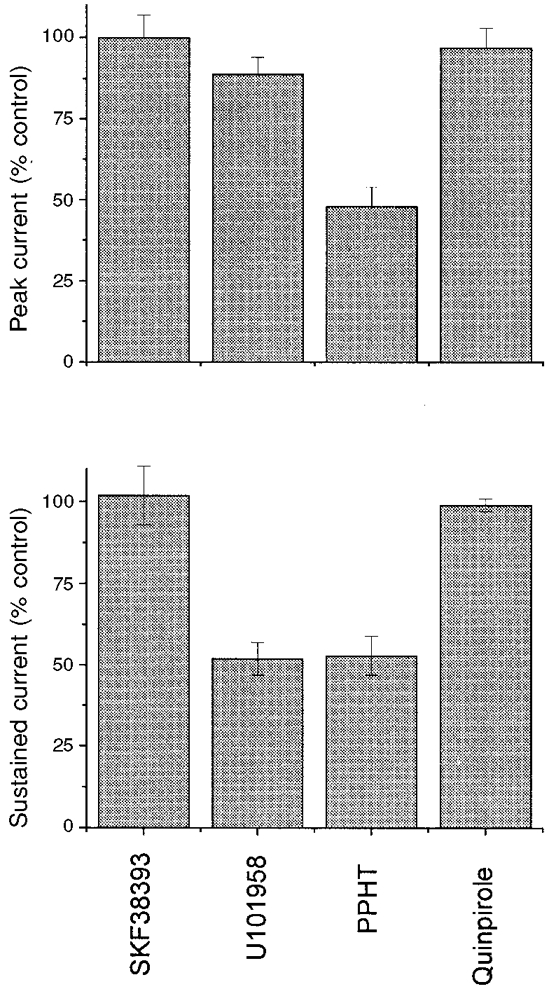
Sustained neurohypophysial IK was monitored in the presence of various dopamine receptor agonists. IK was evoked using the same pulse protocol as employed in Fig. 1. After establishing a predrug baseline, nerve terminals were superfused with 100 μm SKF38393 (D1/D5), 30 μm U101958 (D4), 30 μm PPHT (D2/D3/D4) or 100 μm quinpirole (D2/D3/D4) for 3 min. Sustained IK at that time was normalized to the predrug control IK for each nerve terminal. Data represent means ±s.e.m. for 3-6 experiments.
Actions of dopamine receptor ligands
To gain insight into the pharmacological identity of the receptors that mediate these responses, two additional dopamine receptor ligands were evaluated for their effects on peak and sustained IK (Fig. 2). To test the possible involvement of D1 and D5 receptors, we employed SKF38393, an agonist that binds to these two closely related receptors with high affinity (Kebabian et al. 1997). At 100 μm this drug had no effect on either peak or sustained IK (Fig. 2), suggesting that D1 and D5 receptors do not modulate K+ channels in this preparation. D2, D3 and D4 receptors are closely related and exhibit a considerable amount of overlap in their ability to bind various ligands (Civelli et al. 1993; Sibley et al. 1993). However, quinpirole, an agonist known to activate each of these three receptors, had no effect at 100 μm on either peak or sustained IK in the mouse neurohypophysis (Fig. 2). This observation is somewhat surprising, since quinpirole is thought to activate the same dopamine receptors as PPHT (Kebabian et al. 1997). Thus, the responses to these drugs are not consistent with known dopamine receptors.
We also tested the D4 receptor antagonist L745870 (Patel et al. 1997), against the inhibition of IK by PPHT. Although this compound has a very high affinity for D4 receptors (Ki, 0.43 nM), 30 μm L745870 failed to alter the inhibition of IK by 30 μm PPHT (EC50, 1.8 μm; Wilke et al. 1998) in three nerve terminals tested (Fig. 3A). These disparate results with U101958 and L745870 suggest that either the receptors are not D4, or the drugs are not as selective as previously thought.
Figure 3. Actions of L745870 and dopamine.
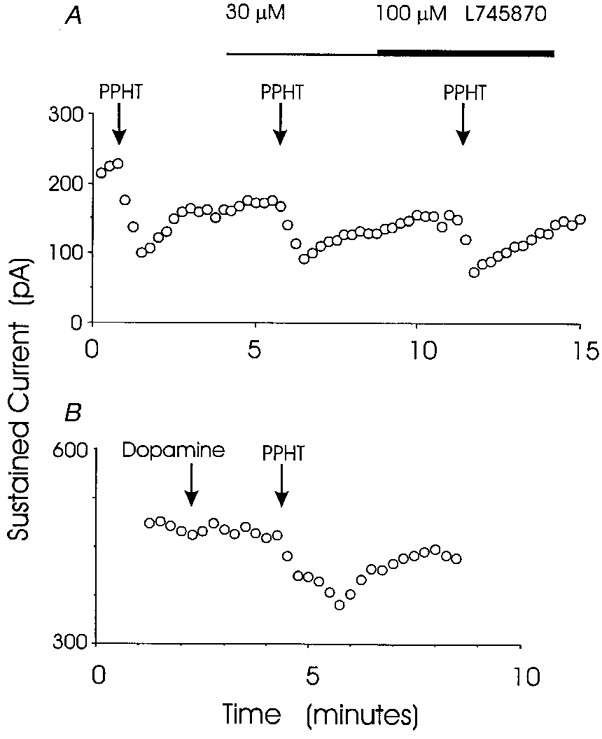
A, the D4 receptor-specific antagonist L745870 had no effect on the reduction of sustained IK by 30 μm PPHT. In this experiment no inhibition of the PPHT response was seen with either 30 or 100 μm L745870. Similar results were obtained in 3 nerve terminals. In the presence of 30 μm L745870, PPHT reduced the sustained IK to 62 ± 8% of control, which is similar to the reduction of sustained IK in Fig. 2. B, dopamine (100 μm) failed to reduce sustained IK. In the same nerve terminal 10 μm PPHT reduced IK. These experiments followed the protocol used in Fig. 2. Neither peak nor sustained IK were reduced by dopamine in 4 nerve terminals (see text).
As a final test for dopamine receptors, we applied 100 μm dopamine to four nerve terminals. The result of one of these experiments is shown in Fig. 3B. Dopamine had no effect on peak or sustained IK, while 10 μm PPHT reduced both components in the same recording. In the presence of dopamine, peak IK was 96 ± 6% of control and sustained IK was 98 ± 8% of control (n = 4). When taken together, these results suggest that the responses in mouse neurohypophysial nerve terminals to putative dopamine receptor-specific ligands are not mediated by dopamine receptors.
Genetic analysis of dopamine receptors
The pharmacological experiments described above gave results inconsistent with known dopamine receptor properties. For this reason we performed experiments in transgenic mice lacking specific types of dopamine receptors. The different molecular forms of dopamine receptors are encoded by five distinct genes (Civelli et al. 1993; Sibley et al. 1993). Three of these, denoted D2, D3, and D4, are structurally, as well as pharmacologically, closely related and are known to respond to PPHT. Therefore these three receptors were selected for genetic analysis. In mice lacking the D4 receptor, U101958 was found to inhibit sustained IK as effectively as in wild-type mice. The concentration- response curves for this agonist in nerve terminals from these two mouse strains are shown in Fig. 4. These plots of sustained IKversus U101958 concentration were fitted to the following equation:
| (2) |
where E denotes IK as percentage of control, C denotes the concentration of U101958, Emax denotes the maximal inhibitory effect, and EC50 denotes the value of C producing a half-maximal response. Fitting to this expression yielded EC50 values of 34.1 ± 3.5 μm in wild-type and 26.4 ± 5.6 μm in D4 receptor-deficient mice. These values are not significantly different (P = 0.14), indicating that the receptors are indistinguishable in terms of sensitivity to U101958. Emax values in each case were essentially zero, indicating that the activation of this receptor completely eliminated sustained IK seen at the end of 500 ms voltage pulses. The similar action of U101958 in wild-type and D4 receptor deficient-mice indicates that the response to U101958 is not mediated by a D4 receptor.
