

Abstract
The effects on contractility of three peptides reported to inhibit protein kinase C (PKC) translocation in an isozyme-specific manner were studied: a peptide from the C2 domain of conventional PKCs (C2-2), a peptide from the N-terminal variable domain of εPKC (εV1-2) and a peptide (ABP) from the actin-binding domain of εPKC (ε(223–228)).
Isometric force was directly recorded from individual hyperpermeable ferret portal vein or aortic smooth muscle cells.
Phenylephrine contracted permeabilized portal vein cells at pCa 6.7 but not at pCa 7.0. However, phenylephrine did contract aortic cells at pCa 7.0.
C2-2 inhibited phenylephrine-induced contraction, but did not affect resting tension, in portal vein cells at pCa 6.7. In aortic cells at either pCa 6.7 or 7.0, C2-2 had no effect on either basal tension or phenylephrine-induced contraction.
ABP did not evoke any changes in phenylephrine-induced contraction or baseline tension in either portal vein or aortic cells.
εV1-2 inhibited phenylephrine-induced contraction and decreased resting tension in aortic cells at pCa 7.0, but not in portal vein cells at pCa 6.7.
Western blots indicated that portal vein cells contained substantially more αPKC than aortic cells. Portal vein cells also contained small amounts of βPKC, which was undetectable in aortic cells. In contrast, aortic cells contained more εPKC than portal vein cells. Even though εPKC was expressed in portal vein and αPKC in aorta, imaging studies indicated that they were not translocated in these cell types.
These results suggest that the Ca2+-dependent isozymes of PKC (α and/or β) play a major role in contraction of the portal vein but not of the aorta. In contrast, the results are consistent with εPKC, but not Ca2+-dependent PKC isozymes, regulating contractility of the aorta.
Activation of protein kinase C (PKC) isozymes is generally associated with translocation from the soluble to the particulate cell fraction (Kraft et al. 1982). Translocation of PKC to the particulate fraction was initially thought to reflect direct association of the enzyme with lipids at the plasma membrane. However, data from several laboratories indicate that PKC interacts with specific target proteins at the sites of translocation (Mochly-Rosen et al. 1991; Chapline et al. 1993; Hyatt et al. 1994; Liou & Morgan, 1994; Ron et al. 1994). Activated PKC isozymes are thought to bind anchoring proteins referred to as RACKs (receptors for activated C-kinase) (Mochly-Rosen et al. 1991; Ron et al. 1994; Mochly-Rosen, 1995) or PICKs (proteins that interact with C-kinase) (Liao et al. 1994; Staudinger et al. 1995). It has been suggested that the functional specificity of each PKC isozyme is determined, in part, by the differential localization of the isozyme-specific RACKs (Ron et al. 1995).
A RACK for βPKC, RACK1, has been cloned, and at least part of its binding site on βPKC has been mapped to a short sequence within the C2 domain (Ron et al. 1994). C2-2, a short synthetic peptide derived from this region, inhibits phorbol ester-induced translocation of the C2-containing isozymes in cardiac myocytes and insulin-induced βPKC translocation and function in Xenopus oocytes (Ron et al. 1995). The C2 domain is present in all ‘conventional’ PKCs and thus it would be expected that the C2-2 peptide would inhibit the function of all members of this class. In contrast, εV1-2, a short peptide derived from the V1 region of εPKC, was shown to inhibit translocation of the novel PKC, εPKC (Johnson et al. 1996). Therefore, εV1-2 and C2-2 peptides are expected to inhibit the functions of different classes of PKC isozymes.
In addition, actin filaments may represent a new class of PKC-binding proteins; a binding site for actin has been identified between the first and second cysteine-rich regions within the regulatory domain of εPKC (Prekeris et al. 1996). εPKC (223–228) is a synthetic hexapeptide that corresponds to the putative actin-binding domain of εPKC, and has been shown to compete with native εPKC for binding to purified actin.
We have previously demonstrated that phenylephrine causes contraction of single cells of ferret aorta and portal vein at constant [Ca2+], and that PKC activation plays an important role in the maintenance of phenylephrine-induced contraction in both cell types (Khalil & Morgan, 1992; Horowitz et al. 1996). We suggested that the phenylephrine-induced contraction involves activation of a Ca2+-dependent PKC isozyme in portal vein but a Ca2+-independent isozyme in aorta (Khalil & Morgan, 1992). If a RACK does play a critical role in PKC-dependent contractility, isozyme-specific translocation inhibitors should inhibit PKC-induced contraction in an isozyme-specific manner. To test this hypothesis, we have determined the effects of these peptides on phenylephrine-induced contraction of single permeabilized portal vein and aortic smooth muscle cells.
We have previously recorded isometric force from saponin-permeabilized single cells of the ferret aorta (Collins et al. 1992; Katsuyama et al. 1992) and demonstrated that these cells retain receptor-coupled responses (Brozovich et al. 1990; Collins et al. 1992). In the present study, we used this method to directly investigate the effects of RACK-binding peptides on phenylephrine-induced contraction of single permeabilized portal vein or aortic smooth muscle cells, and report that these inhibitors are effective and tissue-specific inhibitors of contractility.
METHODS
Preparation of portal vein and aortic smooth muscle tissues
All procedures were performed according to protocols approved by the Boston Biomedical Research Institute Animal Care and Use Committee. Ferrets were killed by an overdose of the anaesthetic chloroform, and the aorta or portal vein was quickly removed to a dissection dish filled with oxygenated Krebs solution (see below for composition of solutions). The tissue was cleaned of connective tissue, opened longitudinally and the endothelium was removed by gentle abrasion of the inner surface of the tissue with a rubber policeman. The tissue was cut into 2 mm-wide strips, and wet weight was determined. In some experiments, lung tissue, for use as a positive control in immunoblots, was also removed from the ferrets.
Immunoblotting
Tissue samples were homogenized in a buffer containing protease inhibitors as described previously (Menice et al. 1997). Protein-matched samples were subjected to electrophoresis on sodium dodecyl sulphate-8 % polyacrylamide gels and transferred electrophoretically to poly (vinylidene difluoride) membranes. The membranes were blocked with 5 % dried milk in phosphate-buffered saline (PBS)-Tween and incubated in the primary antibody solution at 4°C overnight. Mouse monoclonal anti-αPKC and anti-βI-IIPKC were obtained from Transduction Laboratories. Rabbit polyclonal antibodies to ε- and ηPKC were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). The blots were visualized with enhanced chemiluminescence using the Supersignal CL-HRP Substrate System (Pierce Chemical Co., Rockford, IL, USA) as described by the manufacturer.
