

Abstract
It is well established that the activity of Na+,K+-ATPase (NKA) is regulated by protein kinases A (PKA) and C (PKC), but results on their effects have been conflicting. The aim of this study was to examine if this is ascribed to the intracellular concentration of Ca2+ ([Ca2+]i).
Rat renal NKA was stably expressed in COS cells (green monkey kidney cells). Increases in [Ca2+]i were achieved with the Ca2+ ionophore A23187 and verified by direct measurements of [Ca2+]i using fura-2 AM as an indicator. The activity of NKA was measured as ouabain-sensitive 86Rb+ uptake and the state of phosphorylation of NKA was monitored with two site-directed phosphorylation state-specific antibodies.
Activation of PKA with forskolin decreased NKA activity by 45.5 ± 8.9 % at low [Ca2+]i (120 nM) and increased it by 40.5 ± 6.4 % at high [Ca2+]i (420 nM). The change in NKA activity by forskolin correlated with the level of increase in [Ca2+]i.
The effect of 1-oleoyl-2-acetoyl-sn-glycerol (OAG), a specific PKC activator, on the activity of NKA was also Ca2+ dependent, being inhibitory when [Ca2+]i was low (29.3 ± 3.6 % decrease at 120 nM Ca2+) and stimulatory when [Ca2+]i was high (36.6 ± 10.1 % increase at 420 nM Ca2+).
The α subunit of NKA was phosphorylated under both low and high [Ca2+]i conditions upon PKA or PKC activation. PKA phosphorylates Ser943. PKC phosphorylates Ser23.
To see if the observed effects on NKA activity are secondary to changes in Na+ entry, we measured NKA hydrolytic activity using permeabilized membranes isolated from cells under controlled Na+ conditions. A decreased activity at low [Ca2+]i and no change in activity at high [Ca2+]i were observed following forskolin or OAG treatment.
Purified NKA from rat renal cortex was phosphorylated and inhibited by PKC. This phosphorylation-associated inhibition of NKA was neither affected by Ca2+ nor by calmodulin, tested alone or together.
We conclude that effect of PKA/PKC on NKA activity is dependent on [Ca2+]i. This Ca2+ dependence may provide an explanation for the diversity of responses of NKA to activation of either PKA or PKC.
Na+,K+-ATPase (NKA) is an ubiquitous membrane bound protein that transports Na+ and K+ across the plasma membrane against their electrochemical gradients (Skou & Esmann, 1992). It plays a pivotal role for many basic cellular functions such as active transport of certain solutes, regulation of cell volume, and restoration of the membrane potential. An altered enzyme activity occurs in a number of clinical disorders, such as hypertension and diabetes (for a review see Laski & Kurtzman, 1996). Therefore, elucidating the regulatory mechanisms that underlie the control of NKA activity has become an important issue.
There is overwhelming evidence that the activity of NKA can be modulated by first messengers as well as by signalling pathways that involve activation of cAMP-dependent protein kinase (PKA) and/or protein kinase C (PKC) (for a review see Ewart & Klip, 1995). It is well established that NKA from several species can be phosphorylated by PKA (Bertorello et al. 1991; Fisone et al. 1994; Lopina et al. 1995) and that rat NKA can be phosphorylated by PKC (Bertorello et al. 1991; Feschenko & Sweadner, 1994; Logvinenko et al. 1996). Phosphorylation by PKA and PKC has been demonstrated both in vitro and in vivo (Middleton et al. 1993; Beguin et al. 1994; Fisone et al. 1995; Carranza et al. 1996a; Feschenko & Sweadner, 1997; Cheng et al. 1997b,d; Li et al. 1998). Phosphorylation sites for both PKA (Beguin et al. 1994; Feschenko & Sweadner, 1994; Fisone et al. 1994) and PKC (Beguin et al. 1994; Feschenko & Sweadner, 1995; Logvinenko et al. 1996) have been identified.
However, studies on the functional effects of PKA/PKC phosphorylation have led to contradictory results. Studies on microsomal NKA preparations from various tissues and species (Limas et al. 1973; Tria et al. 1974; Braughler & Corder, 1978; Lingham & Sen, 1982; Tung et al. 1990; Bertorello et al. 1991; Lopina et al. 1995) have revealed an inhibitory effect of cAMP or PKA on NKA. Incubation of HeLa cells (Middleton et al. 1990) or mouse LTK− cells (Horiuchi et al. 1993) with cAMP is accompanied by a decrease in NKA enzymatic activity. In contrast, injection or superfusion of Xenopus oocytes with cAMP has a clear stimulatory effect on the pump current (Vasilets & Schwarz, 1992). In rat renal proximal tubules, activation of PKA has been reported to cause both a decrease (Hussian et al. 1997; Holtbäck et al. 1998) and an increase (Carranza et al. 1996b) in pump activity. Similar conflicting results have been reported for PKC activation, including inhibitory effects (Shahedi et al. 1992; Beron et al. 1997; Singh & Linas, 1997), no change (Feschenko & Sweadner, 1997) and stimulatory effects (Mito & Delamere, 1993; Sampson et al. 1994). Activation of PKC in isolated rat renal proximal tubules (Bertorello & Aperia, 1989; Satoh et al. 1993; Feraille et al. 1995) as well as in OK (opossum kidney) cells (Middleton et al. 1993; Pedemonte et al. 1997) has also been reported to cause both inhibitory and stimulatory effects.
Since the discrepancies in results cannot be explained only by species differences, it is likely that differences in experimental conditions have also played a role. One such condition may be the level of intracellular concentration of Ca2+ ([Ca2+]i), which is highly dependent on buffer conditions and on the preparation of tissues and cells. There is evidence that Ca2+ can modulate the activity of NKA (for review see Yingst, 1988). Studies performed on isolated guinea-pig ventricular myocytes have indicated that the effect of the β-adrenergic agonist isoproterenol (isoprenaline) on the NKA current is [Ca2+]i dependent (Gao et al. 1992). Here we show that [Ca2+]i does indeed modulate the response of rat renal NKA to PKA and PKC activation. Most of the protocols in this study were performed on green monky kidney COS cells stably expressing rat renal NKA. We have previously shown that in this cell system rat NKA is inhibited upon PKA or PKC activation (Belusa et al. 1997; Cheng et al. 1997b,d). We now show that the inhibitory effects of PKA/PKC phosphorylation is reversed by increases in [Ca2+]i. A portion of this work has been presented at the 14th International Congress of Nephrology ‘97, Sydney, Australia (Cheng et al. 1997c) and at the 30th American Society of Nephrology Annual meeting ‘97, San Antonio, TX, USA (Cheng et al. 1997a).
METHODS
Purification of rat renal NKA
NKA was purified from rat renal cortex as described (Jorgensen, 1974). Forty-day-old Sprague-Dawley rats were deeply anaesthetized with pentobarbital (80 mg (kg body weight)−1i.p.). The aorta was cut to empty the organs of blood and the kidneys rapidly removed. The preparation was analysed by SDS-PAGE, stained with Coomassie Brilliant Blue. Densitometric analysis of the stained gels indicated that NKA α subunit constituted 55-60 % of total protein. The specific activity of NKA in the preparation used was calculated as ∼700 μmol of ATP hydrolysed (mg protein)−1 h−1. The ouabain (5 mM)-insensitive ATPase activity was less than 3 % of total ATPase activity.