Figure 4. U101958 concentration-response curves in nerve terminals from wild-type and D4 receptor-deficient mice.
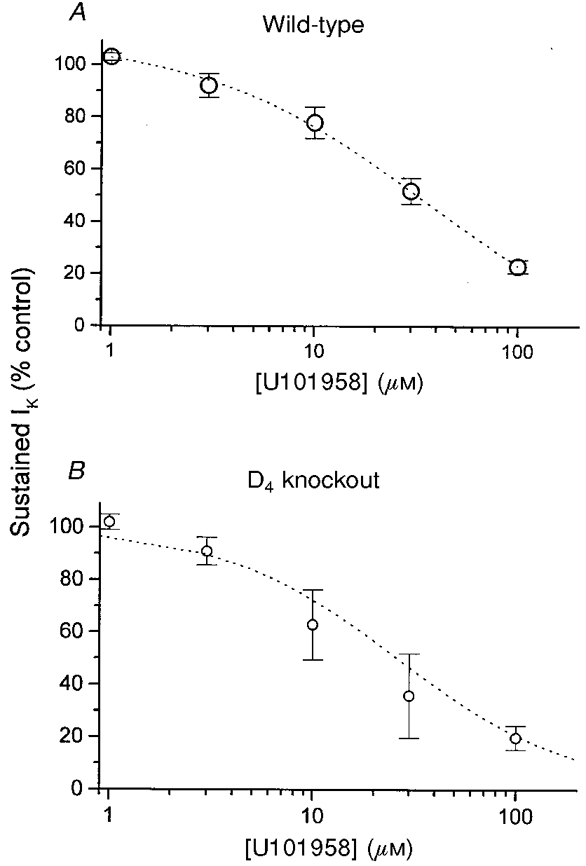
At each concentration, IK was recorded before and after addition of the putative D4 receptor-selective ligand U101958. The voltage pulse protocol for activating IK was identical to that used in Fig. 1. Sustained IK in the presence of U101958 was normalized to the predrug control IK for each nerve terminal. Data are expressed as means ±s.e.m. for experiments from 3 wild-type (A) and 3 D4 receptor-deficient mice (D4 knockout, B). In each case, the best fit to eqn (2) is plotted (dotted curve) (see text for values).
Since PPHT reduced both rapidly and slowly inactivating components of IK in mouse neurohypophysial nerve terminals (Fig. 1A), the response to this drug provides the broadest test for the presence of receptors coupled to K+ channels. Like U101958, PPHT inhibited IK in mice lacking the gene for the D4 receptor (Fig. 5). The reduction of the two exponential components of IK was similar to that seen in wild-type mice (Fig. 1). As noted above, the reduction of the fast component of IK was to 50% of control and the reduction of the slow component was to 56% of control. In three recordings from D4 receptor-deficient mice fast and slow components were reduced to 41 ± 13 and 57 ± 14% of control, respectively. To determine whether the reduction of IK by PPHT was mediated by related dopamine receptors, we tested the actions of this drug in D2 and D3 receptor-deficient mice. Both peak and sustained IK were reduced in each type of animal by amounts similar to that seen in wild-type mice. Fits of eqn (1) showed that in D2 receptor-deficient mice PPHT reduced the fast and slow components of IK to 53 ± 6 and 61 ± 7% of control, respectively (n = 3; Fig. 5). In three recordings from D3 receptor-deficient mice the fast and slow components were reduced to 55 ± 7 and 66 ± 4% of control, respectively. Thus, the receptors mediating the inhibition of the two components of IK are not among the known PPHT-sensitive dopamine receptors. It should be noted that in all three strains of dopamine receptor-deficient mice, the two components of IK decayed with similar time constants, indicating that K+ channel properties were not altered by these genetic manipulations.
Figure 5. The response to PPHT was unaltered in nerve terminals from dopamine receptor-deficient mice.
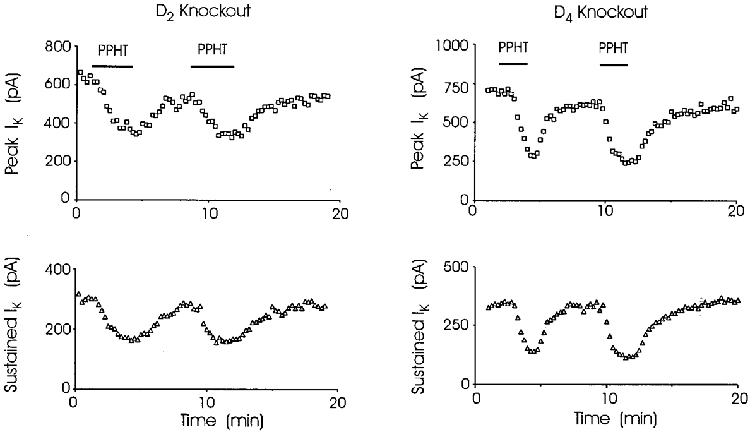
IK was recorded at 15 s intervals before, during and after repeated challenge with agonist in D2 or D4 receptor-deficient mice. Superfusion of nerve terminals with 30 μm PPHT reduced both peak (□) and sustained (▵) IK. Each time the effect was reversed when PPHT was washed from the medium. Reduction of rapidly and slowly inactivating components of IK in dopamine receptor-deficient mice was indistinguishable from wild-type (see text). Bars indicating application of PPHT in upper panels apply also to lower panels.
Pharmacological analysis of sigma receptors in rat neurohypophysis
With plausible candidate dopamine receptors eliminated by both pharmacological and genetic experiments, we turned to the hypothesis that sigma receptors mediate the modulation of K+ channels. The above experiments were conducted in mice to enable use of transgenic animals. The remaining experiments were conducted in rats. Sigma receptor ligands reduced IK in rat neurohypophysial nerve terminals, and two examples are shown in Fig. 6. DTG is a highly selective sigma receptor ligand, exhibiting no cross-reactivity with other known receptor types; haloperidol is a commonly used antipsychotic agent that binds to both sigma and dopamine receptors (Su, 1993). Figure 6 demonstrates that both of these compounds reduced the peak and sustained components of neurohypophysial IK. Figure 7 demonstrates that the reduction of IK by DTG was reversed by drug removal. In no instance did a known sigma receptor ligand fail to reduce IK, and more than 100 nerve terminals were tested. Thus, sigma receptors are present on both oxytocin and vasopressin nerve terminals.
Figure 6. Sigma receptor ligands inhibit rat neurohypophysial IK.
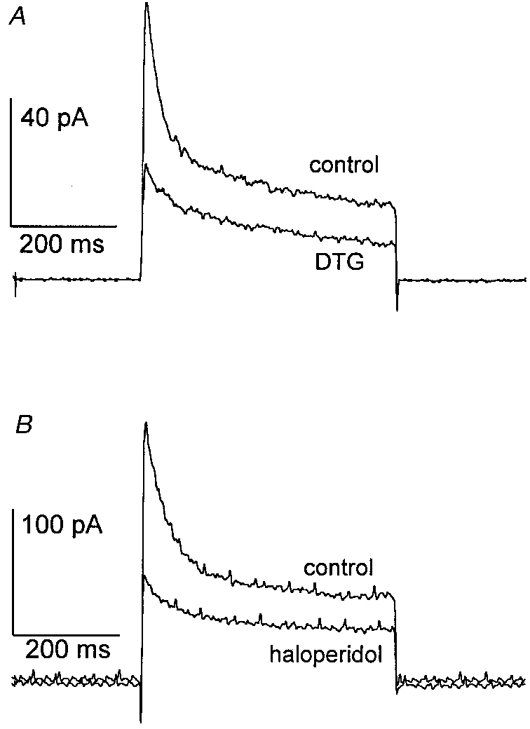
The pulse protocol used to activate IK was the same as in Fig. 1. A, during exposure to 100 μm DTG, a marked reduction in both peak and sustained IK was observed. B, exposure to 100 μm haloperidol produced a similar reduction in peak and sustained IK.