Preparation of single cells
Single vascular smooth muscle cells from aorta or portal vein were isolated using a modification of a previously published method (DeFeo & Morgan, 1985; Collins et al. 1992). The aorta and portal vein were cut into small pieces (2 mm × 2 mm) and placed in a siliconized flask containing digestion medium. For each 50 mg of aorta (wet weight), the digestion medium A consisted of 2 mg CLS 2 collagenase (Type II, 228 U mg−1; Worthington Biochemical, Freehold, NJ, USA) and 5 mg elastase (Grade II, 3.65 U mg−1; Boehringer Mannheim, Indianapolis, IN, USA) in 7.5 ml of Ca2+-Mg2+-free Hanks' balanced salt solution (HBSS) and 1 % bovine serum albumin (Gibco BRL, Gaithersburg, MD, USA). The tissue pieces were incubated in a shaking water bath at 34°C under an atmosphere of 95 % O2-5 % CO2 for 70 min. The pieces were then filtered on a nylon mesh, rinsed with 10 ml Ca2+-Mg2+-free HBSS, and reincubated for 20 min in digestion medium B, i.e. the same digestion solution except for a decrease in the amount of collagenase to 1 mg and addition of 5000 U soybean trypsin inhibitor (Type II-S; Sigma, St Louis, MO, USA). The tissue pieces were filtered and rinsed in 10 ml Ca2+-Mg2+-free HBSS. The filtrate containing the dissociated cells was poured over unsiliconized glass coverslips, and further incubated for 20 min in digestion medium B. After filtering and rinsing with 10 ml Ca2+-Mg2+-free HBSS, the dissociated cells were plated onto another set of coverslips. For isolation of portal vein cells, the same protocol was followed, but the first incubation was shortened to 40 min. Coverslips were stored on ice for 1–5 h until use. Cells were not centrifuged or aspirated by pipette. The isolated cells were assayed daily to confirm that they shortened in response to phenylephrine in 2.5 mM Ca2+-HBSS.
Permeabilization and tension measurement
The coverslips were placed on the movable stage of a Nikon inverted microscope. Aorta or portal vein cells were exposed to a relaxing solution (pCa 9) containing 30 μg ml−1 saponin for 5 min, and then the solution was changed to saponin-free pCa 7.0 or 6.7 solution. Permeabilized cells were chosen according to the following criteria: diminished phase-luscence under phase-contrast optics; visible nucleus; length ≥ 50 μm; and firm attachment to the coverslip. Microtools (Glass 1BRL, W/FIL 1.0 mm, 1B100F-4, 12773-08H; World Precision Instruments, Sarasota, FL, USA) were prepared by the use of a micropipette puller (Industrial Science Associates, Inc., Ridgewood, NY, USA); the tip diameters of the microtools were < 5 μm. The microtools were placed with their tips touching the top surface of the cell (microtools approached the coverslip at an angle of approximately 45 deg) and the cells were left undisturbed for 2–3 min. We have found that mammalian vascular smooth muscle cells prepared in this manner have the important advantage of sticking to glass and, therefore, readily attach to the glass microelectrodes. The microtool attached to the transducer (Cambridge model 400A) was lifted so that the end of the cell was raised off the coverslip. The other microtool was gently pressed down to immobilize the other end of the cell, which was stretched to approximately 110 % of its original length. Control recordings confirmed that the drift of the transducer was negligible over the course of the recording. At least one recording was made daily with a cell attached to the microtool in the absence of added drugs to confirm that the baseline drifted ≤ 50 μg over the time course of an experiment. Baseline recordings of 3–5 min were routinely made before experimental manipulations were performed. If baseline recordings displayed any detectable evidence of slow baseline fluctuations, experimental manipulations were not performed. Increases or decreases in force were assumed to have reached a plateau when no further increase or decrease occurred over a 2–3 min period. Force measurements were made manually by placing a ruler on the trace through the mid-point of the noise level. All experiments were performed at room temperature.
Digital imaging
Cells were fixed with 4 % paraformaldehyde either in the resting state or after 10 min of stimulation with 10−5 M phenylephrine. Subsequently the cells were permeabilized with 0.1 % Triton X-100, blocked with 10 % goat serum and reacted with either mouse monoclonal anti-αPKC (Transduction Laboratories) or rabbit polyclonal anti-εPKC (Santa Cruz Biotechnologies) followed by Rhodamine Red-X secondary antibody (Molecular Probes) and mounted with Fluorosave (Calbiochem, San Diego, CA, USA) before analysis.
Images were obtained using a Nikon Diaphot 300 inverted microscope equipped with a Nikon × 40 oil-immersion objective lens (NA, 1.3). Filters used were 560 ± 20 nm (excitation), 595 nm (dichroic) and 630 ± 30 nm (emission) for Rhodamine Red-X. Images were recorded with a liquid-cooled CCD camera (photonetrucs CH250) via Photometrics Microsoft compatible image-processing software (PMIS). A modified version of a previously described ratio analysis (Khalil & Morgan, 1992) was performed to determine the relative distribution of PKC isozymes within each cell and to normalize for possible differences in staining efficiency between cells. The part of the section containing the nuclear area was avoided when obtaining ratio values.
Solutions
Ca2+ buffers were prepared according to an iterative computer program that calculates the amounts of stock solutions required for a given set of free ion concentrations, taking into consideration the binding constants for the ionic species present, temperature and ionic strength (Brozovich et al. 1988). Ionic strength was set at 0.2 M and Mops was used to buffer the pH at 7.0. The concentrations of the constituents were: 1 nm to 0.1 mM Ca2+; 1 mM Mg2+; 135 mM K+; 3 mM MgATP; 15 mM EGTA; > 15 mM Mops; 15 mM phosphocreatine; and creatine kinase (approximately 20 U ml−1), added daily before the experiments. The major anion was propionate. Krebs solution (used only for dissecting the tissue) contained (mM): 120 NaCl, 5.9 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4 and 11.5 dextrose; at pH 7.4 when bubbled with 95 % O2-5 % CO2. The Ca2+-Mg2+-free HBSS used for cell isolation contained (mM): 137 NaCl, 5.4 KCl, 0.44 KH2PO4, 0.42 NaH2PO4, 4.17 NaHCO3, 5.55 glucose and 10 Hepes; pH 7.4. PBS-Tween solution for immunoblotting contained (mM): 80 Na2HPO4, 20 NaH2PO4, 100 NaCl and 0.05 % (v/v) Tween.