Phosphorylation of purified NKA by PKC in vitro
Phosphorylation of purified NKA was carried out in a reaction volume of 50 μl containing: 50 mM Hepes (pH 7.5), 10 mM MgCl2, 1.2 mM CaCl2, 1.25 μg purified NKA, and 4 μU PKC (final concentration ∼80 ng ml−1). In control samples, PKC was omitted and an equal volume of bovine serum albumin (BSA) (final concentration 80 ng ml−1) dissolved in PKC storage buffer (50 % glycerol (v/v), 0.02 % Tween 20 (w/v), 20 mM Tris-HCl, 0.5 mM EGTA, 5 mM dithioerythritol, pH 7.5) was added to the reaction mix. The reaction was initiated by the addition of ATP to a final concentration of 0.1 mM. In some experiments tracer amounts of [γ-32P]ATP were included in the reaction mix to confirm that NKA was phosphorylated. The incubation proceeded at 25°C for 1 h and was stopped by cooling on ice. The phosphorylation of NKA was verified by detecting the radioactivity of 32P incorporated into the NKA α subunit as described (Fisone et al. 1994) or by detecting the changes in the immunoreactivity to a phosphorylation state-specific antibody (Feschenko & Sweadner, 1997) of NKA that had been subjected to SDS-PAGE and transferred to nitrocellulose membranes.
Expression of NKA α subunit in COS cells
The entire cDNA coding for wild-type rat NKA α1 was excised from a Bluescript vector and subcloned into the eukaryotic expression vector pXM as previously described (Cheng et al. 1997b). The vector pXM contains the adenovirus major late promoter and the SV40 origin, early gene enhancer and polyadenylation sequence. To obtain cell lines with the NKA cDNA stably integrated into chromosomes, plasmid DNA was linearized with NdeI and transfected into COS-7 cells using the calcium-phosphate-DNA precipitation method. We used ouabain sensitivity to select those COS cells in which the cDNA encoding the rat α1 subunit had been stably transfected (Cheng et al. 1997b). Due to a difference in ouabain binding affinity between monkey and rat NKA, monkey cells expressing the rat NKA survive concentrations of ouabain that kill wild-type monkey cells (COS-7 cells). Cells were cultured in Dulbecco's modified Eagle's (DME) medium supplemented with 10 % fetal calf serum (FCS). Sixty hours following cell transfection, ouabain was added to the medium to a final concentration of 10−5 M. After about 10 days, hundreds of individual ouabain-resistant colonies appeared. These colonies were pooled and propagated. The method has been described in detail previously (Cheng et al. 1997b). In the surviving cells, the transfected NKA represents approximately 50-60 % of total NKA.
Cell culture
Cells were grown at 37°C, 5 % CO2, and 95 % humidified air on 90 mm culture dishes or 6-well culture plates in DME medium supplemented with 10 % fetal calf serum (FCS) and 1 % Pen-Strep (1 % penicillin-streptomycin). A low concentration of ouabain (10−5 M) was present in the medium to block endogenous NKA activity (Cheng et al. 1997b). The number of cells was selected to seed on the culture plates so that they would reach confluency in 4 or 5 days. Media were changed 16 h before the cells were used for experiments.
Increases in intracellular Ca2+
To increase the level of [Ca2+]i, cells were preincubated with varying amounts of Ca2+ ionophore A23187 (from 10−8 to 10−5 M) to obtain [Ca2+]i ranging between 120 and 420 nM. The calcium concentration in incubation solution was 1 mM. The levels of [Ca2+]i were determined by direct measurements of intracellular Ca2+ as described below. In the continuous presence of A23187, an increase in [Ca2+]i occurred within 1 min following the addition of the drug, which reached a plateau after about 5 min that lasted for at least 15 min (Fig. 1). Thus, to test the effect of Ca2+, cells were preincubated with the Ca2+ ionophore 5 min before PKA or PKC was activated. The kinetics of the [Ca2+]i change was similar with different concentrations of A23187 used.
Figure 1. The kinetic change in [Ca2+]i induced by A23187.
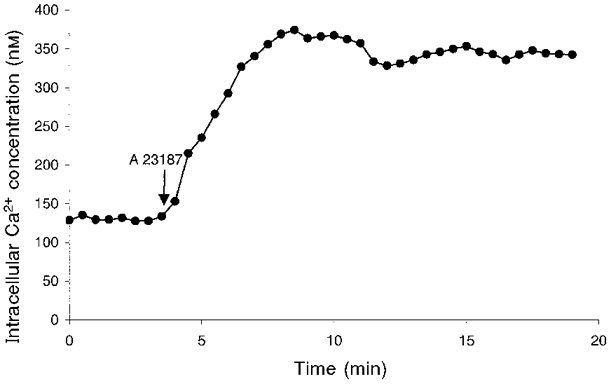
[Ca2+]i was measured using fura-2 AM as an indicator on SPEX dual-beam excitation spectrofluometer as described in Methods. Shown here is an example of changes in [Ca2+]i induced by A23187, which was added at 10−6 M. Similar kinetic changes induced by different concentrations of A23187 were also observed. The data represent mean values obtained from 12 individual cells in one experiment.
Activation of PKA and PKC
To activate PKA, confluent cells were incubated with forskolin (10−5 M), used in combination with the phosphodiesterase inhibitor IBMX (5 × 10−4 M). To activate PKC, OAG (10−8 M), the diacyglycerol analogue, was used. Unless otherwise specified, all the incubations were performed for 15 min at 37°C in a physiological salt solution (PSS) containing (mM): 110 NaCl, 5 KCl, 1.0 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.0 NaH2PO4, 20 Hepes (pH 7.4 at 37°C), and 10 glucose. All the cell incubation solutions were prewarmed at 37°C and equilibrated with 5 % CO2 and 95 % O2 before applied to the cell cultures.
Cell lysate preparation
Following drug treatment, cells were incubated for 15 min on ice in a lysis buffer containing: 130 mM NaCl, 5 mM EDTA, 2 mM EGTA, 20 mM Tris-HCl (pH 8.0), 50 mM NaF, 1 % (w/w) Triton X-100, 0.1 % (w/w) SDS, 100 μM phenylmethylsulfonyl fluoride (PMSF), 25 mM benzamidine, 20 μg ml−1 leupeptin, 20 μg ml−1 antipain, 5 μg ml−1 pepstatin A, 5 μg ml−1 chymostatin, and 0.2 % (w/v) BSA. Cells were then scraped and briefly sonicated. Protein concentrations were determined using a kit from Bio-Rad and BSA as a standard. Samples containing equal amounts of protein were subjected to SDS-PAGE and immunoblotting.
Western blot analysis and determination of phosphorylation
Electrophoresis was performed using an 8 % polyacrylamide gel. Protein transfer, immunoblotting and phosphorylation determination were performed as previously described (Cheng et al. 1997b).
Two different site-selective phosphorylation state-specific antibodies were used in this study: monoclonal antibody Mck1, which specifically binds to the Ser23-dephosphorylated, but not the phosphorylated, form of rat NKA α1 subunit (Feschenko & Sweadner, 1997); and polyclonal antibody 471, which selectively detects the Ser943-phosphorylated, but not the dephosphorylated, form of NKA α subunit (Fisone et al. 1994). For comparison, the abundance of NKA protein was also examined. This was achieved by using an antibody (McAb 6F, 1:100 dilution) that is not sensitive to changes in phosphorylation (Feschenko & Sweadner, 1997).
Measurement of intracellular Ca2+
Single cell-based [Ca2+]i measurements were made in a SPEX dual-beam excitation spectrofluometer using fura-2 as an indicator. Cells grown on 25 mm glass coverslips were loaded with a membrane-permeable acetoxymethyl (AM) ester of fura-2 (2 μM) at 37°C for 1 h under standard culture conditions as described above. The cells were then washed and subsequently superfused with a PSS solution. The Petri dish was placed on the thermostable stage of a fluorescence microscope (Zeiss Inc., Thornwood, NY, USA) equipped with a fluorescence detection and quantification system. Cytosolic free Ca2+ concentration was calculated from the ratio of fluorescence detected at 510 nm using excitation wavelengths of 345 and 380 nm. The fluorescence ratio signal was calibrated at the end of each experiment by adding 1 μM ionomycin to equilibrate the cytoplasm with the external PSS solution where the Ca2+ concentration was 1 mM. This solution was then replaced with a PSS solution containing 5 mM EGTA. [Ca2+]i was calculated according to the equation described by Grynkiewicz et al. (1985) assuming a Kd of fura-2 for Ca2+ of 224 nM. All experimental solutions were kept at 37°C and equilibrated with 5 % CO2 and 95 % O2.
