

Abstract
Openers of the ATP-sensitive potassium channel (KATP channel) increase and blockers decrease renin secretion. Here we report the effects of levcromakalim (LCRK, a channel opener) and glibenclamide (GBC, a blocker) on membrane potential, whole-cell current and the cytoplasmic Ca2+ concentration of renin-secreting cells (RSC). Studies were performed on afferent arterioles from the kidney of Na+-depleted rats.
As monitored with the fluorescent oxonol dye DiBAC4(3), LCRK (0.3 and 1 μm) induced a hyperpolarization of ≈15 mV which was abolished by GBC (1 μm).
Whole-cell current-clamp experiments showed that RSC had a membrane potential of −61 ± 1 mV (n = 16). LCRK (1 μm) induced a hyperpolarization of 9.9 ± 0.2 mV (n = 16) which, in the majority of cells, decreased slowly with time.
Capacitance measurements showed a strong electrical coupling of the cells in the preparation.
At −60 mV, LCRK induced a hyperpolarizing current in a concentration-dependent manner with an EC50 of 152 ± 31 nm and a maximum current of about 200 pA.
Application of GBC (1 μm) produced no effect; however, when applied after LCRK (300 nm), GBC inhibited the opener-induced hyperpolarizing current with an IC50 of 103 ± 36 nm.
LCRK (0.3 and 1 μm) did not significantly affect the cytoplasmic Ca2+ concentration either at rest or after stimulation by angiotensin II.
The data show that LCRK induces a GBC-sensitive hyperpolarizing current in rat RSC. This current presumably originates from the activation of KATP channels which pharmacologically resemble those in vascular smooth muscle cells. The stimulatory effect of KATP channel opening on renin secretion is not mediated by a decrease in intracellular Ca2+ concentration.
Renin, an aspartyl protease, controls the activity of the renin-angiotensin-aldosterone system, which, in turn, plays a dominant role in the control of blood pressure and electrolyte balance of the organism. The major source of renin in mammals is the juxtaglomerular myoepitheloid cells (renin-secreting cells, RSC) in the media of the afferent arteriole close to the entrance into the glomerulus (for review see Hackenthal et al. 1990). Control of renin secretion from the RSC is complex, involving intrarenal mechanisms (e.g. the macula densa and the baroreceptor mechanisms) and extrarenal signals (e.g. from sympathetic nerves and circulating hormones; for review see Kurtz, 1989; Hackenthal et al. 1990; Osswald & Quast, 1995). An intriguing feature of the control of renin secretion is the observation that manoeuvres that decrease the cytoplasmic Ca2+ concentration ([Ca2+]i) increase the rate of renin secretion from RSC and vice versa, termed the ‘Ca2+ paradox’ (Kurtz, 1989; Hackenthal et al. 1990). Despite the physiological importance of renin secretion there have only been a few electrophysiological studies of RSC (reviewed in Osswald & Quast, 1995). These studies were conducted in multicellular preparations from the mouse using either isolated glomeruli with a remnant of the afferent arteriole attached or hydronephrotic kidney halves. Subsequent to microelectrode studies of the effects of hormones on membrane potential (Fishman, 1976; Bührle et al. 1984, 1985), Kurtz & Penner (1989) examined the electrophysiological properties of RSC in the afferent arteriole using the whole-cell voltage-clamp technique combined with Ca2+ measurements by fura-2 fluorescence. Despite strong electrical coupling of the cells in this preparation, the authors were able to show the presence of inwardly and outwardly rectifying K+ conductances and of a major Ca2+-dependent Cl− conductance.
In several species including man, openers of the KATP channel increase plasma renin activity (Ferrier et al. 1989; Richer et al. 1990; Pratz et al. 1991), and the sulphonylurea glibenclamide (GBC), the classical inhibitor of these channels, decreases it (Richer et al. 1990; Pratz et al. 1991). In cultured or freshly isolated RSC (Ferrier et al. 1989; Jensen et al. 1998; Vallon et al. 1998) and in glomeruli with attached afferent arterioles (Jensen et al. 1998) prepared from rat or mouse kidney, KATP channel openers also increase renin secretion; this suggests the existence of KATP channels in RSC. A plausible working hypothesis for these effects of the openers is that KATP channel opening hyperpolarizes the RSC and that this hyperpolarization decreases resting [Ca2+]i and thereby increases renin secretion (Oswald & Quast, 1995). In vascular smooth muscle cells (from which the RSC are derived, see below), it has indeed been shown that the hyperpolarization induced by KATP channel openers reduces resting [Ca2+]i (Ito et al. 1991).
Up to now there have been no electrophysiological studies on KATP channels in renin-secreting cells. Here we have examined the effects of the selective KATP channel opener levcromakalim (LCRK) and of the inhibitor GBC on membrane potential, whole-cell current and [Ca2+]i of RSC. Since the effects of KATP channel modulators on renin secretion have generally been studied in the rat (in vivo: Richer et al. 1990; Pratz et al. 1991; cell culture: Ferrier et al. 1989; Vallon et al. 1998) this study was conducted in afferent arterioles from rat kidney. Following the example of Fishman (1976) we used sodium-depleted animals. Under this diet, the renin-angiotensin-aldosterone system is upregulated and smooth muscle cells in the distal part of the afferent arteriole are metaplastically transformed into renin-secreting myoepithelial cells (Cantin et al. 1977; Bührle et al. 1984; Hackenthal et al. 1987; Wurfer et al. 1988).
METHODS
Preparation of rat glomeruli
All animal experimentation described here was conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the German Law on the Protection of Animals. Sprague-Dawley rats (250–500 g) were treated with furosemide (frusemide) (10 mg kg−1, i.p.) and kept on a NaCl-depleted diet (C1036, Altromin, Lage, Germany) for at least 2 weeks. Animals were killed by cervical dislocation, exsanguinated and the kidneys removed. Glomeruli were prepared at 37°C by a modified version of published methods (Kurtz & Penner, 1989; Metzger & Quast, 1996). A freshly isolated kidney was transferred in a Hepes-buffered physiological salt solution (PSS) containing (mM): NaCl, 142; KCl, 2.8; MgCl2, 1; CaCl2, 1; and D(+)-glucose, 11; buffered with Hepes (10 mM) and titrated to pH 7.4 with NaOH at 37°C. The kidney was decapsulated, cut longitudinally into two halves and the cortex isolated. For fluorescence experiments a cortex half was passed successively through two stainless steel sieves of 280 and 160 μm mesh size and the glomeruli were collected on a 63 μm sieve. For patch-clamp experiments, cortex was minced with a razor blade and incubated for 50 min in 20 ml PSS with 20 mg collagenase A (Boehringer Mannheim) at 37°C under gentle shaking. The suspension was then passed through stainless steel sieves of mesh size 200 and 125 μm and the final material collected on a 63 μm sieve. Microscopic inspection showed that the preparations consisted mainly of glomeruli with some contamination by tubular fragments; about 10 % of the glomeruli contained a remnant of the afferent arteriole up to 100 μm in length (Fig. 1). Glomeruli were transferred to the recording chamber equipped with a poly-L-lysine-coated coverslip. Fluorescence measurements were performed by selecting an area of 30 μm × 30 μm from the afferent arteriole near the entrance into the glomerulus (Fig. 1). Electrophysiological recordings were taken from cells in the afferent arteriole at a distance between 5 and 70 μm from the glomerulus (mean distance, 38 ± 15 μm; determined from 36 out of 49 cells used for the experiments shown in Figs 4 and 5); cells were approached from the outside. For both types of measurement, only bulgy roundish cells were used, which are fully transformed RSC (Fig. 1). These cells are clearly distinguishable from smooth muscle cells which have a spindle-like appearance and a spiral arrangement around the vessel (Bührle et al. 1984; Hackenthal et al. 1990). The visual classification of the cells was performed with a × 40 objective (LD-ACHROPLAN; numerical aperture, 0.6) and differential interference contrast microscopy (Zeiss, Oberkochen, Germany).