Peptides and drugs
Peptide C2-2 (MDPNGLSDPYVKL; βPKC (186–198)), scrambled C2-2 (GYSKMPLPNDDLV), εV1-2 (EAVSLKPT; εPKC (14–21)), scrambled εV1-2 (LSETKPAV), actin-binding peptide (LKKQET; εPKC (223–228)) and PSSI (RFARKGALRQKNV; αPKC (19–31)) were synthesized in The University of Calgary Peptide Synthesis Core Facility using a Beckman model 990B automated peptide synthesizer and purified by preparative reverse-phase HPLC. All peptides were shown to be > 95 % pure by analytical HPLC and their structures were verified by amino acid composition analysis. The following drugs were used: PGF2α, phosphocreatine, creatine kinase and phenylephrine (all from Sigma). General laboratory reagents were of analytical grade or better and were purchased from Sigma or Fisher Scientific (NJ, USA).
Statistics
All values given in the text are means ±s.e.m. Differences between means were evaluated using Student's t test. Significant differences were taken at the P < 0.05 level. The values (n) given represent numbers of cells used in each experiment.
RESULTS
Effect of C2-2 peptide on phenylephrine-induced contraction of single permeabilized portal vein and aortic smooth muscle cells
The effects of phenylephrine on contractility of single permeabilized portal vein or aortic smooth muscle cells are illustrated in Figs 1 and 2. At pCa 7.0 (approximately resting [Ca2+]i) 10−5 M phenylephrine evoked a gradual but sustained contraction of aortic (Fig. 2A) but not portal vein cells (not shown). Both cell types, however, contracted in response to phenylephrine at pCa 6.7 (Figs 1A and 3). The mean steady-state increase in force in response to phenylephrine at pCa 6.7 was 272.1 ± 20.1 μg (n = 18) in portal vein cells and 242.6 ± 10.8 μg (n = 6) in aorta (Fig. 3). The mean steady-state amplitude of contraction in aorta at pCa 7.0 was 252.9 ± 14.4 μg (n = 19) and was not significantly different from that at pCa 6.7 (Fig. 3). We have previously reported that the phenylephrine-induced contraction of aortic cells is not Ca2+ dependent over this range of [Ca2+] (Collins et al. 1992).
Figure 1. Effect of C2-2 peptide on phenylephrine-induced contraction and resting tension in single permeabilized portal vein cells at pCa 6.7.

A, force recording in response to phenylephrine (10−5 M). B, effect of C2-2 peptide (10 μm) on phenylephrine-induced contraction. C, effect of C2-2 peptide on resting tension. D, effect of pre-treatment with C2-2 peptide on phenylephrine-induced contraction. PGF2α (100 μm) was added after the phenylephrine-induced contraction had stabilized. E, control trace. Dashed lines indicate baseline force at pCa 6.7 or, in B, the level of the phenylephrine-induced contraction.
Figure 2. Effect of C2-2 peptide on phenylephrine-induced contraction and resting tension in single permeabilized aortic cells at pCa 7.0.

A, force recording in response to phenylephrine (10−5 M). B, effect of C2-2 peptide (10 μm) on phenylephrine-induced contraction. C, effect of PSSI (3 μm) on phenylephrine-induced contraction. D, effect of C2-2 peptide on resting tension. E, effect of pre-treatment with C2-2 peptide on phenylephrine-induced contraction. F, control trace. Dashed lines indicate baseline force at pCa 7.0.
Figure 3. Statistical analysis of the effects of C2-2 peptide on single permeabilized portal vein and aortic cells.
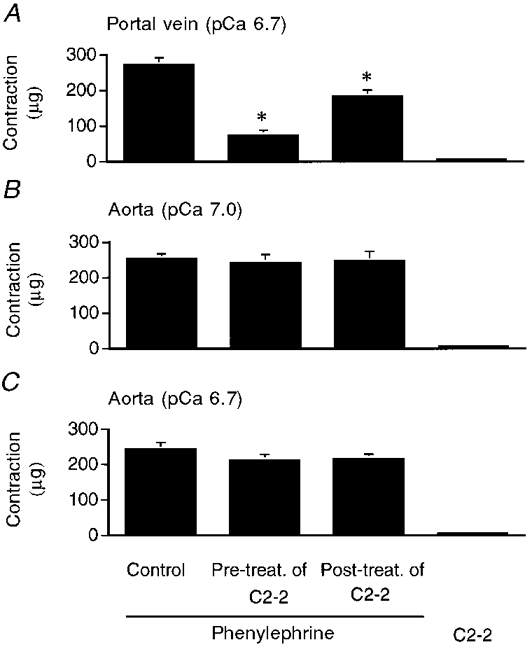
A, analysis of the effects of C2-2 peptide on single permeabilized portal vein cells at pCa 6.7. B and C, analysis of the effects of C2-2 peptide on single permeabilized aortic cells at pCa 7.0 (B) and pCa 6.7 (C). Control, force induced by phenylephrine (10−5 M); Pre-treat. of C2-2, effect of 20 min of pre-treatment with C2-2 peptide (10 μm) on phenylephrine-induced contraction; Post-treat. of C2-2, effect of C2-2 peptide on phenylephrine-induced contraction; C2-2, force response to C2-2 peptide alone. *P < 0.05 compared with contraction induced by phenylephrine alone.
Since it was uncertain whether these peptides could compete with PKC after translocation, we investigated the effect of C2-2 peptide on phenylephrine-induced contraction using both pre- and post-treatment protocols. As shown in Figs 1B and 3A, when portal vein cells were stimulated by phenylephrine, the subsequent addition of 10 μm C2-2 peptide decreased the extent of phenylephrine-induced contraction from 272.1 ± 20.1 to 182 ± 19.1 μg (n = 5). The scrambled version of C2-2 (10 μm) had no effect. The amplitudes of contraction in response to phenylephrine without and with scrambled C2-2 peptide were 272.1 ± 20.1 and 275 ± 18.9 μg (n = 3), respectively. In contrast, the peptide did not cause any change in the contraction of aortic cells (Fig. 2B). In aortic cells, the mean steady-state amplitudes of contraction in response to phenylephrine at pCa 7.0 without and with C2-2 peptide were 252.9 ± 14.4 and 246 ± 28.2 μg (n = 5), respectively (Fig. 3B). Treatment with C2-2 peptide alone did not affect resting tension in portal vein (Fig. 1C) or aortic (Fig. 2D) cells.