Measurement of 86Rb+ uptake
The transport activity of NKA was determined in intact cells by measuring ouabain-sensitive 86Rb+ uptake under linear conditions. The experimental details have been given elsewhere (Cheng et al. 1997b). Experiments were performed at 37°C under a humidified atmosphere with 5 % CO2-95 % O2 in the PSS solution. The incubation buffer was prewarmed at 37°C and equilibrated with 5 % CO2-95 % O2. The uptake of 86Rb+ from the medium was calculated on the basis of the specific activity of the incubation medium. Ouabain-sensitive 86Rb+ uptake was determined as the difference between 86Rb+ uptake in the absence and presence of 5 mM ouabain.
Determination of NKA activity
Isolation and permeabilization of cell membranes and subsequent determination of NKA activity were performed as described (Fisone et al. 1994; Cheng et al. 1997b). In brief, following preincubation of cells with drugs, cell membranes were isolated, resuspended in TME buffer (75 mM Tris (pH 7.5), 12.5 mM MgCl2, 1.5 mM EDTA) and quickly frozen on dry ice and thawed at room temperature to open vesicles formed during the membrane preparation. Aliquots of membrane fragments were incubated for 15 min at 37°C in 100 μl of a solution containing (mM): 20 KCl, 5 MgCl2, 30 Tris-HCl (pH 7.4), 1 EGTA, 3 Tris-ATP, and tracer amounts of [γ-32P]-ATP. NaCl was added to 10 or 70 mM. To maintain a constant osmolarity, choline chloride was also added, so that the sum of NaCl and choline chloride was kept constant at 130 mM. To prevent dephosphorylation of Na+,K+-ATPase, the buffer was complemented with the protein phosphatase inhibitors okadaic acid (250 nM) and FK506 (25 nM). An amount of enzyme was selected so that total ATP hydrolysis did not exceed 20 %, and ATP hydrolysis was linear with time. The reaction was stopped by the addition of 700 μl of activated charcoal. The 32Pi liberated was determined in the supernatant after centrifugation. For the determination of ouabain-insensitive rat ATPase activity, NaCl and KCl were omitted, and 5 mM ouabain was added. The protein content of cell membranes was determined as described above.
The determination of NKA activity in purified enzyme preparations was essentially made using the same protocol as described above except that the reaction was made in a buffer containing (mM): 100 NaCl, 5 KCl, 5 MgCl2, 50 Tris-HCl (pH 7.4 at 37°C), 3 Tris-ATP, and tracer amounts of [γ-32P]ATP. To study the effect of Ca2+ and calmodulin on the NKA activity, Ca2+ present in the reaction mix was removed by dilution and centrifugation using a Centricon concentrator (Amicon Inc., Beverly, MA, USA). The concentrated sample was washed twice with ice-cold SH buffer (250 mM sucrose and 30 mM histidine, pH 7.2), and the pellet was resuspended in 50 μl SH buffer for the determination of NKA activity. Control and phosphorylated enzymes were treated identically. The different concentrations of Ca2+ in the assay medium were obtained using a combination of CaCl2 and EGTA. The concentrations of free Ca2+ in buffers were calculated as described (Bers et al. 1994) using a computer program (MAXCHELATOR, Stanford University: http://www.stanford.edu/~cpatton) in which all the substances that could possibly influence the binding of Ca2+ to EGTA were taken into account.
Chemicals
Forskolin, IBMX, monensin and EGTA were purchased from Sigma; 1-oleoyl-2-acetoyl-sn-glycerol (OAG) was from Avanti Polar Lipids, Inc., Alabaster, AL, USA; the bisindolyl maleimide GF109203x and A23187 were from Calbiochem; fura-2 AM and SBFI were from Molecular Probes. All drugs were stored as stock solutions in DMSO at -20°C. The final concentration of DMSO in the working solutions was 0.1 % (v/v), which was always added as vehicle to each control solution. Ouabain was purchased from Merck; PKC from rat brain and calmodulin were purchased from Boehringer-Mannheim; 86RbCl was from Amersham International; [γ-32P]-ATP was from New England Nuclear; and DMEM, fetal calf serum (FCS) and 1 % Pen-Strep were from Life Technologies Inc.
Statistics
Values are given as means ±s.e.m. Statistical comparisons between two groups were performed by Student's t test. P < 0.05 was considered significant.
RESULTS
Effect of forskolin on the activity and Ser943 phosphorylation of NKA at both low and high [Ca2+]i
To assess the possible role of [Ca2+]i in the regulation of NKA activity by PKA, cells were pretreated with the Ca2+ ionophore A23187. An amount of the Ca2+ ionophore was selected so that different levels of increase in [Ca2+]i were produced. The increases in [Ca2+]i were verified by direct intracellular Ca2+ measurement using fura-2 as an indicator. Following A23187 pretreatment, cells were incubated for 15 min at 37°C with forskolin (10−5 M) plus the phosphodiesterase inhibitor IBMX (5 × 10−4 M) to activate PKA. The activity of NKA was measured by 86Rb+ uptake and the state of phosphorylation of Ser943, the site of the NKA α subunit phosphorylated by PKA, was assessed. At low [Ca2+]i (120 ± 14 nM, n = 21), activation of PKA by forskolin and IBMX caused a significant decrease (45.5 ± 8.9 %) in ouabain-sensitive 86Rb+ uptake (Fig. 2A). In contrast, at high [Ca2+]i (420 ± 32 nM, n = 16), forskolin and IBMX caused a significant increase (40.5 ± 6.4 %) in ouabain-sensitive 86Rb+ uptake. The basal NKA activity (in nmol mg−1 min−1) was 18.75 ± 2.10 at low [Ca2+]i and 20.62 ± 2.53 at high [Ca2+]i, values which are not statistically different (P > 0.05).
Figure 2. The effects of forskolin and IBMX on the activity and Ser943 phosphorylation of NKA at both low and high [Ca2+]i.
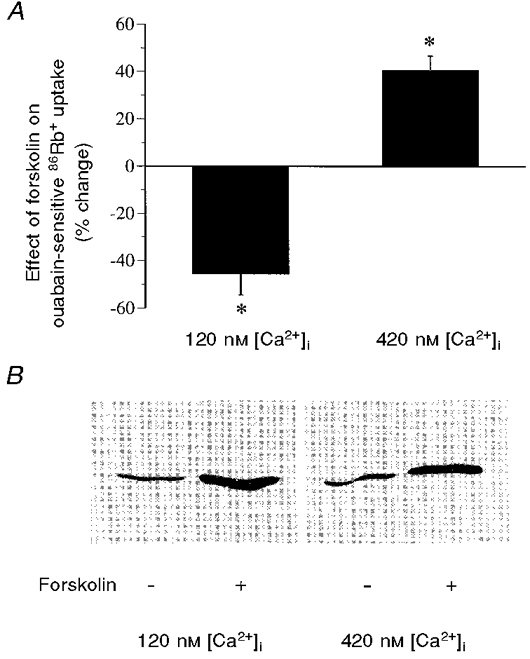
Confluent cultures of COS cells expressing rat renal NKA were treated with forskolin (10−5 M) and IBMX (5 × 10−4 M) for 15 min at 37 °C. Following drug treatment, both the activity and Ser943 phosphorylation of NKA were assayed. The levels of [Ca2+]i were measured in a SPEX dual-beam excitation spectrofluometer using fura-2 AM as an indicator. An increase in [Ca2+]i was achieved by preincubation of cells with the Ca2+ ionophore A23187 (10−5 M). The [Ca2+]i values shown here represent the mean values of the [Ca2+]i from 16-21 individual cells in four separate experiments, which were calculated based on an in vivo calibration system (see Methods). A, the effect of forskolin and IBMX on the activity of NKA, which was measured as ouabain-sensitive 86Rb+ uptake. Values are means ±s.e.m. of 5 separate experiments. The basal NKA activity (in nmol mg−1 min−1) was 18.75 ± 2.10 at low [Ca2+]i and 20.62 ± 2.53 at high [Ca2+]i. B, the effect of forskolin and IBMX on the state of Ser943 phosphorylation, which was determined by Western blot analysis with an antibody specifically recognizing the Ser943-phosphorylated, but not the dephosphorylated, form of NKA. A blot representative of 3 experiments is shown. *P < 0.01.