Figure 1. Glomerular preparation from the kidney of a Na+-depleted rat.
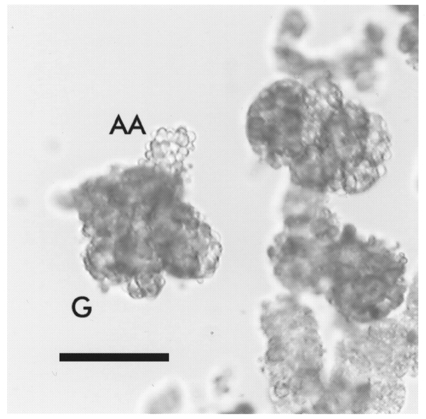
The preparation was exposed to collagenase to allow seal formation for patch-clamp experiments. This treatment completely removed Bowman's capsule around the glomeruli. RSC in the afferent arteriole appear like a bunch of grapes which is typical for salt-depleted rats. Only vessels with these bulgy roundish cells were used for experiments. G denotes a glomerulus and AA the afferent arteriole. The scale bar represents 100 μm.
Figure 4. LCRK-induced current (IK(LCRK)) in RSC.
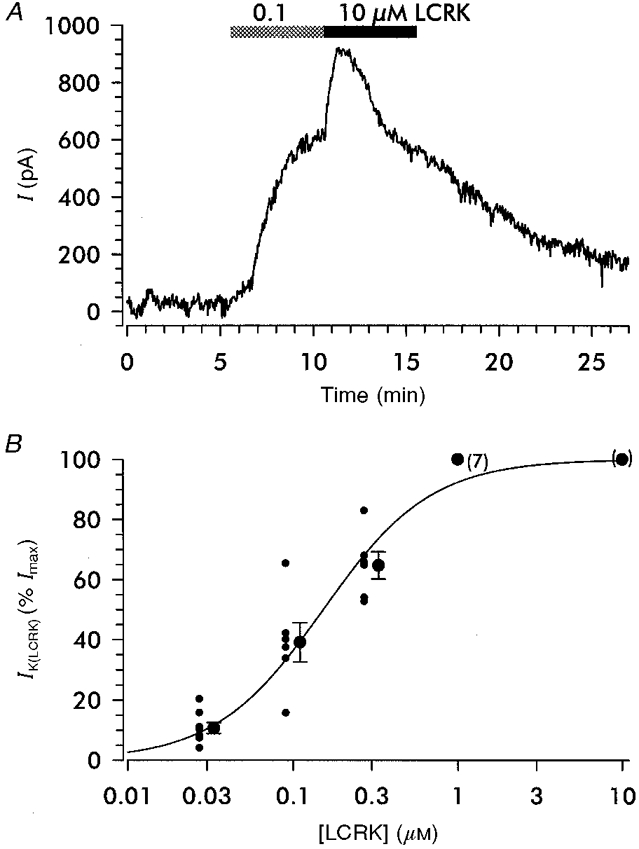
A, whole-cell recording at physiological K+ concentrations and a temperature of 37 °C; holding potential was −60 mV. Application of 0.1 and 10 μm LCRK induced concentration-dependent outward currents (IK(LCRK)). Note the rapid fading of the current in the continued presence of 10 μm LCRK. B, concentration dependence of IK(LCRK). Currents were normalized with respect to the maximal current (Imax) with 10 μm LCRK; Imax was 189 pA (median with 95 % confidence intervals of 161 and 368 pA; n = 20). The normalized values determined in individual experiments (small circles) and the means (larger circles) and s.e.m. are shown; for clarity, symbols are shifted to the left and right, respectively. The curve shows the Hill fit to the individual values giving a mid-point (EC50) of 152 ± 31 nm and a Hill coefficient of 1.32 ± 0.25.
Figure 5. Inhibition of IK(LCRK) by GBC.
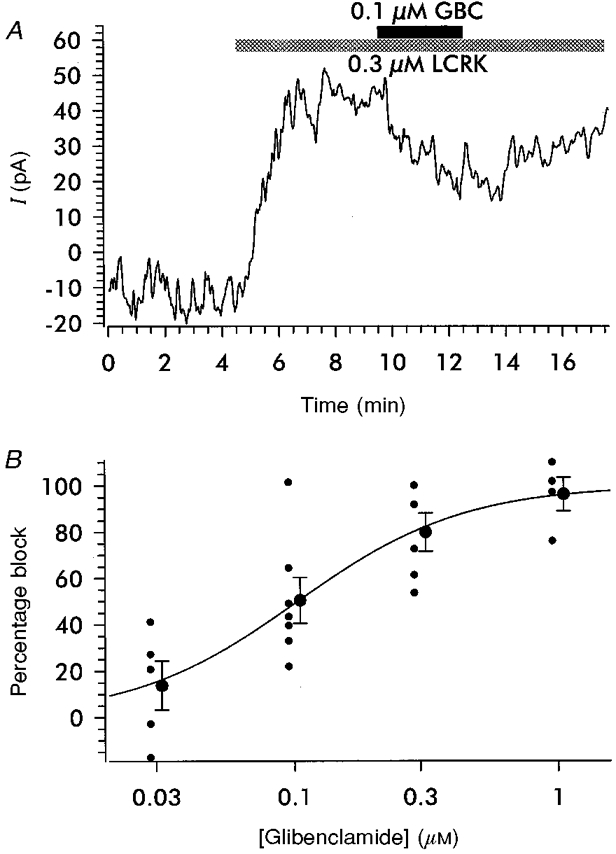
A, original trace showing the reduction of the current induced by 0.3 μm LCRK by GBC (0.1 μm). Within the time course of the experiment, the effect of GBC could be washed out only partially. Data were recorded in the whole-cell configuration under a physiological K+ gradient; holding potential was −60 mV and temperature was 37 °C. B, concentration-dependent inhibition of IK(LCRK) by GBC. The degree of inhibition was normalized with respect to the current induced by 0.3 μm LCRK prior to the addition of GBC. Normalized individual values and means ±s.e.m. are shown; for clarity, symbols are shifted to the left and right, respectively. The curve shows the Hill fit to the individual values giving an IC50 value of 103 ± 36 nm and a Hill coefficient of 1.38 ± 0.58.