Pre-treatment of portal vein cells with C2-2 peptide for 20 min had more dramatic effects; the phenylephrine-induced contraction was reduced from 272.1 ± 20.1 to 73.3 ± 15.4 μg (n = 6) (Figs 1D and 3A). Aortic cells, however, showed no effect of the peptide (Fig. 2E). In contrast, in portal vein, the scrambled C2-2 peptide did not cause any change in the phenylephrine-induced contraction and the amplitudes of contraction in response to phenylephrine without and with scrambled C2-2 peptide were 272.1 ± 20.1 and 270 ± 17.3 μg (n = 3), respectively. Furthermore, to confirm that the same portal vein cells pre-treated with peptide exhibited normal contractility, we added 100 μm PGF2α after the phenylephrine-induced contraction in the presence of C2-2 peptide had stabilized. PGF2α was previously shown to induce contraction of single permeabilized cells at constant, low [Ca2+]i due to the combined inhibition of myosin light chain phosphatase and activation of PKC (Suematsu et al. 1991; Katsuyama & Morgan, 1993). As shown in Fig. 1D, PGF2α caused a further increase in force generation above that induced by phenylephrine in the presence of C2-2 peptide.
Since C2-2 peptide had no effect on aortic cells, we performed additional control experiments to verify that the aortic cells were adequately permeabilized. The effect of a peptide inhibitor of PKC (PSSI), of similar size to C2-2, on the phenylephrine-induced contraction of aortic cells is shown in Fig. 2C. At the plateau of force generation by phenylephrine, 3 μm PSSI was added and resulted in complete inhibition of the phenylephrine-induced contraction. Figures 1E and 2F show control recordings to illustrate that the drift of the transducer was negligible over the time course of the recordings.
We also tested the effect of C2-2 peptide on single permeabilized aortic cells at pCa 6.7. As shown in Fig. 3C, C2-2 peptide did not evoke a significant change in phenylephrine-induced contraction or resting tension. Furthermore, pre-treatment of the cells with the peptide for 20 min did not inhibit the subsequent phenylephrine-induced contraction.
Effect of εPKC (223–228), an actin-binding peptide, on phenylephrine-induced contraction of single permeabilized portal vein and aortic smooth muscle cells
Residues 223–228 of εPKC have the interesting property of binding actin (Prekeris et al. 1996). The effects of pre- and post-treatment with 20 μmεPKC actin-binding peptide (ABP) on the phenylephrine-induced contraction of portal vein and aortic smooth muscle cells are shown in Figs 4 and 5, respectively. ABP did not evoke any change in the phenylephrine-induced contraction of portal vein (Fig. 4A) or aortic (Fig. 5A) cells. In Figs 4 and 5, the apparent slowing in tension development by phenylephrine in the presence of ABP was not a consistent finding, but may have been due to differences in the diffusion distance for the applied drug. The mean steady-state amplitudes of contraction in response to phenylephrine without and with ABP were, respectively, 272.1 ± 20.1 and 269 ± 11.7 μg (n = 5) in portal vein and 252.9 ± 14.4 and 244 ± 6.8 μg (n = 5) in aortic cells (Fig. 6). Treatment with ABP alone had no effect on resting tension in portal vein (Fig. 4B) or aortic (Fig. 5B) cells. We also tested the effect of pre-treatment with ABP on phenylephrine-induced contraction. Pre-treatment of the cells with ABP for 20 min did not affect phenylephrine-induced contraction of portal vein (Fig. 4C) or aortic (Fig. 5C) cells. The mean steady-state amplitudes of contraction in response to phenylephrine without and with ABP, respectively, were 272.1 ± 20.1 and 268 ± 15.6 μg (n = 5) in portal vein and 252.9 ± 14.4 and 244 ± 12.1 μg (n = 5) in aortic cells (Fig. 6).
Figure 4. Effect of actin-binding peptide on phenylephrine-induced contraction and resting tension in single permeabilized portal vein cells at pCa 6.7.

A, effect of actin-binding peptide (ABP; 20 μm) on phenylephrine (10−5 M)-induced contraction. B, effect of ABP on resting tension. C, effect of pre-treatment with ABP on phenylephrine-induced contraction. D, control recording. Dashed lines indicate baseline force at pCa 6.7.
Figure 5. Effect of actin-binding peptide on phenylephrine-induced contraction and resting tension in single permeabilized aortic cells at pCa 7.0.

A, effect of ABP (20 μm) on phenylephrine (10−5 M)-induced contraction. B, effect of ABP on resting tension. C, effect of pre-treatment with ABP on phenylephrine-induced contraction. D, control recording. Dashed lines indicate baseline force at pCa 7.0.
Figure 6. Statistical analysis of the effects of actin-binding peptide on single permeabilized portal vein cells at pCa 6.7 (A) and aortic smooth muscle cells at pCa 7.0 (B).

Control, force induced by phenylephrine (10−5 M); Pre-treat. of ABP, effect of 20 min of pre-treatment with ABP (20 μm) on phenylephrine-induced contraction; Post-treat. of ABP, effect of ABP on phenylephrine-induced contraction; ABP, force generated by ABP alone.
Effect of εV1-2 peptide on phenylephrine-induced contraction of single permeabilized portal vein and aortic smooth muscle cells
We next investigated the effect of pre- and post-treatment with 5 μmεV1-2 peptide on the phenylephrine-induced contraction of portal vein and aortic cells. As shown in Fig. 7A, addition of εV1-2 peptide did not affect the amplitude of the phenylephrine-induced contraction of portal vein cells. The mean steady-state amplitudes of contraction in response to phenylephrine without and with εV1-2 peptide were 272.1 ± 20.1 and 270 ± 14.8 μg (n = 5), respectively (Fig. 9A). However, εV1-2 peptide significantly inhibited the phenylephrine-induced contraction of aortic smooth muscle cells (Fig. 8A), from 252.9 ± 14.4 to 132 ± 18.3 μg (n = 5) (Fig. 9B). The scrambled version of εV1-2 had no effect. The mean steady-state amplitudes of contraction in response to phenylephrine without and with scrambled εV1-2 peptide were 252.9 ± 14.4 and 256.6 ± 8.8 μg (n = 3), respectively. The apparent delay in the response to εV1-2 peptide (Fig. 8A) was not a consistent finding. Treatment with εV1-2 peptide alone did not affect resting tension in portal vein cells (Fig. 7B), but decreased resting tension in aortic cells (Fig. 8B). In aortic cells, the mean steady-state decrease in resting tension by εV1-2 peptide was 100 ± 24.7 μg (n = 5) (Fig. 9B). In contrast, scrambled εV1-2 peptide did not affect resting tension. It is of interest that we have previously shown a similar decrease in basal (i.e. ‘intrinsic’) tone of ferret aortic cells with PSSI (Collins et al. 1992).