The phosphorylation of Ser943 was analysed by Western blot using an antibody that selectively recognizes the Ser943-phosphorylated, but not the dephosphorylated, form of NKA (Fisone et al. 1994; Cheng et al. 1997b) (Fig. 2B). For comparison, the abundance of NKA protein was also examined using an antibody that is not sensitive to changes in phosphorylation (Feschenko & Sweadner, 1997). At both low and high [Ca2+]i, activation of PKA by forskolin and IBMX caused a significant increase in phosphorylation of NKA. The percentage increase in immunoreactivity by forskolin and IBMX was 191.3 ± 23.2 % at low [Ca2+]i (P < 0.01) and 186.1 ± 34.6 % at high [Ca2+]i (P < 0.01). Under both [Ca2+]i conditions, the abundance of NKA was not significantly affected by forskolin and IBMX treatment. The percentage change in immunoreactivity was 103.3 ± 4.4 % at low [Ca2+]i (P > 0.05) and 106.1 ± 4.7 % at high [Ca2+]i (P > 0.05).
To examine whether there was a quantitative relationship between [Ca2+]i and the effect of forskolin on NKA activity, we performed a series of experiments in which various amounts of A23187 were used (Fig. 3). Increasing [Ca2+]i initially attenuated and then reversed the inhibitory effect of forskolin and IBMX on NKA activity in an approximately linear fashion.
Figure 3. The relationship between the levels of [Ca2+]i and changes in NKA activity induced by forskolin and IBMX.
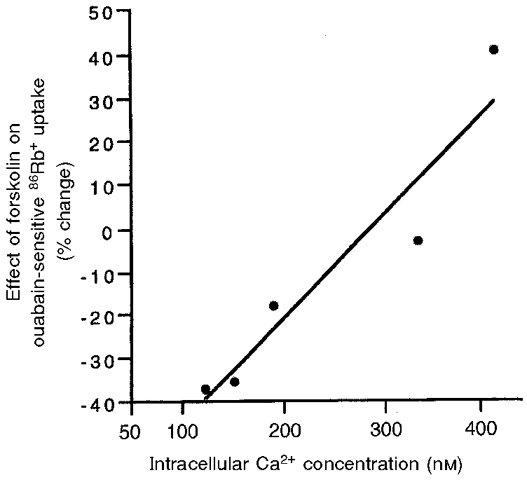
The percentage changes in NKA activity induced by forskolin (10−5 M) and IBMX (5 × 10−4 M) (mean values of 2-4 separate experiments) were plotted as a function of the levels of [Ca2+]i at which the regulation of NKA activity was performed. The activity of NKA in both control and forskolin- and IBMX-incubated cells was measured as ouabain-sensitive 86Rb+ uptake. The different levels of [Ca2+]i were produced by the application of different concentrations of A23187 and were measured in a SPEX dual-beam excitation spectrofluometer as described in the Methods. The [Ca2+]i values shown here represent the mean values of the [Ca2+]i from 16-21 individual cells in 4 separate experiments. These five [Ca2+]i values are (mean ±s.e.m.) 120 ± 14, 151 ± 15, 188 ± 23, 331 ± 24 and 420 ± 32 nM, which represent the levels of [Ca2+]i of control cells and cells treated with A23187 at 10−8, 10−7, 10−6 and 10−5 M, respectively.
Effect of OAG on the activity and Ser23 phosphorylation of NKA at both low and high [Ca2+]i
To assess the possible role of [Ca2+]i in the regulation of NKA activity by PKC, cells were treated as above with A23187 and the effects of OAG, an activator of the classic and novel isoforms of PKC (Nishizuka, 1984), were measured (Fig. 4A). At low [Ca2+]i, activation of PKC by OAG caused a significant decrease (29.3 ± 3.6 %) in ouabain-sensitive 86Rb+ uptake. In contrast, OAG caused a significant increase (36.6 ± 10.1 %) in ouabain-sensitive 86Rb+ uptake at high [Ca2+]i.
Figure 4. The effects of OAG on the activity and Ser23 phosphorylation of NKA at both low and high [Ca2+]i.
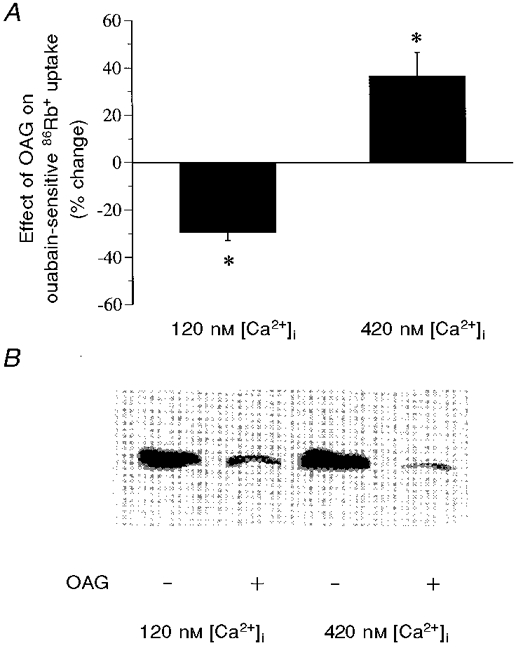
Confluent cultures of COS cells expressing rat renal NKA were treated with OAG (10−8 M) for 15 min at 37 °C under low and high [Ca2+]i conditions. Following drug treatment, both the activity and Ser23 phosphorylation of NKA were assayed. A, the effect of OAG on the activity of NKA. Values are means ±s.e.m. of 5 separate experiments. The basal NKA activity (in nmol mg−1 min−1) was 18.75 ± 2.10 at low [Ca2+]i and 20.62 ± 2.53 at high [Ca2+]i. B, the effect of OAG on the state of Ser23 phosphorylation, which was determined by Western blot analysis with an antibody specifically recognizing the Ser23-dephosphorylated, but not the phosphorylated, form of NKA. A blot representative of 3 experiments is shown. *P < 0.01.
The effect of OAG on the state of phosphorylation of Ser23, the site phosphorylated by PKC, was measured (Fig. 4B). In this study, the phosphorylation of Ser23 was also monitored by Western blot analysis using a phosphorylation state-specific antibody. Unlike the antibody used for detection of the Ser943 phosphorylation, this antibody selectively reacts with the dephosphorylated, but not the phosphorylated Ser23 of the NKA α subunit (Feschenko & Sweadner, 1997). Under basal conditions, an immunoreactive band corresponding to the α subunit of NKA (molecular mass ∼110 kDa; Fig. 4B, lanes 1 and 3) was detected in cell lysates. Activation of PKC by OAG significantly decreased the immunoreactive signal at both low and high [Ca2+]i (Fig. 4B, lanes 2 and 4). The percentage decrease in immunoreactivity was 56.7 ± 3.6 % at low [Ca2+]i (P < 0.01) and 64.9 ± 7.3 % at high [Ca2+]i (P < 0.01). Under both [Ca2+]i conditions, the abundance of NKA was not significantly affected by OAG treatment. The percentage change in immunoreactivity was 98.2 ± 10.1 % at low [Ca2+]i (P > 0.05) and 101.6 ± 8.9 % at high [Ca2+]i (P > 0.05).