Figure 7. Reversibility of sigma receptor-mediated inhibition.
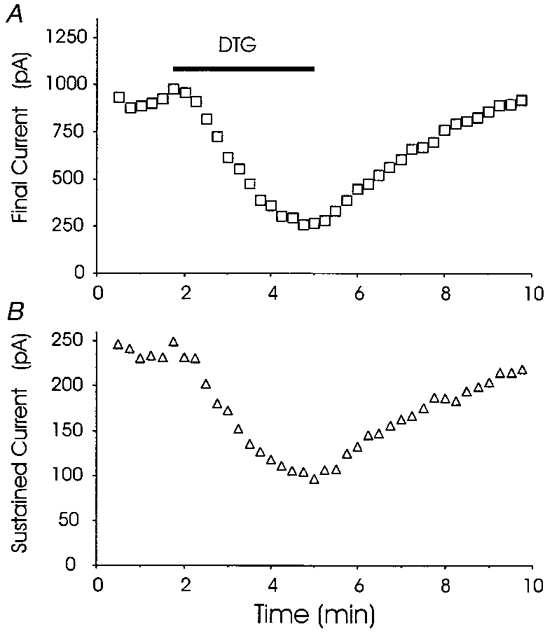
Peak (A) and sustained (B) IK were recorded in rat nerve terminal at 15 s intervals before, during and after challenge with 100 μm DTG. The timing of DTG application is indicated by the bar in A (applies also to B). The stimulation protocol for each voltage pulse was identical to that described in Fig. 1. Superfusion with 100 μm DTG reversibly reduced both peak and sustained IK.
To evaluate the effect of sigma receptor ligands on different K+ channel subtypes, time constants and individual current amplitudes were determined by fitting current decay to a double exponential function (eqn (1)). As in mouse, the time constants remained essentially unchanged, and the amplitudes of both the fast and slow components were reduced by 100 μm haloperidol (fast component reduced to 50 ± 5% of control, slow component reduced to 70 ± 5% of control; n = 7). As noted above, these two components correspond well with the transient IA and the Ca2+-dependent IBK (Bielefeldt et al. 1992). Thus, in rat as in mouse both of these K+ channel subtypes were modulated.
The effect of haloperidol is concentration dependent. Using an experimental protocol similar to that employed in Fig. 6, current was recorded before and after the addition of various concentrations of haloperidol. As described above for experiments in mouse, changes in IA and IBK were estimated by monitoring peak and sustained components of whole-terminal IK. These were plotted as a function of haloperidol concentration and fitted to eqn (2) (Fig. 8). Reduction of both peak and sustained IK showed similar dependencies on haloperidol concentration. Parameter values for peak and sustained IK, respectively, obtained from these fits were: Emax, 58 ± 4 and 66 ± 3%; EC50, 42 ± 9 and 64 ± 12 μm. The similar EC50 values (P = 0.10) suggest that a single sigma receptor simultaneously modulates both IA and IBK.
Figure 8. Haloperidol concentration-response curves in rat.
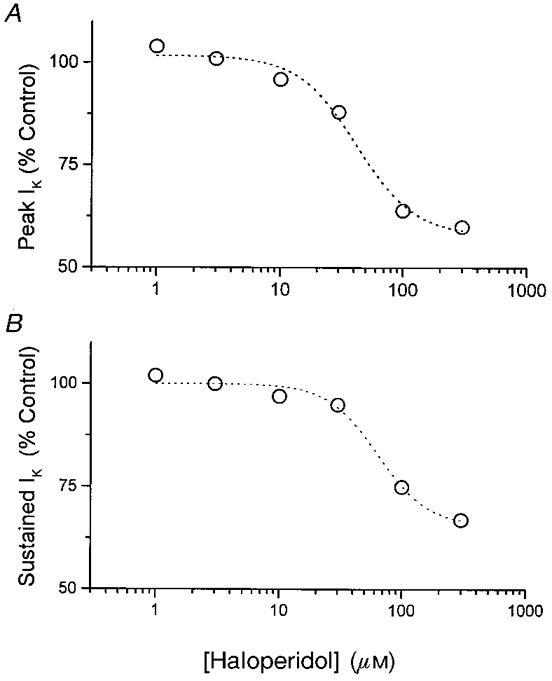
Peak (A) and sustained (B) IK were measured in the presence of haloperidol, and normalized to predrug controls for each nerve terminal. Data are expressed as means for 3-6 nerve terminals. Dotted curves are best fits to eqn (2) (see text for parameter values).
To characterize the pharmacology of this neurohypophysial receptor more completely, a range of sigma receptor ligands was screened for the ability to reduce peak and sustained IK (Fig. 9). All of the standard sigma receptor ligands tested (DTG, SKF10047, pentazocine, and haloperidol) reduced both components of IK. Binding studies have demonstrated that the non-selective dopamine receptor agonist apomorphine also interacts with sigma receptors (Su, 1993), and a reduction in IK in response to this drug was seen here (Fig. 9). Because of its common clinical utility in a variety of neuroendocrine disorders, we also tested the effect of another dopaminergic agent bromocriptine on these peptidergic nerve terminals. At concentrations of up to 100 μm this agent had no effect on rat neurohypophysial IK (Fig. 9), and the same concentrations of bromocriptine failed to alter PPHT-induced inhibition of IK (n = 2; data not shown), suggesting that it does not interact with the neurohypophysial sigma receptor.
Figure 9. Sigma receptor ligand efficacies in rat.
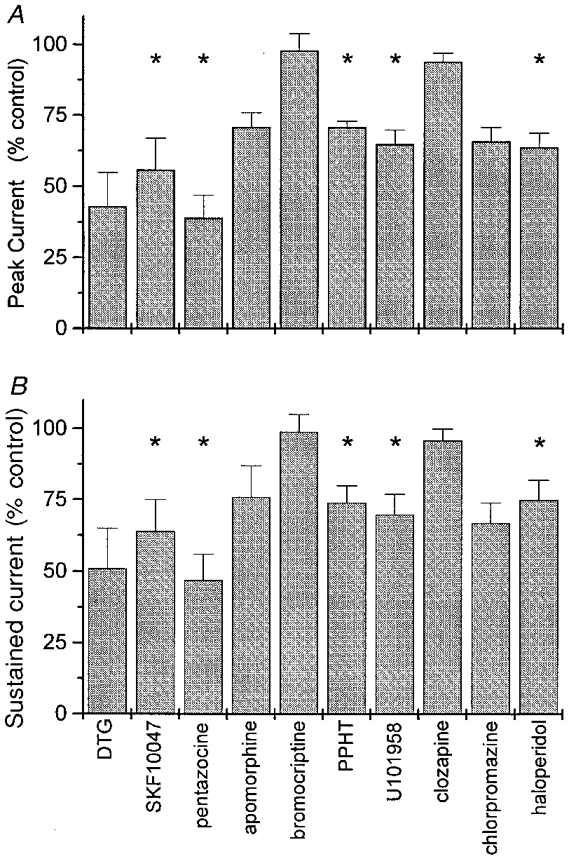
Peak (A) and sustained (B) IK were measured in the presence of various sigma receptor ligands, using the pulse protocol of Fig. 1. After establishing a stable baseline, nerve terminals were superfused with various drugs (all at 100 μm) for 3 min and reassessed. Peak and sustained IK were normalized to the predrug control for each nerve terminal. Data represent means ±s.e.m.; *P < 0.05 for the probability that the mean is 100%. Only 3 nerve terminals were tested with DTG, apomorphine, and chlorpromazine, so statistical significance was not tested even though the effects were generally large. For all other recordings 4-6 nerve terminals were tested.