Figure 7. Effect of εV1-2 peptide on phenylephrine-induced contraction and resting tension in single permeabilized portal vein cells at pCa 6.7.

A, effect of εV1-2 peptide (5 μm) on phenylephrine (10−5 M)-induced contraction. B, effect of εV1-2 peptide on resting tension. C, effect of 20 min of pre-treatment with εV1-2 peptide on phenylephrine-induced contraction. D, control recording. Dashed lines indicate baseline force at pCa 6.7.
Figure 9. Statistical analysis of the effects of εV1-2 peptide on single permeabilized portal vein cells at pCa 6.7 (A) and aortic smooth muscle cells at pCa 7.0 (B).

Control, force induced by phenylephrine (10−5 M); Pre-treat. of εV1-2, effect of 20 min of pre-treatment with εV1-2 peptide (5 μm) on phenylephrine-induced contraction; Post-treat. of εV1-2, effect of εV1-2 peptide on phenylephrine-induced contraction; εV1-2, force generated by εV1-2 peptide alone. *P < 0.05 compared with contraction induced by phenylephrine alone.
Figure 8. Effect of εV1-2 peptide on phenylephrine-induced contraction and resting tension in single permeabilized aortic cells at pCa 7.0.
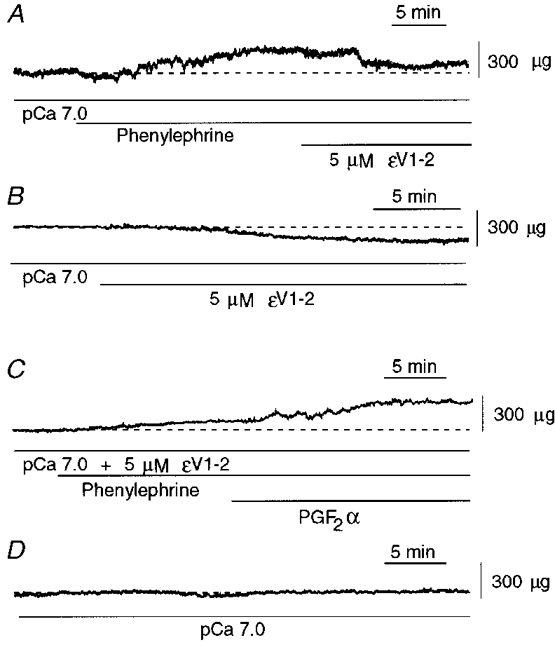
A, effect of εV1-2 peptide (5 μm) on phenylephrine (10−5 M)-induced contraction. B, effect of εV1-2 peptide on resting tension. C, effect of 20 min of pre-treatment with εV1-2 peptide on phenylephrine-induced contraction. PGF2α was added after the phenylephrine-induced contraction had stabilized. D, control recording. Dashed lines indicate baseline force at pCa 7.0.
We also tested the effect of pre-treatment with εV1-2 peptide on phenylephrine-induced contraction. Pre-treatment of the cells with εV1-2 peptide for 20 min did not affect subsequent phenylephrine-induced contraction of portal vein cells (Fig. 7C), but inhibited phenylephrine-induced contraction of aortic smooth muscle cells (Fig. 8C), from 252.9 ± 14.4 to 68 ± 18.5 μg (n = 5) (Fig. 9B). In contrast, scrambled εV1-2 peptide did not affect the phenylephrine-induced contraction; the mean steady-state amplitudes of contraction in response to phenylephrine without and with scrambled εV1-2 peptide were 252.9 ± 14.4 and 258.3 ± 13.6 μg (n = 3), respectively. We confirmed that the same population of cells was capable of maximally contracting; addition of PGF2α at the plateau of force generation by phenylephrine in the presence of εV1-2 peptide resulted in a significant increase in force (Fig. 8C).
Immunoblotting of PKC isoenzymes in portal vein and aorta
To investigate the expression of PKC isozymes in portal vein and aorta, immunoblots were performed using isozyme-specific antibodies. As shown in Fig. 10, anti-αPKC detected a prominent band of 82 kDa in portal vein and a weak band in protein-matched aortic extracts. Portal vein extracts also contained βPKC (80 kDa), but none was detectable in aorta. εPKC (90 kDa) was detected in both tissues and the signal was consistently stronger in aorta compared with portal vein extracts. Since ηPKC contains a sequence (residues 18–25: EAVGLQPT) similar to that of the εV1-2 peptide (Osada et al. 1990), we also screened both tissues for the presence of ηPKC (Fig. 10) and found a faint signal in portal vein but no detectable signal in aorta. Ferret lung was used as a positive control. Thus, the effectiveness of the εV1-2 peptide in the aorta cells cannot be explained by the presence of ηPKC.
Figure 10. Immunoblots of αPKC, βI-IIPKC, εPKC and ηPKC in portal vein and aorta.

FA, ferret aorta; FPV, ferret portal vein; FL, ferret lung.
Digital imaging of αPKC and εPKC
Since the immunoblots indicated the presence of small but detectable amounts of αPKC and εPKC in aorta and portal vein, respectively, even though the peptide translocation inhibitors had no effect, we investigated whether these isozymes are normally translocated in these cell types. As shown in Fig. 11, and as has been reported previously (Khalil et al. 1994), αPKC translocates from the cytosol to the vicinity of the surface membrane upon addition of phenylephrine to portal vein cells. On average, the surface to cytosol fluorescence ratio significantly (P = 0.0001) increased from 0.75 ± 0.3 (n = 8) in resting cells to 2.06 ± 0.16 (n = 6) in stimulated cells. As expected from the immunoblots, the brightness of aortic cells stained for αPKC (Fig. 11) was far less than that for portal vein cells. When surface to cytosol ratios were calculated from αPKC-stained aortic cells, a resting ratio of 0.77 ± 0.03 (n = 6) was obtained, in comparison with a stimulated ratio of 0.81 ± 0.04 (n = 6), which was not significantly different (P = 0.1293).
Figure 11.

Typical images of aortic and portal vein cells either at rest or after 10 min stimulation with 10−5 M phenylephrine (PE) and then stained to locate αPKC (top) or εPKC (bottom). Scale bar, 5 μm.