Role of Na+ entry in the regulation of the NKA activity by forskolin or OAG
To investigate if the observed inhibition or stimulation of NKA activity induced by forskolin or OAG is secondary to changes in [Na+]i brought about by putative changes in Na+ entry, we measured NKA activity using permeabilized membranes isolated from these cells under controlled Na+ conditions. The results are summarized in Table 1. In this study, cells were treated with forskolin or OAG as described above under both low and high [Ca2+]i conditions. Following drug treatment, cells were lysed and crude cell membranes were prepared in the presence of protein phosphatase inhibitors okadaic acid and FK506. The activity of NKA was assayed as ouabain-sensitive ATP hydrolysis in 10 and 70 mM Na+, respectively. At low [Ca2+]i, activation of PKA with forskolin and IBMX resulted in a significant decrease in NKA activity, measured under both non-saturating (i.e. 10 mM) and saturating (i.e. 70 mM) Na+ conditions. The results are consistent with the effects on NKA-dependent Rb+ fluxes assayed in intact cells. At high [Ca2+]i, no change in NKA activity was observed following PKA activation. Similar changes in NKA activity were also observed for OAG-induced PKC activation (Table 1). The basal activity of NKA was lower at high [Ca2+]i than at low [Ca2+]i but the difference was not statistically significant.
Table 1.
NKA activity in cell membranes isolated from cells pretreated with forskolin or OAG under low or high [Ca2+]i conditions
| NKA activity (nmol mg−1 h−1) | ||
|---|---|---|
| 10 mM Na+ | 70 mM Na+ | |
| Low [Ca2+]i | ||
| Control | 2285 ± 195 | 5211 ± 329 |
| FSK + IBMX | 1452 ± 217* | 4228 ± 477* |
| OAG | 1419 ± 162* | 4044 ± 388* |
| High [Ca2+]i | ||
| Control | 2132 ± 218 | 5043 ± 585 |
| FSK + IBMX | 2136 ± 376 | 5588 ± 877 |
| OAG | 2529 ± 492 | 5302 ± 954 |
COS cells stably expressing rat renal NKA were incubated with forskolin (FSK; 10−6 M) and IBMX (5 ± 10−4 M) or OAG (10−8 M) for 15 min at 37 °C at low (120 nM) or high (420 nM) [Ca2+]i. Following incubation, membranes were isolated and permeabilized, and Na+, K+-ATPase activity was measured as ouabain-sensitive ATP hydrolysis in the presence of protein phosphatase inhibitors okadaic acid and FK506 at non-saturating (i.e. 10 mM) and saturating (i.e. 70 mM) levels of Na2+ as described in Methods. Values represent means ± S.E.M. of 7 separate experiments.
Significantly different from control, P < 0.01, Student's paired t test.
Effects of Ca2+-calmodulin on PKC-mediated regulation of purified NKA
To examine whether Ca2+ exerts its effect on NKA via a direct interaction with the enzyme, we phosphorylated NKA in vitro and compared the phosphorylation-induced changes in NKA activity in the absence or presence of Ca2+ and/or its binding protein, calmodulin. The activity of NKA was measured as ouabain-sensitive ATP hydrolysis.
Phosphorylation of NKA by PKC resulted in a significant decrease in NKA activity (Table 2). When the specific PKC inhibitor bisindolyl maleimide GF109203x was present, both phosphorylation and inhibition of NKA were abolished (data not shown). To examine the effect of Ca2+, we added to the assay buffer various concentrations of Ca2+ and/or calmodulin. As shown in Table 2, the inhibition of NKA activity by PKC phosphorylation was not affected by Ca2+ or by calmodulin, tested alone or together.
Table 2.
Inhibition of the activity of rat renal Na+,K+-ATPase by PKC phosphorylation: effects of Ca2+ and calmodulin (CaM)
| Na+,K+-ATPase activity | ||||
|---|---|---|---|---|
| Ca2+(μM) | CaM(μM) | Control(μmol P1 mg−1 h−1) | PKC phosphorylation (μmol P1 mg−1 h−1) | % control |
| 0 | 0 | 610 ± 7 | 345 ± 28* | 56.7 ± 5.0 |
| 10 | 0 | 577 ± 17 | 327 ± 32* | 56.9 ± 6.6 |
| 10 | 1 | 621 ± 13 | 347 ± 31* | 56.2 ± 6.0 |
| 1000 | 0 | 513 ± 14 | 297 ± 28* | 58.1 ± 5.8 |
| 1000 | 1 | 656 ± 38 | 365 ± 43* | 55.9 ± 7.4 |
Na+,K+-ATPase purified from rat renal cortex was phosphorylated in vitro by PKC as described under the Methods. The hydrolytic activity of Na+,K+-ATPase was assayed in 100 μl of reaction mixture containing (mM): 100 NaCl, 5 KCl, 5 MgCl2, 50 Tris-HCl (pH 7.4 at 37 °C), 3 Tris-ATP, and tracer amounts of [γ-32P]ATP in the absence or presence of listed additives (i.e. Ca2+ and CaM). Values are means ± S.E.M. of 5 separate experiments.
Significantly different from control, P < 0.01.
DISCUSSION
Regulation of NKA activity by hormones and second messengers has been extensively studied (for a review see Ewart & Klip, 1995). Although it is generally agreed that rapid changes in NKA activity can be achieved by agents that affect the activity of PKA and/or PKC, the direction in which these substances affect NKA varies significantly. The present study shows that the level of [Ca2+]i is one important determinant in the response of NKA to second messenger activation, and suggests that future studies of the role of PKA/PKC activation in the regulation of NKA activity should be performed under controlled [Ca2+]i conditions.
Table 3 summarizes some of the most relevant studies on short-term regulation of NKA activity by PKA/PKC. Notably, most of the studies in which an inhibitory effect was observed were performed using Ca2+-free conditions. On the other hand, a stimulatory effect was often observed using conditions where the [Ca2+]i had been high or might have been rendered high by experimental manipulation. This may, for instance, have been the case when cells were subjected to pretreatment with a K+-free solution. K+-free pretreatment will lead to an inhibition of NKA activity and a subsequent increase in intracellular Na+ ([Na+]i). The latter might in turn lead to an inhibition of the Na+-Ca2+ exchanger and an increase in [Ca2+]i (Blaustein, 1993). Another example in which [Ca2+]i might have been increased is the study in which a high osmolarity solution (400 mosmol kg−1) was used. A decrease in cell volume resulting from the increase in osmolarity of the medium may trigger an increase in [Ca2+]i (Watson, 1991).
Table 3.