Butyrophenones such as haloperidol are highly efficacious in the clinical treatment of various psychotic disorders (Creese et al. 1996), and this antipsychotic activity has been attributed to antagonist activity at neuronal dopamine receptors (Kebabian et al. 1997). We therefore screened two additional antipsychotic agents for activity at this receptor. Chlorpromazine (an older phenothiazine antipsychotic) had agonist efficacy similar to that of haloperidol (Fig. 9). Clozapine (a newer atypical antipsychotic agent) had no measurable effect when given alone (Fig. 9), and it failed to block the sigma receptor-mediated inhibition of IK induced by 30 μm PPHT (63 ± 5%, n = 3, data not shown, versus 55 ± 12% in the absence of clozapine).
To provide an additional test of the hypothesis that both IA and IBK are modulated by the same population of sigma receptors in the rat neurohypophysis, we compared the reduction in peak current with the reduction in sustained current for a number of sigma receptor ligands (Fig. 10). Linear regression revealed that these data are highly correlated (r = 0.97), indicating that the parallel inhibition of IA and IBK observed with haloperidol (Fig. 8) can be extended to the entire panel of sigma receptor ligands tested in this preparation.
Figure 10. Plot of reduction in peak IKversus reduction in sustained IK.
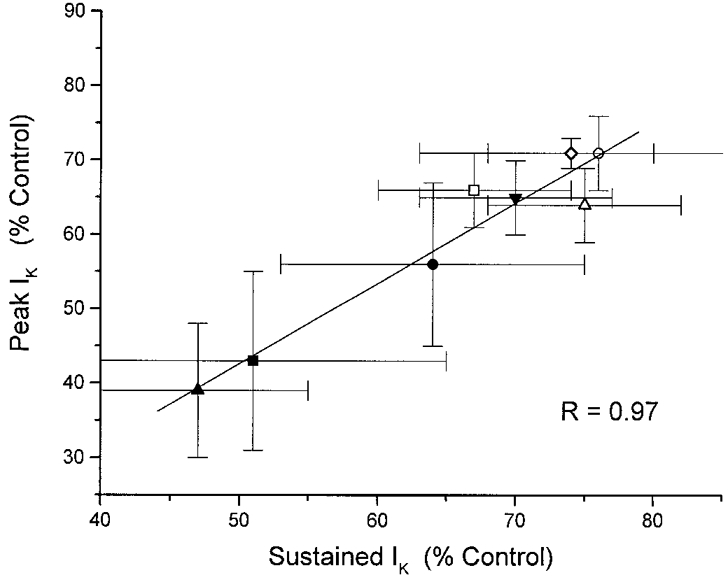
For each of the ligands shown in Fig. 9, peak IK is here plotted against sustained IK. Symbols: •, SKF10047; ○, apomorphine; ▪, DTG; □, chlorpromazine; ▴, pentazocine; ▵, haloperidol; ▾, U101958; ⋄, PPHT. Linear regression analysis revealed a statistically significant correlation (r = 0.97, which for 8 points gives P < 0.001 for the probability of a random sample), suggesting that the same population of sigma receptors is responsible for the modulation of both IA and IBK.
Tests of endogenous sigma receptor ligands
Studies conducted in the placenta have suggested that progesterone could be an endogenous ligand for the sigma receptor (Su et al. 1988; Ramamoorthy et al. 1995). This ovarian steroid has been shown to bind to sigma receptors with an apparent KD in the low micromolar range (Su et al. 1988). However, adrenal and ovarian steroids failed to produce an observable change in IK (Table 1). Progesterone alone (100 μm) had no effect on either peak (90 ± 3%) or sustained (98 ± 4%) neurohypophysial IK (n = 6). Furthermore, it had no effect on the current reduction induced by haloperidol (Fig. 11). Corticosterone, hydrocortisone, and dehydroepiandrosterone sulphate were also without effect (Table 1).
Table 1.
Endogenous substances tested which had no effect on peak and sustained IK
| Drug | Concentration | n |
|---|---|---|
| Dopamine | 100 μm | 3 |
| 1 mm | 1 | |
| Norepinephrine | 100 μm | 2 |
| Epinephrine | 100 μm | 2 |
| Serotonin | 100 μm | 3 |
| Progesterone | 100 μm | 6 |
| 1 mm | 1 | |
| DHEA-S | 100 μm | 2 |
| Hydrocortisone | 10 μm | 2 |
| Corticosterone | 10 μm | 2 |
| CRH | 10 nm | 3 |
| 30 nm | 4 | |
| 100 nm | 3 | |
| ANP | 100 nm | 2 |
| Angiotensin-II | 10 μm | 10 |
| Endothelin-I | 100 nm | 4 |
| Endothelin-III | 2 nm | 2 |
| 20 nm | 4 | |
| Interleukin-6 | 10 ng ml−1 | 5 |
| Neuropeptide Y | 50 μm | 3 |
Each compound was tested using the pulse protocol shown in Fig. 6 and found to have no effect on either peak or sustained IK. ANP, atrial natriuretic peptide; CRH, corticotrophin-releasing hormone; DHEA-S, dehydroepiandrosterone sulphate.
Figure 11. Progesterone had no pharmacological activity on rat neurohypophysial IK.
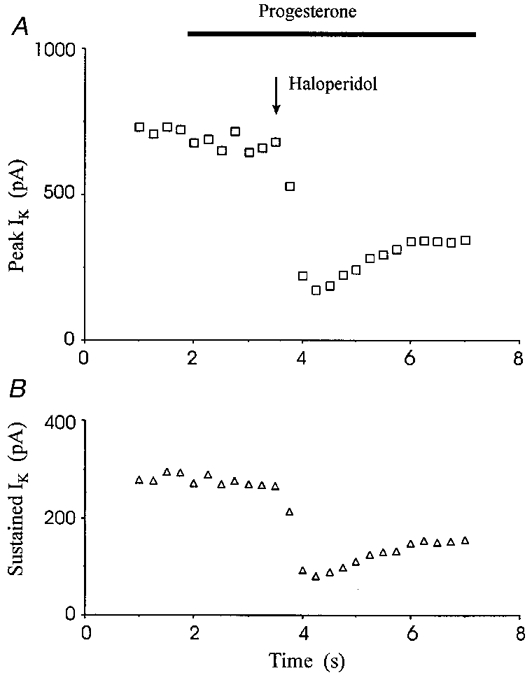
IK activated by the pulse protocol of Fig. 1 was recorded at 15 s intervals. Adding progesterone (100 μm) failed to alter peak or sustained IK, and in the presence of progesterone, haloperidol (100 μm) still reduced both components of IK.
Given the number of dopamine receptor ligands that interact with sigma receptors, we tested dopamine again in rat (at concentrations up to 1 mM), and saw no measurable effect (Table 1). Serotonin, norepinephrine and epinephrine were also tested (Table 1), along with the β-adrenergic receptor agonist isoproterenol (at 10 μm, 100 μm, and 1 mM); all were without significant effect.