With respect to εPKC, we have previously reported that phenylephrine causes a translocation to the surface of the cell (Khalil et al. 1992) and have confirmed those findings here. Resting aortic cells stained for εPKC had a mean surface to cytosol ratio of 0.87 ± 0.02 (n = 6) which was increased significantly (P = 0.0002) in the presence of phenylephrine to 2.02 ± 0.11 (n = 6) (Fig. 11). Again, as predicted by the immunoblots, portal vein cells stained for εPKC were far less bright than were aorta cells (Fig. 11). Furthermore, no significant translocation upon stimulation was detected. The surface to cytosol fluorescence ratio was 0.79 ± 0.01 (n = 6) for resting cells and 0.81 ± 0.05 (n = 6) for stimulated cells (P = 0.103). In all cases, it was confirmed that cells stained with the secondary antibody alone had no measurable fluorescence.
DISCUSSION
The main purpose of the present study was to use peptides designed to block isozyme-specific targeting sequences to determine which PKC isozymes are necessary for phenylephrine-induced contraction of single permeabilized portal vein and aortic smooth muscle cells of the ferret. We have previously found that brief saponin treatment of single smooth muscle cells results in a hyperpermeable preparation which retains receptor-coupled function (Collins et al. 1992). We have also previously reported that, with this method, addition of calmodulin in the buffers is not necessary to produce Ca2+-tension relationships that mimic those reported for intact cells loaded with Ca2+ indicators. Furthermore, the amplitude of the phenylephrine-induced contraction in permeabilized single cells made by this method is not different from that in intact cells (Brozovich et al. 1990). This implies that our permeabilization method does not cause a significant loss of small molecules such as calmodulin that might affect contractility. We have also previously presented evidence that the phenylephrine-induced contraction of permeabilized aortic cells at constant [Ca2+] is the result of activation of a Ca2+-independent PKC isozyme (Collins et al. 1992).
In the present study, phenylephrine evoked contraction of portal vein cells at pCa 6.7 but not at pCa 7.0; in contrast, it did evoke contraction of aortic cells at pCa 7.0. Thus, although the phenylephrine-induced contraction of ferret aortic cells has previously been shown to be Ca2+ independent (Collins et al. 1992), in portal vein cells this contraction requires at least pCa 6.7. Although the contraction in portal vein cells is Ca2+ dependent, the results indicate a ‘Ca2+ sensitization’ since, at constant [Ca2+], the addition of phenylephrine increases force.
In imaging studies using Bodipy Phorbol, a non-isozyme-specific PKC probe, we showed that phenylephrine-induced shortening and translocation of PKC to the surface membrane are dependent on extracellular Ca2+ in portal vein but not aortic smooth muscle cells (Khalil & Morgan, 1992). We suggested that this is related to the presence of Ca2+-dependent PKC isozymes in portal vein, but Ca2+-independent isozymes in aorta (Khalil & Morgan, 1992). We have also previously reported that translocation of αPKC requires ∼150 nm[Ca2+]i and reaches a maximal level at ∼200 nm[Ca2+]i (Khalil et al. 1994). Thus, the fact that pCa 6.7 is required for phenylephrine-induced contraction of portal vein cells is consistent with the in situ Ca2+ dependence of αPKC translocation.
It has been reported that PKC isozymes translocate to unique subcellular sites following activation and that translocation is required for their function. Translocation has been suggested to involve binding of activated isozymes to specific anchoring proteins, RACKs (Mochly-Rosen et al. 1991; Ron & Mochly-Rosen, 1994, 1995; Mochly-Rosen, 1995; Johnson et al. 1996). Therefore, we postulated that synthetic peptides derived from RACK-binding sequences of PKC isozymes may inhibit the translocation and function of PKC isozymes in permeabilized smooth muscle cells.
As described in the Introduction, C2-2 is a peptide derived from the C2 domain of βPKC and has been reported to be a translocation inhibitor of Ca2+-dependent PKC isozymes (Ron et al. 1994). εV1-2 is a short peptide derived from the V1 region of εPKC and is reported to be a translocation inhibitor of εPKC (Johnson et al. 1996); ηPKC contains a similar sequence (EAVGLQPT, residues 18–25) (Osada et al. 1990) and so this peptide could be expected to inhibit translocation of ηPKC as well as εPKC. However, even though ηPKC was detected in portal vein, it was not detectable in aorta and, therefore, cannot explain the effects of the εV1-2 peptide. If stimulation-induced translocation of PKC isozymes is required for their function, then introduction of these isozyme-specific translocation inhibitors into single permeabilized portal vein and aortic cells should inhibit their functions. We focused on the effects of C2-2 and εV1-2 peptides on phenylephrine-induced contraction of portal vein and aortic cells at constant [Ca2+].
A major finding of the present study was that C2-2 peptide inhibited phenylephrine-induced contraction of portal vein, but not of aorta. We also confirmed the expression of α and βPKC isozymes in the portal vein. These results suggest that phenylephrine-induced contraction of the portal vein may be caused by activation of Ca2+-dependent PKC isozymes containing the C2 domain. Furthermore, these results suggest that the function of the activated PKC isozymes in smooth muscle cells is dependent on interaction with a RACK. On the other hand, relatively small amounts of αPKC, and no βPKC, were detected in the aorta. The failure of C2-2 peptide to inhibit phenylephrine-induced contraction of aortic smooth muscle cells could be due to the very low level of αPKC expression or to a lack of intact signalling cascades linking αPKC to contraction in these cells, e.g. absence of an appropriate RACK. Indeed, the imaging data indicate that the small amount of αPKC present in the aorta does not translocate in the presence of phenylephrine.
Inhibition of phenylephrine-induced contraction of portal vein cells by pre-treatment with C2-2 peptide was more effective than inhibition by post-treatment with the peptide. Previous studies investigating the effects of PKC translocation inhibitors have generally been performed using pre-treatment with the peptides (Smith & Mochly-Rosen, 1992; Ron & Mochly-Rosen, 1994; Ron et al. 1995; Johnson et al. 1996; Yedovitzky et al. 1997). In the case of exposure of the cell to the peptide prior to activation of PKC, the translocation inhibitor presumably binds to a RACK and, following stimulation with phenylephrine, prevents the interaction of PKC with the RACK. In the case of post-treatment with the peptide, RACKs should be pre-occupied by activated PKC which may have already phosphorylated its substrate, necessitating the action of a phosphatase for reversal of the contraction and a lesser effect of the peptide would therefore be expected.