Fluorescence experiments
To monitor changes in membrane potential, the fluorescent oxonol dye DiBAC4(3) (bis-(1,3-dibutylbarbituric acid)trimethine oxonol; Molecular Probes) was used (Bräuner et al. 1984; Langheinrich & Daut, 1997). Epifluorescence was measured with an inverted microscope (DIAPHOT 300, Nikon, Japan), equipped with a × 40 fluor oil-immersion objective (numerical aperture, 1.3; Nikon), a dichroic mirror (DM 505, Nikon) and a long wave pass barrier filter (BA 520, Nikon). Excitation light was generated by a 75 W xenon lamp and the wavelength was set to 488 nm with a monochromator (Photon Technology International (PTI), NJ, USA). Emitted light (wavelength > 520 nm) was collected by a photomultiplier (PTI) connected to an acquisition computer. Fluorescence was recorded at a sampling rate of 0.5 points s−1 using FeliX software (version 1.01, PTI). Glomeruli were continuously superfused with PSS containing 1 μm DiBAC4(3) at a flow rate of 1 ml min−1 at 37°C. After 80 min incubation, fluorescence reached a stable value and drug application was started. Autofluorescence amounted to about 15 % of the total signal and was not subtracted.
The fluorescence signal was calibrated using an approach similar to that described by Langheinrich & Daut (1997). Cells were rendered permeable to monovalent cations using gramicidin (1 μm) and Na+ was replaced by the impermeant cation N-methyl-D-glucamine (NMDG). In the presence of DiBAC4(3) (1 μm), the K+ concentration was varied and the NMDG concentration was adjusted accordingly in order to maintain a constant osmolarity. [K+]o was increased from 2.8 to 40 mM and [K+]i was assumed to be 140 mM, so membrane potential was calculated to vary between −102 and −33 mV. Correlation of the change in fluorescence (ΔF, %) with the calculated membrane potential showed that ΔF =± 1 % corresponded to a change of 4.0 ± 0.3 mV.
For measurement of [Ca2+]i, the dye fura-2 was used. Excitation wavelengths were set to 340 nm and 380 nm and the microscope was equipped with a dichroic mirror (430DCLPO2, Omega Optical, VT, USA) and an interference barrier filter (510WB40, Omega Optical). Cells were loaded with the indicator by incubation of glomeruli in PSS with 8 or 16 μm fura-2 AM (Molecular Probes), the membrane-permeable acetoxymethyl (AM) ester of fura-2, and 0.01–0.04 % (v/v) Pluronic F127 dissolved in DMSO for 60 min at room temperature or 35°C. After washing with PSS for 30 min fluorescence was evaluated using the same equipment as described above. The fura-2 fluorescence ratio (R) was calibrated following Grynkiewicz et al. (1985). In accordance with other groups (Fowler et al. 1996; Kornfeld et al. 1997) calibration was performed in the absence of glomeruli using 1 μm fura-2 pentapotassium salt. The following parameters were obtained (Grynkiewicz et al. 1985): Rmin= 0.27 ± 0.01, Rmax= 15.5 ± 0.7, β= 20.3 ± 1.4, and the KD value of Ca2+ binding to fura-2 was determined to be 192 ± 18 nm (n = 3).
Patch-clamp experiments
The patch-clamp technique was used in the whole-cell configuration as described by Hamill et al. (1981) with PSS in the bath at 37°C. Patch pipettes were drawn from filament borosilicate glass capillaries (GC 150F-15, Clark Electromedical Instruments) and heat polished using a horizontal microelectrode puller (Zeitz, Augsburg, Germany). After filling with (mM): potassium glutamate, 135; NaCl, 10; MgCl2, 1; Hepes, 10; EGTA, 1; and Na2ATP, 0.3; titrated to pH 7.2 with NaOH, pipettes had a resistance of 3–5 MΩ. All potentials given were corrected for a liquid junction potential of 10 mV (Neher, 1992). Data were recorded with an EPC 9 amplifier using Pulse software (HEKA, Lambrecht, Germany).
GBC (Sigma) and LCRK (SmithKline-Beecham, Harlow, UK) were prepared as 1 mM stock solutions in DMSO/ethanol (50/50, v/v). Solvent concentration (≤ 0.2 per thousand) was without effect on membrane potential or current.
Calculations and statistics
Results are expressed as means ±s.e.m. Concentration-response curves were fitted to the Hill equation:
where f(x) is the LCRK-induced current, IK(LCRK) (%Imax), or percentage block of this current, x is the concentration of LCRK or GBC, C is the mid-point of the curve (EC50 or IC50) and n is the Hill coefficient. Fitting was done according to the Marquardt- Levenberg algorithm using the program SigmaPlot 4.01 (Statistical Product & Service Solutions Inc., IL, USA). Errors in the fitting parameters were estimated by this software on the basis of the residual sum of squares in the univariate approximation. When performing calculations involving numbers with errors, the laws of error propagation were applied.
RESULTS
Membrane potential measurements in RSC
Figure 2A shows an original trace of DiBAC4(3) fluorescence. Superfusion of LCRK (0.3 μm) induced a decrease in fluorescence by 4.5 % corresponding to a hyperpolarization of about 18 mV. The effect was slow in onset, reflecting the slow response rate of the dye (Bräuner et al. 1984), and was well maintained during the presence of the agonist (20 min). The mean hyperpolarization produced by 0.3 and 1 μm LCRK was 18 ± 10 mV (n = 10) and 15 ± 5 mV (n = 9), respectively. Figure 2B shows that the hyperpolarization (∼8 mV) produced by 0.3 μm LCRK was reversed by addition of GBC (1 μm). When GBC (1 μm) was given alone or simultaneously with LCRK (0.3 μm), fluorescence remained unchanged.
Figure 2. Changes in DiBAC4(3) fluorescence induced by levcromakalim.