Effects of PKA or PKC activation on the activity of NKA in relation to Ca2+ conditions
| NKA preparations | Experiment condition | Treatment | Effects on NKA | References |
|---|---|---|---|---|
| Microsomes or membranes from liver, kidney, heart, brain or pancreatic islets of rat, human and dog | Ca2+-free buffers | cAMP or PKA | ↓ATP hydrolysis | Limas et al. 1973; Tria et al. 1974; Braughler & Corder, 1978; Lingham & Sen 1982; Tugg et al. 1990 |
| Membranes from rabbit kidney or ciliary processes | Ca2+-free buffer | cAMP or PKA | ↓ATP hydrolysis | Delamere et al. 1990 |
| Rat renal tubule suspension | Ca2+-free buffer | Forskolin | ↓ATP hydrolysis | Hussian et al. 1997 |
| Isolated rabbit iris–ciliary body | Ca2+-free buffer | cAMP | ↓Rb+ uptake | Delamere et al. 1990 |
| Isolated rat renal tubules | Buffer 0.25 mM Ca2+ | cAMP | ↓ATP hydrolysis* | Fryckstedt & Aperia, 1992; Satoh et al. 1993; Holtbäck et al. 1998 |
| Mouse fibroblast LTK− cells | [Ca2+]i not increased | cAMP | ↓ATP hudrolysis | Horiuchi et al. 1993 |
| HeLa cells | [Ca2+]i~nM | cAMP | ↓Rb+ uptake* | Middleton et al. 1990 |
| Isolated guinea-pig myocytes | [Ca2+]i<150 nM | PKA | ↓pump current | Gao et al. 1992 |
| Isolated guinea-pig myocytes | [Ca2+]i>150 nM | PKA | ↑pump current | Gao et al. 1992 |
| Isolated rabbit renal tubules | Buffer 1.8 nM Ca2+; low K+ (0.1 mM) pretreatment | cAMP or forskolin | ↑transmembrane potential | Breton et al. 1994 |
| Isolated shark rectal gland cells | Buffer 2.5 mM Ca2+ | cAMP | ↑Rb+ uptake | Marver et al. 1986 |
| Xenopus oocytes | Buffer Ca2+ 2mM; K+-free pretreatment and oocyte injection | cAMP | ↑pump current | Vasiloets et al. 1992 |
| Purified NKA from rat kidney and shark rectal gland | Ca2+-free buffers | PKC | ↓ATP hydrolysis | Bertorello et al. 1991; Logvinenko et al. 1996 |
| MDCK cells | Ca2+-free buffer | PMA/OAG | ↓ATP hydrolysis | Shahedi et al. 1992 |
| Isolated rat renal tubules | Buffer 0.25 mM Ca2+ | PDBu | ↓ATP hydrolysis | Bertorello & Aperia, 1989; Satoh et al. 1993 |
| Isolated rat choroid plexus | Buffer 0.25 mM Ca2+ | PDBu | ↓ATP hydrolysis | Fisone et al. 1995 |
| Cultured rat tubule cells | Buffer 0.4 mM Ca2+ | PMA | ↓Rb+ uptake | Sinfh & Linas, 1997 |
| A6 cells | Buffer 1.0 mM Ca2+ | PMA | ↑pump current | Beron et al. 1997 |
| Isolated rat hepatocytes | [Ca2+]i∼250 nM | PMA | ↑Rb+ uptake | Lynch et al. 1986 |
| Cultured skeletal muscle | Buffer 0.7 mM Ca2+; K+- free pretreatment | Phorbol esters | ↑Rb+ uptake | Sampson et al. 1994 |
| Isolated rat renal tubules | Buffer 1.0 mM Ca2+; osmolarity 400mosmol kg−1; k+-free and low Rb+ pretreatment | PDBu | ↑or no change in Rb+ uptake/ATP hydrolysis | Feraille et al. 1995 |
| Cultured human ciliary epithelium | Buffer 2.6 mM Ca2+ | PDBu | ↑Rb+ uptake | Mito & Delamere, 1993 |
| Cultured rabbit non-pigmented ciliary epithelium | Buffer 2.5 mM Ca2+ | PDBu | ↑Rb+ uptake | Delamere et al. 1997 |
The decreased NKA activity was abolished in the presence of agents that increase [Ca2+]i PDBu, 4-phorbol 12,13-dibutyrate.
In this study, we employed two different methods to measure the functional effect of NKA, i.e. 86Rb+ fluxes which measure the transport activity of NKA under in vivo conditions, and ATP hydrolysis which was performed in permeabilized cell preparations under controlled ionic conditions. While the former provides the best evaluation of the contribution of NKA to cation transport of intact cells, the latter allows us to discriminate primary active cation transport (through the pump) from secondary active transport (such as that mediated by changes in Na+ entry). With these two methods, we found that activation of PKA or PKC led to inhibition of NKA activity at low [Ca2+]i, as reflected by a decrease in ATP hydrolysis and 86Rb+ uptake, and stimulation of 86Rb+ uptake but no change in ATP hydrolysis at high [Ca2+]i. The results suggest that the PKA/PKC inhibition of NKA at low [Ca2+]i is direct. The stimulatory effect of forskolin or OAG on ouabain-sensitive Rb+ uptake at high [Ca2+]i may be due to a combined effect, i.e. release of PKA/PKC-induced inhibition of NKA by Ca2+ modulation and secondary stimulation of NKA by an increase in Na+ entry. In accordance with this hypothesis, we have found in ongoing studies that activation of PKA by forskolin and IBMX causes an increase in [Na+]i under low [Ca2+]i conditions but no change in [Na+]i when high [Ca2+]i conditions are used. Similar changes in [Na+]i were also observed for OAG-induced PKC activation (also see Belusa et al. 1997).
The mechanism by which Ca2+ modulates the regulation of NKA by PKA/PKC remains to be elucidated. This study shows that the state of phosphorylation of the α subunit by PKA/PKC is not influenced by variations in [Ca2+]i. It has also been shown that the inhibitory effect of PKC phosphorylation on purified NKA activity is neither influenced by Ca2+ nor by calmodulin. It is therefore likely that the modulatory effect of Ca2+ on phosphorylated NKA activity is effected via one or more Ca2+-dependent intracellular signalling pathway(s).
The finding that [Ca2+]i modulates the regulation of NKA activity by PKA and PKC has several important physiological implications. First, it implies that hormones activating PKA/PKC without concomitant increases in [Ca2+]i may have an inhibitory effect on NKA activity, while hormones that both activate PKA/PKC and increase [Ca2+]i may have a stimulatory effect. Second, in the active state of some excitable tissues such as the beating heart, the exercising skeletal muscles and the firing neurons, the average [Ca2+]i is expected to be quite high. Application of hormones or neurotransmitters to those tissues may be expected to lead to an increased activity of NKA, which has implications both for the intracellular ion homeostasis and the transmembrane potential. Third, the [Ca2+]i-dependent characteristic of regulation of NKA by PKA/PKC may be important in the feedback control of NKA activity. Inhibition of NKA leads to an increase in [Na+]i (Wong & Foskett, 1991; Blaustein, 1993). Increased [Na+]i will attenuate the Na+-Ca2+ exchanger (Blaustein, 1993), which results in increased [Ca2+]i. The resulting increase in [Ca2+]i should counteract PKA/PKC-induced inhibition of NKA, allowing NKA activity to return toward resting levels, or even turning the direction to a stimulation or an oscillation (Wong & Foskett, 1991).
Acknowledgments
We wish to thank Mona Agrén and Ann-Christine Eklöf for preparation of purified NKA, Lill-Britt Svensson for assistance in the measurement of ATP hydrolysis, and Eivor Zettergren for assistance in cell culture. The antibody used for detection of PKC-mediated phosphorylation of NKA was kindly provided by Dr Kathleen J. Sweadner at Massachusetts General Hospital, Charlestown, MA 02129, USA. This study was supported by grants from the Swedish Medical Research Council (03644) and the Swedish Heart Lung Foundation (A.A.) and by USPHS grant MH-40899 (P.G.).