Another major finding of the present study was that εV1-2 peptide inhibited phenylephrine-induced contraction of aortic but not portal vein cells. Furthermore, the level of expression of εPKC in aorta was significantly greater than that in portal vein cells, qualitatively confirming our previous studies in which εPKC was detected in aorta but, at equivalent exposure times in both Western blot and single cell imaging, was not detectable in portal vein (Khalil et al. 1992). Similarly, Walker et al. (1998) have reported the presence of greater quantities of εPKC in ferret aorta than in ferret portal vein or ferret femoral artery. εV1-2 peptide has been shown to inhibit the translocation of εPKC, but not α, β or δPKC isozymes (Johnson et al. 1996). Furthermore, the εV1 domain fragment binds to the εPKC-specific RACK when introduced into cells and thus inhibits phorbol ester or hormone-induced εPKC translocation and binding to its RACK. Our results, therefore, indicate that interaction of activated εPKC with this RACK is required for phenylephrine-induced contraction of the aorta. The εV1-2 peptide also reduced resting tension in aortic cells at pCa 7.0. These results suggest that basal active tension involves partial activation of εPKC in aortic cells. We previously showed that the addition of PSSI to unstimulated permeabilized cells caused a significant drop in basal tension at pCa 7.0, and suggested that a fraction of the basal tension is the result of activation of a Ca2+-independent PKC isozyme (Collins et al. 1992).
On the other hand, the εV1-2 peptide failed to inhibit phenylephrine-induced contraction or reduce basal tension in portal vein cells at pCa 6.7. This was surprising given the expression of εPKC in portal vein, albeit at a lower level than that in the aorta. At pCa 6.7, both Ca2+-dependent and Ca2+-independent PKC isozymes may be activated by phenylephrine. The fact that phenylephrine did not evoke contraction of portal vein cells at pCa 7.0 and that the translocation inhibitor of εPKC did not affect the amplitude of the phenylephrine-induced contraction, strongly suggests that members of the signalling cascade linking εPKC to contraction are missing from the portal vein. Again, this could be due, for example, to the absence of a specific RACK. Imaging of εPKC in portal vein cells showed no detectable translocation of εPKC upon addition of phenylephrine. It is of interest that Walker et al. (1998) have similarly reported that, despite the presence of the Ca2+-independent εPKC in intact portal vein, this tissue fails to contract in response to a phorbol ester in the absence of extracellular Ca2+.
Activation of PKC by phenylephrine has been shown to induce translocation of cytosolic PKC to the surface membrane in a previous (Khalil & Morgan, 1991) and in the present study, and to stimulate binding to and phosphorylation of a variety of cytosolic regulatory proteins (Mochly-Rosen et al. 1991). The plasmalemmal location of activated PKC has been difficult to reconcile with the distant intracellular location of contractile proteins. However, it has been reported previously that the phenylephrine-induced translocation of cytosolic PKC to the surface membrane is associated with a transient redistribution of cytosolic mitogen-activated protein (MAP) kinase to the surface membrane before cell contraction (Khalil & Morgan, 1993). Coincident with cell contraction, MAP kinase undergoes a second redistribution away from the plasmalemma and towards the vicinity of the contractile filaments (Khalil & Morgan, 1993). It has also been reported that PD-098059, a specific inhibitor of MAP kinase kinase, inhibits contraction, MAP kinase activation and caldesmon phosphorylation by phenylephrine (Dessy et al. 1998). Therefore, translocation of PKC appears to trigger a phosphorylation cascade resulting in activation of MAP kinase (as well as other kinases), phosphorylation of the thin filament-associated protein caldesmon and, finally, contraction.
It has been suggested by others that an isozyme-specific interaction between εPKC and filamentous actin may serve as a necessary prelude to glutamate-induced exocytosis from nerve terminals, implying that actin is a principal anchoring protein or scaffolding protein for εPKC within nerve endings (Prekeris et al. 1996). The binding site for actin is located between the first and second cysteine-rich regions within the regulatory C1 domain of εPKC (Prekeris et al. 1996). We examined the effects of a synthetic actin-binding peptide corresponding to the putative actin-binding domain of εPKC (εPKC (223–228)), which has been shown to compete with native εPKC for binding to purified actin in vitro, on phenylephrine-induced contraction of portal vein and aortic smooth muscle cells. The peptide failed to have any effect on phenylephrine-induced contraction or resting tension in either cell type.
In conclusion, our results support the involvement of a Ca2+-dependent PKC isozyme (α and/or βPKC) in the signalling cascade leading to contraction of portal vein but not aortic smooth muscle cells. On the other hand, our results suggest the involvement of εPKC (but not acting via its actin-binding domain) in contraction of aortic but not portal vein smooth muscle cells.
Acknowledgments
The authors are grateful for the expert assistance of Carrie Sougnez and Samantha Matson in performing the Western blot analysis of tissues reported here. This work was supported by KOSEF (to Y.-H. L.), a grant from the Alberta Heart and Stroke Foundation (to M. P. W.) and Public Health Service Grants HL42293 and HL31704 (to K. G. M.).