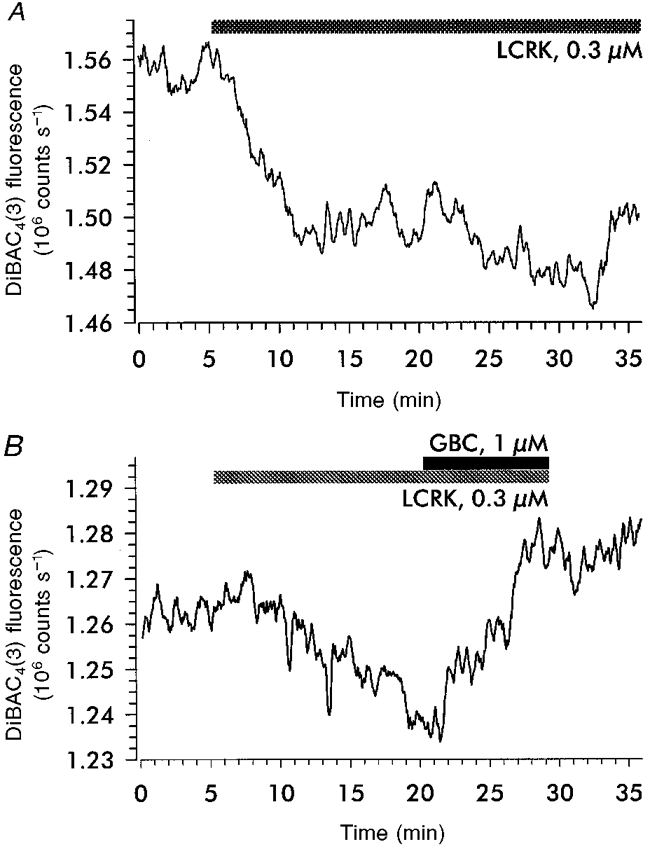
Glomeruli were incubated with the membrane potential-sensitive oxonol dye DiBAC4(3) and the epifluorescence from the afferent arteriole near the entrance into the glomerulus was monitored. A, levcromakalim (LCRK, 0.3 μm) induced a sustained decrease in fluorescence by 4.5 % corresponding to a hyperpolarization of ≈18 mV. B, reversal of the LCRK (0.3 μm)-induced decrease in fluorescence (2 %, corresponding to a hyperpolarization of ≈8 mV) by glibenclamide (GBC, 1 μm). The slight increase in fluorescence during the time period shown probably reflects continued dye uptake.
To obtain more direct measurements of membrane potential, experiments were performed in the current-clamp mode of the patch-clamp technique. Initially, using glomeruli from rats on a normal diet, the whole-cell configuration was established in < 5 % of attempts; with glomeruli from Na+-depleted rats, the success rate increased to 15–20 %. We assume that the increase of the number of RSC accompanied with the reorganization of the vessel led to a reduction of the extracellular matrix, making contact of the pipette to the cell membrane easier. Only results obtained in the latter preparation are presented. RSC had a membrane potential of −61 ± 1 mV (n = 16); two cells had a membrane potential negative to −85 mV and were not used for this study. Figure 3 shows three typical traces illustrating the effect of LCRK and GBC on membrane potential. About 50 % of the cells showed transient depolarization of about 20 mV of variable duration (1–30 s) and frequency, which appears as spikes in the recordings (Fig. 3B and C). LCRK (1 μm) induced hyperpolarizations up to ∼20 mV (mean, 9.9 ± 0.2 mV; n = 16) within 2–3 min (Fig. 3A-C). GBC (1 μm) alone had no effect on membrane potential (Fig. 3B); however, when given after application of LCRK (1 μm), it reversed the LCRK-induced hyperpolarization (Fig. 3B and C; n = 5). When given simultaneously with 1 μm LCRK, GBC (1 μm) prevented the hyperpolarizing effect of LCRK alone (Fig. 3C; n = 5).
Figure 3. Effects of LCRK and GBC on membrane potential in RSC.
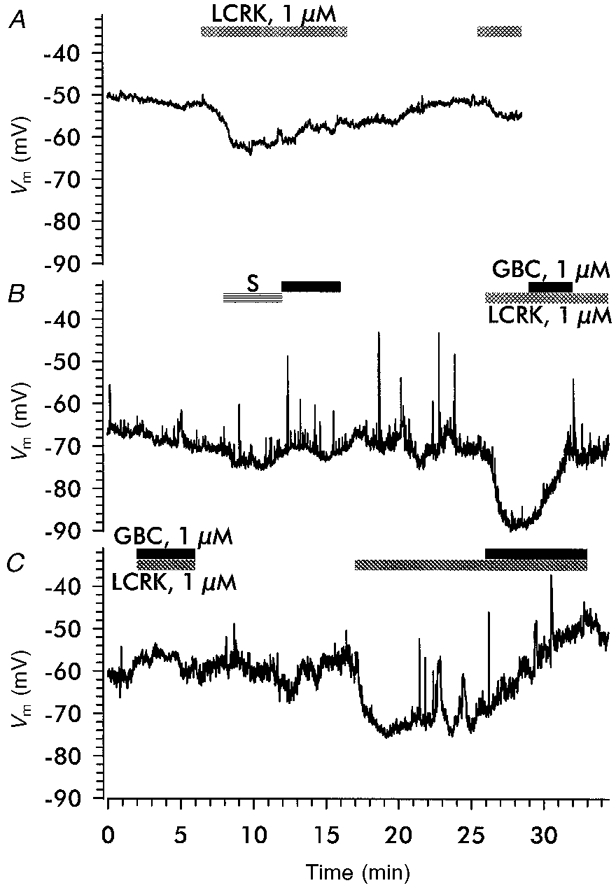
Membrane potential was measured with the whole-cell patch-clamp technique in the current-clamp mode. A, trace showing the effect of LCRK (1 μm) on membrane potential. Note the fading of the effect during prolonged application of the agonist (−50 % within 5 min after reaching the maximum) and the small response to a second challenge with LCRK (1 μm) 10 min after washout. B and C, superfusion of solvent (S, 0.1 per thousand ethanol + 0.1 per thousand DMSO), GBC (1 μm) and LCRK (1 μm). Note the spiking activity in these traces; see text for details.
In the majority of cells, the response to a prolonged application of the opener (1 μm) was not sustained. Figure 3A shows a trace where the hyperpolarization induced by 1 μm LCRK (10 mV) decayed by 30–40 % within 3 min after reaching the maximum. A second challenge with 1 μm LCRK, 10 min after washout of the first, induced a hyperpolarization of only 3 mV, indicating tachyphylaxis of the response to this concentration of agonist.