Various neuropeptides have also been implicated as potential endogenous sigma receptor agonists (Ramamoorthy et al. 1995; Ault et al. 1998). One such ligand, neuropeptide Y (NPY), is known to modulate intestinal ion currents by acting at a haloperidol-sensitive binding site (Riviere et al. 1990; Colmers & Bleakman, 1991). Nonetheless, 100 μm NPY left both peak and sustained IK at 99 ± 4% and 101 ± 4%, respectively, of their pre-drug control values (n = 3). Additional peptide hormones and autocoids were tested for their ability to modulate neurohypophysial IK (Table 1). As with the steroids and amines discussed above, these peptides were without effect.
Voltage-clamp analysis
To investigate the mechanism by which sigma receptors modulate neurohypophysial IK, we studied the effect of haloperidol on the voltage dependence of K+ channel activation. Peak current was recorded before and after a 3 min exposure to 100 μm haloperidol. A current-voltage plot was then constructed using test pulses from -100 to 20 mV, (Fig. 12A). At all voltages tested, haloperidol reduced IK by proportional amounts, indicating that sigma receptor agonists do not produce their inhibitory effect on neurohypophysial IK by shifting the voltage dependence of channel activation.
Figure 12. Voltage-independent modulation of IK.
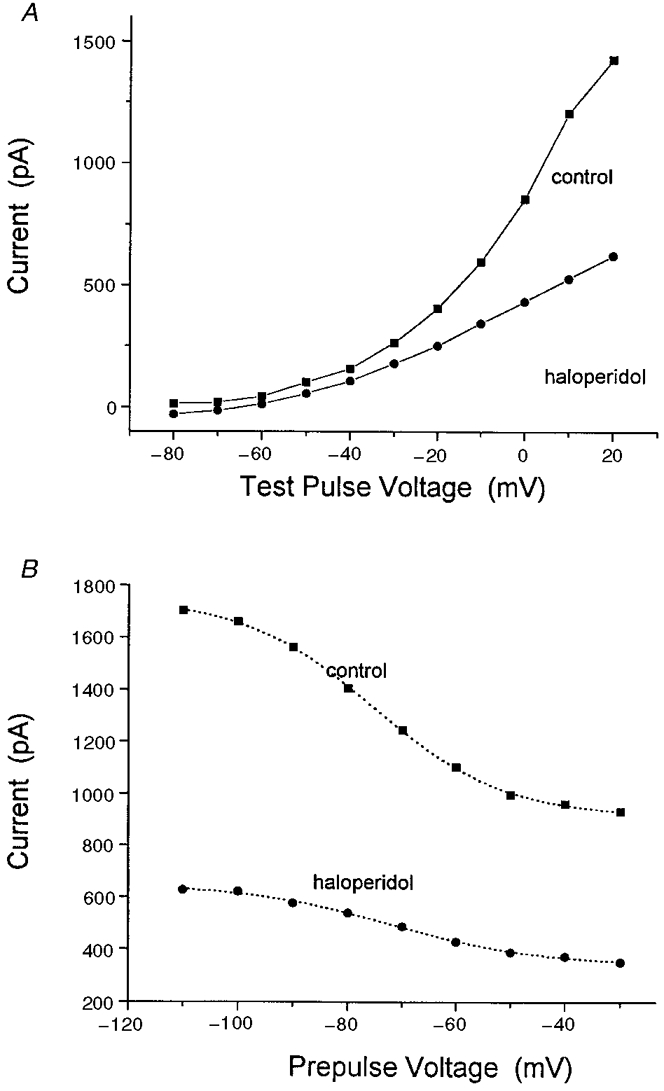
A, plot of neurohypophysial IKversus voltage. Nerve terminals were held at -80 mV and stepped to various voltages for 500 ms at 5 s intervals. Peak current was then plotted as a function of voltage (▪). After a 3 min exposure to 100 μm haloperidol (•), the same nerve terminals were again subjected to a repeat series of voltage pulses. Data points represent mean current from 3 separate experiments. B, voltage dependence of inactivation. Nerve terminals were held at various prepulse potentials (from -110 to -30 mV) for 500 ms, then stepped to 10 mV for an additional 500 ms. Peak current at 10 mV was then plotted as a function of prepulse voltage (▪) to assess the voltage-dependent inactivation of neurohypophysial K+ channels. After a 3 min exposure to 100 μm haloperidol each terminal was then subjected to the same test series, and peak current was again plotted against prepulse voltage (•). Data represent mean values from 3 separate experiments. Best fitting Boltzmann functions (eqn (3)) were drawn as dotted curves. Haloperidol failed to alter the voltage dependence of activation or inactivation (see text for parameter values).
The effect of haloperidol on channel inactivation was also studied. Following prepulse potentials varying from -110 to -30 mV, IK was activated by depolarizing voltage steps to 10 mV. Again, peak IK was recorded before and after a 3 min exposure to 100 μm haloperidol. The data were then averaged from three experiments, plotted as a function of prepulse voltage, and fitted to the following Boltzmann function:
| (3) |
All four parameters, Imin, Imax, κ and V½ were varied to achieve the best fit. Imin and Imax represent the current asymptotes at negative and positive prepulse potentials, κ is the steepness factor and V½ is the mid-point for voltage dependence. Under control conditions, κ= 12.1 ± 0.5 mV and V½= -75.4 ± 0.5 mV (Fig. 12B). After a 3 min exposure to 100 μm haloperidol, neither the steepness factor (κ= 13.7 ± 1.5 mV) nor the voltage mid-point (V½= -71.9 ± 1.3 mV) of inactivation had been significantly altered. Similar results were obtained previously with PPHT (Wilke et al. 1998). In summary, sigma receptor ligands appear to reduce current through neurohypophysial K+ channels without shifting the voltage dependence of activation or inactivation.
DISCUSSION
This study focused on the identity of a receptor coupled to K+ channels in rodent neurohypophysis. The initial phase of this work ruled out the involvement of dopamine receptors, and the second phase showed that IK is modulated by sigma receptors.
Lack of involvement of dopamine receptors
Earlier work from this laboratory showed that a number of ligands thought to be selective for dopamine receptors are active in the modulation of IK in the rodent neurohypophysis. PPHT and U101958 both inhibited neurohypophysial IK. Furthermore, eticlopride and RBI257 blocked the response (Wilke et al. 1998). The actions of PPHT and eticlopride suggested a role for one of the closely related D2, D3, or D4 receptors. Because U101958 and RBI257 bind to D4 receptors (Schlachter et al. 1995; Kebabian et al. 1997), these results were initially interpreted as evidence that D4 receptors modulate neurohypophysial IK. However, this view was clouded by the fact that another drug that binds to D4 receptors, quinpirole (100 μm), had no effect on rat neurohypophysial IK (Wilke et al. 1998). For this reason the hypothesis of dopamine receptor modulation of neurohypophysial IK was subjected to more critical tests, and those experiments described here led to a different conclusion. The present study showed that nerve terminals of transgenic mice lacking D2, D3 and D4 receptors had the same responses to PPHT as nerve terminals from wild-type mice. The similar sensitivity of IK to U101958 in wild-type and D4 receptor-deficient mice (Fig. 4) made it very unlikely that the expression of an alternative receptor allele is enhanced to substitute for the deficit. Furthermore, the broad-spectrum agonist quinpirole failed to elicit a response, the D4 receptor-specific antagonist L745870 failed to block responses to PPHT, and the modulation of IK by PPHT and U101958 could not be reproduced by dopamine itself. Thus, both pharmacological and genetic experiments ruled out the involvement of three dopamine receptors of known molecular structure in the modulation of neurohypophysial IK. The enhancement and inhibition of oxytocin release by dopamine (Bridges et al. 1976; Barnes & Dyball, 1982) therefore probably does not result from the modulation of neurohypophysial IK.