References
- Beguin P, Beggah AT, Chibalin AV, Burgener-Kairuz P, Jassier F, Mathews PM, Rossier BC, Geering K. Phosphorylation of the Na, K-ATPase α subunit by protein kinase A and C (PKC) in vitro and in intact cells: identification of a novel motif for PKC-mediated phosphorylation. Journal of Biological Chemistry. 1994;269:24437–24445. [PubMed] [Google Scholar]
- Belusa R, Wang ZM, Matsubara T, Sahlgren B, Dulubova I, Nairn AC, Ruoslahti E, Greengard P, Aperia A. Mutation of protein kinase C phosphorylation site on rat-α1 Na+,K+-ATPase alters regulation of intracellular Na+ and pH, and influences cell shape and adhesiveness. Journal of Biological Chemistry. 1997;272:20179–20184. doi: 10.1074/jbc.272.32.20179. [DOI] [PubMed] [Google Scholar]
- Beron J, Forster I, Beguin P, Geering K, Verrey F. Phorbol 12-myristate 13-acetate down-regulates Na,K-ATPase independent of its protein kinase C site: decrease in basolateral cell surface area. Molecular Biology of the Cell. 1997;8:387–398. doi: 10.1091/mbc.8.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca2+ buffers (Review) Methods in Cell Biology. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]
- Bertorello A, Aperia A. Na+-K+-ATPase is an effector protein for protein kinase C in renal proximal tubule cells. American Journal of Physiology. 1989;256:F370–373. doi: 10.1152/ajprenal.1989.256.2.F370. [DOI] [PubMed] [Google Scholar]
- Bertorello AM, Aperia A, Walaas SI, Nairn AC, Greengard P. Phosphorylation of the catalytic subunit of Na+,K+-ATPase inhibits the activity of the enzyme. Proceedings of the National Academy of Sciences of the USA. 1991;88:11359–11362. doi: 10.1073/pnas.88.24.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP. Physiological effects of endogenous ouabain: control of intracellular Ca2+ stores and cell responsiveness. American Journal of Physiology. 1993;264:C1367–1387. doi: 10.1152/ajpcell.1993.264.6.C1367. [DOI] [PubMed] [Google Scholar]
- Braughler J, Corder C. Reversible in activation of purified (Na++ K+)-ATPase from human renal tissue by cyclic AMP-dependent protein kinase. Biochimica et Biophysica Acta. 1978;424:455–465. doi: 10.1016/0005-2744(78)90184-5. [DOI] [PubMed] [Google Scholar]
- Breton S, Beck JS, Laprade R. cAMP stimulates proximal convoluted tubule Na+,K+-ATPase activity. American Journal of Physiology. 1994;266:F400–410. doi: 10.1152/ajprenal.1994.266.3.F400. [DOI] [PubMed] [Google Scholar]
- Carranza M-L, Feraille E, Favre H. Protein kinase C-dependent phosphorylation of Na+,K+-ATPase-α subunit in rat kidney cortical tubules. American Journal of Physiology. 1996a;271:C136–143. doi: 10.1152/ajpcell.1996.271.1.C136. [DOI] [PubMed] [Google Scholar]
- Carranza M-L, Feraille E, Kiroytcheva M, Rousselot M, Favre H. Stimulation of ouabain-sensitive 86Rb+ uptake and Na+,K+-ATPase α subunit phosphorylation by a cAMP-dependent signalling pathway in intact cells from rat kidney cortex. FEBS Letters. 1996b;396:309–314. doi: 10.1016/0014-5793(96)01121-0. [DOI] [PubMed] [Google Scholar]
- Cheng XJ, Aizman O, Aperia A, Greengard P, Nairn AC. [Ca2+]i modulates the effects of PKA and PKC activation on renal Na+,K+-ATPase. Journal of American Society of Nephrology. 1997a;8:31A. [Google Scholar]
- Cheng XJ, Fisone G, Aizman O, Aizman R, Levenson R, Greengard P, Aperia A. PKA mediated phosphorylation and inhibition of Na+,K+-ATPase in response to β adrenergic hormone. American Journal of Physiology. 1997b;273:C893–901. doi: 10.1152/ajpcell.1997.273.3.C893. [DOI] [PubMed] [Google Scholar]
- Cheng XJ, Holtbäck U, Belusa R, Aperia A. Bi-directional regulation of Na+,K+-ATPase activity; the role of Ca2+ Nephrology. 1997c;3:S13. [Google Scholar]
- Cheng XJ, Höög JO, Nairn AC, Greengard P, Aperia A. Regulation of rat Na+,K+-ATPase activity by PKC is modulated by state of phosphorylation of Ser-943 by PKA. American Journal of Physiology. 1997d;273:C1981–1986. doi: 10.1152/ajpcell.1997.273.6.C1981. [DOI] [PubMed] [Google Scholar]
- Delamere NA, Parkerson J, Hou Y. Indomethacin alters the Na, K-ATPase response to protein kinase C activation in cultured rabbit nonpigmented ciliary epithelium. Investigative Ophthalmology & Vision Science. 1997;38:866–875. [PubMed] [Google Scholar]
- Delamere NA, Robin RS, King KL. Alteration of sodium, potassium-adenosine triphosphatase activity in rabbit ciliary processes by cyclic adenosine monophosphate-dependent protein kinase. Investigative Ophthalmology & Vision Science. 1990;31:2164–2170. [PubMed] [Google Scholar]
- Ewart HS, Klip A. Hormonal regulation of the Na+,K+-ATPase: mechanisms underlying rapid and sustained changes in pump activity. American Journal of Physiology. 1995;269:C295–311. doi: 10.1152/ajpcell.1995.269.2.C295. [DOI] [PubMed] [Google Scholar]
- Feraille E, Carranza M-L, Buffin-Meyer B, Rousselot M, Doucet A, Favre H. Protein kinase C-dependent stimulation of Na+,K+-ATPase in proximal convoluted tubules. American Journal of Physiology. 1995;268:C1277–1283. doi: 10.1152/ajpcell.1995.268.5.C1277. [DOI] [PubMed] [Google Scholar]
- Feschenko MS, Sweadner KJ. Conformation-dependent phosphorylation of Na, K-ATPase by protein kinase A and protein kinase C. Journal of Biological Chemistry. 1994;269:30436–30444. [PubMed] [Google Scholar]
- Feschenko MS, Sweadner KJ. Structural basis species-specific differences in the phosphorylation of Na,K-ATPase by protein kinase C. Journal of Biological Chemistry. 1995;270:14072–14077. doi: 10.1074/jbc.270.23.14072. 10.1074/jbc.270.23.14072. [DOI] [PubMed] [Google Scholar]
- Feschenko MS, Sweadner KJ. Phosphorylation of Na, K-ATPase by protein kinase C at Ser18 occurs in intact cells but does not result in direct inhibition of ATP hydrolysis. Journal of Biological Chemistry. 1997;272:17726–17733. doi: 10.1074/jbc.272.28.17726. 10.1074/jbc.272.28.17726. [DOI] [PubMed] [Google Scholar]
- Fisone G, Cheng SX-J, Nairn AC, Czernik AJ, Hemmings HC, Jr, Höög J-O, Bertorello AM, Kaiser R, Bergman T, Jörnvall H, Aperia A, Greengard P. Identification of the phosphorylation site for cAMP-dependent protein kinase on Na+,K+-ATPase and effects of site-directed mutagenesis. Journal of Biological Chemistry. 1994;269:9368–9373. [PubMed] [Google Scholar]
- Fisone G, Snyder GL, Fryckstedt J, Caplan MJ, Aperia A, Greengard P. Na+,K+-ATPase in the choroid plexus. Regulation by serotonin/protein kinase C pathway. Journal of Biological Chemistry. 1995;270:2427–2430. doi: 10.1074/jbc.270.6.2427. 10.1074/jbc.270.6.2427. [DOI] [PubMed] [Google Scholar]