Whole-cell current measurements
Capacitive currents were large (> 20 pF, corresponding to membrane areas > 2000 μm2). Assuming a spherical cell shape, this corresponds to a diameter of > 25 μm which is about 3 times larger than the individual cells visible in Fig. 1. In addition, these currents decayed slowly with several time constants (τ from 1 to 100 ms). This indicated strong electrical coupling of the RSC in the vessel. Capacitive currents could not be compensated by the compensation unit of the amplifier; hence, capacitance compensation was switched off. Due to this cell-cell coupling it was impossible to clamp the preparation reliably at voltages deviating significantly from the zero current potential. Voltage ramps evoked conductances resembling delayed and inward rectifier potassium currents; however, these responses were variable and could not be reliably quantified (data not shown).
At a holding potential of −60 mV, LCRK (0.1 and 10 μm shown in Fig. 4A) induced hyperpolarizing currents (IK(LCRK)). In agreement with the current-clamp experiments this current was not maintained at LCRK concentrations ≥ 1 μm (see Fig. 4A for 10 μm LCRK). The peak current induced by 10 μm LCRK (Imax) was determined to be 189 pA (median) and showed large variations from experiment to experiment, reflected by the 95 % confidence intervals of 161 and 368 pA (n = 20). Attempts to determine the current-voltage relationship of IK(LCRK) were not successful since the responses of the preparation to voltage ramps were variable due to cell-cell coupling (see above) and IK(LCRK) was dominated by the much larger contributions of inward and delayed rectifier K+ currents. For evaluation of the concentration dependence of IK(LCRK), the current responses to LCRK were normalized with respect to the peak current obtained with 10 μm LCRK, applied directly after the test concentration of LCRK (Fig. 4A). The fit to the normalized data gave an EC50 for LCRK of 152 ± 31 nm and a Hill coefficient of 1.32 ± 0.25 (Fig. 4B).
The concentration-dependent inhibition of IK(LCRK) by GBC was studied in preparations challenged with 0.3 μm LCRK; the response to this concentration of agonist was well maintained and large enough to allow the measurement of a graded inhibition by GBC (Fig. 5A). The original trace (Fig. 5A) shows that 0.1 μm GBC induced a reduction of the current by 40 %; upon washout of GBC, the inhibition was partially reversed. The fit of the results from this series of experiments to the Hill function gave an IC50 value for GBC of 103 ± 36 nm and a Hill coefficient of 1.38 ± 0.58 (Fig. 5B).
[Ca2+]i measurements
The cytoplasmic free Ca2+ concentration in RSC at rest was 114 ± 4 nm (n = 56) and did not change with the distance from the glomerulus using a measuring window of 30 μm × 30 μm and arterioles composed exclusively of protruding roundish cells. [Ca2+]i responded to changes in [Ca2+]o. Figure 6 shows original traces depicting the exposure to low (0 mM Ca2++ 5 mM EGTA; Fig. 6A) and high (20 mM Ca2+; Fig. 6B) extracellular Ca2+. The presence of EGTA reduced [Ca2+]i by 59 ± 11 nm (n = 10); with 20 mM Ca2+ in the medium [Ca2+]i increased by 64 ± 18 nm (n = 6). A short application of angiotensin II (ANG II, 3 nm) induced a transient increase of [Ca2+]i by 21 ± 4 nm (n = 15; Fig. 6C); after a recovery period of several minutes, the stimulus could be repeated giving an identical result (data not shown). Increasing the K+ concentration in the bath induced small elevations of [Ca2+]i (20 mM K+: 18 ± 4 nm, n = 3; 30 mM K+: 29 ± 3 nm, n = 23; 60 mM K+: 38 ± 4 nm, n = 53; Fig. 6D); these changes were essentially abolished in the presence of the dihydropyridine Ca2+ antagonist isradipine (0.1 μm; not shown).
Figure 6. [Ca2+]i in RSC loaded with fura-2.
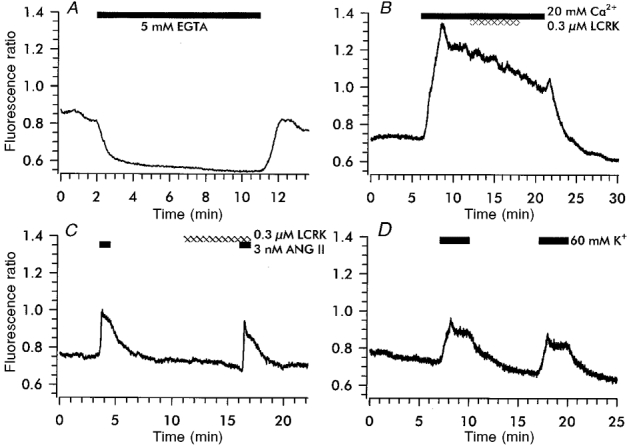
A, removal of Ca2+ from the bath solution by EGTA induced a decrease in [Ca2+]i by 75 nm, which was reversible upon washout of EGTA. B, an increase of [Ca2+] in the bath from 1 to 20 mM increased [Ca2+]i by 137 nm; LCRK (0.3 μm) was without affect on [Ca2+]i. C, double stimulation of the preparation with angiotensin II (ANG II) increased [Ca2+]i by 64 and 69 nm. Application of LCRK (0.3 μm) prior to and during the second challenge did not modify the response. D, superfusion of 60 mM K+ increased [Ca2+]i by 43 and 40 nm.
LCRK did not affect [Ca2+]i under various conditions, either at rest (0.3 or 1 μm; n = 4 each; data not shown) or with elevated [Ca2+]i (0.3 μm LCRK, 20 mM Ca2+ in the bath; n = 6; Fig. 6B). In addition, the response to the second ANG II stimulus was not modified by LCRK (0.3 μm; n = 4; Fig. 6C). Glibenclamide (1 μm) affected neither resting [Ca2+]i (n = 4) nor the ANG II-induced increase in [Ca2+]i (data not shown).
DISCUSSION
This study has shown that LCRK induces a GBC-sensitive hyperpolarizing current in RSC in the afferent arteriole from rat kidney; surprisingly, however, LCRK did not change [Ca2+]i at rest or during stimulation with ANG II.
The membrane potential of these cells was determined to be −61 ± 1 mV which is in good agreement with values published by others for RSC in the afferent arteriole of the mouse (−35 and −70 mV, Fishman, 1976; −55 mV, Bührle et al. 1984; −58 mV, Bührle et al. 1985; −62 mV, Kurtz & Penner, 1989; −69 mV, Kurtz & Penner, 1990).
LCRK, applied at 1 μm (see Fig. 3) produced a hyperpolarization by 9.9 ± 0.2 and 15 ± 5 mV in the current clamp and the fluorescence measurements, respectively. The large error in the result of the fluorescence experiments reflects the difficulties with this method which uses a slowly reacting dye (Bräuner et al. 1984). The limited size of the hyperpolarization may be due to a high electrical leakiness of the cells compared with the LCRK-induced current due to a paucity of channels or incomplete activation.