Involvement of sigma receptors
With genetic and pharmacological evidence against dopamine receptors, we explored the possibility that sigma receptors mediate the modulation of IK (Su, 1993). This receptor has been defined on the basis of binding properties, and has a high affinity for many dopamine receptor-specific ligands (Su, 1993). The pituitary has been shown to contain a high density of binding sites for sigma receptor-specific ligands (Wolfe et al. 1989). Sigma receptor ligands include compounds from several different biochemical and pharmacological categories. Apomorphine is a non-selective dopamine receptor agonist (Su, 1993). Haloperidol is a neuronal dopamine receptor antagonist used clinically in the treatment of psychosis (Creese et al. 1996). Both apomorphine and haloperidol exhibit nanomolar affinity for sigma receptors (Quirion et al. 1992; Seth et al. 1997). DTG and pentazocine bind to sigma receptors with a KD in the nanomolar range, but binding to dopamine receptors cannot be detected; SKF10047 also binds selectively to sigma receptors in preference to dopamine receptors (Su, 1993). The observation that all of these agents elicited a physiological response in the posterior pituitary, together with the studies eliminating dopamine receptors, thus identified the receptor responsible for the modulation of neurohypophysial IK as a sigma receptor.
These results indicate that responses to dopamine receptor-specific ligands must be interpreted with caution. For example, U101958 appears to be highly efficacious at a non-dopamine receptor. This observation will have to be taken into account in interpreting the effects of U101958 and other structurally related ligands in other preparations. It may mean that some results previously attributed to dopamine receptors are actually mediated by sigma receptors.
K+ channel modulation
Haloperidol has previously been shown to reduce a tonic outward current in NCB-20 cells, a hybrid neuronal cell line known to express a low affinity sigma receptor (Wu et al. 1991). Furthermore, both haloperidol and chlorpromazine reversibly inhibited voltage-dependent K+ current in PC12 cells (Nakazawa et al. 1995). Our observation that sigma receptor ligands modulate IK in neurohypophysial slices demonstrates that a similar sigma receptor-mediated response can be seen in native tissue.
The peptidergic nerve terminals of the rat neurohypophysis contain at least three distinct types of K+ channel (Bielefeldt et al. 1992), each of which could play a role in the regulation of neuropeptide release (Jackson et al. 1991; Bielefeldt & Jackson, 1993, 1994; Jackson, 1995). The data obtained in the present study demonstrated that sigma receptor ligands inhibit current through two of these channel types, an A-current channel (IA) and a Ca2+-dependent K+ channel (IBK). Various ligands had different efficacies in the inhibition of IK, but in rat the effects on IA and IBK were proportional (Fig. 10). As in our previous work with PPHT and U101958 (Wilke et al. 1998), haloperidol reduced both IA and IBK in the rat neurohypophysis. Both effects were concentration dependent with EC50 values that were similar. These results suggest that in rat the modulation of both types of K+ channel is mediated by a single molecular species of receptor.
While IA and IBK were reduced proportionally by sigma receptor ligands in the rat, a similar analysis in the mouse neurohypophysis produced a different result. As in the rat (Bielefeldt et al. 1992), mouse neurohypophysial IK can be resolved into two distinct kinetic components with different inactivation kinetics. However, in the mouse, IA and IBK were modulated to different degrees by PPHT and U101958. Both components of mouse neurohypophysial IK were reversibly inhibited by PPHT, but U101958 inhibited only the Ca2+-activated K+ channel. This differential regulation of IK by PPHT and U101958 may reflect the presence of two distinct sigma receptors within the mouse neurohypophysis, one of which is activated by both PPHT and U101958, and the other of which is activated only by PPHT. However, the different responses to these two drugs could reflect agonist-directed coupling to different transduction pathways. PACAP receptors provide a precedent for such differential coupling (Spengler et al. 1993), and a three-state receptor model has been proposed to account for this result (Leff et al. 1997). These results suggest that there are subtle differences in the expression of sigma receptors between rat and mouse.
Endogenous ligands
Given the abundance of dopamine and the presence of dopaminergic nerve terminals in the neurohypophysis (Baumgarten et al. 1972; Holzbauer & Racke, 1985), one would expect to find dopamine receptors in this tissue, and tests on the effects of dopamine and dopamine receptor-specific ligands on other membrane properties would help in addressing this issue. However, dopamine is clearly not capable of activating sigma receptors, and an endogenous ligand for the sigma receptor has yet to be identified.
Although the sigma binding site was originally thought to be an opioid receptor (Martin et al. 1976), subsequent studies showed that this is not the case (Su, 1993). The observation that these receptors are preferentially expressed by cells of neuroendocrine lineage led some investigators to consider steroid hormones as potential endogenous activators of the sigma receptor (Su, 1993). Neurosteroids, such as allopregnanolone, are known to modulate membrane excitability in nerve cell bodies (Paul & Purdy, 1992) and in the peptidergic nerve terminals of the neurohypophysis (Zhang & Jackson, 1994). Although binding of progesterone by sigma receptors has been reported (Su et al. 1988), progesterone exhibited neither agonist nor antagonist activity in the present system (Fig. 11). DHEA-S, corticosterone, and hydrocortisone were also without agonist activity (Table 1).
Based upon the CNS distribution of sigma receptors, investigators have suggested that the endogenous sigma receptor ligand may actually be a biogenic amine or a neuropeptide (Su, 1993; Ault et al. 1998). While binding data can be found to support many of these claims (Ramamoorthy et al. 1995), functional studies have thus far failed to ascertain the identity of an endogenous sigma receptor ligand. In the present analysis, we screened 15 ligands as plausible candidates for activity at the neurohypophysial sigma receptor and found that none had any measurable effect. Further tests using this preparation may ultimately lead to the identification of endogenous molecules that activate this receptor.
Modulation of neuropeptide secretion
Previous work on the neurohypophysis has indicated important roles for K+ channels in the regulation of neuropeptide secretion. T he inactivation of K+ channels during repetitive action potential activity enhances Ca2+ entry (Jackson et al. 1991) and facilitates release (Gainer et al. 1986). Increasing neurohypophysial Ca2+-activated K+ channels by either Ca2+ entry or phosphorylation leads to action potential failure, which would block release; reducing the current though the Ca2+-activated K+ channel has the opposite effect (Bielefeldt & Jackson, 1993, 1994). Thus, we would expect that the sigma receptor-mediated inhibition of neurohypophysial IK would have similar consequences, broadening action potentials to enhance evoked release, and making action potential failure less likely to reduce use-dependent depression.
Roughly half the nerve terminals of the neurohypophysis release oxytocin and the other half release vasopressin. These two closely related peptide hormones have very different physiological functions and therefore would be expected to be under different forms of regulation. However, we found that every nerve terminal tested responded to the presentation of a sigma receptor ligand with a reduction in K+ current (in a total of more than 100 recordings). This would suggest that both types of nerve terminals have sigma receptors, and that sigma receptor activation can modulate the release of both oxytocin and vasopressin. However, without identifying the peptide within nerve terminals undergoing recording, we are not in a position to say whether there are quantitative differences in the magnitude of the response. Aside from possible differences in sigma receptor responsiveness, selective modulation of oxytocin or vasopressin secretion could occur through highly localized release targeted at the appropriate nerve terminal. Another possibility is that enhancement could be selective because of the differences in optimal electrical stimuli for the release of oxytocin and vasopressin (Bicknell et al. 1984; Bondy et al. 1987). Similar forms of IK modulation could have a different impact depending on the nature of the electrical drive from the hypothalamus. In summary, the findings presented here generate a number of interesting hypotheses that can be tested by assaying the effects of sigma receptor ligands on electrically evoked secretion from the neurohypophysis.