- Fryckstedt J, Aperia A. Sodium-dependent regulation of sodium, potassium, adenosine-tri-phosphatase (Na+,K+-ATPase) activity in medullary thick ascending limb of Henle segments. Effect of cyclic adenosine-monophosphate, guanosine-nucleotide-binding-protein activity and arginine vasopressin. Acta Physiologica Scandinavica. 1992;144:185–190. doi: 10.1111/j.1748-1716.1992.tb09284.x. [DOI] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Baldo GJ. Isoprenaline, Ca2+ and the Na+-K+ pump in guinea-pig ventricular myocytes. The Journal of Physiology. 1992;449:689–704. doi: 10.1113/jphysiol.1992.sp019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien R. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Holtbäck U, Ohtomo Y, Förberg P, Sahlgren B, Aperia A. Neuropeptide Y shifts the equilibrium between the α- and β-adrenergic tonus in proximal tubule cells. American Journal of Physiology. 1998;275:F1–7. doi: 10.1152/ajprenal.1998.275.1.F1. [DOI] [PubMed] [Google Scholar]
- Horiuchi A, Takeyasu K, Mouradian MM, Jose PA, Felder RA. D1a dopamine receptor stimulation inhibits Na+/K+-ATPase activity through protein kinase A. Molecular Pharmacology. 1993;43:281–285. [PubMed] [Google Scholar]
- Hussian T, Abdul-Wahab R, Lokhandwala MF. Bromocriptine stimulates Na+,K+-ATPase in renal proximal tubules via the cAMP pathway. European Journal of Pharmacology. 1997;321:259–263. doi: 10.1016/s0014-2999(97)00039-3. 10.1016/S0014-2999(97)00039-3. [DOI] [PubMed] [Google Scholar]
- Jorgensen PL. Isolation of (Na+ plus K+)-ATPase. Methods in Enzymology. 1974;32:277–290. [PubMed] [Google Scholar]
- Laski ME, Kurtzman NA. The renal adenosine triphosphatase: functional integration and clinical significance. Mineral and Electrolyte Metabolism. 1996;22:410–422. [PubMed] [Google Scholar]
- Li D, Cheng SXJ, Fisone G, Caplan MJ, Ohtomo Y, Aperia A. Effects of okadaic acid, calyculin A and PDBu on state of phosphorylation of rat renal Na, K-ATPase. American Journal of Physiology. 1998;275:F863–869. doi: 10.1152/ajprenal.1998.275.6.F863. [DOI] [PubMed] [Google Scholar]
- Limas C, Natargiacomo V, Cohen J. Effect of cyclic 3′, 5′-AMP on the (Na+K)-ATPase of myocardial sarcolemma. Cardiovascular Research. 1973;7:477–481. doi: 10.1093/cvr/7.4.477. [DOI] [PubMed] [Google Scholar]
- Lingham RB, Sen AK. Regulation of rat brain (Na++ K+)-ATPase activity by cyclic AMP. Biochimica et Biophysica Acta. 1982;688:475–485. doi: 10.1016/0005-2736(82)90359-5. [DOI] [PubMed] [Google Scholar]
- Logvinenko NS, Dulubova I, Fedosova N, Larsson SH, Nairn AC, Esmann M, Greengard P, Aperia A. Phosphorylation by protein kinase C of serine-23 of the α1 subunit of rat Na+,K+ ATPase affects its conformational equilibrium. Proceedings of the National Academy of Sciences of the USA. 1996;93:9132–9137. doi: 10.1073/pnas.93.17.9132. 10.1073/pnas.93.17.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopina OD, Robtsov AM, Mollakova IB, Petrukhov SP, Chibalin AV, Nickashin AV. Phosphorylation of alpha-subunit of Na, K-ATPase from duck salt gland and dog kidney by protein kinase A and C changes kinetic properties of the enzyme. Biophysical Journal. 1995;68:A308. [Google Scholar]
- Lynch CP, Wilson PB, Blackmore PF, Exton JH. The hormone-sensitive hepatic Na+-pump: Evidence for regulation by diacylglycerol and tumor promoters. Journal of Biological Chemistry. 1986;261:14551–14556. [PubMed] [Google Scholar]
- Marver D, Lear S, Marver LT, Silva P, Epstein FH. Cyclic AMP-dependent stimulation of Na, K-ATPase in shark rectal gland. Journal of Membrane Biology. 1986;94:205–215. doi: 10.1007/BF01869716. [DOI] [PubMed] [Google Scholar]
- Middleton JM, Khan WA, Collinsworth G, Hannun YA, Medford RM. Heterogeneity of protein kinase C-mediated rapid regulation of Na/K-ATPase in kidney epithelial cells. Journal of Biological Chemistry. 1993;268:15958–15964. [PubMed] [Google Scholar]
- Middleton JP, Raymond JR, Whorton AR, Dennis VW. Short-term regulation of Na+/K+ adenosine triphosphatase by recombinant human serotonin 5-HT1A receptor expressed in Hela cells. Journal of Clinical Investigation. 1990;86:1799–1805. doi: 10.1172/JCI114909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito T, Delamere NA. Alteration of active Na-K transport on protein kinase C activation in cultured ciliary epithelium. Investigative Ophthalmology & Vision Science. 1993;34:539–546. [PubMed] [Google Scholar]
- Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumor promotion. Nature. 1984;308:693–698. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- Pedemonte CH, Pressley TA, Lokhandwala MF, Cinelli AR. Regulation of Na+,K+-ATPase transport activity by protein kinase C. Journal of Membrane Biology. 1997;155:219–227. doi: 10.1007/s002329900174. 10.1007/s002329900174. [DOI] [PubMed] [Google Scholar]
- Sampson SR, Brodie C, Alboim SV. Role of protein kinase C in insulin activation of the Na-K pump in cultured skeletal muscle. American Journal of Physiology. 1994;266:C751–758. doi: 10.1152/ajpcell.1994.266.3.C751. [DOI] [PubMed] [Google Scholar]
- Satoh T, Cohen HT, Katz AI. Different mechanisms of renal Na-K-ATPase regulation by protein kinases in proximal and distal nephron. American Journal of Physiology. 1993;265:F399–405. doi: 10.1152/ajprenal.1993.265.3.F399. [DOI] [PubMed] [Google Scholar]
- Shahedi M, Laborde K, Bussieres L, Dechaux M, Sachs C. Protein kinase C activation causes inhibition of Na/K-ATPase activity in Madin-Darby canine kidney epithelial (MDCK) cells. Pflügers Archiv. 1992;420:269–274. doi: 10.1007/BF00374458. [DOI] [PubMed] [Google Scholar]
- Singh H, Linas SL. Role of proein kinase C in β2-adrenoceptor function in cultured rat proximal tubule epithelial cells. Americam The Journal of Physiology. 1997;273:F193–199. doi: 10.1152/ajprenal.1997.273.2.F193. [DOI] [PubMed] [Google Scholar]
- Skou JC, Esmann M. The Na,K-ATPase (Review) Journal of Bioenergetics & Biomembranes. 1992;24:249–261. doi: 10.1007/BF00768846. [DOI] [PubMed] [Google Scholar]
- Tria E, Luly P, Tomasi V, Trevisani A, Barnabei O. Modulation by cyclic AMP in vitro of liver plasma membrane (Na+-K+)-ATPase and protein kinases. Biochimica et Biophysica Acta. 1974;343:297–306. doi: 10.1016/0304-4165(74)90094-4. [DOI] [PubMed] [Google Scholar]
- Tung P, Pai G, Johnson DG, Punzalan R, Levin SR. Relationships between adenylate cyclase and Na+,K+-ATPase in rat pancreatic islets. Journal of Biological Chemistry. 1990;265:3936–3939. [PubMed] [Google Scholar]
- Vasilets LA, Schwarz W. Regulation of endogenous and expressed Na/K-pumps in Xenopus oocytes by membrane potential and stimulation of protein kinases. Journal of Membrane Biology. 1992;125:119–132. doi: 10.1007/BF00233352. [DOI] [PubMed] [Google Scholar]
- Watson PA. Function follows form: generation of intracellular signals by cell deformation. FASEB Journal. 1991;5:2013–2019. doi: 10.1096/fasebj.5.7.1707019. [DOI] [PubMed] [Google Scholar]
- Wong MMY, Foskett JK. Oscillations of cytosolic sodium during calcium oscillations in exocrine acinar cells. Science. 1991;254:1014–1016. doi: 10.1126/science.1948071. [DOI] [PubMed] [Google Scholar]
- Yingst DR. Modulation of the Na, K-ATPase by Ca and intracellular proteins. Annual Review of Physiology. 1988;50:291–303. doi: 10.1146/annurev.ph.50.030188.001451. [DOI] [PubMed] [Google Scholar]