More precise information came from voltage-clamp experiments which showed that LCRK induced an outward current with an EC50 value of 0.15 ± 0.03 μm. This value is lower than those observed in other preparations (rat portal vein: 0.5 μm, Noack et al. 1992; rabbit portal vein: 1.3 μm, Russel et al. 1992; rabbit mesenteric artery: 1.9 μm (value for racemic cromakalim, of which levcromakalim is the active enantiomer), Quayle et al. 1995). In view of the strong inactivation of the current observed at high concentrations of LCRK (see Fig. 4) it is possible that the true maximum of the current response could not be reached and that the apparent EC50 value determined here is indeed too low. The transient nature of the responses to higher concentrations (≥ 1 μm) of LCRK observed here in the current and membrane potential measurements is also evident in current traces obtained in rabbit portal vein by Russel et al. (1992) and Beech et al. (1993b), and in pig urethra by Teramoto et al. (1997). Comparable inactivation has been shown too for 86Rb+ efflux in rat aorta (Quast et al. 1993). In other cells, however, the currents induced by high concentrations of LCRK were well maintained over minutes (rabbit mesenteric artery: Quayle et al. 1995; A10 cells: Russ et al. 1997). The reason for these differences is not clear, and they may depend on special regulatory mechanisms of the KATP channel in these cells.
GBC (1 μm) completely inhibited the hyperpolarization and current induced by LCRK (0.3 and 1 μm); the current produced by LCRK (0.3 μm) was inhibited by GBC with an IC50 value of 100 nm. This value is in excellent agreement with the IC50 of GBC for inhibition of cromakalim (1 μm)-induced 86Rb+ efflux from rat aortic strips (100 nm, Quast & Cook, 1989). The vascular KATP channel opened by Mg2+ salts of nucleoside diphosphates or depletion of ATP is closed by GBC with an IC50 value of ∼20 nm (Beech et al. 1993a; Xu & Lee, 1994), a value in excellent agreement with that determined in [3H]GBC binding studies in rat aortic rings (Ki= 20 nm, Löffler & Quast, 1997). In the presence of an opener, however, higher concentrations of GBC are needed to close the channel reflecting the quasi-competitive negative allosteric coupling of the sites for openers and sulphonylureas (Bray & Quast, 1992).
Collectively, these observations provide evidence that rat RSC are endowed with KATP channels; in their sensitivity to LCRK and GBC they resemble vascular KATP channels (Ashcroft & Ashcroft, 1990; Quast, 1996; Quayle et al. 1997). The direct demonstration and further characterization of these channels was prevented by the inherent complexities and instabilities of preparations with strongly coupled cells. The existence of such channels on RSC is corroborated by the following observations. First, high affinity binding sites for the KATP channel opener [3H]P1075 have recently been demonstrated in this preparation (Metzger & Quast, 1996). These sites, which show the typical pharmacological profile of the vascular KATP channel, are apparently localized on the remnant of the afferent arteriole adhering to the glomerulus. Second, KATP channel openers of different chemical classes (LCRK, pinacidil and diazoxide) increase renin secretion in cultured RSC (Ferrier et al. 1989; Jensen et al. 1998; Vallon et al. 1998), and, in isolated afferent arterioles, the LCRK effect is inhibited by GBC (Jensen et al. 1998). Finally, the fact that the ‘myoepithelial’ RSC are metaplastically transformed smooth muscle cells (Cantin et al. 1977) fits well with the observation that these cells express KATP channels of the vascular subtype.
What could be the physiological function of KATP channels on RSC? The fact that GBC alone altered neither membrane currents nor membrane potential shows that these channels were closed under the experimental conditions. This is in agreement with the observation that GBC did not attenuate basal renin secretion from isolated afferent arterioles or isolated RSC (Linseman et al. 1995; Jensen et al. 1998). It is well known that in several vascular beds KATP channels are opened by neurotransmitters and hormones that increase cAMP levels (reviewed in Quayle et al. 1997); however, these hormonal stimuli were absent in our preparation as was renal perfusion pressure, which is an important regulator of renin secretion (Kurtz, 1989; Jensen et al. 1998).
A major objective of this study was to test the hypothesis that KATP channel openers might increase renin secretion by reducing [Ca2+]i in RSC (Osswald & Quast, 1995). The fura-2 measurements showed that [Ca2+]i responded to changes in [Ca2+]o, and to stimulation by ANG II and increases in extracellular K+. The response to high [K+]o was inhibited by isradipine, suggesting the presence of voltage-operated Ca2+ channels on RSC. However, the changes in [Ca2+]i elicited by KCl (60 mM) were small, arguing against a major importance of such channels in these cells. This is essentially in line with the work of Kurtz and colleagues, who have provided solid evidence against the existence of voltage-operated Ca2+ channels on RSC (Kurtz & Penner, 1990; Scholz & Kurtz, 1995). The potential pathways which control Ca2+ entry into RSC, including the contribution of the Na+-Ca2+ exchanger, are under investigation.
Surprisingly, LCRK, at concentrations that induced maximum effects on current and membrane potential, did not affect [Ca2+]i, either at rest or at levels elevated by high [Ca2+]o or ANG II (Fig. 6). In view of the minor importance of voltage-operated Ca2+ channels in the ‘myoepithelial’ RSC, one would classify these cells as ‘non-excitable’. In non-excitable cells, e.g. endothelial cells, Ca2+ entry pathways are active under hyperpolarizing conditions and the increased driving force for Ca2+ entry upon hyperpolarization leads to an increase in [Ca2+]i (for reviews see Choquet & Korn, 1988; Nilius et al. 1997). This was not observed here. If, on the other hand, the Ca2+ handling in RSC resembles that of smooth muscle cells, the hyperpolarizing action of the KATP channel openers should decrease [Ca2+]i at rest (Ito et al. 1991). In addition, LCRK should reduce the Ca2+ increase mediated by contractile agonists as described for pinacidil and LCRK in rabbit mesenteric artery (Ito et al. 1991, 1992; Yamagishi et al. 1992); conversely, depolarization increases the formation of the Ca2+-releasing second messenger inositol 1,4,5,-trisphosphate (Yamagishi et al. 1992; Ganitkevich & Isenberg, 1993). In this study, LCRK did not change resting [Ca2+]i or the response to ANG II. A possible explanation is that the hyperpolarization produced by LCRK (approximately −10 mV) was too small to elicit these effects (see also above); alternatively, the Ca2+ handling of RSC could be very unusual.