Clinically, the long-term use of haloperidol and chlorpromazine as antipsychotic agents has been associated with a drug-induced syndrome of inappropriate antidiuretic hormone release (SIADH) (Peck & Shenkman, 1979). The results obtained in the present study demonstrate that both haloperidol and chlorpromazine are capable of directly modulating membrane excitability within the neurohypophysial nerve terminals that store and release this hormone. The present study therefore also provides a potential mechanistic explanation for the drug-induced SIADH associated with the chronic use of these agents. Interestingly, clozapine (a newer atypical antipsychotic agent) had no measurable effect on IK within the neurohypophysis. To date, there have been no reports of SIADH associated with the long-term administration of clozapine.
Acknowledgments
R. A. W. is a trainee in the Clinical Investigator Pathway of the University of Wisconsin Department of Medicine. Support for this research was provided by a Hilldale Fellowship to P. J. L., and by NIH Grant NS30016 to M. B. J. We thank Drs Gerard Ahern and David Sibley for valuable discussions, and J. L. Larson for outstanding technical assistance.
References
- Ault DT, Radeff JM, Werling JJ. Modulation of [3H]-dopamine release from rat nucleus accumbens by neuropeptide Y may involve a sigma1-like receptor. Journal of Pharmacology and Experimental Therapeutics. 1998;284:553–560. [PubMed] [Google Scholar]
- Barnes PRJ, Dyball REJ. Inhibition of neurohypophysial hormone release by dopamine in the rat. The Journal of Physiology. 1982;327:85–86. P P. [Google Scholar]
- Baumgarten HG, Bjorklund A, Holstein AF, Nobin A. Organization and ultrastructural identification of the catecholamine nerve terminals in the neural lobe and pars intermedia of the rat pituitary. Zeitschrift für Zellforschung und Mikroskopische Anatomie. 1972;126:483–517. doi: 10.1007/BF00306908. [DOI] [PubMed] [Google Scholar]
- Bicknell RJ, Brown D, Chapman C, Hancock PD, Leng G. Reversible fatigue of stimulus-secretion coupling in the rat neurohypophysis. The Journal of Physiology. 1984;348:601–613. doi: 10.1113/jphysiol.1984.sp015128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielefeldt K, Jackson MB. A calcium-activated potassium channel causes frequency-dependent action-potential failures in a mammalian nerve terminal. Journal of Neurophysiology. 1993;70:284–298. doi: 10.1152/jn.1993.70.1.284. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Jackson MB. Phosphorylation and dephosphorylation modulate a Ca2+-activated K+ channel in rat peptidergic nerve terminals. The Journal of Physiology. 1994;475:241–254. doi: 10.1113/jphysiol.1994.sp020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielefeldt K, Rotter JL, Jackson MB. Three potassium channels in rat posterior pituitary nerve endings. The Journal of Physiology. 1992;458:41–67. doi: 10.1113/jphysiol.1992.sp019405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy CA, Gainer H, Russell JT. Effects of stimulus frequency and potassium channel blockage on the secretion of vasopressin and oxytocin from the neurohypophysis. Neuroendocrinology. 1987;46:258–267. doi: 10.1159/000124829. [DOI] [PubMed] [Google Scholar]
- Bondy CA, Whitnall MH, Brady LS, Gainer H. Coexisting peptides in hypothalamic endocrine systems: Some functional implications. Cellular and Molecular Neurobiology. 1989;9:427–446. doi: 10.1007/BF00712791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque CW. Intraterminal recordings from the rat neurohypophysis in vitro. The Journal of Physiology. 1990;421:247–262. doi: 10.1113/jphysiol.1990.sp017943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges TE, Hillhouse EW, Jones MT. The effect of dopamine on neurohypophysial hormone release in vivo and from the rat neural lobe and hypothalamus in vitro. The Journal of Physiology. 1976;260:647–666. doi: 10.1113/jphysiol.1976.sp011537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabert C, Cavegn C, Bernard A, Mills A. Characterization of the functional activity of dopamine ligands at human recombinant dopamine D4 receptors. Journal of Neurochemistry. 1994;63:62–65. doi: 10.1046/j.1471-4159.1994.63010062.x. [DOI] [PubMed] [Google Scholar]
- Civelli O, Bunzow JR, Grandy DK. Molecular diversity of the dopamine receptors. Annual Review of Pharmacology and Toxicology. 1993;33:281–307. doi: 10.1146/annurev.pa.33.040193.001433. [DOI] [PubMed] [Google Scholar]
- Colmers W, Bleakman D. Effects of neuropeptide Y on the electrical properties of neurons. Trends in Neurosciences. 1991;17:373–379. doi: 10.1016/0166-2236(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Journal of Neuropsychiatry and Clinical Neuroscience. 1996;8:223–226. doi: 10.1176/jnp.8.2.223. [DOI] [PubMed] [Google Scholar]
- Crowley WR, Armstrong WE. Neurochemical regulation of oxytocin secretion in lactation. Endocrinological Reviews. 1992;13:33–65. doi: 10.1210/edrv-13-1-33. [DOI] [PubMed] [Google Scholar]
- Crowley WR, Parker SL, Armstrong WE, Wang W, Grosvenor CE. Excitatory and inhibitory dopaminergic regulation of oxytocin secretion in the lactating rat: Evidence for respective mediation by D-1 and D-2 dopamine receptor subtypes. Neuroendocrinology. 1991;53:493–502. doi: 10.1159/000125763. [DOI] [PubMed] [Google Scholar]
- Gainer H, Wolfe S, Jr, Obaid AL, Salzberg BM. Action potentials and frequency-dependent secretion in the mouse neurohypophysis. Neuroendocrinology. 1986;43:557–563. doi: 10.1159/000124582. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Dopamine. In: Rogawski MA, Barker JL, editors. Neurotransmitter Actions in the Vertebrate Nervous System. New York: Plenum Press; 1985. pp. 285–319. [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Holzbauer M, Racke K. The dopaminergic innervation of the intermediate lobe and of the neural lobe of the pituitary gland. Medical Biology. 1985;63:97–116. [PubMed] [Google Scholar]
- Hsu S-F, Jackson MB. Rapid exocytosis and endocytosis in nerve terminal of the rat posterior pituitary. The Journal of Physiology. 1996;494:539–553. doi: 10.1113/jphysiol.1996.sp021512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB. Passive current flow and morphology in the nerve terminal arborizations of the posterior pituitary. Journal of Neurophysiology. 1993;69:692–702. doi: 10.1152/jn.1993.69.3.692. [DOI] [PubMed] [Google Scholar]
- Jackson MB. Presynaptic excitability. International Review of Neurobiology. 1995;38:201–251. doi: 10.1016/s0074-7742(08)60527-9. [DOI] [PubMed] [Google Scholar]
- Jackson MB, Konnerth A, Augustine GJ. Action potential broadening and frequency-dependent facilitation of calcium signals in pituitary nerve terminals. Proceedings of the National Academy of Sciences of the USA. 1991;88:380–384. doi: 10.1073/pnas.88.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebabian JW, Tarazi FI, Kula NS, Baldessarini RJ. Compounds selective for dopamine receptor subtypes. Drug Discovery Today. 1997;2:333–340. [Google Scholar]