References
- Brozovich FV, Walsh MP, Morgan KG. Regulation of force in skinned, single cells of ferret aortic smooth muscle. Pflügers Archiv. 1990;416:742–749. doi: 10.1007/BF00370624. [DOI] [PubMed] [Google Scholar]
- Brozovich FV, Yates LD, Gordon AM. Muscle force and stiffness during activation and relaxation. Implications for the actomyosin ATPase. Journal of General Physiology. 1988;91:399–420. doi: 10.1085/jgp.91.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapline C, Ramsay K, Klauck T, Jaken S. Interaction cloning of protein kinase C substrates. Journal of Biological Chemistry. 1993;268:6858–6861. [PubMed] [Google Scholar]
- Collins EM, Walsh MP, Morgan KG. Contraction of single vascular smooth muscle cells by phenylephrine at constant [Ca2+]i. American Journal of Physiology. 1992;262:H754–762. doi: 10.1152/ajpheart.1992.262.3.H754. [DOI] [PubMed] [Google Scholar]
- DeFeo TT, Morgan KG. Responses of enzymatically isolated mammalian vascular smooth muscle cells to pharmacological and electrical stimuli. Pflügers Archiv. 1985;404:100–102. doi: 10.1007/BF00581502. [DOI] [PubMed] [Google Scholar]
- Dessy C, Kim I, Sougnez CL, Laporte R, Morgan KG. A role for MAP kinase in differentiated smooth muscle contraction evoked by α-adrenoceptor stimulation. American Journal of Physiology. 1998;275:C1081–1086. doi: 10.1152/ajpcell.1998.275.4.C1081. [DOI] [PubMed] [Google Scholar]
- Horowitz A, Clément-Chomienne O, Walsh MP, Morgan KG. ε-Isoenzyme of protein kinase C induces a Ca2+-independent contraction in vascular smooth muscle. American Journal of Physiology. 1996;271:C589–594. doi: 10.1152/ajpcell.1996.271.2.C589. [DOI] [PubMed] [Google Scholar]
- Hyatt SL, Liao L, Chapline C, Jaken S. Identification and characterization of α-protein kinase C binding proteins in normal and transformed REF52 cells. Biochemistry. 1994;33:1223–1228. doi: 10.1021/bi00171a023. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. Journal of Biological Chemistry. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- Katsuyama H, Morgan KG. Mechanisms of Ca2+-independent contraction in single permeabilized ferret aorta cells. Circulation Research. 1993;72:651–657. doi: 10.1161/01.res.72.3.651. [DOI] [PubMed] [Google Scholar]
- Katsuyama H, Wang CL, Morgan KG. Regulation of vascular smooth muscle tone by caldesmon. Journal of Biological Chemistry. 1992;267:14555–14558. [PubMed] [Google Scholar]
- Khalil RA, Lajoie C, Morgan KG. In situ determination of [Ca2+]i threshold for translocation of the α-protein kinase C isoform. American Journal of Physiology. 1994;266:C1544–1551. doi: 10.1152/ajpcell.1994.266.6.C1544. [DOI] [PubMed] [Google Scholar]
- Khalil RA, Lajoie C, Resnick MS, Morgan KG. Ca2+-independent isoforms of protein kinase C differentially translocate in smooth muscle. American Journal of Physiology. 1992;263:C714–719. doi: 10.1152/ajpcell.1992.263.3.C714. [DOI] [PubMed] [Google Scholar]
- Khalil RA, Morgan KG. Imaging of protein kinase C distribution and translocation in living vascular smooth muscle cells. Circulation Research. 1991;69:1626–1631. doi: 10.1161/01.res.69.6.1626. [DOI] [PubMed] [Google Scholar]
- Khalil RA, Morgan KG. Phenylephrine-induced translocation of protein kinase C and shortening of two types of vascular cells of the ferret. The Journal of Physiology. 1992;455:585–599. doi: 10.1113/jphysiol.1992.sp019317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil RA, Morgan KG. PKC-mediated redistribution of mitogen-activated protein kinase during smooth muscle cell activation. American Journal of Physiology. 1993;265:C406–411. doi: 10.1152/ajpcell.1993.265.2.C406. [DOI] [PubMed] [Google Scholar]
- Kraft AS, Anderson WB, Cooper HL, Sando JJ. Decrease in cytosolic calcium/phospholipid-dependent protein kinase activity following phorbol ester treatment of EL4 thymoma cells. Journal of Biological Chemistry. 1982;257:13193–13196. [PubMed] [Google Scholar]
- Liao L, Hyatt SL, Chapline C, Jaken S. Protein kinase C domains involved in interactions with other proteins. Biochemistry. 1994;33:1229–1233. doi: 10.1021/bi00171a024. [DOI] [PubMed] [Google Scholar]
- Liou YM, Morgan KG. Redistribution of protein kinase C isoforms in association with vascular hypertrophy of rat aorta. American Journal of Physiology. 1994;267:C980–989. doi: 10.1152/ajpcell.1994.267.4.C980. [DOI] [PubMed] [Google Scholar]
- Menice CB, Hulvershorn J, Adam LP, Wang C-L A, Morgan KG. Calponin and mitogen-activated protein kinase signaling in differentiated vascular smooth muscle. Journal of Biological Chemistry. 1997;272:25157–25161. doi: 10.1074/jbc.272.40.25157. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D, Khaner H, Lopez J. Identification of intracellular receptor proteins for activated protein kinase C. Proceedings of the National Academy of Sciences of the USA. 1991;88:3997–4000. doi: 10.1073/pnas.88.9.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada S, Mizuno K, Saido TC, Akita Y, Suzuki K, Kuroki T, Ohno S. A phorbol ester receptor/protein kinase, nPKη, a new member of the protein kinase C family predominantly expressed in lung and skin. Journal of Biological Chemistry. 1990;265:22434–22440. [PubMed] [Google Scholar]
- Prekeris R, Mayhew MW, Cooper JB, Terrian DM. Identification and localization of an actin-binding motif that is unique to the epsilon isoform of protein kinase C and participates in the regulation of synaptic function. Journal of Cell Biology. 1996;132:77–90. doi: 10.1083/jcb.132.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Chen C-H, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: a homolog of the β subunit of G proteins. Proceedings of the National Academy of Sciences of the USA. 1994;91:839–843. doi: 10.1073/pnas.91.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Luo J, Mochly-Rosen D. C2 region-derived peptides inhibit translocation and function of β protein kinase C in vivo. Journal of Biological Chemistry. 1995;270:24180–24187. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- Ron D, Mochly-Rosen D. Agonists and antagonists of protein kinase C function, derived from its binding proteins. Journal of Biological Chemistry. 1994;269:21395–21398. [PubMed] [Google Scholar]
- Ron D, Mochly-Rosen D. An autoregulatory region in protein kinase C: the pseudoanchoring site. Proceedings of the National Academy of Sciences of the USA. 1995;92:492–496. doi: 10.1073/pnas.92.2.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BL, Mochly-Rosen D. Inhibition of protein kinase C function by injection of intracellular receptors for the enzyme. Biochemical and Biophysical Research Communications. 1992;188:1235–1240. doi: 10.1016/0006-291x(92)91363-u. [DOI] [PubMed] [Google Scholar]
- Staudinger J, Zhou J, Burgess R, Elledge SJ, Olson EN. PICK1: a perinuclear binding protein and substrate for protein kinase C isolated by the yeast two-hybrid system. Journal of Cell Biology. 1995;128:263–271. doi: 10.1083/jcb.128.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suematsu E, Resnick M, Morgan KG. Change of Ca2+ requirement for myosin phosphorylation by prostaglandin F2α. American Journal of Physiology. 1991;261:C253–258. doi: 10.1152/ajpcell.1991.261.2.C253. [DOI] [PubMed] [Google Scholar]
- Walker LA, Gailly P, Jensen PE, Somlyo AV, Somlyo AP. The unimportance of being (protein kinase C) epsilon. FASEB Journal. 1998;12:813–821. doi: 10.1096/fasebj.12.10.813. [DOI] [PubMed] [Google Scholar]
- Yedovitzky M, Mochly-Rosen D, Johnson JA, Gray MO, Ron D, Abramovitch E, Cerasi E, Nesher R. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic β-cells. Journal of Biological Chemistry. 1997;272:1417–1420. doi: 10.1074/jbc.272.3.1417. [DOI] [PubMed] [Google Scholar]