The mechanism by which the KATP channel openers stimulate renin secretion remains unknown. Since LCRK does not decrease [Ca2+]i, one has to look for other possibilities. For instance, the hyperpolarization produced by the opener could affect the secretory machinery of the RSC, the cell volume or another cellular parameter to increase renin secretion. Another possibility is that the opener could act on mitochondrial KATP channels (Inoue et al. 1991; Garlid et al. 1996) and that the increase in renin secretion is unrelated to the hyperpolarization of the plasma membrane. This would then be analogous to the opener-induced preconditioning of the heart (Garlid et al. 1997). The mechanism for the opener effect on renin secretion is under active investigation.
Acknowledgments
We are grateful to Dr M. Albinus and Dr F. Metzger for helpful discussions and for introducing us to the glomeruli isolation technique. We also thank SmithKline-Beecham for LCRK. This study was supported by the Deutsche Forschungsgemeinschaft, grant Qu 100/2-1 and Qu 100/2-2 (U. Russ and U. Quast) and by a grant from the Federal Ministry of Education, Science, Research and Technology, and the Interdisciplinary Centre for Clinical Research (IZKF) Tübingen (Fö. 01KS 9602, U. Rauch).
References
- Ashcroft SJH, Ashcroft FM. Properties and functions of ATP-sensitive K-channels. Cellular Signalling. 1990;2:197–214. doi: 10.1016/0898-6568(90)90048-f. 10.1016/0898-6568(90)90048-F. [DOI] [PubMed] [Google Scholar]
- Beech DJ, Zhang H, Nakao K, Bolton TB. K channel activation by nucleotide diphosphates and its inhibition by glibenclamide in vascular smooth muscle cells. British Journal of Pharmacology. 1993a;110:573–582. doi: 10.1111/j.1476-5381.1993.tb13849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ, Zhang H, Nakao K, Bolton TB. Single channel and whole-cell K-currents evoked by levcromakalim in smooth muscle cells from the rabbit portal vein. British Journal of Pharmacology. 1993b;110:583–590. doi: 10.1111/j.1476-5381.1993.tb13850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräuner T, Hülser DF, Strasser RJ. Comparative measurements of membrane potentials with microelectrodes and voltage-sensitive dyes. Biochimica et Biophysica Acta. 1984;771:208–216. doi: 10.1016/0005-2736(84)90535-2. [DOI] [PubMed] [Google Scholar]
- Bray KM, Quast U. A specific binding site for K+ channel openers in rat aorta. Journal of Biological Chemistry. 1992;267:11689–11692. [PubMed] [Google Scholar]
- Bührle CP, Nobiling R, Mannek E, Schneider D, Hackenthal E, Taugner R. The afferent glomerular arteriole: immunocytochemical and electrophysiological investigations. Journal of Cardiovascular Pharmacology. 1984;6:S383–S393. [PubMed] [Google Scholar]
- Bührle CP, Nobiling R, Taugner R. Intracellular recordings from renin-positive cells of the afferent glomerular arteriole. American Journal of Physiology. 1985;249:F272–281. doi: 10.1152/ajprenal.1985.249.2.F272. [DOI] [PubMed] [Google Scholar]
- Cantin M, Araujo-Nascimento M-F, Benchimol S, Desormeaux Y. Metaplasia of smooth muscle cells into juxtaglomerular cells in the juxtaglomerular apparatus, arteries, and arterioles of the ischemic (endocrine) kidney. An ultrastructural-cytochemical and autoradiographic study. American Journal of Pathology. 1977;87:581–602. [PMC free article] [PubMed] [Google Scholar]
- Choquet D, Korn H. Modulation of voltage-dependent potassium channels in B lymphocytes. Biochemical Pharmacology. 1988;37:3797–3802. doi: 10.1016/0006-2952(88)90058-5. [DOI] [PubMed] [Google Scholar]
- Ferrier CP, Kurtz A, Lehner P, Shaw SG, Pusterla C, Saxenhofer H, Weidmann P. Stimulation of renin secretion by potassium-channel activation with cromakalim. European Journal of Clinical Pharmacology. 1989;36:443–447. doi: 10.1007/BF00558067. [DOI] [PubMed] [Google Scholar]
- Fishman MC. Membrane potential of juxtaglomerular cells. Nature. 1976;260:542–544. doi: 10.1038/260542a0. [DOI] [PubMed] [Google Scholar]
- Fowler BC, Carmines PK, Nelson LD, Bell PD. Characterization of sodium-calcium exchange in rabbit renal arterioles. Kidney International. 1996;50:1856–1862. doi: 10.1038/ki.1996.506. [DOI] [PubMed] [Google Scholar]
- Ganitkevich VY, Isenberg G. Membrane potential modulates inositol 1,4,5-trisphosphate-mediated Ca2+ transients in guinea-pig coronary myocytes. The Journal of Physiology. 1993;470:35–44. doi: 10.1113/jphysiol.1993.sp019845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D'Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circulation Research. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial KATP channel as a receptor for potassium channel openers. Journal of Biological Chemistry. 1996;271:8796–8799. doi: 10.1074/jbc.271.15.8796. 10.1074/jbc.271.15.8796. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hackenthal E, Metz R, Bührle CP, Taugner R. Intrarenal and intracellular distribution of renin and angiotensin. Kidney International. 1987;31(suppl. 20):S4–S17. [PubMed] [Google Scholar]
- Hackenthal E, Paul M, Ganten D, Taugner R. Morphology, physiology, and molecular biology of renin secretion. Physiological Reviews. 1990;70:1067–1116. doi: 10.1152/physrev.1990.70.4.1067. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Inoue I, Nagase H, Kishi K, Higuti T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352:244–247. doi: 10.1038/352244a0. [DOI] [PubMed] [Google Scholar]
- Ito S, Kajikuri J, Itoh T, Kuriyama H. Effects of lemakalim on changes in Ca2+ concentration and mechanical activity induced by noradrenaline in the rabbit mesenteric artery. British Journal of Pharmacology. 1991;104:227–233. doi: 10.1111/j.1476-5381.1991.tb12411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Seki N, Suzuki S, Ito S, Kajikuri J, Kuriyama H. Membrane hyperpolarization inhibits agonist-induced synthesis of inositol 1,4,5-trisphosphate in rabbit mesenteric artery. The Journal of Physiology. 1992;451:307–328. doi: 10.1113/jphysiol.1992.sp019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BL, Gambaryan S, Scholz H, Kurtz A. KATP channels are not essential for pressure-dependent control of renin secretion. Pflügers Archiv. 1998;435:670–677. doi: 10.1007/s004240050568. [DOI] [PubMed] [Google Scholar]