- Kelly MA, Rubinstein M, Asa SL, Zhang G, Saez C, Bunzow JR, Allen RG, Hnasko R, Ben-Jonathan N, Grandy DK, Low MJ. Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron. 1997;19:103–113. doi: 10.1016/s0896-6273(00)80351-7. [DOI] [PubMed] [Google Scholar]
- Leff P, Scaramellini C, Law C, McKechnie K. A three-state receptor model of agonist action. Trends in Pharmacological Sciences. 1997;18:355–362. doi: 10.1016/s0165-6147(97)01105-x. [DOI] [PubMed] [Google Scholar]
- Lemos JR, Nowycky MC. Two types of calcium channels coexist in peptide-releasing vertebrate nerve terminals. Neuron. 1989;2:1419–1426. doi: 10.1016/0896-6273(89)90187-6. [DOI] [PubMed] [Google Scholar]
- Liu L, Momsma FL, Sibley DR, Chioda LA. D2L, D2S, and D3 dopamine receptors stably transfected into NG108–15 cells couple to a voltage dependent potassium channel via distinct G-protein mechanisms. Synapse. 1996;24:156–164. doi: 10.1002/(SICI)1098-2396(199610)24:2<156::AID-SYN7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine and nalorphine like drugs in the nondependent and morphine-dependent chronic spinal dog. Journal of Pharmacology and Experimental Therapeutics. 1976;197:517–532. [PubMed] [Google Scholar]
- Melis MR, Argiolas A, Stancampiano R, Gessa GL. Effect of apomorphine on oxytocin concentrations in different brain areas and plasma of male rats. European Journal of Pharmacology. 1990;182:101–107. doi: 10.1016/0014-2999(90)90497-t. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, Ito K, Koizumi S, Ohno Y, Inoue K. Characterization of inhibition by haloperidol and chlorpromazine of a voltage-activated K+ current in rat phaeochromocytoma cells. British Journal of Pharmacology. 1995;116:2603–2610. doi: 10.1111/j.1476-5381.1995.tb17214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Freedman S, Chapman KL, Emms F, Fletcher AE, Knowles M, Marwood R, McAllister G, Myers J, Patel S, Curtis N, Kulagowski JJ, Leeson PD, Ridgill M, Graham M, Matheson S, Rathbone D, Watt AP, Bristow LJ, Rupniak NMJ, Baskin E, Lynch JJ, Ragan CI. Biological profile of L-745,870, a selective antagonist with high affinity for the dopamine D4 receptor. Journal of Pharmacology and Experimental Therapeutics. 1997;283:636–647. [PubMed] [Google Scholar]
- Paul SM, Purdy RH. Neuroactive steroids. FASEB Journal. 1992;6:2311–2322. [PubMed] [Google Scholar]
- Peck V, Shenkman L. Haloperidol-induced syndrome of inappropriate secretion of antidiuretic hormone. Clinical Pharmacology and Therapeutics. 1979;26:442–444. doi: 10.1002/cpt1979264442. [DOI] [PubMed] [Google Scholar]
- Quirion R, Bowen WD, Itzhak Y, Junien JL, Musacchio LM, Rothman RB, Su T-P, Tam SW, Taylor DP. A proposal for the classification of sigma binding sites. Trends in Pharmacological Science. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-a. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy JD, Ramamoorthy S, Mahesh VB, Leibach FH, Ganapathy V. Cocaine-sensitive sigma receptor and its interaction with steroid hormones in the human placental syncytiotrophoblast and in choriocarcinoma cells. Endocrinology. 1995;136:924–932. doi: 10.1210/endo.136.3.7867601. [DOI] [PubMed] [Google Scholar]
- Riviere PJM, Pascaud X, Junien JL, Porreca F. Neuropeptide Y and Jo1784, a selective sigma ligand, alter intestinal ion transport through a common haloperidol-sensitive site. European Journal of Pharmacology. 1990;187:557–559. doi: 10.1016/0014-2999(90)90388-m. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Phillips TJ, Dziewczapolski G, Zhang G, Fang Y, Larsen JL, McDougall JA, Chester JA, Saez C, Pugsley TA, Gershanik O, Low MJ, Grandy DK. Mice lacking dopamine D4 receptors are supersensitive to ethanol, cocaine, and methamphetamine. Cell. 1997;90:991–1001. doi: 10.1016/s0092-8674(00)80365-7. [DOI] [PubMed] [Google Scholar]
- Schlachter SK, Poel TJ, Lawson CF, Dinh DM, Lajiness ME, Romero AG, Palmer JR, Carter DB, Smith MW. Substitute aminopiperidines as selective and potent D4 dopamine receptor antagonists. Society for Neuroscience Abstracts. 1995;21:252.7. [Google Scholar]
- Seth P, Leibach FH, Ganapathy V. Cloning and structural analysis of the cDNA and the gene encoding the murine type 1 sigma receptor. Biochemical and Biophysical Research Communications. 1997;241:535–540. doi: 10.1006/bbrc.1997.7840. [DOI] [PubMed] [Google Scholar]
- Sibley DR, Monsma FJ, Shen Y. Molecular neurobiology of dopamine receptors. International Review of Neurobiology. 1993;35:391–415. doi: 10.1016/s0074-7742(08)60573-5. [DOI] [PubMed] [Google Scholar]
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Boeckert J, Seeburg PH, Journot L. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993;365:170–175. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]
- Su T-P. Evidence for sigma opioid receptor: binding of [3H]SKF-10047 to etorphine-inaccessible sites in guinea pig brain. Journal of Pharmacology and Experimental Therapeutics. 1982;223:284–290. [PubMed] [Google Scholar]
- Su T-P. Delineating biochemical and functional properties of sigma receptors: Emerging concepts. Critical Reviews in Neurobiology. 1993;7:187–203. [PubMed] [Google Scholar]
- Su T-P, London ED, Jaffe JH. Steroid binding at sigma receptors suggests a link between endocrine, nervous and immune systems. Science. 1988;240:219–221. doi: 10.1126/science.2832949. [DOI] [PubMed] [Google Scholar]
- TenBrink RE, Bergh CL, Duncan JN, Harris DW, Huff RM, Lahti RA, Lawson CF, Lutzke BF, Martin IJ, Rees SA, Schlachter SK, Sih JC, Smith MW. (S)-(-)-4-[4-[2-(isochroman-1-yl)ethyl]-piperazin-1-yl]-benzenesulfonamide, a selective D4 antagonist. Journal of Medicinal Chemistry. 1996;39:2435–7. doi: 10.1021/jm960084f. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Capogna M, Scanziani M. Presynaptic inhibition in the hippocampus. Trends in Neurosciences. 1993;16:222–226. doi: 10.1016/0166-2236(93)90160-n. [DOI] [PubMed] [Google Scholar]
- Watt VJ, Neve KA. Activation of type II adenylate cyclase by D2, and D4, but not D3 dopamine receptors. Molecular Pharmacology. 1998;52:181–186. doi: 10.1124/mol.52.2.181. [DOI] [PubMed] [Google Scholar]
- Wilke RA, Hsu S-F, Jackson MB. Dopamine D4 receptor mediated inhibition of potassium current in neurohypophysial nerve terminals. Journal of Pharmacology and Experimental Therapeutics. 1998;284:542–548. [PubMed] [Google Scholar]
- Wolfe SA, Culp SG, De Souza EB. σ-Receptors in endocrine organs: identification, characterization, and autoradiographic localization in rat pituitary, adrenal, testis, and ovary. Endocrinology. 1989;124:1160–1172. doi: 10.1210/endo-124-3-1160. [DOI] [PubMed] [Google Scholar]
- Wu X-Z, Bell JA, Spivak CE, London ED, Su T-P. Electrophysiological and binding studies on intact NCB-20 cells suggest the presence of a low affinity sigma receptor. Journal of Pharmacology and Experimental Therapeutics. 1991;257:351–359. [PubMed] [Google Scholar]
- Zhang SJ, Jackson MB. Neuroactive steroids modulate GABAA receptors in peptidergic nerve terminals. Journal of Neuroendocrinology. 1994;6:533–538. doi: 10.1111/j.1365-2826.1994.tb00616.x. [DOI] [PubMed] [Google Scholar]