- Kornfeld M, Gutiérrez AM, Persson AEG, Salomonsson M. Angiotensin II induces a tachyphylactic calcium response in the rabbit afferent arteriole. Acta Physiologica Scandinavica. 1997;160:165–173. doi: 10.1046/j.1365-201X.1997.00153.x. [DOI] [PubMed] [Google Scholar]
- Kurtz A. Cellular control of renin secretion. Reviews of Physiology, Biochemistry and Pharmacology. 1989;113:1–40. doi: 10.1007/BFb0032674. [DOI] [PubMed] [Google Scholar]
- Kurtz A, Penner R. Angiotensin II induces oscillations of intracellular calcium and blocks anomalous inward rectifying potassium current in mouse renal juxtaglomerular cells. Proceedings of the National Academy of Sciences of the USA. 1989;86:3423–3427. doi: 10.1073/pnas.86.9.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz A, Penner R. Effects of angiotensin II on intracellular calcium and electrical function of mouse renal juxtaglomerular cells. Kidney International. 1990;38(suppl. 30):S51–S54. [PubMed] [Google Scholar]
- Langheinrich U, Daut J. Hyperpolarization of isolated capillaries from guinea-pig heart induced by K+ channel openers and glucose deprivation. The Journal of Physiology. 1997;502:397–408. doi: 10.1111/j.1469-7793.1997.397bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Lawson JA, Jones DA, Ludens JH. Glyburide attenuates calmodulin antagonist-stimulated renin release from isolated mouse juxtaglomerular cells. American Journal of Physiology. 1995;269:F242–247. doi: 10.1152/ajprenal.1995.269.2.F242. [DOI] [PubMed] [Google Scholar]
- Löffler C, Quast U. Pharmacological characterization of the sulphonylurea receptor in rat isolated aorta. British Journal of Pharmacology. 1997;120:476–480. doi: 10.1038/sj.bjp.0700919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger F, Quast U. Binding of [3H]-P1075, an opener of ATP-sensitive K+ channels, to rat glomerular preparations. Naunyn-Schmiedeberg's Archives of Pharmacology. 1996;354:452–459. doi: 10.1007/BF00168436. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods in Enzymology. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Nilius B, Viana F, Droogmans G. Ion channels in vascular endothelium. Annual Review of Physiology. 1997;59:145–170. doi: 10.1146/annurev.physiol.59.1.145. [DOI] [PubMed] [Google Scholar]
- Noack T, Deitmer P, Edwards G, Weston AH. Characterization of potassium currents modulated by BRL 38227 in rat portal vein. British Journal of Pharmacology. 1992;106:717–726. doi: 10.1111/j.1476-5381.1992.tb14400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osswald H, Quast U. Ion channels and renin secretion from juxtaglomerular cells. In: Scherübl H, Hescheler J, editors. The Electrophysiology of Neuroendocrine Cells. Boca Raton: CRC Press; 1995. pp. 301–314. [Google Scholar]
- Pratz J, Mondat S, Montier F, Cavero I. Effects of the K+ channel avtivators, RP 52891, cromakalim and diazoxide, on the plasma insulin level, plasma renin activity and blood pressure in rats. Journal of Pharmacology and Experimental Therapeutics. 1991;258:216–222. [PubMed] [Google Scholar]
- Quast U. ATP-sensitive K+ channels in the kidney. Naunyn-Schmiedeberg's Archives of Pharmacology. 1996;354:213–225. doi: 10.1007/BF00171051. [DOI] [PubMed] [Google Scholar]
- Quast U, Bray KM, Andres H, Manley PW, Baumlin Y, Dosogne J. Binding of the K+ channel opener [3H]P1075 in rat isolated aorta: relationship to functional effects of openers and blockers. Molecular Pharmacology. 1993;43:474–481. [PubMed] [Google Scholar]
- Quast U, Cook NS. Moving together: K+ channel openers and ATP-sensitive K+ channels. Trends in Pharmacological Sciences. 1989;10:431–435. doi: 10.1016/S0165-6147(89)80003-3. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Bonev AD, Brayden JE, Nelson MT. Pharmacology of ATP-sensitive K+ currents in smooth muscle cells from rabbit mesenteric artery. American Journal of Physiology. 1995;269:C1112–1118. doi: 10.1152/ajpcell.1995.269.5.C1112. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiological Reviews. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Richer C, Pratz J, Mulder P, Mondot S, Giudicelli JF, Cavero I. Cardiovascular and biological effects of K+ channel openers, a class of drugs with vasorelaxant and cardioprotective properties. Life Sciences. 1990;47:1693–1705. doi: 10.1016/0024-3205(90)90342-o. [DOI] [PubMed] [Google Scholar]
- Russ U, Metzger F, Kickenweiz E, Hambrock A, Krippeit-Drews P, Quast U. Binding and effects of KATP channel openers in the vascular smooth muscle cell line, A10. British Journal of Pharmacology. 1997;122:1119–1126. doi: 10.1038/sj.bjp.0701514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel SN, Smirnov SV, Aaronson PI. Effects of BRL 38227 on potassium currents in smooth muscle cells isolated from rabbit portal vein and human mesenteric artery. British Journal of Pharmacology. 1992;105:549–556. doi: 10.1111/j.1476-5381.1992.tb09017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz H, Kurtz A. Differential regulation of cytosolic calcium between afferent arteriolar smooth muscle cells from mouse kidney. Pflügers Archiv. 1995;431:46–51. doi: 10.1007/BF00374376. [DOI] [PubMed] [Google Scholar]
- Teramoto N, McMurray G, Brading AF. Effects of levcromakalim and nucleoside diphosphates on glibenclamide-sensitive K+ channels in pig urethral myocytes. British Journal of Pharmacology. 1997;120:1229–1240. doi: 10.1038/sj.bjp.0701033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V, Kirschenmann D, Brenner I, Albinus M, Osswald H. Potassium diet as a determinant for the renal response to systemic potassium channel modulation in anesthetized rats. Naunyn-Schmiedeberg's Archives of Pharmacology. 1998;358:245–252. doi: 10.1007/pl00005249. [DOI] [PubMed] [Google Scholar]
- Wurfer K, Hackenthal E, Metz R, Nobiling R, Simon T, Taugner R. Interzonal and intrazonal heterogeneities in the renin status of the preglomerular arterioles in five species. Histochemistry. 1988;89:283–287. doi: 10.1007/BF00493153. [DOI] [PubMed] [Google Scholar]
- Xu X, Lee KS. Characterization of the ATP-inhibited K+ current in canine coronary smooth muscle cells. Pflügers Archiv. 1994;427:110–120. doi: 10.1007/BF00585949. [DOI] [PubMed] [Google Scholar]
- Yamagishi T, Yanagisawa T, Taira N. K+ channel openers, cromakalim and Ki4032, inhibit agonist-induced Ca2+ release in canine coronary artery. Naunyn-Schmiedeberg's Archives of Pharmacology. 1992;346:691–700. doi: 10.1007/BF00168744. [DOI] [PubMed] [Google Scholar]